

Abstract
The anti-Parkinsonian drug rasagiline is a selective, irreversible inhibitor of monoamine oxidase and is used in the treatment of Parkinson’s disease (PD). Its postulated neuroprotective effects may be attributed to MAO inhibition, or to its propargylamine moiety. The major metabolite of rasagiline, aminoindan, has shown promising neuroprotective properties in vitro but there is a paucity of studies investigating in vivo effects of this compound. Therefore, we examined neuroprotective effects of rasagiline and its metabolite aminoindan in a double lesion model of PD. Male Fisher 344 rats received i.p. injections of the noradrenergic neurotoxin DSP-4 and intrastriatal stereotaxic microinjections of the dopamine neurotoxin 6-OHDA. Saline, rasagiline or aminoindan (3 mg/kg/day s.c.) were delivered via Alzet minipumps for 4 weeks. Rats were then tested for spontaneous locomotion and a novel object recognition task. Following behavioral testing, brain tissue was processed for ELISA measurements of growth factors and immunohistochemistry. Double-lesioned rats treated with rasagiline or aminoindan had reduced behavioral deficits, both in motor and cognitive tasks compared to saline-treated double-lesioned rats. BDNF levels were significantly increased in the hippocampus and striatum of the rasagiline- and aminoindan-lesioned groups compared to the saline-treated lesioned group. Double-lesioned rats treated with rasagiline or aminoindan exhibited a sparing in the mitochondrial marker Hsp60, suggesting mitochondrial involvement in neuroprotection. Tyrosine hydroxylase (TH) immunohistochemistry revealed a sparing of TH-immunoreactive terminals in double-lesioned rats treated with rasagiline or aminoindan in the striatum, hippocampus, and substantia nigra.
These data provide evidence of neuroprotection by aminoindan and rasagiline via their ability to enhance BDNF levels.
Keywords: Parkinson’s disease, Dopamine, Norepinephrine, Monoamine oxidase inhibitors, Cognition, Neuroprotection
1. Introduction
Parkinson’s disease (PD) is clinically characterized by tremor, bradykinesia, and rigidity (Connolly and Lang, 2014). These motor symptoms develop when about 50% of dopaminergic nigrostriatal neurons and about 80% striatal dopamine terminals are lost (Braak et al., 2003). The disease is also characterized by non-motor symptoms, including depression, sleep disturbance, apathy, anxiety, olfactory dysfunction, sweating, and constipation (Garcia-Ruiz et al., 2014), associated with a system-wide and progressive degeneration of several neuronal populations. Cognitive impairments are also common in PD, and the functional impact for the patient and caregivers is significant (Pessoa-Rocha et al., 2014; Kehagia et al., 2010). Cognitive impairment often represents a barrier for successful treatment of PD and an unmet therapeutic need in this large patient population.
The loss of noradrenergic (NE) neurons in the locus coeruleus (LC) is significant and progressive in PD, and may play a role for early clinical signs of the disease (Buchman et al., 2012). In many cases, the degeneration of LC–NE neurons occurs earlier than the loss of dopamine neurons in PD (German et al., 1992; Del Tredici and Braak, 2013) and is part of a progressive, system-wide degeneration that starts in the peripheral nervous system (Hawkes et al., 2010; Meissner, 2012). Studies suggest that dopamine neurons in the SN (SN-DA) become more sensitive to various insults in animals with previous lesions of the NE innervation (Mavridis et al., 1991). We have shown that systemic treatment with the selective NE neurotoxin DSP-4 (N-(2-chloroethyl)-N-ethyl-bromo-benzylamine) leads to accelerated neuropathology and cognitive impairment in a mouse model of Down syndrome (Lockrow et al., 2011). Our studies suggest that the detrimental effects of a DSP-4 lesion may be due to a reduction in brain-derived neurotrophic factor (BDNF) (Lockrow et al., 2011), which is important for survival of both LC and SN monoamine neurons, and for learning and memory function (Fumagalli et al., 2006). DSP-4 also potentiates memory deficits in conjunction with DA lesions caused by the neurotoxin 6-hydroxydopamine (6-OHDA) (Marin et al., 2008), and increases inflammatory and neuronal response to beta-amyloid in Alzheimer’s disease models (Wenk et al., 2003). Recent studies indicate that NE denervation may play a role in the development of dyskinesia in PD, providing yet another important functional consequence of LC–NE neurodegeneration (Shin et al., 2014). An LC–SN double lesion model may therefore represent a more accurate model for PD, since it captures both motor and cognitive symptoms of the disease (Perez et al., 2009).
Oxidative stress is thought to be one of the underlying mechanisms leading to cellular dysfunction and subsequent loss of neuronal cells in PD (Hwang, 2013). Mitochondrial dysfunction and increased oxidative stress could also affect protein degradation, therefore leading to accumulation of misfolded or damaged proteins (Henchcliffe and Beal, 2008).
Monoamine oxidase (MAO) catalyzes the oxidation of biogenic monoamine neurotransmitters (Ou et al., 2006). Inhibiting MAO-B results in increased dopamine levels, which alleviates the symptoms of PD (Shih et al., 1999). Propargylamine-derived MAO-B inhibitors such as rasagiline have neuroprotective and anti-apoptotic properties in vitro (Maruyama et al., 2002). In contrast to the first-generation MAO-B inhibitor selegiline which metabolizes in the liver to neurotoxic L-methamphetamine (Shin, 1997), rasagiline (N-propargyl-1-(R)-aminoindan) metabolizes to 1-(R)-aminoindan (Youdim et al., 2001), a non-toxic compound devoid of amphetamine-like activity (Chen and Swope, 2005).
In addition to its anti-apoptotic activities (e.g. Weinreb et al., 2004; Bar-Am et al., 2005; Naoi et al., 2013b), rasagiline has been shown to upregulate GDNF mRNA and protein levels in SH-SY5Y neuroblastoma cell culture by activating nuclear transcription factor kappa B (NF-кB) (Maruyama et al., 2004), which is involved in the control of a large number of cellular processes, such as immune and inflammatory responses, developmental processes, cellular growth, and apoptosis. Rasagiline has also been shown to upregulate BDNF mRNA expression in PC12 pheochromocytoma rat cells deprived of serum (Weinreb et al., 2004). In a genomic study, Weinreb et al. (2009) reported that rasagiline increased mRNA expression of BDNF and GDNF by 5–10 fold in the rat midbrain compared to non-treated controls. Since a lack of trophic support has been proposed as a cause of neurodegenerative diseases including PD and Alzheimer’s disease (e.g. Weissmiller and Wu, 2012), these findings support the idea that rasagiline may act in a disease-modifying fashion (Naoi and Maruyama, 2010; Teo and Ho, 2013). Rasagiline has also been shown to exert neuroprotective effects in rodent models of PD exposed to the nigrostriatal neurotoxins MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), 6-OHDA (6-hydroxydopamine) and lactacystin (Blandini et al., 2004; Sagi et al., 2007; Zhu et al., 2008). Rasagiline was recently shown in a large clinical trial (Olanow et al., 2009) to delay disease progression in patients with early PD.
Interestingly, 1-(R)-aminoindan, while being a weak reversible inhibitor of MAO and lacking the propargylamine moiety (Bar-Am et al., 2004), appears to contribute to the neuroprotective activity of the parent compound rasagiline. Indeed, in vitro aminoindan prevented cell death in a cytotoxic model of high density human neuroblastoma SK-N-SH cells and exhibited neuroprotective activities against 6-OHDA in rat pheochromocytoma PC12 cells (Bar-Am et al., 2007). However, there are few studies investigating the in vivo effects of aminoindan.
The overall aim of the current study was to compare the effects of rasagiline and aminoindan on locomotor and cognitive performance in a double lesion model of PD, which takes into consideration both dopaminergic and noradrenergic involvements in the pathophysiology of the disease. Since mitochondrial dysfunction is one main cause of increased oxidative stress and apoptosis in PD, we sought to investigate specific changes in the mitochondrial marker Hsp60. In addition, we assessed the effects of both compounds on growth factor levels, in particular BDNF, to investigate potential mechanisms of neuroprotection in double-lesioned PD rats.
2. Results
2.1. Behavioral assessment
A preliminary study showed that rasagiline or aminoindan had no effect on the behavior of non-lesioned rats. Student’s t-tests between non-lesioned control rats and aminoindan- or rasagiline-treated non-lesioned rats showed no significant difference on novel object recognition or spontaneous locomotion tasks (p>0.05). Therefore, our study is focused on investigating the effects of both rasagiline and aminoindan in double-lesioned animals.
2.1.1. Novel object recognition task
Novel object recognition is indicative of exploratory behavior as well as memory function. When analyzing the discrimination index (Fig. 1A), a one-way ANOVA revealed an overall difference between the treatment groups [F3,21=3.935, p=0.023]. In post-hoc testing (Tukey’s), saline-treated double-lesioned rats performed worse on this task compared to non-lesioned controls (p=0.024; Fig. 1A), suggesting that these rats were not able to discriminate between a familiar object and a novel object. The double-lesioned rats treated with either aminoindan or rasagiline performed significantly better than the saline-treated double-lesioned rats (p=0.036 and p=0.028, respectively), suggesting that aminoindan and rasagiline treatments protected against lesion-induced decline in novel object recognition memory performance. In addition to reduced capability to discriminate between objects, the saline-treated double-lesioned rats spent less time in the center of a spontaneous locomotor open field box (one-way ANOVA: F3,34=4.453, p=0.010; followed by Tukey’s post-hoc test, p=0.043) compared to control rats (Fig. 1B), an indication of anxiety or a reduction of exploratory behavior. However, the double-lesioned rats treated with either aminoindan or rasagiline spent the same amount of time in the center of the chamber as controls animals, therefore showing less evidence of anxiety compared to their saline-treated double-lesioned counterparts (p=0.025 and p=0.014, respectively).
Fig. 1 –
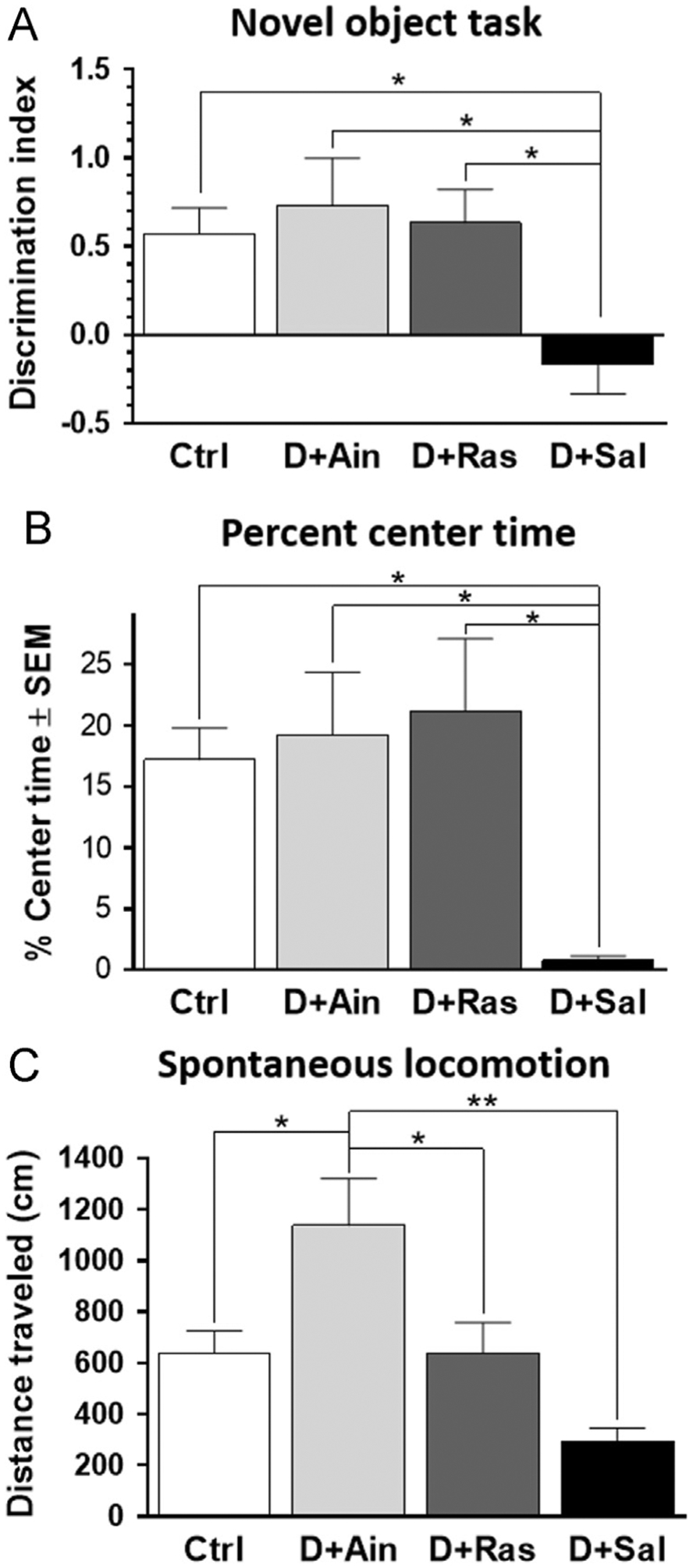
(A) Novel object recognition task. Saline-treated double-lesioned rats did not discriminate object novelty, showing no preference for novel object and a reduced discrimination index compared to all other groups. This was improved by treatment with aminoindan or rasagiline. (B) Percent center time. Saline-treated double-lesioned rats spent significantly less time in the center of the platform, suggesting anxiety or loss of exploratory behavior. This was reversed by rasagiline or aminoindan treatments. (C) Spontaneous locomotion. Total distance traveled (cm) over a 60-min session. Both rasagiline- and aminoindan-treated double-lesioned rats exhibited an increase in spontaneous locomotion, compared to the saline-treated double-lesioned rats. The aminoindan-treated double-lesioned rats exhibited the greatest locomotor activity of all the treatment groups. In (A)–(C), significant differences were tested with a one-way ANOVA followed by Tukey’s post-hoc test (*p<0.05 and **p<0.01).
2.1.2. Spontaneous locomotion
Saline-treated double-lesioned rats exhibited a significant deficit in total distance traveled during the 60-min testing period (Fig. 1C). An one-way ANOVA showed a significant lesion effect (F3,32=3.175, p=0.005). Tukey’s post-hoc analysis revealed that double-lesioned rats treated with aminoindan exhibited the greatest level of locomotor activity, when compared to non-lesioned controls (p=0.030), rasagiline-treated double-lesioned rats (p=0.040), and saline-treated double-lesioned rats (p=0.005).
2.2. ELISA for neurotrophic factors
In order to further explore biological mechanisms for rasagiline and aminoindan neuroprotection, BDNF (Fig. 2) and GDNF (data not shown) levels were assessed in frontal cortex, striatum, and hippocampus by ELISA. Both aminoindan and rasagiline treatments in double-lesioned rats gave rise to increased BDNF but not GDNF levels in the frontal cortex, striatum and hippocampus, compared to saline-treated double-lesioned rats. One-way ANOVA revealed overall significant differences for BDNF levels in the frontal cortex [F3,30=3.551, p=0.026], striatum [F3,29=4.406, p=0.012], and hippocampus [F3,30=3.154, p=0.039]. Tukey post-hoc tests showed that the BDNF levels in the frontal cortex and striatum (Fig. 2A and B) were significantly increased in the aminoindan-treated double-lesioned rats compared to the saline-treated double-lesioned rats (p=0.028 and p=0.019, respectively). BDNF levels were also significantly increased in the striatum of the aminoindan-treated group compared to non-lesioned control rats (p<0.05). The BDNF levels in the hippocampus (Fig. 2C) of the rasagiline-treated double-lesioned rats were significantly increased compared to the saline-treated double-lesioned rats (p=0.030).
Fig. 2 –
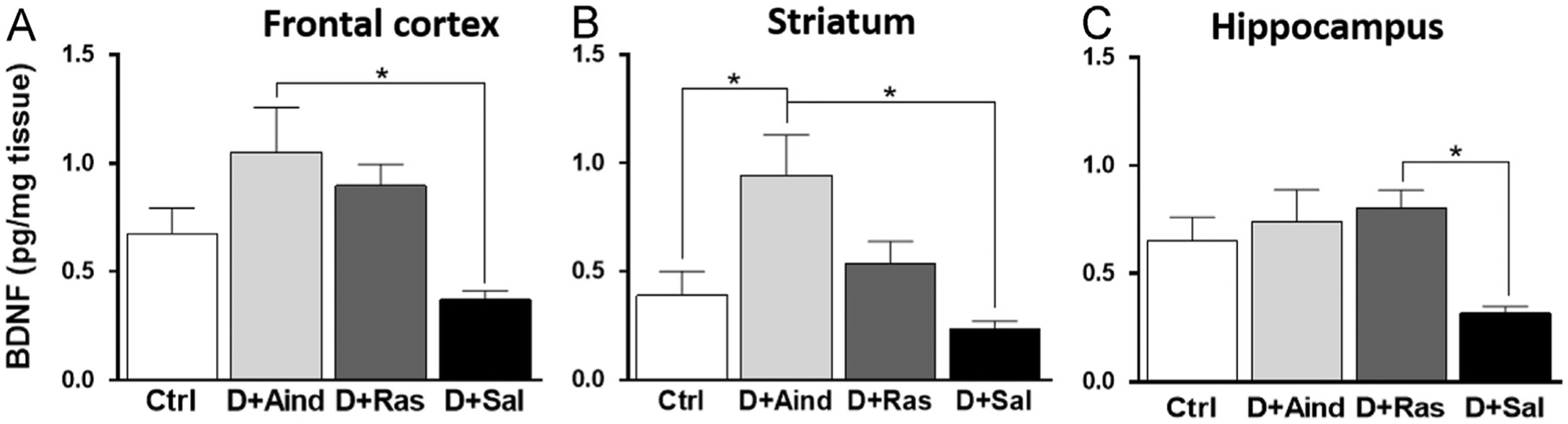
BDNF levels. There were overall group differences for BDNF levels in the frontal cortex (A), striatum (B) and hippocampus (C). Significant differences were tested with a one-way ANOVA followed by Tukey’s post-hoc test (*p<0.05).
2.3. Hsp60 immunostaining
To examine potential biological mechanisms for rasagiline and aminoindan neuroprotection, we stained for the mitochondrial marker Hsp60. Hsp60 staining was markedly reduced in the SN pars compacta (SNpc) and pars reticulata (SNpr) in saline-treated double-lesioned rats (Fig. 3), compared to all other treatment groups. Insets in Fig. 3A and B show a lower neuronal staining of Hsp60 in SN neurons in the pars compacta of the saline-treated double-lesioned rats compared to the control rats. A one-way ANOVA on Hsp60-immunoreactivity in the SN, including both the pars compacta and the pars reticulata regions (Fig. 3E), gave a close to significance result [F3,21=3.003; p=0.052]. Tukey’s post-hoc analysis revealed that aminoindan-treated double-lesioned rats had significantly increased staining densities of Hsp60-ir compared to saline-treated double-lesioned rats (Fig. 3E; p=0.033). These data suggest that the potential neuroprotective properties of aminoindan and rasagiline may be mediated by mitochondrial protection. Interestingly, as with the BDNF levels, aminoindan treatment gave rise to a higher increase in Hsp60 staining than the rasagiline treatment, suggesting better neuroprotective properties for aminoindan.
Fig. 3 –
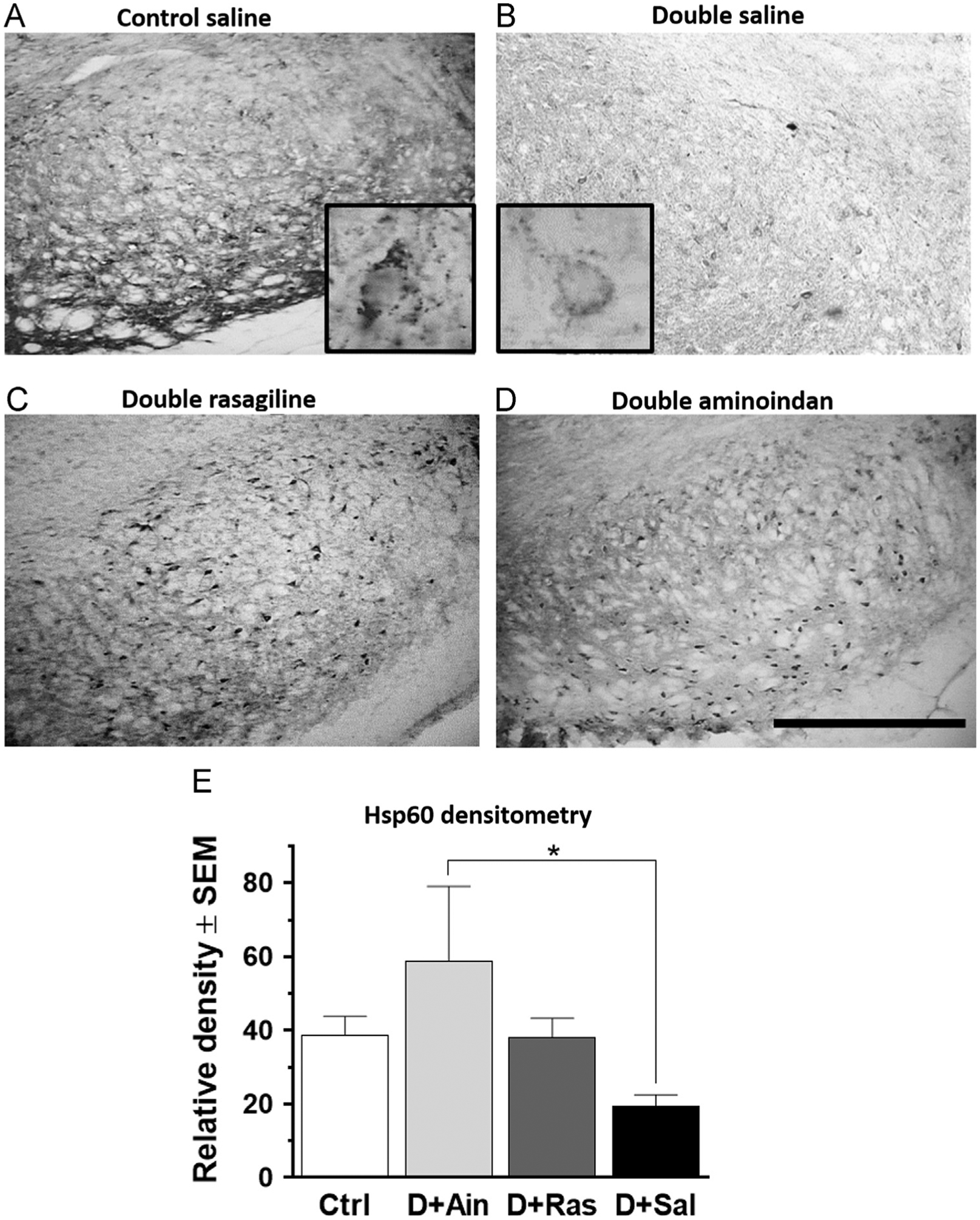
Hsp60 immunostaining in the SN (A–D) and densitometry (E). Hsp60-ir was significantly reduced in the neurons in the SN pars compacta and SN pars reticulata in saline-treated double-lesioned rats (B) compared to control rats (A). Both rasagiline (C) and aminoindan (D) treatments prevented this loss. Insets in (A) and (B) show a higher magnification (60×) of the neuronal morphology for the mitochondrial marker. Bar graph (E) shows densitometry results for the SN and confirms a higher immunoreactivity of Hsp60 in the aminoindan-treated double-lesioned rats (p<0.05) compared to the saline-treated double-lesioned rats. Scale bar in (D) represents 500 μm. Significant differences were tested with a one-way ANOVA followed by Tukey’s post-hoc test (*p<0.05).
To investigate which neuronal populations expressed Hsp60, double immunofluorescence labeling with Hsp60 and TH or Hsp60 and NeuN were performed on SN sections. Hsp60 co-labeled with TH in dopaminergic neuronal cell bodies of the SN pars compacta (Fig. 4A) as well as with interneurons labeled with the neuron specific antigen NeuN in the SN pars reticulata (Fig. 4B) in an aminoindan-treated double-lesioned animal. These data suggest that mitochondrial function was preserved in the dopaminergic neurons as well as in the interneurons of the SN pars reticulata when lesioned rats were treated with aminoindan or rasagiline.
Fig. 4 –
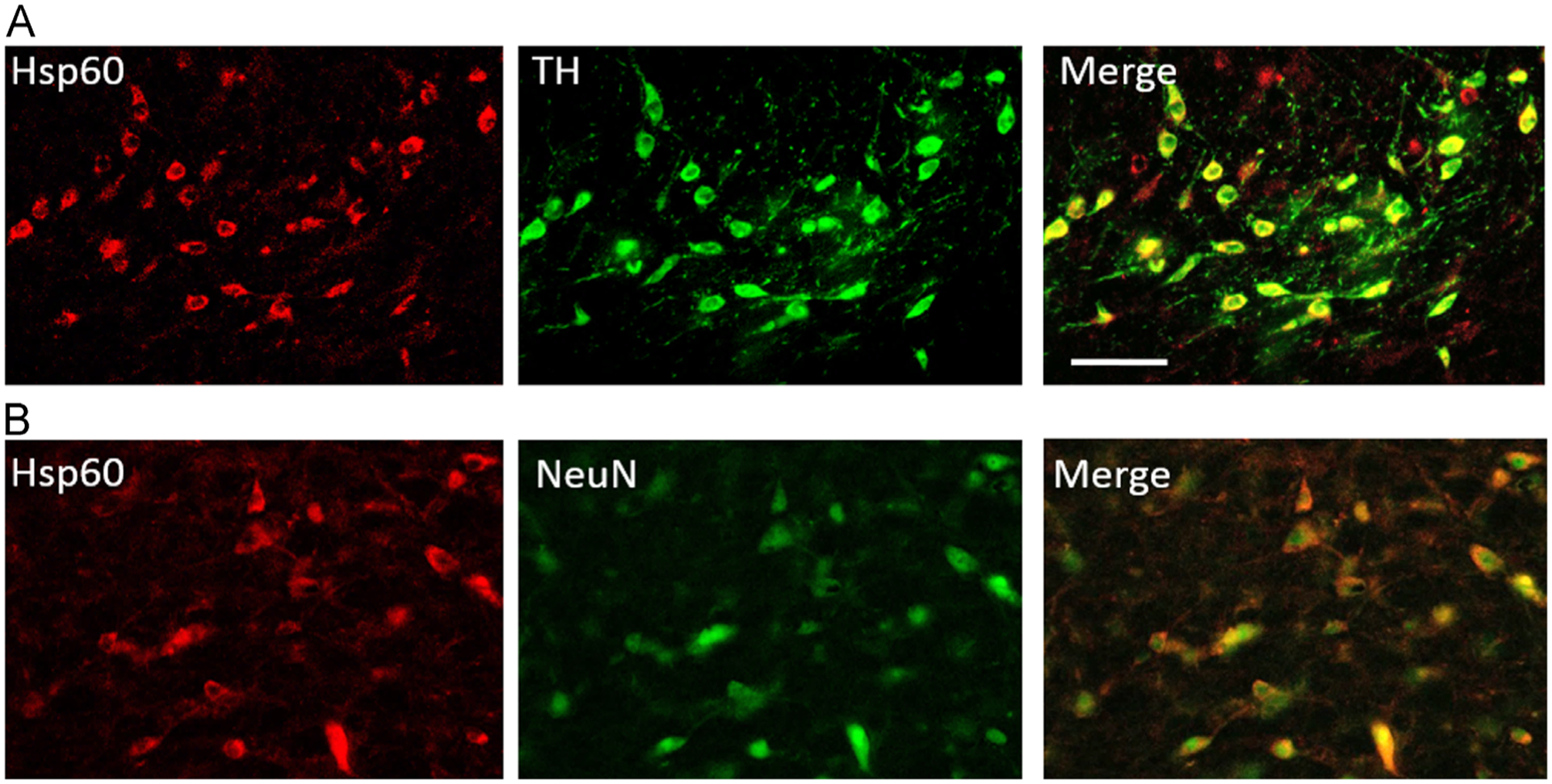
Representative double immunofluorescence staining of the substantia nigra of an aminoindan-treated double- lesioned rat. (A) Hsp60 and TH: Dopaminergic neurons in the pars compacta observed with TH staining co-expressed Hsp60. TH labeling shown in green on the middle panel, Hsp60 shown in red in the left panel, and overlay shown on the right panel. (B) Hsp60 and NeuN: In the SN pars reticulata, interneurons were visualized with NeuN (green, center panel) and co-labeling for Hsp60 (red, left panel) was observed. Overlay is shown on the right panel. Scale bar in (C) represents 100 μm.
2.4. TH immunohistochemistry
In the dentate gyrus of hippocampus, double lesions gave rise to a loss of TH-immunoreactive (-ir) fibers (Fig. 5). A one-way ANOVA revealed that there was a significant difference between the treatment groups (F3,32=11.48, p<0.0001, Fig. 5E). Saline-treated double-lesioned rats had a significant loss of TH-ir compared to control rats (p<0.0001). Both aminoindan and rasagiline treatments were protective against the loss of TH-ir observed in saline-treated double-lesioned rats (p=0.021 and p=0.024, respectively).
Fig. 5 –
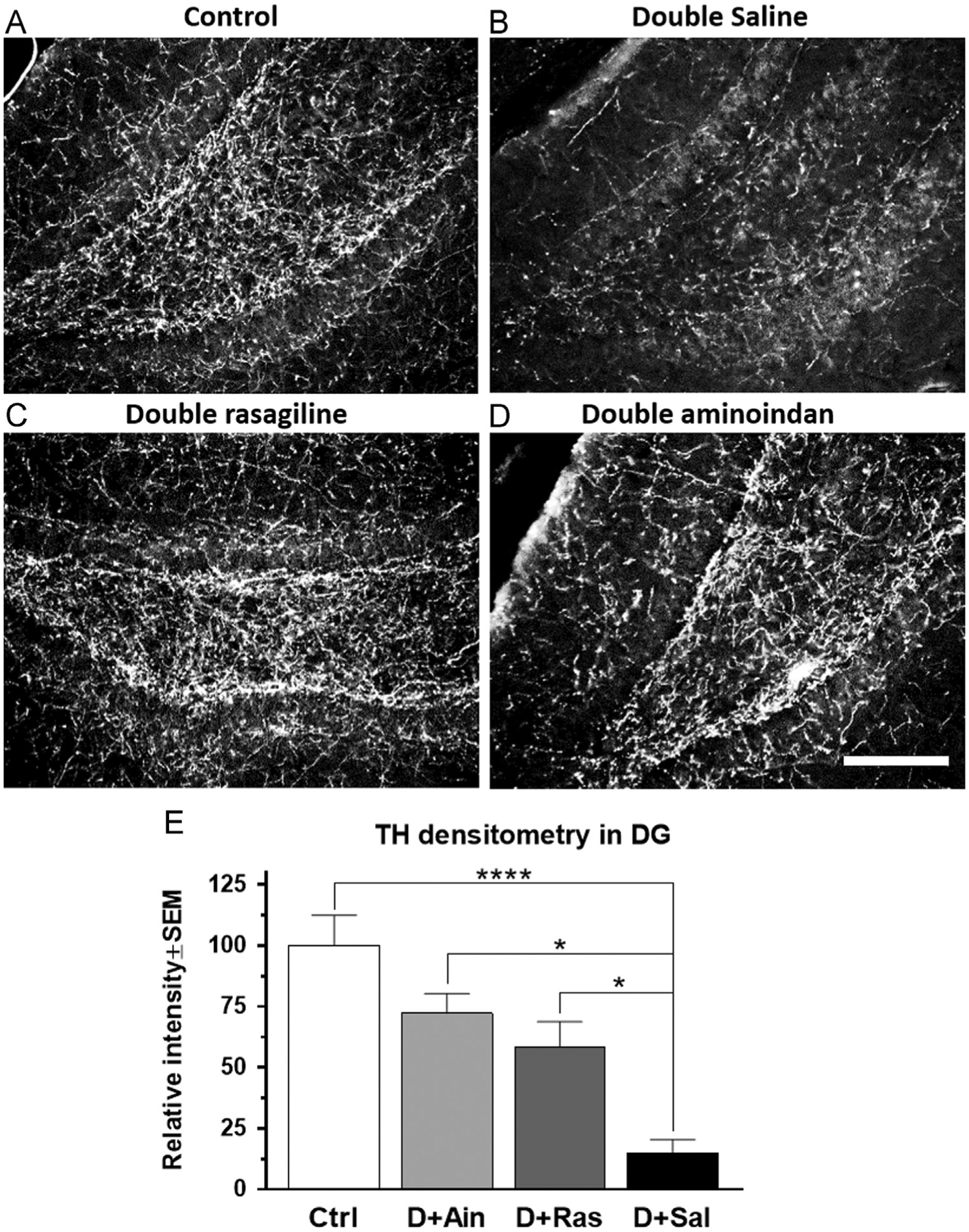
TH immunofluorescence staining in the hippocampal dentate gyrus (A–D) and densitometry (E). Fibers in the dentate gyrus of saline-treated double-lesioned rats exhibited a marked loss of TH-ir (B). Both rasagiline (C) and aminoindan (D) treatments attenuated the double-lesioned effect on TH-ir (E). Scale bar in (D) represents 250 μm. Significant differences were tested with a one-way ANOVA followed by Tukey’s post-hoc test (*p<0.05 and ****p<0.0001).
A significant loss of TH-ir was also observed in the SN pars reticulata of saline-treated double-lesioned rats, compared to other treatment groups (Fig. 6). The drug treatment significantly affected the TH-ir in the SN [F3,23=5.745, p=0.004]. Both rasagiline- and aminoindan-treated double-lesioned rats displayed an increase in TH-positive fibers of the pars reticulata (Fig. 6C and D), as indicated by Tukey’s post-hoc analyses showing that TH-ir in the SN was significantly greater in the double-lesioned rasagiline-treated group compared to the saline-treated double-lesioned rats (p=0.002, Fig. 6E). In order to further explore neuroprotective effects of the two compounds, subjective counting of TH-immunoreactive cell bodies in the SN pars compacta was performed by an unbiased investigator, and expressed as percent of non-lesioned controls. Further evidence for a significant sparing of the number of TH neurons observed in the pars compacta region of SN (F3,21=8.44, p=0.0007, Fig. 6F), with both rasagiline- and aminoindan-treated double-lesioned rats exhibiting significantly increased numbers of TH-immunoreactive neurons compared to the saline-treated double-lesioned group (p=0.039 and p=0.048, respectively).
Fig. 6 –
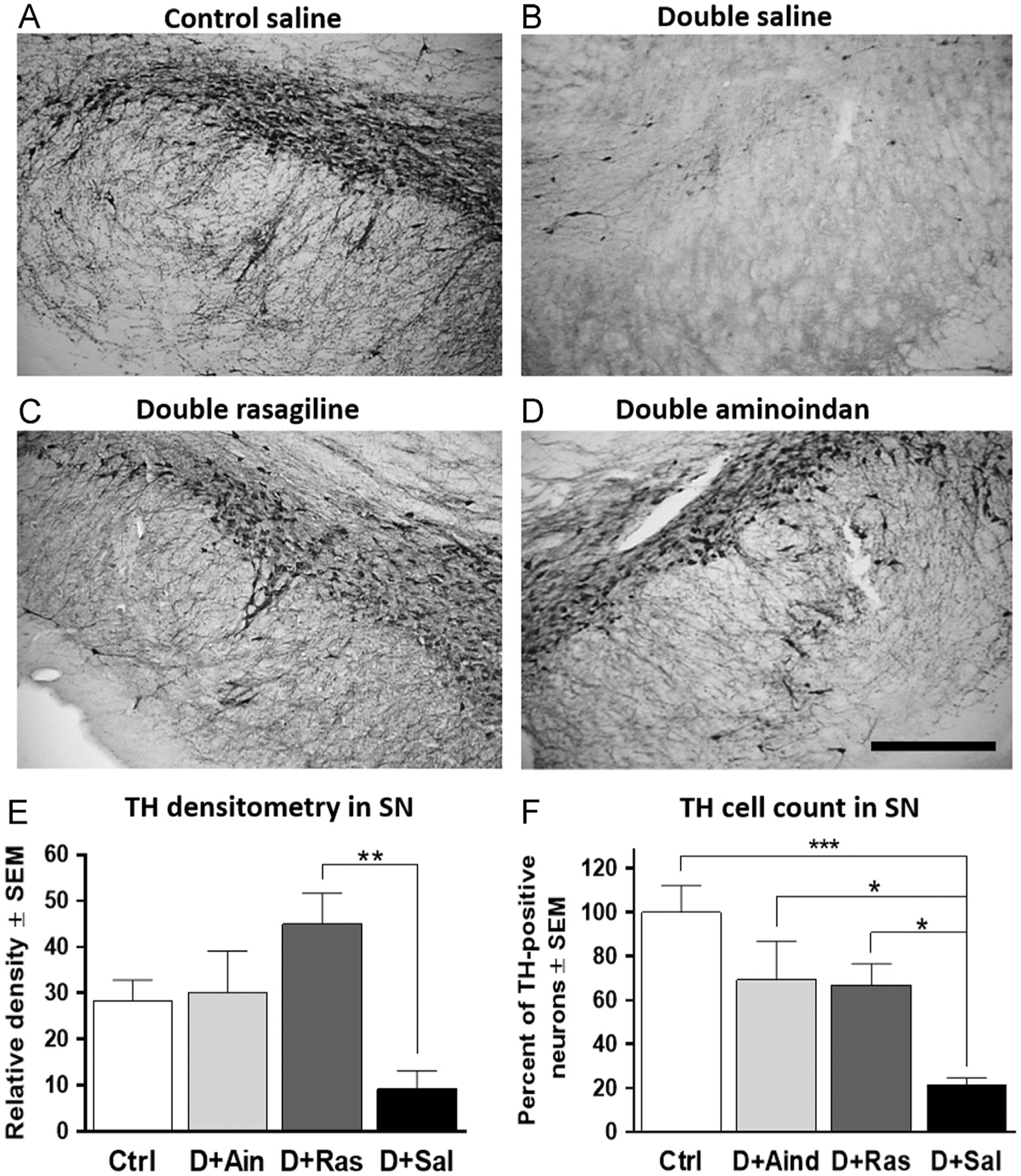
TH immunostaining (A–D), densitometry (E), and relative cell count (F) in SN. The saline-treated double-lesioned rats had a significant loss of TH-ir in the SN (B), compared to controls (A). Both rasagiline and aminoindan prevented DAergic degeneration in the SN (C, D, E and F). Scale bar in (D) represents 250 μm. Significant differences were tested with a one-way ANOVA followed by Tukey’s post-hoc test (*p<0.05, **p<0.01 and ***p<0.001).
In the striatum (Fig. 7A–D), saline-treated double-lesioned rats had a significant loss of TH-ir, especially in proximity to the injection site (see arrows in Fig. 7). Striatal staining for TH terminals was protected in double-lesioned rats treated with rasagiline or aminoindan (Fig. 7C and D). One-way ANOVA analysis suggested that TH densitometry in the striatum was significantly affected by the treatment groups [F3,29=6.599, p=0.002]. Tukey’s post-hoc test showed a significant increase of TH-ir staining in double-lesioned rats treated with aminoindan (p=0.028) and rasagiline (p=0.006) (Fig. 7E). Thus, TH densitometry data strongly suggest that aminoindan and rasagiline prevented degeneration of dopamine neurons in the SN and the innervation around the injection site in the striatum.
Fig. 7 –
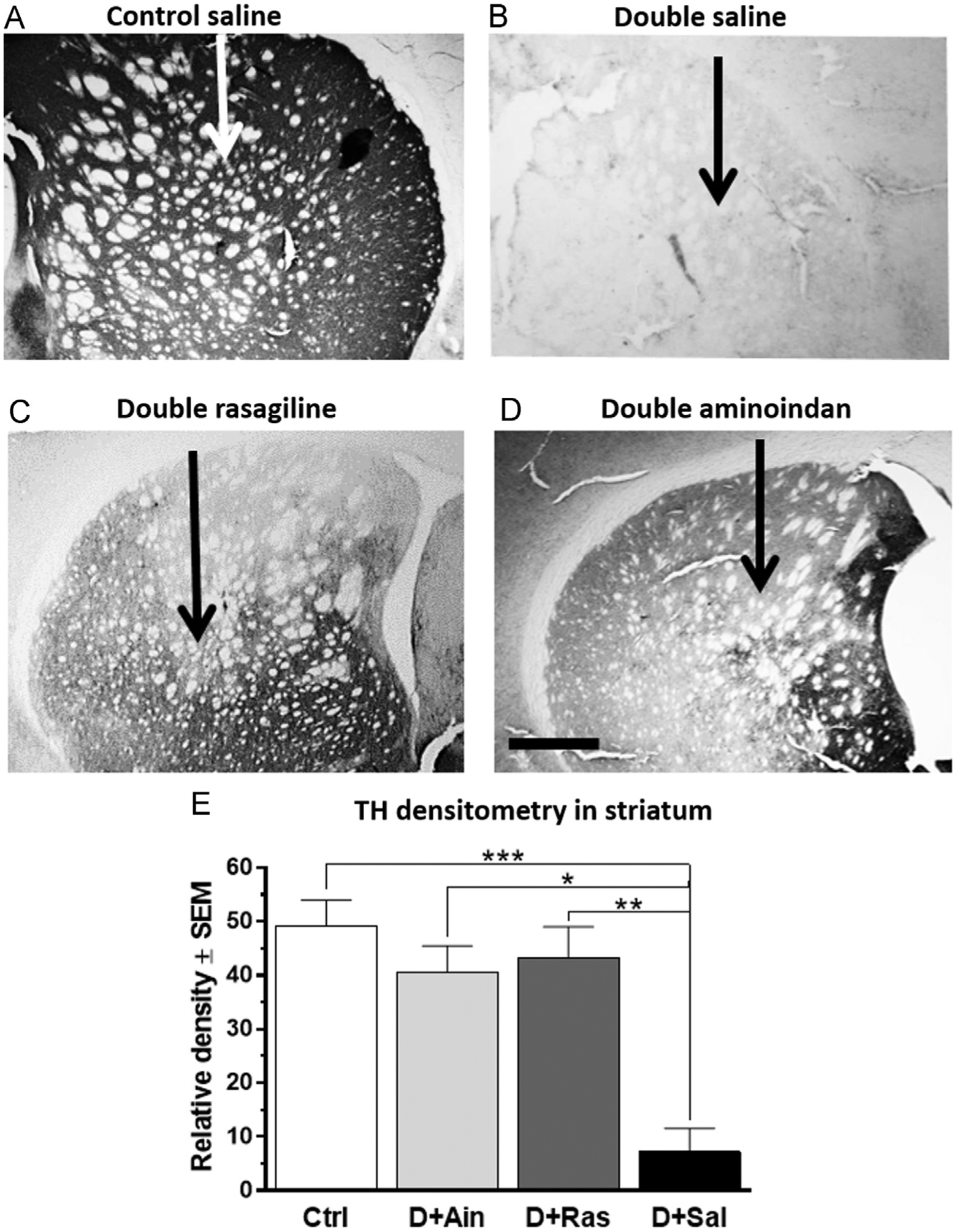
TH immunostaining (A–D) and densitometry (E) in the striatum. The double lesion lead to an almost complete loss of TH-ir in the striatum (B) when compared to the control rats (A). The loss of striatal TH-ir was prevented in aminoindan- and rasagiline-treated double-lesioned rats (C and D). This was confirmed by densitometry measurements (E). Scale bar in (D) represents 500 μm. Significant differences were tested with a one-way ANOVA followed by Tukey’s post-hoc test (*p<0.05, **p<0.01 and ***p<0.001).
In contrast to dopamine neuron loss seen in the SN, we saw no evidence for loss of noradrenergic neuronal cell bodies in the LC after the double lesion (data not shown).
3. Discussion
While rasagiline has demonstrated potent neuroprotective activities both in vitro and in vivo experimental studies (Weinreb et al., 2010), the corresponding properties of its potentially active metabolite aminoindan have mostly been examined in in vitro models (Bar-Am et al., 2010). A recent study by Weinreb et al. (2011) demonstrated in unilateral 6-OHDA lesioned rats that aminoindan reversed behavioral asymmetry and spared catecholamine concentration in the striatum. However, experiments had not been conducted in vivo showing both cognitive and motor improvements in a combined lesion model, nor had the effectiveness of the two compounds been explored behaviorally. The data from our current study demonstrate for the first time that aminoindan significantly increased BDNF levels in the frontal cortex, and striatum, while rasagiline treatment only elevated BDNF levels in the hippocampus. The increased levels of BDNF with these treatments occurred in conjunction with protection against deficits in working memory in a double lesion model of PD. These results suggest that the propargylamine moiety is not required for the neuroprotective properties. Aminoindan and rasagiline both protected striatum, SN, and hippocampus from loss of dopaminergic and noradrenergic innervation caused by the double lesion, as evidenced by TH densitometry, and prevented TH-immunoreactive cell loss in the SN pars compacta, as evidenced by cell counts. It should be noted that no observable TH changes were observed in the LC in any of the four groups. This is consistent with other studies using the same dose and time period (Fritschy and Grzanna, 1991). The administration of rasagiline or aminoindan improved motor activity in double-lesioned rats compared to saline-treated double-lesioned rats, suggesting prevention of motor symptoms by both of these drugs following the double lesion.
Double-lesioned saline-treated rats exhibited a marked decrease in Hsp60 immunoreactivity in the SN while aminoindan, and to a lesser extent rasagiline, appeared to protect against Hsp60 loss of immunoreactivity. Collectively, our findings suggest that prolonged treatment with the MAO-B inhibitor rasagiline and its metabolite, aminoindan, both are neuroprotective for motor and cognitive consequences in a double-lesion model of PD, and that these effects seem to be mediated by sparing of neuronal mitochondria, and by increased BDNF levels in target regions via neuroprotection of mitochondria.
There is increasing evidence that neurodegeneration associated with PD is closely linked to a lack of trophic support (see Rodrigues et al. (2014) for review), such that BDNF and GDNF are considered promising for the treatment of neurodegenerative diseases (Tiwari and Chaturvedi, 2014). Prolonged systemic treatment with rasagiline or aminoindan in our study resulted in increased BDNF levels in three different brain areas, with significantly higher levels of BDNF observed in the frontal cortex and striatum in the aminoindan-treated double-lesioned group compared to all other groups. Our findings are consistent with previous in vitro studies showing that aminoindan enhanced the levels of PKC (protein kinase C), a downstream signaling protein of BDNF involved in cell survival (Bar-Am et al., 2007). Thus, the increased BDNF levels reported in this study and in previous work (Weinreb et al., 2009) may not be primarily due to the propargylamine moiety. Previous in vitro and in vivo studies also described increased mRNA levels of BDNF following exposure to rasagiline (Bar-Am et al., 2005; Naoi et al., 2013a), as well as increased BDNF levels in CSF of PD patients following rasagiline administration (Maruyama et al., 2002), but BDNF levels have not been examined following aminoindan treatment in vivo in rodent studies previously. We examined, for the first time, the neuroprotective properties of aminoindan in a double lesion model of PD, and our results indicate that aminoindan has neuroprotective properties, suggesting a translational potential for aminoindan in the clinic. Interestingly, a recent study poses that both rasagiline and aminoindan bind to α-synuclein, making this protein less likely to aggregate or form fibrils (Kakish et al., 2015), suggesting a possible interesting mechanistic goal for future studies.
Since mitochondrial dysfunction is a hallmark of PD pathology, enhancing mitochondrial function or increasing degradation of defective mitochondria has become a focus of numerous drug trials (Schapira et al., 2014). The mitochondrial chaperone protein Hsp60 assists with folding of matrix proteins and specific deletion of this protein leads to motor neuron disease (Magnoni et al., 2013). Heat shock proteins were found to be protective in PD and could be a possible therapeutic target (Luo et al., 2006). Rasagiline was shown to prevent apoptosis in SH-SY5Y cells through suppression of cytochrome c release from mitochondria, suggesting that rasagiline may exert its protective effects by preventing mitochondrial damage (Naoi et al., 2013b). Our Hsp60 data support this theory, in that Hsp60 staining was reduced in SN pars reticulata interneurons of saline-treated double-lesioned rats, but that rasagiline or aminoindan treatment resulted in a significant increase in Hsp60 staining. This drug effect appeared to not be restricted to the progargylamine moiety since aminoindan appeared to exert a greater increase in Hsp60 immunostaining when compared to rasagiline treatment. The data obtained from the Hsp60 immunostaining may provide a biological substrate for rasagiline-mediated effects and therefore guide future development of disease modifying targets in PD.
Taken together, our data suggest that the neuroprotective effects observed with aminoindan are similar or even greater than those with rasagiline, depending on the outcome measured. Further in vitro studies are needed to compare, under the same experimental conditions, if the beneficial effects observed with rasagiline and aminoindan are all attributable to aminoindan and if the relatively lower efficacy observed with rasagiline is due to a lower concentration of aminoindan resulting from the metabolism of the administered rasagiline drug. In particular, since oxidative stress is thought to be one of the underlying mechanisms leading to cellular dysfunction and subsequent neuronal damage in PD, comparison of the protective effects of rasagiline and aminoindan in in vitro oxidative stress models would be of interest.
In summary, our results suggest that rasagiline and aminoindan protect dopaminergic cells in the nigrostriatal pathway and, noradrenergic terminals in the hippocampus, leading to improved cognitive and motor performance. Exploring biological mechanisms for aminoindan and rasagiline effects in a complex PD model, such as the one described in this study, may lead to further modification or expanded use of MAO-B inhibitors for patients with PD, and may lead to development of effective therapies for cognitive impairment associated with PD.
4. Experimental procedures
Animal protocols were approved by the Medical University of South Carolina Institutional Care and Use Committee and carried out according to guidelines from the National Institutes of Health.
4.1. Lesion procedures and animals
Male Fisher 344 rats (175–200 g, Harlan Laboratories) were housed in an AAALAC accredited Animal Care Facility at MUSC. The treatment groups consisted of 10 rats in the control non-lesioned group (Ctrl), 7 rats in the aminoindan-treated double-lesioned group (D+Aind), 12 rats in the rasagiline-treated double-lesioned group (D+Ras), and 9 in the saline-treated double-lesioned group (D+Sal). The rats were injected with DSP-4 (N-(2-chloroethyl)-N-ethyl-2-bro-mobenzylamine, Sigma; 50 mg/kg, i.p.) known to be toxic for NE neurons, and capable of crossing the blood–brain barrier (Kostrzewa, 2009). One week following the DSP-4 injection, rats were deeply anesthetized using chloral hydrate (400 mg/kg, i.p.) and injected with desipramine (20 mg/kg i.p.; Sigma-Aldrich, MO) to increase the selectivity and efficacy of the 6-OHDA induced-lesion of the nigrostriatal DA neurons. At the same time, the rats were administered bupivacaine, a long-acting local anesthetic (2.5% 0.1 mL, s.c., Sigma). Rats were placed in a stereotaxic apparatus (Stoelting) and six burr holes were drilled in the skull using a Dremel electric drill at the following coordinates: Hole 1: AP: +1.0, ML: ±3.0, DV: −5.0 from dura; Hole 2: AP: −0.1, ML: ±3.7, DV: −5.0; Hole 3: AP: −1.2, ML: ±4.5, DV: −5.0, based on previous studies (Winkler et al., 2002). 6-OHDA (2,4,5-trihydroxyphenethylamine, Sigma-Aldrich, St. Louis, MO; 5 μg/μL per injection site, containing 0.9% NaCl and 0.02% ascorbate) was injected into the striatum at each site via Hamilton syringe. The syringe was left in place for 10 min after the infusion and then retracted slowly.
4.2. Drug treatment
Three weeks following the double lesion procedure, rats were lightly anesthetized using isoflurane and a small incision was made in the scruff of the neck, following a bupivacaine local anesthetic injection (see above). Alzet minipumps (Durect Corporation, Cupertino, CA) were inserted subcutaneously, and the incision was closed using 3–0 sutures. The Alzet pumps were calibrated to deliver sterile saline (vehicle), 3 mg/kg/day rasagiline (TEVA Neuroscience) or 3 mg/kg/day aminoindan (TEVA Neuroscience), based on recent studies in rats (Eliash et al., 2009). The pumps were manufactured to deliver a steady dose of the compounds for a minimum of four weeks.
4.3. Novel object recognition
Behavioral studies started 3 weeks after the implantation of the Alzet minipumps. Following two days of habituation to the testing area, rats were exposed first to two identical objects for three minutes (trial 1). After 90 min (trial 2), one object was replaced with a new object and rats were free to explore for another three minutes. The proportion of time spent investigating the novel object relative to old object, divided by total object time was calculated, i.e. the discrimination index, according to previous studies from our laboratory (Lockrow et al., 2011).
4.4. Spontaneous locomotion
One day following the NORT task, we utilized an automated system (Digiscan locomotion boxes) to examine spontaneous locomotion (Boger et al., 2007). The rats were placed in Accuscan photocell chambers (Accuscan Instruments, Inc. Columbus, OH) containing 16 photocell beams measuring horizontal distance traveled and 8 photobeams measuring vertical activity. Beam breaks were recorded every five minutes for 60 min using VersaMax/Digiscan System Software (Accuscan Instruments, Inc).
4.5. BDNF and GDNF tissue levels
Following behavioral assessment, the animals were sacrificed by an overdose of isoflurane, and the brains were rapidly extracted and dissected. The left striatum, hippocampus, and frontal cortex were dissected and homogenized in 300 μL of lysis buffer (pH 7.3) at 4 °C (Lockrow et al., 2011) and centrifuged at 10,000 g for 15 min at 4 °C. The levels of BDNF and GDNF in the supernatant were assayed using commercially available kits (Promega, Madison, WI) according to the manufacturer’s instructions. In these assays, the captured BDNF or GDNF was bound by a second specific antibody, incubated with chromagenic substrate, and color change was measured in an ELISA plate reader at 450 nm. The assays afford quantification in the range of 7.8–500 pg/mL for BDNF and 15.6–1000 pg/mL for GDNF, with a cross-reactivity of <2–3%. Duplicate samples were analyzed for each data point.
4.6. Immunohistochemistry
The right brain hemisphere was dissected and processed for immunohistochemistry or immunofluorescence. Tissue was immersion-fixed in 4% paraformaldehyde for 48 h and transferred to 30% sucrose in phosphate buffered saline (PBS) at 4 °C. Sections (40 μm) of striatum, SN, and LC were prepared for immunohistochemistry according to our previously published methods (Granholm et al., 1997) using a rabbit polyclonal tyrosine hydroxylase antibody (TH, 1:1000, Pel-Freeze, Rogers, AK) or a rabbit polyclonal heat-shock protein 60 antibody, a mitochondrial protein implicated in mitochondrial protein import and macromolecular assembly (Hsp60, 1:1000, Abcam, Cambridge, MA). Briefly, primary antibodies were applied to serial free-floating sections taken from the striatum, hippocampus, SN, or LC based on our previous protocols (Boger et al., 2006). Endogenous peroxidase activity was quenched by treating sections with 10% H2O2, 20% methanol in 0.01 M Tris buffer saline (TBS, pH=7.4) for 15 min. Sections were then permeabilized in TBST (TBS with 0.25% Triton X-100) following treatment for 20 min with 0.1 M sodium metaperiodate in TBS. Non-specific binding was controlled by incubation in 10% normal goat serum (NGS) in TBST for 1 h. Sections were then incubated for 48 h in TBST with 3% NGS with the primary antibody at 4 °C. Sections were then washed and incubated for 1 h with an appropriate secondary antibody (1:200, Vector Laboratories) and 1 h with avidin–biotin complex (ABC kit, Vector Laboratories). The reaction was developed using DAB peroxidase substrate kit (Vector Laboratories) to enhance the reaction and produce a color stain. This reaction was stopped using 0.1 M phosphate buffer, and the sections were mounted on glass slides, dehydrated, and cover-slipped with DPX (Sigma). To control for staining intensity, staining of all sections for each antibody was conducted on the same day, and developed with DAB for the same amount of time. To allow for confocal microscopy (Fluoview BX61, Olympus), sections from hippocampus immunostained for TH were incubated with Alexa 594 (1:250, Life Technologies) secondary antibody.
Densitometry measurements of TH and Hsp60 immunoreactivity were determined using Nikon NIS-Elements (Nikon Instruments Inc. Americas, Melville, NY) to measure a gray scale value with the range 0–256, where 0 represents white and 256 black. Images were captured with a Nikon Eclipse 80i microscope equipped with a QImaging Fast 1394 digital camera. Staining density was obtained when background staining was subtracted from mean staining intensities for each section. Measurements were performed blinded and 3–4 sections per brain were averaged to obtain one value per subject. In the SN, the region of interest (ROI) for densitometry included both the pars compacta and dendritic processes of the pars reticulata. In order to confirm the results generated by densitometry measurements, TH-immunoreactive neuronal cell bodies were manually counted in the SN pars compacta. The user was blinded to experimental groups, and three sections per brain were averaged to give one value per animal, and values were expressed as percent cell bodies compared to cell numbers in non-lesioned subjects, since the preparation of the sections did not permit stereological measurements to generate total numbers of neurons.
Double immunofluorescence staining of SN sections was conducted with TH and Hsp60 or NeuN and Hsp60 in order to determine which cell types expressed Hsp60. Briefly, sections were incubated with rabbit polyclonal Hsp60 (1:200, Abcam) overnight at 4 °C, washed several times and then incubated with secondary antibody Alexa 594 for 1 h at room temperature. Sections were then washed in TBS and incubated with either mouse NeuN antibody conjugated to Alexa 488 (1:100, Millipore), or TH antibody (1:250, Abcam) followed by secondary Alexa 488. Photomicrographs were generated using a Nikon Eclipse 80i microscope.
4.7. Statistical analysis
Data were represented as mean±standard error of the mean (SEM). They were analyzed using one-way ANOVA followed by Tukey’s post-hoc tests. All statistical analyses were performed with GraphPad Prism version 6.00 (La Jolla, CA), and statistical significance was set at p<0.05.
Acknowledgments
This work was made possible by a Grant from TEVA Pharmaceuticals, and a Medical Accuracy Review was conducted by Teva. The authors would like to thank Mr. Alfred Moore, Ms. Claudia Umphlet, and Ms. Laura Columbo for expert technical support for this project.
REFERANCES
- Bar-Am O, Amit T, Youdim MBH, 2004. Contrasting neuroprotective and neurotoxic actions of respective metabolites of anti-Parkinson drugs rasagiline ans selegiline. Neurosci. Lett 355, 169–172. [DOI] [PubMed] [Google Scholar]
- Bar-Am O, Weinreb O, Amit T, Youdim MBH, 2005. Regulation of Bcl-2 family proteins, neurotrophic factors, and APP processing in the neurorescue activity of propargylamine. FASEB J 19, 1899–1901. [DOI] [PubMed] [Google Scholar]
- Bar-Am O, Amit T, Youdim MB, 2007. Aminoindan and hydroxyaminoindan, metabolites of rasagiline and ladostigil, respectively, exert neuroprotective properties in vitro. J. Neurochem 103, 500–508. [DOI] [PubMed] [Google Scholar]
- Bar-Am O, Weinreb O, Amit T, Youdim MB, 2010. The neuroprotective mechanism of 1-(R)-aminoindan, the major metabolite of the anti-parkinsonian drug rasagiline. J. Neurochem 112, 1131–1137. [DOI] [PubMed] [Google Scholar]
- Blandini F, Armentero MT, Fancellu R, Blaugrund E, Nappi G, 2004. Neuroprotective effect of rasagiline in a rodent model of Parkinson’s disease. Exp. Neurol 187, 455–459. [DOI] [PubMed] [Google Scholar]
- Boger HA, Middaugh LD, Huang P, Zaman V, Smith AC, Hoffer BJ, Tomac AC, Granholm AC, 2006. A partial GDNF depletion leads to earlier age-related deterioration of motor function and tyrosine hydroxylase expression in the substantia nigra. Exp. Neurol 202, 336–347. [DOI] [PubMed] [Google Scholar]
- Boger HA, Middaugh LD, Patrick KS, Ramamoorth S, Denehy ED, Zhu H, Pacchioni AM, Granholm AC, McGinty JF, 2007. Long-term consequences of methamphetamine exposure in young adults are exacerbated in glial cell line-derived neurotrophic factor heterozygous mice. J. Neurosci 27, 8816–8825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E, 2003. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. [DOI] [PubMed] [Google Scholar]
- Buchman AS, Nag S, Shulman JM, Lim AS, VanderHorst VG, Leurgans SE, Schneider JA, Bennett DA, 2012. Locus coeruleus neuron density and parkinsonism in older adults without Parkinson’s disease. Mov. Disord 27, 1625–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, Swope DM, 2005. Clinical pharmacology of rasagiline: a novel, second-generation propargylamine for the treatment of Parkinson disease. J. Clin. Pharmacol 45, 878–894. [DOI] [PubMed] [Google Scholar]
- Connolly BS, Lang AE, 2014. Pharmacological treatment of Parkinson disease: a review. J. Am. Med. Assoc 311, 1670–1683. [DOI] [PubMed] [Google Scholar]
- Del Tredici K, Braak H, 2013. Dysfunction of the locus coeruleus-norepinephrine system and related circuitry in Parkinson’s disease-related dementia. J. Neurol. Neurosurg. Psychiatry 84, 774–783. [DOI] [PubMed] [Google Scholar]
- Eliash S, Dror V, Cohen S, Rehavi M, 2009. Neuroprotection by rasagiline in thiamine deficient rats. Brain Res 1256, 138–148. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Grzanna R, 1991. Experimentally-induced neuron loss in the locus coeruleus of adult rats. Exp. Neurol 111, 123–127. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Racagni G, Riva MA, 2006. Shedding light into the role of BDNF in the pharmacotherapy of Parkinson’s disease. Pharmacogenom. J 6, 95–104. [DOI] [PubMed] [Google Scholar]
- Garcia-Ruiz PJ, Chaudhuri KR, Martinez-Martin P, 2014. Non-motor symptoms of Parkinson’s disease: a review…from the past. J. Neurol. Sci 338, 30–33. [DOI] [PubMed] [Google Scholar]
- German DC, Manaye KF, White CL 3rd, Woodward DJ, McIntire DD, Smith WK, Kalaria RN, Mann DM, 1992. Disease-specific patterns of locus coeruleus cell loss. Ann. Neurol 32, 667–676. [DOI] [PubMed] [Google Scholar]
- Granholm AC, Mott JL, Bowenkamp K, Eken S, Henry S, Hoffer BJ, Lapchak PA, Palmer MR, van Horne C, Gerhardt GA, 1997. Glial cell line-derived neurotrophic factor improves survival of ventral mesencephalic grafts to the 6-hydroxydopamine lesioned striatum. Exp. Brain Res 116, 29–38. [DOI] [PubMed] [Google Scholar]
- Hawkes CH, Del Tredici K, Braak H, 2010. A timeline for Parkinson’s disease. Park. Relat. Disord 16, 79–84. [DOI] [PubMed] [Google Scholar]
- Henchcliffe C, Beal MF, 2008. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol 4, 600–609. [DOI] [PubMed] [Google Scholar]
- Hwang O, 2013. Role of oxidative stress in Parkinson’s disease. Exp. Neurobiol 22, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakish J, Tavassoly O, Lee JS, 2015. Rasagiline, a suicide inhibitor of monoamine oxidase, binds reversibly to alphasynuclein. ACS Chem. Neurosci 6, 347–355. [DOI] [PubMed] [Google Scholar]
- Kehagia AA, Barker RA, Robbins TW, 2010. Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson’s disease. Lancet Neurol 9, 1200–1213. [DOI] [PubMed] [Google Scholar]
- Kostrzewa RM, 2009. Evolution of neurotoxins: from research modalities to clinical realities. Curr. Protoc. Neurosci 46 1.18.1–1.18.10. [DOI] [PubMed] [Google Scholar]
- Lockrow J, Boger H, Gerhardt G, Aston-Jones G, Bachman D, Granholm AC, 2011. A noradrenergic lesion exacerbates neurodegeneration in a Down syndrome mouse model. J. Alzheimers Dis 23, 471–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo GR, Chen S, Le WD, 2006. Are heat shock proteins therapeutic target for Parkinson’s disease? Int. J. Biol. Sci 3, 20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnoni R, Palmfeldt J, Christensen JH, Sand M, Maltecca F, Corydon TJ, West M, Casari G, Bross P, 2013. Late onset motoneuron disorder caused by mitochondrial Hsp60 chaperone deficiency in mice. Neurobiol. Dis 54, 12–23. [DOI] [PubMed] [Google Scholar]
- Marin C, Aguilar E, Bonastre M, 2008. Effect of locus coeruleus denervation on levodopa-induced motor fluctuations in hemiparkinsonian rats. J. Neural Transm 115, 1133–1139. [DOI] [PubMed] [Google Scholar]
- Maruyama W, Akao Y, Carrillo MC, Kitani K, Youdim MB, Naoi M, 2002. Neuroprotection by propargylamines in Parkinson’s disease: suppression of apoptosis and induction of prosurvival genes. Neurotoxicol. Teratol 24, 675–682. [DOI] [PubMed] [Google Scholar]
- Maruyama W, Nitta A, Shamoto-Nagai M, Hirata Y, Akao Y, Youdim MBH, Furukawa S, Nabeshima T, Naoi M, 2004. N-propargyl-1 (R)-aminoindan, rasagiline, increases glial cell-derived neurotrophic factor (GDNF) in neuroblastoma SH-SY5Y cells through activation of NK-кB transcription factor. Neurochem. Int 44, 393–400. [DOI] [PubMed] [Google Scholar]
- Mavridis M, Degryse AD, Lategan AJ, Marien MR, Colpaert FC, 1991. Effects of locus coeruleus lesions on parkinsonian signs, striatal dopamine and substantia nigra cell loss after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in monkeys: a possible role for the locus coeruleus in the progression of Parkinson’s disease. Neuroscience 41, 507–523. [DOI] [PubMed] [Google Scholar]
- Meissner WG, 2012. When does Parkinson’s disease begin? From prodromal disease to motor signs. Rev. Neurol 168, 809–814. [DOI] [PubMed] [Google Scholar]
- Naoi M, Maruyama W, 2010. Monoamine oxidase inhibitors as neuroprotective agents in age-dependent neurodegenerative disorders. Curr. Pharm. Des 16, 2799–2817. [DOI] [PubMed] [Google Scholar]
- Naoi M, Maruyama W, Inaba-Hasegawa K, 2013a. Revelation in the neuroprotective functions of rasagiline and selegiline: the induction of distinct genes by different mechanisms. Expert Rev. Neurother 13, 671–684. [DOI] [PubMed] [Google Scholar]
- Naoi M, Maruyama W, Yi H, 2013b. Rasagiline prevents apoptosis induced by PK11195, a ligand of the outer membrane translocator protein (18 kDa), in SH-SY5Y cells through suppression of cytochrome c release from mitochondria. J. Neural Transm 120, 1539–1551. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Rascol O, Hauser R, Feigin PD, Jankovic J, Lang A, Langston W, Melamed E, Poewe W, Stocchi F, Tolosa E, 2009. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl. J. Med 361, 1268–1278. [DOI] [PubMed] [Google Scholar]
- Ou XM, Chen K, Shih JC, 2006. Monoamine oxidase A and repressor R1 are involved in apoptotic signaling pathway. Proc. Natl. Acad. Sci. USA 103, 10923–10928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez V, Marin C, Rubio A, Aguilar E, Barbanoj M, Kulisevsky J, 2009. Effect of the additional noradrenergic neurodegeneration to 6-OHDA-lesioned rats in levodopa-induced dyskinesias and in cognitive disturbances. J. Neural Transm 116, 1257–1266. [DOI] [PubMed] [Google Scholar]
- Pessoa-Rocha N, Reis HJ, Vanden Berghe P, Cirillo C, 2014. Depression and cognitive impairment in Parkinson’s disease: a role for inflammation and immunomodulation?. Neuroimmunomodulation 21, 88–94. [DOI] [PubMed] [Google Scholar]
- Rodrigues TM, Jeronimo-Santos A, Outeiro TF, Sebastiao AM, Diogenes MJ, 2014. Challenges and promises in the development of neurotrophic factor-based therapies for Parkinson’s disease. Drugs Aging 31, 239–261. [DOI] [PubMed] [Google Scholar]
- Sagi Y, Mandel S, Amit T, Youdim MBH, 2007. Activation of tyrosine kinase receptor signaling pathway by rasagiline facilitates neurorescue and restoration of nigrostriatal dopamine neurons in post-MPTP-induced parkinsonism. Neurobiol. Dis 25, 35–44. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Olanow CW, Greenamyre JT, Bezard E, 2014. Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: future therapeutic perspectives. Lancet 384, 545–555. [DOI] [PubMed] [Google Scholar]
- Shih JC, Chen K, Ridd MJ, 1999. Monoamine oxidase: from genes to behavior. Annu. Rev. Neurosci 22, 197–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin E, Rogers JT, Devoto P, Bjorklund A, Carta M, 2014. Noradrenaline neuron degeneration contributes to motor impairments and development of L-DOPA-induced dyskinesia in a rat model of Parkinson’s disease. Exp. Neurol 257, 25–38. [DOI] [PubMed] [Google Scholar]
- Shin HS, 1997. Metabolism of selegiline in humans. Identification, excretion, and stereochemistry of urine metabolites. Drug. Metab. Dispos 25, 657–662. [PubMed] [Google Scholar]
- Teo KC, Ho SL, 2013. Monoamine oxidase-B (MAO-B) inhibitors: implications for disease-modification in Parkinson’s disease. Transl. Neurodegener 2, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari SK, Chaturvedi RK, 2014. Peptide therapeutics in neurodegenerative disorders. Curr. Med. Chem 21, 2610–2631. [DOI] [PubMed] [Google Scholar]
- Weinreb O, Bar-Am O, Amit T, Chillag-Talmor O, Youdim MBH, 2004. Neuroprotection via pro-survival protein kinase C isoforms associated with Bcl-2 family members. FASEB J 18, 1471–1473. [DOI] [PubMed] [Google Scholar]
- Weinreb O, Amit T, Sagi Y, Drigues N, Youdim MBH, 2009. Genomic and proteomic study to survey the mechanism of action of the anti-Parkinson’s disease drug, rasagiline compared with selegiline, in the rat midbrain. J. Neural Transm 166, 1457–1472. [DOI] [PubMed] [Google Scholar]
- Weinreb O, Amit T, Bar-Am O, Youdim MBH, 2010. Rasagiline: a novel anti-Parkinsonian monoamine oxidase-B inhibitor with neuroprotective activity. Prog. Neurobiol 92, 330–344. [DOI] [PubMed] [Google Scholar]
- Weinreb O, Bar-Am O, Prosolovich K, Amit T, Youdim MBH, 2011. Does 1-(R)-aminoindan possess neuroprtective properties against experimental Parkinson’s disease?. Antioxid. Redox Signal 14, 767–775. [DOI] [PubMed] [Google Scholar]
- Weissmiller AM, Wu C, 2012. Current advances in using neurotrophic factors to treat neurodegenerative didorders. Transl. Neurodegener 1, 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk GL, McGann K, Hauss-Wegrzyniak B, Rosi S, 2003. The toxicity of tumor necrosis factor-alpha upon cholinergic neurons within the nucleus basalis and the role of norepinephrine in the regulation of inflammation: implications for Alzheimer’s disease. Neuroscience 121, 719–729. [DOI] [PubMed] [Google Scholar]
- Winkler C, Kirik D, Bjorklund A, Cenci MA, 2002. L-DOPA-induced dyskinesia in the intrastriatal 6-hydroxydopamine model of parkinson’s disease: relation to motor and cellular parameters of nigrostriatal function. Neurobiol. Dis 10, 165–186. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Gross A, Finberg JP, 2001. Rasagiline [N-propargyl-1R(+)-aminoindan], a selective and potent inhibitor of mitochondrial monoamine oxidase B. Br. J. Pharmacol 132, 500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Xie W, Pan T, Jankovic J, Li J, Youdim MB, Le W, 2008. Comparison of neuroprotective and neurorestorative capabilities of rasagiline and selegine against lactacystin-induced nigrostriatal dopaminergic degeneration. J. Neurochem 105, 1970–1978. [DOI] [PubMed] [Google Scholar]