

Abstract
Background
Epilepsy remains a significant public health concern in Sub-Saharan Africa (SSA) where diverse etiological factors contribute to its prevalence. Among these factors are conditions originating from the neuroectoderm, such as tuberous sclerosis. Insufficient medical attention and a lack of comprehensive multidisciplinary care contribute to its under-recognition.
Materials and methods
We conducted a retrospective descriptive study, involving 12 patients admitted to the neurology and pediatric departments of the University Hospital Ignace Deen between 2010 and 2022 due to recurring epileptic seizures. Subsequently, these patients were diagnosed with Tuberous sclerosis using the Schwartz 2007 criteria. The aim of this study is to reassess this condition from a clinical and paraclinical point of view in a tropical environment.
Results
Tuberous sclerosis, also known as Bourneville disease, was diagnosed in 12 patients exhibiting focal motor seizures and complex focal seizures likely associated with cortical and subcortical tubers detectable by EEG and neuroimaging, including CT and MRI. Delayed treatment resulted in varying degrees of mental decline. Additionally, some patients displayed cardiac hamartomas and intracranial posterior and anterior aneurysms as minor diagnostic indicators.
Conclusion
The study reveals a consistent clinical presentation accompanied by deteriorating neurological and psychological symptoms attributed to delayed multidisciplinary management. These findings are utilized to assess therapeutic strategies and prognostic outcomes.
Keywords: Epilepsy, Cortical and subcortical tubers, Bourneville sclerosis, Republic of Guinea
Highlights
-
•
Epileptic seizures prompted all consultations, frequent status epilepticus noted.
-
•
Neurodevelopmental disorders in 9 patients: autism, delayed psychomotor development.
-
•
Two cases of intracranial aneurysms found.
-
•
Neuroradiology: cortical and subcortical tubers, subependymal giant cell astrocytoma.
-
•
EEG: Type III tracings, hypsarrhythmia in West syndrome cases.
1. Introduction
In Guinea, a collaborative study with Harvard Medical School investigates epilepsy, shedding light on its clinical and paraclinical evaluation and management [[1], [2], [3], [4]]. It identifies diverse etio-clinical and therapeutic aspects, notably highlighting the overlooked status of phacomatosis like tuberous sclerosis, known for recurrent seizures and mental deterioration, yet lacking adequate attention in sub-Saharan Africa [[5], [6], [7], [8]].
The presence of motor epilepsies and complex focal seizures, such as West's syndrome and Lennox Gastaut syndrome, accompanied by progressive cognitive decline is firmly established in both early and recent publications [[9], [10], [11], [12], [13], [14]] and in various reviewed series [[15], [16], [17], [18], [19], [20]]. The introduction of magnetic resonance imaging and the development of electroencephalography techniques have enabled a better diagnostic approach to neurocutaneous syndromes in particular tuberous sclerosis, which occurs in populations of all ethnic and geographical origins with a prevalence varying between 1/5000 and 1/10000 [[21], [22], [23], [24], [25], [26], [27], [28]]. The significance of this study lies in its illustration of tuberous sclerosis and the challenges it poses in diagnosis, particularly when distinguishing it from other neurodegenerative conditions associated with epilepsy. These conditions include Klippel-Trenaunay syndrome [29], Parkes Weber syndrome [30], Nova syndrome [31], Rendu Osler syndrome [32], Sturge Weber syndrome, Krabbe disease [33], Von Hippel Lindau disease [34], Fabry disease [35], Meningocerebral Angiomatosis [36], and Ivry-Van Bogaert syndrome [37].
2. Material and methods
We conducted a retrospective cross-sectional study of 518 patients hospitalized for recurrent epileptic seizures between 2010 and 2022. Twelve (12) patients (7 boys and 5 girls) were diagnosed with Tuberous Sclerosis based on the Schwartz 2007 criteria [38]. Each patient underwent a clinical assessment conducted by a neurologist, a pediatrician, and a child psychiatrist. This included determining whether seizures were focal, investigating family histories of patients with seizures or related illnesses, reviewing the patient's medical and neonatal history, as well as their gestational history.
The inclusion criteria were those revised for the diagnosis of tuberous sclerosis by Schwartz et al. 2007 [38] based on the major and minor criteria listed by Roach et al. [39] and Hyman et al. [40]. All patients underwent a series of additional tests, including CBC, ESR, fasting plasma glucose, 24-hour proteinuria, serum calcium, serum iron, SGPT, and SGOT transaminases, CPK, and CRP. Lumbar puncture enabled cytological and biochemical evaluation of cerebrospinal fluid. PCR tests for various viruses, including HSV1 and 2, EBV, CMV, HIV, HSV, VZV, were supplemented by measles antibody assays in serum and CSF. Each patient underwent at least two EEG tracings using a NIPPON Neurofax IE 910A device, along with ophthalmological examinations using an ophthalmoscope 1ECLR6 3000 HEINE mini for fundus assessment and visual acuity. Neuroradiological MRI/CT scans were performed to detect sub ependymal nodules, tubers, and other abnormalities using a CT Scanner 1-slice-spinal Toshiba. Additional investigations, such as repetitive neurodevelopmental assessment, echocardiogram, and abdominal and renal ultrasounds, were conducted based on symptoms. EEG tracings were categorized into three types using an EEG Nihon Neurofax machine.
Type I:
-
-
EEG with dominance of alpha rhythms of parieto-occipital topography whose amplitude is greater than 40 μvolts without pathological rhythms.
-
-
EEG with dominance of alpha rhythms of small amplitudes up to 25 μvolts with a tendency to flattening.
Type II:
-
-
EEG without dominance as such with existence of irregular alpha rhythms without presence of pathological waves.
-
-
EEG with theta rhythms of 4 to 6 Hz mainly of parieto-temporal topography of low amplitude of 30 to 40 μvolts, isolated or sometimes grouped in the forms of paroxysmal puffs.
Type III: - EEG with theta and delta rhythm showing abnormal patterns.
-
-
EEG with slowing alpha rhythms associated with puffs of theta and delta waves.
3. Results
During the study period, twelve cases were observed, involving seven boys and five girls, aged between 13 months and 14 years at the time of their initial consultation. Epileptic seizures prompted all consultations, particularly complaints suggestive of frequent status epilepticus due to lack of appropriate treatment. Family history was investigated in all patients, with only four cases having parents reporting paroxysmal manifestations, albeit without precise details. Additionally, dermatological lesions, including facial angiofibroma in two patients and shagreen patches in one, were noted.
In the history of the disease, all patients initially sought traditional medicine. In five (5) patients, West syndrome was diagnosed based on spasms with brief axial movements, often in flexion of the upper limbs and extension of the lower limbs. Lennox Gastaut syndrome was identified in three (3) patients, in the four (4) remaining cases partial epileptic seizures were secondarily generalized with the character of tonico-clonic seizures. Neurodevelopmental disorders in the form of autism, delayed psychomotor development and specific learning disorders were identified in nine (9) patients. In one patient, thirty-five (35) days after birth, a cardiac hamartoma [Fig. 1] was diagnosed by cardiac Echo-Doppler, a pathology that usually falls into the category of Rhabdomyomas, which are often diagnosed antenatal. (See Table 1.)
Fig. 1.
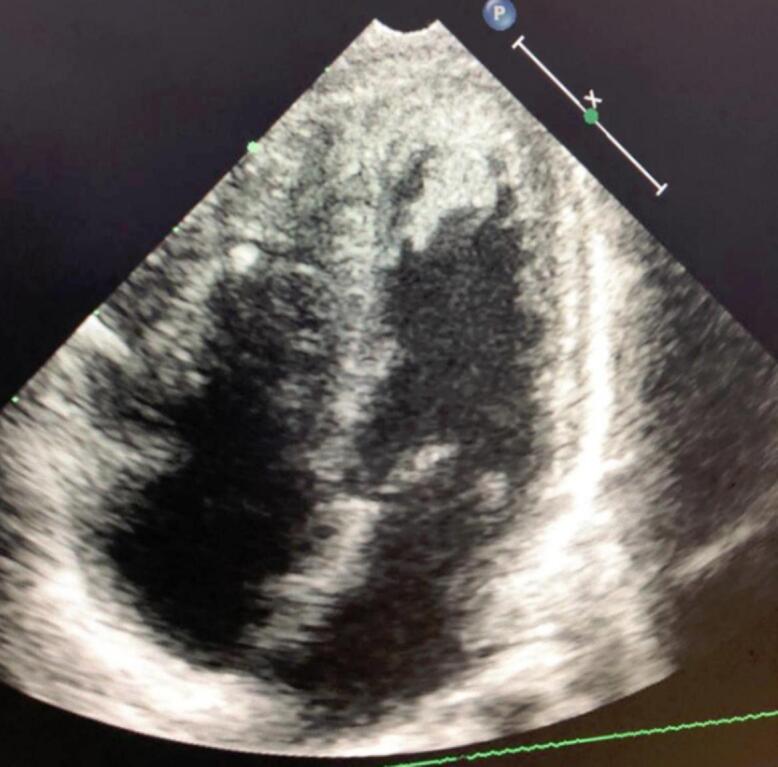
Apical four-cavity section showing hamartoma in the right and left ventricle.
Table 1.
Clinical signs in the state phase.
| No | Clinical signs | Number of patients | Proportion |
|---|---|---|---|
| 1 |
Epilepsy Focal seizures and generalized seizures (West syndrome, Lennox Gastaut), |
12 | 100 |
| 2 |
Psychiatric manifestations Psychomotor retardation, autism, specific learning disorders, language delay, visual-spatial disorders |
8 | 67 |
| 3 |
Dermatological manifestations Facial angiofibromas, Koenen's tumors, shagreen patches |
7 | 58 |
| 4 |
Eye disease Retinal hamartomas |
2 | 17 |
| 5 |
Oral diseases Fibromas of the gums and holes in dental enamel |
2 | 17 |
| 6 |
Kidney disease Renal cysts, malignant renal tumors |
1 | 17 |
| 7 |
Cardiac diseases Cardiac hamartomas |
1 | 8 |
| 8 | Anterior and Posterior intracranial aneurysms | 2 | 8 |
| 9 |
Neuroradiological signs Sub-ependymal tubers and nodules |
12 |
100 |
In two (2) cases, two intracranial aneurysms were found [Fig. 3, Fig. 4]. Cognitive disorders in these patients were initially discrete before becoming intense, aggravated by the delay in medical management. Autism was identified in four (4) patients, mainly characterized by indifference to stimuli, unstable behavior, and psycho-intellectual disorders characterized by mental retardation, with psychomotor agitation in two (2) patients aged 8 to 14 years. In this study, we observed one case of apparently normal development during the study. Associations with aneurysms and cortical hamartomas were noted in two (2) patients.
Fig. 3.
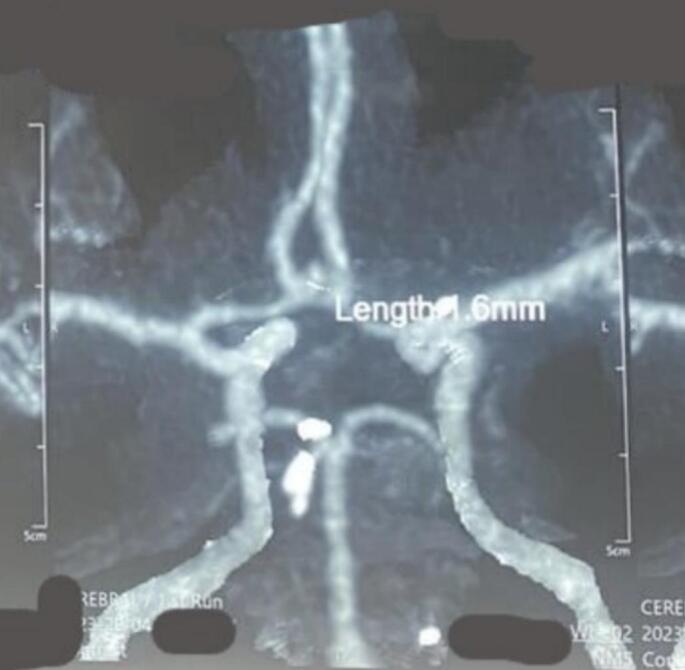
Aneurysm on the right posterior communicator.
Fig. 4.
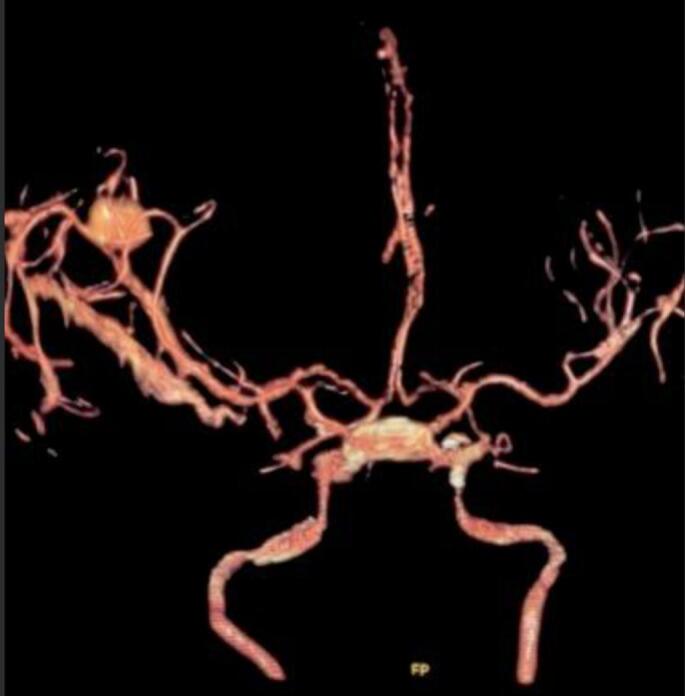
Polylobed saccular aneurysm on the upper surface of the junction between the anterior cerebral and right sylvian arteries, measuring 6.5 mm with a narrow neck of 1.4 mm.
Biological data: The biological data in this study shows a normal CBC and Sedimentation rate in all patients except for one patient who had normochromic anemia with hemoglobin 7 g/dl and positive plasmodium test as a comorbidity. In the CSF, the cell count and total protein were normal in all patients.
Neuroradiological data: All twelve patients underwent neuroradiological scans of the brain and magnetic resonance imaging (MRI), which showed cortical tubers in the parietal and occipital areas, as well as subcortical tubers (Fig. 7). On MRI, the cortical tubers were hypointense in T1 and hyperintense in T2 and sub-ependymal giant cell astrocytoma were found in two (2) patients located in the region of the Foramen of Monroe (Fig. 8).
Fig. 7.
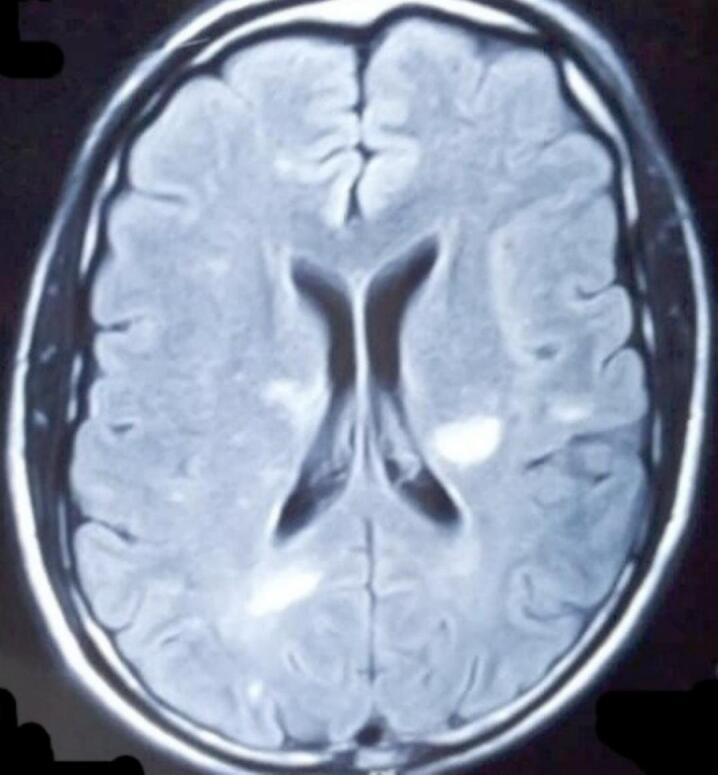
Cerebral MRI shows a subcortical hypersignal on the FLAIR sequence compatible with tubercles.
Fig. 8.
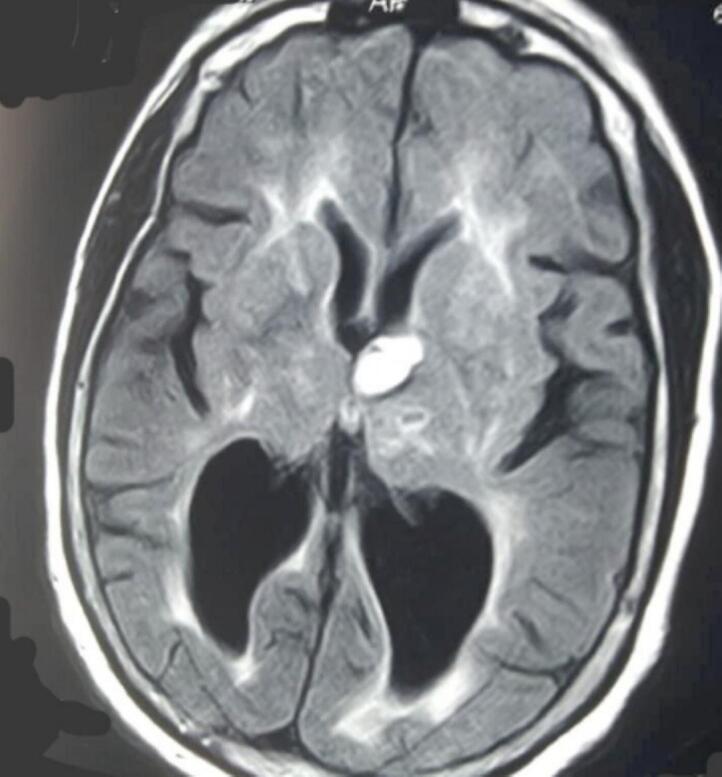
Cerebral MRI in flair sequence shows a nodular hypersignal, subependymal opposite Monroe's foramen on the left, compatible with a tubercle.
Electroencephalographic data: Electroencephalographic data show type III tracings in all patients expressing diffuse distress (Fig. 5). In cases of West syndrome, the tracing reflects a hypsarrhythmia characterized by slow waves and irregular peaks with changes in duration and topography (Fig. 6).
Fig. 5.

Wake trace only, showing numerous hypsarrhythmias with a diffuse and bilateral appearance.
Fig. 6.

Wake trace only, showing numerous hypsarrhythmia's with a diffuse and bilateral appearance.
4. Discussion
Twelve cases of epileptic manifestations associated with tuberous sclerosis also known as the disease of Bourneville were collected in this retrospective study of 518 patients with epileptic seizures over a period of ten years. Since the Vogt's triad was first described, comprising mental deficiency, epilepsy, and cutaneous lesions such as achromic angiofibromas and Konen's tumors [23], several authors [41,42] have noted its presence in only 30% of observations. Consequently, additional manifestations have been recognized and included, leading to the establishment of major and minor criterias [[19], [20], [21], [22], [23], [24], [25], [26]] accepted by most authors [[27], [28], [29], [30], [31], [32], [33], [34]]. The diagnosis was enriched by the presence of hamartomas in multiple organs including the brain, skin, kidneys and heart [[35], [36], [37]] and biologically by the presence of two notified genes [29,30].
Thus, in general, the clinical pictures observed in this study do not differ fundamentally from those described in the literature, with two entities: epileptic seizures, mainly focal, associated with generalized manifestations, and impairment of psychomotor development and mental retardation, including autism, which affects approximately 50% in our series. The presence of hamartomas in various organs, sometimes associated with dermatological lesions, was identified in our series: a 35-day-old child with a cardiac hamartoma on echocardiography [Fig. 1, Fig. 2] who presented with spasms mimicking West syndrome [Fig. 3, Fig. 4].
Fig. 2.
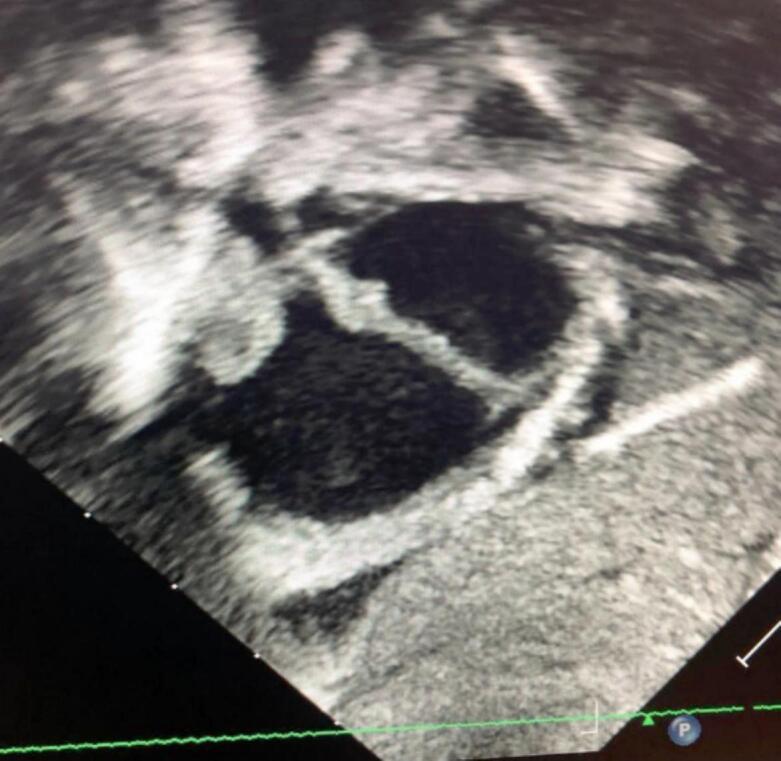
Three-dimensional subcortical section showing a hamartoma in the right ventricle and superior cardiac volume abruption.
The epileptic seizures observed here correspond to the known clinical and electrophysiological characteristics of this condition: West's syndrome with the classic triad of flexion spasm, psychomotor regression and hypsarrhythmia in five (5) patients and Lennox Gastaut (3 cases) and non-systematized focal forms which are secondarily generalized. Nevertheless, it should be noted that there are severe manifestations with autism, abnormal psychomotor development and mental retardation in several cases, which can be explained by the absence of early management of epileptic seizures due to socio-cultural conceptions attributing epilepsy to supernatural forces, as described in our work at Harvard Medical School [30]. Hemangiomas are frequently noted, however the discovery of intracranial aneurysms has only been reported 18 times in the literature [43] and our study mentions two new cases. The same is true for the cardiac hamartomas [Fig. 1, Fig. 2] reported in one case in our series.
On the other hand, under-medicalization due to inadequate care is an additional explanation for the seriousness of the psychiatric manifestations and mental retardation going as far as profound. Elsewhere, most authors [[31], [32], [33], [34]] believe that the onset of mental retardation is often correlated with the number of cortical and subcortical tubers and their location in the frontal and occipital regions [24,32,35,36]. Autism was found in around 50–60% of cases [[37], [39], [40]], as in our study, is the expression of cortical and subcortical dysfunction in the temporal locations of the tubers, as well as dysfunction of the brainstem, cerebellum and caudate nuclei.
This study and others demonstrate that CT/MRI neuroimaging aids in diagnosing tuberous sclerosis complex (TSC) by revealing characteristic lesions, such as tubers and subependymal nodules as shown in figs. [[4], [5], [6], [7]]. These tubers appear as hyperdense calcified lesions on CT and hypointense or isointense in T1 and hyperintense in T2 on MRI. TSC1 or TSC2 mutations, present in 75%–90% of TSC cases, encode hamartin and tuberin, which regulate cell growth via the mTOR signaling pathway [44]. While these genetic markers are important, TSC is primarily diagnosed based on clinical symptoms. Genetic testing is reserved for ambiguous or mild cases to confirm the diagnosis and facilitate targeted therapies. Research has led to the approval of mTOR inhibitors for treating renal angiomyolipoma, brain subependymal giant cell astrocytoma, and pulmonary lymphangioleiomyomatosis, though further studies are needed to expand their use [44,45]. In our report, the clinical symptoms alone were sufficient to diagnose TSC, despite the lack of genetic testing.
Table 2 provides an overview of various epilepsy syndromes, each characterized by distinct clinical presentations, diagnostic methods, treatment modalities, and prognoses. Tuberous Sclerosis Complex (TSC) is typified by seizures, cognitive impairment, and renal angiomyolipomas, diagnosed via EEG and MRI, and managed with antiepileptic drugs, everolimus, and surgery, with prognostic outcomes varying based on symptom severity and comorbidities [[1], [2], [3], [4]]. Klippel-Trenaunay Syndrome and Parkes Weber Syndrome exhibit vascular abnormalities, necessitating imaging for diagnosis and employing interventions such as sclerotherapy and surgery for symptom management, with prognoses influenced by symptom severity and complications like thrombosis or hemorrhage [29,30]. Distinctive features like facial port-wine stains and neurological symptoms characterize Sturge-Weber Syndrome, diagnosed through imaging and treated with antiepileptic drugs, laser therapy, and surgery, with prognoses ranging from seizure control to persistent neurological deficits [33]. Von Hippel-Lindau Disease involves tumor development, requiring imaging for surveillance and employing surgical or embolic interventions, with prognosis linked to tumor burden and malignancy risk [34]. Fabry Disease, marked by systemic complications, necessitates enzyme assays and genetic testing for diagnosis, managed with enzyme replacement and symptom care, with prognosis improved by early intervention but still carrying risks of end-organ damage [35]. Meningioangiomatosis and Ivry-Van Bogaert Syndrome, both presenting with neurological symptoms, require imaging for lesion detection and employ a combination of surgical and symptomatic management, with prognoses varying based on disease severity and treatment response [36,37]. These summaries underscore the diverse clinical profiles and management strategies of each syndrome, emphasizing the importance of early investigation and proper management in optimizing patient outcomes.
Table 2.
Summarizes diverse clinical manifestations, diagnostic procedures, treatment options, and prognostic outlooks for several epilepsy syndromes.
| Epilepsy Syndromes/References | Clinical Symptoms | Paraclinical Investigations | Therapeutic Available | Prognosis and Outcomes |
|---|---|---|---|---|
| Tuberous Sclerosis Complex (TSC) [[1], [2], [3], [4]] | Epileptic seizures, cognitive impairment, behavioral problems, autism spectrum disorder, renal angiomyolipomas, cardiac rhabdomyomas, facial angiofibromas, ash leaf spots | EEG showing hypsarrhythmia or focal epileptiform discharges, MRI revealing cortical tubers, subependymal nodules, and subependymal giant cell astrocytoma | Antiepileptic drugs (AEDs), everolimus for subependymal giant cell astrocytomas, surgical resection of epileptic foci | Variable depending on the severity of symptoms and comorbidities, some individuals may achieve seizure control with treatment while others may have ongoing seizures and cognitive impairment |
| Klippel-Trenaunay Syndrome [29] | Port-wine stains, venous malformations, soft tissue and bone overgrowth, limb hypertrophy | Imaging studies (MRI, CT, Doppler ultrasound) to assess extent of vascular malformations | Symptomatic treatment for pain and cosmetic concerns, sclerotherapy, embolization, surgical intervention | Prognosis depends on severity of symptoms, potential complications from vascular malformations such as thrombosis or hemorrhage |
| Parkes Weber Syndrome [30] | Capillary malformations, soft tissue and bone hypertrophy, arteriovenous malformations | Imaging studies (MRI, CT, Doppler ultrasound) to assess vascular malformations | Symptomatic treatment for pain, compression garments, embolization, surgical intervention | Prognosis depends on severity of symptoms, risk of complications such as bleeding or heart failure from arteriovenous malformations |
| Sturge-Weber Syndrome [33] | Facial port-wine stain, leptomeningeal angiomas, glaucoma, seizures, stroke-like episodes | Imaging studies (MRI, CT) to detect leptomeningeal angiomas | Antiepileptic drugs (AEDs), laser therapy for port-wine stains, glaucoma management, surgical resection of leptomeningeal angiomas | Prognosis varies, some individuals may have seizure control with treatment while others may have refractory epilepsy and neurodevelopmental deficits |
| Von Hippel-Lindau Disease [34] | Hemangioblastomas in CNS and retina, renal cell carcinomas, pheochromocytoma, pancreatic neuroendocrine tumors |
Imaging studies (MRI, CT) to detect CNS and retinal hemangioblastomas | Surgical resection or embolization of hemangioblastomas, surveillance for other tumors | Prognosis depends on tumor burden and complications, risk of malignant transformation in hemangioblastomas |
| Fabry Disease [35] | Pain crises, acroparesthesias, angiokeratomas, gastrointestinal symptoms, renal failure, cardiac complications | Enzyme assay to detect deficient alpha-galactosidase A activity, genetic testing for GLA gene mutations | Enzyme replacement therapy, symptomatic treatment for pain and organ complications | Prognosis improved with early diagnosis and treatment, risk of end-organ damage and complications |
| Meningioangiomatosis [36] | Focal seizures, headache, focal neurological deficits | Imaging studies (MRI) to detect intracranial lesions | Surgical resection of lesions, antiepileptic drugs (AEDs) | Prognosis depends on extent of lesions and response to treatment, risk of seizures and neurological deficits |
| Ivry-Van Bogaert Syndrome [37] | Epilepsy, cerebellar ataxia, cognitive impairment | Imaging studies (MRI) to assess cerebellar abnormalities | Symptomatic treatment for epilepsy and ataxia, physical and occupational therapy | Prognosis variable depending on severity of symptoms, potential for progressive neurological decline |
Comparing the syndromes, it is evident that early investigation is crucial for accurate diagnosis and initiation of appropriate treatment. For instance, in Tuberous Sclerosis Complex (TSC), early identification of characteristic MRI findings such as cortical tubers and sub-ependymal nodules allows for timely intervention, potentially improving seizure control and cognitive outcomes.
Overall, this comparative analysis underscores the significance of early investigation and multidisciplinary management in improving the prognosis and quality of life for individuals with epilepsy syndromes. Timely diagnosis and appropriate treatment interventions can mitigate complications, enhance symptom control, and promote better long-term outcomes.
5. Conclusion
This study shows the presence and persistence of tuberous sclerosis, sometimes associated with cardiac hamartomas and intracranial aneurysms. It remains an under-recognized and under-notified condition in sub-Saharan Africa. Its stereotyped diagnosis is based on classic major and minor criteria, and CT and MRI neuroimaging is helpful in identifying it. Its management requires a multi-disciplinary strategy that includes pediatric neurologists, child psychiatrists, epileptologists and even neurosurgeons.
Disclosures
No previous submission statement: All authors declare that the submitted work has not been published before (neither in English nor in any other language) and that the work is not under consideration for publication elsewhere.
Ethical standards
There were no animal studies involved. All human studies have been approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Other relationships
All authors have affirmed that they possess no additional relationships or engagements that might influence the work submitted.
CRediT authorship contribution statement
Souleymane M'Bara Diallo: Resources, Project administration, Methodology, Investigation, Conceptualization. Mamadou Diallo: Writing – review & editing, Writing – original draft, Resources, Methodology, Investigation, Conceptualization. I.S. Barry: Visualization, Resources, Methodology. M.L. Touré: Project administration, Formal analysis, Data curation, Conceptualization. M.C. Barry: Methodology, Investigation. M.T. Diallo: Project administration, Formal analysis, Data curation, Conceptualization. S.D. Barry: Writing – review & editing, Supervision, Project administration, Methodology, Data curation, Conceptualization. S.Y. Aminou: Methodology, Investigation. G. Carlos Othon: Visualization, Resources, Methodology. B. Diallo: Writing – original draft, Resources. N. Camara: Visualization, Methodology, Investigation. M.B. Diallo: Methodology, Investigation. M. Zoumanigui: Resources, Methodology. E. Lamah: Writing – original draft, Investigation. M. Hinima: Resources, Data curation. Sindu Mukesh: Writing – original draft, Resources. A.K.T. Barry: Resources, Formal analysis, Conceptualization. A. Sacko: Software, Investigation, Formal analysis. Ramit Singla: Writing – review & editing, Supervision. F.A. Cisse Supervision, review and editing. A. Cissé: Methodology, Investigation, Formal analysis, Data curation, Conceptualization.
Declaration of competing interest
On behalf of all authors, the corresponding authors state that there is no conflict of interest. All authors have declared that they have no financial relationships at present with any organizations that might have an interest in the submitted work.
Contributor Information
Souleymane M'Bara Diallo, Email: dialloyali319@gmail.com.
Mamadou Diallo, Email: dr.diallodenka92@gmail.com.
References
- 1.Williams J.A., Cisse F.A., Schaekermann M., et al. Guinea epilepsy project. Smartphone EEG and remote online interpretation for children with epilepsy in the Republic of Guinea: quality, characteristics, and practice implications. Seizure. 2019;71:93–99. doi: 10.1016/j.seizure.2019.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anand P., Othon G.C., Sakadi F., et al. Guinea epilepsy project epilepsy and traditional healers in the Republic of guinea: a mixed methods study. Epilepsy Behav. 2019;92:276–282. doi: 10.1016/j.yebeh.2019.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fitts W., Rahamatou N.T., Abass C.F., et al. School status and its associations among children with epilepsy in the Republic of Guinea. Epilepsy Behav. 2019;97:275–281. doi: 10.1016/j.yebeh.2019.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jang M., Sakadi F., Tassiou N.R., et al. Guinea epilepsy project impact of poorly controlled epilepsy in the Republic of Guinea. Seizure. 2018;61:71–77. doi: 10.1016/j.seizure.2018.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diallo T.M., Cisse A., Morel Y., et al. Premières crises epileptiques tardives. Etude de 42 cas [Late occurrence of frst epileptic seizures: a 42-case series] Med. Trop. (Mars) 2004;64:155–159. [PubMed] [Google Scholar]
- 6.Pitche P., Agbere A.D., Gbadoe A.J., Tatagan A., Tchangaï-Walla K. Sclérose tubéreuse de Bourneville et épilepsie de l’enfant. A propos de quatre observations togolaises. 1998;91(3):235–237. (ref : 16 ref) [PubMed] [Google Scholar]
- 7.Moifo B., Nguefack S., Neossi Guena M., Mah E., Guegang Goudjou E., Mbonda E., Gonsu Fotsing J. Aspects Cliniques Et Scanographiques De La Sclerose Tubereuse De Bourneville: à propos de huit cas pédiatriques révélés par une épilepsie. Mali Med. 2012;1:51–56. [PubMed] [Google Scholar]
- 8.Cissé A., Cissé A.F., Touré A., Souaré I.S., Bah H., Kourouma S., Cissé B., Koulibaly M., Morel Y., Diaby M.M., Koné S., Ka M.L. Doukouré M. Aspects clinique et scannographique de 29 observations de phacomatoses en Guinée [Clinical and tomographic aspects of 29 cases of phakomatosis in Guinea] Med. Trop. (Mars) 2006 Jun;66(3):247–251. French. PMID: 16924815. [PubMed] [Google Scholar]
- 9.Aggoun Y., Hunkeler N., Destephen M., Vial Y., Gudinchet F., Calame A., Payot M. Rhabdomyomatose cardiaque et sclérose tubéreuse de Bourneville chez le foetus. A propos de 2 cas [Cardiac rhabdomyomatosis and Bourneville’s tuberous sclerosis in the fetus. Apropos of 2 cases] Arch. Mal. Coeur Vaiss. 1992 May;85(5):609–613. French. PMID: 1530402. [PubMed] [Google Scholar]
- 10.Muzykewicz D.A., Newberry P., Danforth N., Halpern E.F., Thiele E.A. Conditions psychiatriques comorbides dans une population clinique de 241 patients atteints de complexe de sclérose tubéreuse. Comportement d’épilepsie. 2007;11:506–513. doi: 10.1016/j.yebeh.2007.07.010. [DOI] [Google Scholar]
- 11.Hallett L., Foster T., Liu Z., Blieden M., Valentim J. Fardeau de la maladie et besoins non satisfaits dans le complexe de sclérose tubéreuse avec manifestations neurologiques : revue systématique. Opin. Actuelle de la Rés Méd. 2011;27:1571–1583. doi: 10.1185/03007995.2011.586687. [DOI] [PubMed] [Google Scholar]
- 12.Nabbout R., Belousova E., Benedik M.P., Carter T., Cottin V., Curatolo P., et al. Épilepsie dans le complexe de la sclérose tubéreuse : résultats de l’étude TOSCA. Épilepsie Ouverte. 2018;4:73–84. doi: 10.1002/epi4.12286. [DOI] [Google Scholar]
- 13.Northrup H., Aronow M.E., Bebin E.M., Bissler J., Darling T.N., de Vries P.J., et al. Mise à jour des critères de diagnostic du complexe international de la sclérose tubéreuse et des recommandations de surveillance et de prise en charge. Pediatr. Neurol. 2021;123:50–66. doi: 10.1016/j.pediatrneurol.2021.07.011. [DOI] [PubMed] [Google Scholar]
- 14.Tye C., Mcewen F.S., Liang H., Underwood L., Woodhouse E., Barker E.D., et al. Résultats cognitifs à long terme dans le complexe de la sclérose tubéreuse. Dev. Med. Enfant Neurol. 2020;62:322–329. doi: 10.1111/dmcn.14356. [DOI] [Google Scholar]
- 15.Tritton T., Bennett B., Brohan E., Grant L., Cooper A., Fladrowski C., et al. Services de santé et qualité de vie chez les personnes atteintes du complexe de sclérose tubéreuse (TSC) qui subissent des crises d’épilepsie : une enquête en ligne. Comportement d’épilepsie. 2019;92:213–220. doi: 10.1016/j.yebeh.2018.11.021. [DOI] [Google Scholar]
- 16.Krueger D.A., Care M.M., Holland K., Agricola K., Tudor C., Mangeshkar P., Wilson K.A., Byars A., Sahmoud T., Franz D.N. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N. Engl. J. Med. 2010 Nov 4;363(19):1801–1811. doi: 10.1056/NEJMoa1001671. (PMID: 21047224) [DOI] [PubMed] [Google Scholar]
- 17.Liang S., Zhang J., Yang Z., Zhang S., Cui Z., Cui J., et al. Résultats à long terme de la chirurgie de l’épilepsie dans le complexe de la sclérose tubéreuse. J. Neurol. 2017;264:1146–1154. doi: 10.1007/s00415-017-8507-y. [DOI] [PubMed] [Google Scholar]
- 18.Liu S., Yu T., Guan Y., Zhang K., Ding P., Chen L., et al. Chirurgie répressive de l’épilepsie dans le complexe de la sclérose tubéreuse : une étude rétrospective multicentrique nationale en Chine. Cerveau. 2020;143:570–581. doi: 10.1093/cerveau/awz411. [DOI] [Google Scholar]
- 19.Willems L.M., Schubert-Bast S., Grau J., Hertzberg C., Kurlemann G., Wiemer-Kruel A., et al. Qualité de vie liée à la santé chez les enfants et les adolescents atteints de sclérose tubéreuse et leurs soignants : une étude de cohorte multicentrique en Allemagne. Eur. J. Paediatr. Neurol. 2021;35:111–122. doi: 10.1016/j.ejpn.2021.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Grau J., Zöllner J.P., Schubert-Bast S., Kurlemann G., Hertzberg C., Wiemer-Kruel A., et al. Coûts directs et indirects et facteurs de coût du complexe de la sclérose tubéreuse chez les enfants, les adolescents et les soignants : une étude de cohorte multicentrique. Orphanet J. Rare Dis. 2021;16:282. doi: 10.1186/s13023-021-01899-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skalicky A.M., Rentz A.M., Liu Z., Said Q., Nakagawa J.A., Frost M.D., et al. Fardeau économique, travail et productivité scolaire chez les personnes atteintes de sclérose tubéreuse et leurs familles. J. Med. Econ. 2018;21:953–959. doi: 10.1080/13696998.2018.1487447. [DOI] [PubMed] [Google Scholar]
- 22.Amin S., Mallick A.A., Lux A., O’Callaghan F. Qualité de vie chez les patients atteints de complexe de sclérose tubéreuse (TSC) Eur. J. Paediatr. Neurol. 2019;23:801–807. doi: 10.1016/j.ejpn.2019.09.006. [DOI] [PubMed] [Google Scholar]
- 23.Jansen A.C., Vanclooster S., de Vries P.J., Fladrowski C., Beaure d’Augères G., Carter T., et al. Fardeau de la maladie et qualité de vie dans le complexe de la sclérose tubéreuse : résultats de l’étude TOSCA. Neurol Avant. 2020;11:904. doi: 10.3389/fneur.2020.00904. [DOI] [Google Scholar]
- 24.Marques R., Belousova E., Benedik M.P., Carter T., Cottin V., Curatolo P., et al. Modèles de traitement et utilisation des ressources chez les patients atteints de sclérose tubéreuse : aperçus du registre TOSCA. Neurol Avant. 2019;10:1144. doi: 10.3389/fneur.2019.01144. [DOI] [Google Scholar]
- 25.O’Callaghan F.J., Noakes M.J., Martyn C.N., Osborne J.P. Une étude épidémiologique de la pathologie rénale dans le complexe de la sclérose tubéreuse. BJU Int. 2004;94:853–857. doi: 10.1111/j.1464-410X.2004.05046.x. [DOI] [PubMed] [Google Scholar]
- 26.Osborne J.P., Fryer A., Webb D. Epidemiology of tuberous sclerosis. Ann. N. Y. Acad. Sci. 1991;615:125–127. doi: 10.1111/j.1749-6632.1991.tb37754.x. (PMID: 2039137) [DOI] [PubMed] [Google Scholar]
- 27.Morrison P.J. Tuberous sclerosis: epidemiology, genetics and progress towards treatment. Neuroepidemiology. 2009;33(4):342–343. doi: 10.1159/000254570. Epub 2009 Nov 4. PMID: 19887840. [DOI] [PubMed] [Google Scholar]
- 28.Specchio N., Nabbout R., Aronica E., Auvin S., Benvenuto A., de Palma L., Feucht M., Jansen F., Kotulska K., Sarnat H., Lagae L., Jozwiak S., Curatolo P. Updated clinical recommendations for the management of tuberous sclerosis complex associated epilepsy. Eur. J. Paediatr. Neurol. 2023 Aug 30;47:25–34. doi: 10.1016/j.ejpn.2023.08.005. (Epub ahead of print. PMID: 37669572) [DOI] [PubMed] [Google Scholar]
- 29.Sharma D., Lamba S., Pandita A., Shastri S. Syndrome de Klippel-trénaunay - un syndrome très rare et intéressant. Clin. Med. Insights Circ. Respir. Pulm. Med. 2015;9:1–4. doi: 10.4137/CCRPM.S21645. [ Article gratuit PMC ] [ PubMed ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banzic I., Brankovic M., Maksimović Ž., Davidović L., Marković M., Rančić Z. Syndrome de Parkes Weber – paradigmes de diagnostic et de gestion : une revue systématique. Phlébologie. 2017;32(6):371–383. doi: 10.1177/0268355516664212. [ PubMed ] [ Google Scholar ]. [DOI] [PubMed] [Google Scholar]
- 31.Legrand A., Devriese M., Dupuis-Girod S., Simian C., Venisse A., Mazzella J.M., Auribault K., Adham S., Frank M., Albuisson J., Jeunemaitre X. Frequency of de novo variants and parental mosaicism in vascular Ehlers-Danlos syndrome. Genet. Med. 2019 Jul;21(7):1568–1575. doi: 10.1038/s41436-018-0356-2. Epub 2018 Nov 26. PMID: 30474650. [DOI] [PubMed] [Google Scholar]
- 32.Mahfoudhi M., Khamassi K. Maladie de Rendu-Osler: un diagnostic à ne pas méconnaitre [Hereditary hemorrhagic telangiectasia: a diagnosis not to ignore] Pan Afr. Med. J. 2015 Sep 17;22:40. doi: 10.11604/pamj.2015.22.40.7871. 10.11604/pamj.2015.22.40.7871 French. PMID: 26664541; PMCID: PMC4662539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sudarsanam A., Ardern-Holmes S.L. Sturge-Weber syndrome: from the past to the present. Eur. J. Paediatr. Neurol. 2014 May;18(3):257–266. doi: 10.1016/j.ejpn.2013.10.003. Epub 2013 Nov 7. PMID: 24275166. [DOI] [PubMed] [Google Scholar]
- 34.Richard S., Parker F., Aghakhani N., Allegre G., Portier F., David P., Marsot-Dupuch K. Maladie de von Hippel-Lindau: progrès génétiques et cliniques récents [Von Hippel-Lindau disease: recent advances in genetics and clinical management] J. Neuroradiol. 2005 Jun;32(3):157–167. doi: 10.1016/s0150-9861(05)83133-5. French. PMID: 16134297. [DOI] [PubMed] [Google Scholar]
- 35.Michaud M., Mauhin W., Belmatoug N., Bedreddine N., Garnotel R., Catros F., Lidove O., Gaches F. Maladie de Fabry : quand y penser ? [Fabry disease: a review] Rev. Med. Interne. 2021 Feb;42(2):110–119. doi: 10.1016/j.revmed.2020.08.019. French. Epub 2020 Nov 7. PMID: 33172708. [DOI] [PubMed] [Google Scholar]
- 36.Oyedokun K., Agabna M.M., Israni A., du Plessis D. Meningioangiomatosis: an uncommon cause of focal epilepsy with characteristic neuroimaging and neuropathology. BMJ Case Rep. 2021 Jun 11;14(6) doi: 10.1136/bcr-2021-242953. PMID: 34117000; PMCID: PMC8201970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bersano A., Morbin M., Ciceri E., Bedini G., Berlit P., Herold M., Saccucci S., Fugnanesi V., Nordmeyer H., Faragò G., Savoiardo M., Taroni F., Carriero M., Boncoraglio Giorgio B., Perucca L., Caputi L., Parati Eugenio A., Kraemer M. The diagnostic challenge of Divry van Bogaert and Sneddon syndrome: report of three cases and literature review. J. Neurol. Sci. 2016 May 15;364:77–83. doi: 10.1016/j.jns.2016.03.011. Epub 2016 Mar 5. PMID: 27084221. [DOI] [PubMed] [Google Scholar]
- 38.Schwartz R.A., Fernández G., Kotulska K., Jóźwiak S. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J. Am. Acad. Dermatol. 2007 Aug;57(2):189–202. doi: 10.1016/j.jaad.2007.05.004. (PMID: 17637444) [DOI] [PubMed] [Google Scholar]
- 39.Timotin L., Sarrot-Reynauld F., Lantuejoul S., Pasquier B., Massot C., Ashraf A., Borgel F. Sclérose tubéreuse de Bourneville sans altération intellectuelle, diagnostiquée à l’âge adulte [Tuberous sclerosis without mental impairment, diagnosed in adulthood] Rev. Med. Interne. 2005 Jun;26(6):511–513. doi: 10.1016/j.revmed.2005.01.007. French. Epub 2005 Feb 12. PMID: 15936480. [DOI] [PubMed] [Google Scholar]
- 40.Uysal S.P., Şahin M. Tuberous sclerosis: a review of the past, present, and future. Turk. J. Med. Sci. 2020 Nov 3;50(SI-2):1665–1676. doi: 10.3906/sag-2002-133. PMID: 32222129; PMCID: PMC7672342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dzefi-Tettey K., Edzie E.K., Gorleku P., Piersson A.D., Cudjoe O. Tuberous sclerosis: a case report and review of the literature. Cureus. 2021 Jan 4;13(1) doi: 10.7759/cureus.12481. PMID: 33552794; PMCID: PMC7854338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ballanger F., Quereux G., Barbarot S. Manifestations dermatologiques des maladies d’organes. Springer; Paris: 2012. Sclérose tubéreuse de Bourneville. [DOI] [Google Scholar]
- 43.Aissi M., Younes-Mhenni S., Jerbi-Ommezzine S., Boughammoura-Bouatay A., Frih-Ayed M., Sfar M.H. Anévrismes intracrâniens et sclérose tubéreuse de Bourneville : une association rare [Tuberous sclerosis and intracranial aneurysms: a rare association] Rev. Neurol. (Paris) 2010 Nov;166(11):935–939. doi: 10.1016/j.neurol.2009.12.011. French. Epub 2010 May 15. PMID: 20472258. [DOI] [PubMed] [Google Scholar]
- 44.Caban C., Khan N., Hasbani D.M., Crino P.B. Genetics of tuberous sclerosis complex: implications for clinical practice. Appl. Clin. Genet. 2016 Dec 21;10:1–8. doi: 10.2147/TACG.S90262. PMID: 28053551; PMCID: PMC5189696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henske Elizabeth P., Jóźwiak Sergiusz, Kingswood J. Christopher, Sampson Julian R., Thiele Elizabeth A. Tuberous sclerosis complex. Nat. Rev. Dis. Primers. 2016;2:16035. doi: 10.1038/nrdp.2016.35. [DOI] [PubMed] [Google Scholar]