

Abstract
B-cell malignancies, such as chronic lymphocytic leukemia (CLL) and multiple myeloma (MM), remain incurable, with MM particularly prone to relapse. Our study introduces a novel mouse model with active RANK signaling and the TCL1 oncogene, displaying both CLL and MM phenotypes. In younger mice, TCL1 and RANK expression expands CLL-like B1-lymphocytes, while MM originates from B2-cells, becoming predominant in later stages and leading to severe disease progression and mortality. The induced MM mimics human disease, exhibiting features like clonal plasma cell expansion, paraproteinemia, anemia, and kidney and bone failure, as well as critical immunosurveillance strategies that promote a tumor-supportive microenvironment. This research elucidates the differential impacts of RANK activation in B1- and B2-cells and underscores the distinct roles of single versus combined oncogenes in B-cell malignancies. We also demonstrate that human MM cells express RANK and that inhibiting RANK signaling can reduce MM progression in a xenotransplantation model. Our study provides a rationale for further investigating the effects of RANK signaling in B-cell transformation and the shaping of a tumor-promoting microenvironment.
Subject terms: Cancer models, Lymphoproliferative disorders, Myeloma, Plasma cells
Introduction
B lymphocytes are vital components of the adaptive immune system, orchestrating responses to pathogens through antibody production and memory cell formation [1]. However, their dysregulation can lead to various hematological malignancies, including B-cell lymphomas and multiple myeloma (MM), highlighting the delicate balance between immune competence and oncogenesis [2, 3].
A critical player in B-cell malignancies is the NF-κB signaling pathway, essential for cell survival, differentiation, proliferation, inflammation, and immune regulation. Aberrant NF-κB activation, commonly observed in these malignancies, can result from interactions with the tumor microenvironment or mutations in upstream factors or inhibitors [4]. Within the TNF receptor superfamily (TNFRSF), the receptor activator of NF-κB (RANK; TNFRSF11A) has emerged as a significant factor in B-cell immunopathology [5–7]. Understanding how RANK activation interacts with other oncogenic factors in a lineage-specific manner to promote B-cell transformation remains incomplete, underscoring a broader challenge in deciphering the pathogenesis of B-cell-derived malignancies across different B-cell lineages.
Among these malignancies, chronic lymphocytic leukemia (CLL) and MM are prevalent and have rising incidences in Western countries [8]. Recurrent mutations in the RANK encoding gene TNFRSF11A, specifically RANKK240E, found in diffuse large B-cell lymphoma [5], lead to significant expansion in the B1-cell subset, innate-like, self-renewing B lymphocytes in mice. Upon aging, the RANK-driven B1-cells transform into a CLL-like disease. Furthermore, blocking RANK in both human CLL and the TCL1-driven CLL mouse model reduces disease progression in preclinical models, confirming the relevance of RANK signaling for CLL progression [9]. While murine CLL-like disease origins from B1-cells, MM arises from B2-cells, specifically from the terminally differentiated plasma cells responsible for producing antibodies [10–12]. In myeloma, these cells undergo malignant transformation, leading to uncontrolled proliferation and accumulation within the bone marrow and secretion of abnormal monoclonal antibodies.
The microenvironment of the bone marrow has been shown to play an important role in promoting tumorigenesis in multiple myeloma. Interaction of myeloma cells with bystander cells activates pro-survival and pro-proliferation signaling [13]. Various environmental cues within the bone marrow niche have the capacity to activate canonical and non-canonical NF-κB signaling and promote tumor progression [14]. B-cell activating factor (BAFF), a cytokine that induces non-canonical NF-κB activation, is thought to support multiple myeloma cell survival and to contribute to poor disease progression [15]. RANK signaling is equally able to activate the non-canonical NF-κB pathway, but its role in MM development or propagation is poorly understood to date. Research on myeloma has primarily focused on RANK’s role in promoting bone disease, where RANK ligand (RANKL) is secreted by myeloma and stromal cells to promote osteoclastogenesis [16]. While clinical data on the RANKL-blocking antibody denosumab suggests a potential relevance of RANK signaling in disease progression [17], its direct involvement in myeloma pathogenesis remains to be explored.
Furthermore, while CLL therapies often induce long-term responses by targeting B-cell receptor (BCR) signaling pathways or BCL2 [18, 19], MM treatments frequently face early relapse despite strategies to eliminate malignant plasma cells and modulate the bone marrow microenvironment to restore normal hematopoiesis and immune function [20]. This highlights the need for a deeper understanding of the factors contributing to myeloma formation, drug resistance, and bone marrow niche homing to overcome the limitations of existing therapeutic strategies.
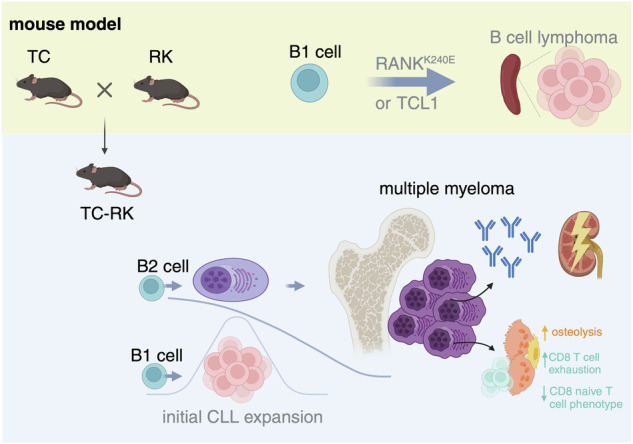
In our previous work we have shown that increased RANK signaling boosts differentiation towards the B1-cell stage which ultimately gives rise to CLL in aged animals [9]. Here we show that the simultaneous activation of RANK signaling and TCL1, a factor known to induce CLL, does not simply accelerate CLL development but enables the outgrowth of aggressive multiple myeloma. We find that mice with a simultaneous increase in RANK signaling and TCL1 expression initially develop B1-cell-derived CLL, which is however later suppressed by a more aggressive B2-cell-derived multiple myeloma outgrowth. The induced disease shows major features of human myeloma. Moreover, we demonstrate that human MM cells express RANK and that RANKL blockade can slow myeloma progression in a xenotransplantation mouse model. Combined, these findings demonstrate that depending on the signaling context RANK activation can give rise to malignancies originating from different B-cell subsets and provides a rationale to further explore possible tumor-promoting roles of RANKL expressing cells in the tumor-microenvironment of MM.
Results
Differential onset and progression of CLL and MM in a genetically induced mouse model
To investigate the impact of active RANK signaling on the acceleration of TCL-1 induced lymphomagenesis in vivo, we generated triple-transgenic Eμ-TCL1 RANKK240E CD19Cre mice (TC-RK) by crossing Eμ-TCL1 mice [21] (TC) with RANKK240E CD19Cre [9] (RK) mice. These TC-RK mice express the diffuse large B-cell lymphoma (DLBCL)-derived hyperactive RANKK240E human variant and enhanced green fluorescent protein (GFP) in B-cells, alongside TCL1, throughout B-cell development from immature to mature stages (Fig. 1a). Considering that both TC and RK mice models develop mature B-cell lymphoma originating from the B1-cell lineage, specifically chronic lymphocytic leukemia (CLL) [9, 21], we assessed B1-cell expansion in peripheral blood using CD19 and CD5 surface markers. As expected, we observed a significant increase in B1/CLL cells in TC-RK mice at 3 and 6 months, compared to mice expressing only a single transgene or the wild-type (including CD19Cre/+ littermate) controls (Fig. 1b).
Fig. 1. Active RANK and TCL1 signaling in B-cells causes a plasma cell disorder.

a Breeding scheme to generate TC-RK mice. Scheme was generated with BioRender.com. b Percentage of CD19+CD5+ of total viable lymphocytes in the peripheral blood of 12- and 24- week-old animals (n = 3–17 per genotype) determined by flow cytometry. Wildtype and CD19Cre animals are pooled as wt. Data was pooled from more than three experiments. c Kaplan-Meier overall survival analysis of TC-RK (n = 14), TC (n = 13), RK (n = 16) and wt (n = 17) control mice. For statistical analysis, p value of the log-rank test is shown. d Macroscopic appearance of representative spleens and lymph nodes (left) and dot plot graph (right) depicts spleen (SP) weight in gram (g) of diseased or aged mice with indicated genotypes (n = 4–11 per genotype). e Percentage of CD19+CD5+ of viable cells in the peripheral blood (PB, left, n = 5–11 per genotype) and SP (right, n = 5–13 per genotype) from animals with indicated genotypes upon signs of disease or aged mice. f Representative flow cytometric analysis of GFP and CD19 expression on viable splenocytes from a diseased TC-RK mouse. g Percentages of GFPhiCD19neg cells of viable cells from BM, SP, PB and LI isolated from diseased TC-RK mice (n = 6–13 per organ) compared to aged RK mice (n = 5–6 per organ). h Percentages of CD138+B220low cells of viable cells from bone marrow (BM) from diseased TC-RK mice and diseased or aged control mice (n = 3–10 per genotype). i Percentages of CD138+B220low cells of viable splenocytes cells from diseased TC-RK mice and diseased or aged control mice (n = 3–9 per genotype). j Percentages of CD138+B220low cells of viable cells from liver (LI) of diseased TC-RK mice and diseased or aged control mice (n = 3–6 genotype). k Representative histograms of forward scatter area (FSC-A), surface RANK, CD138, B220, IgM and IgD, as well as intracellular IRF4 and BLIMP1 from GFPhiCD19neg cells (green) compared to GFP+CD19+ cells (black) from TC-RK mice. l Representative images of CD138 immunohistochemistry from bone of a diseased TC-RK mouse (scale bars: overview = 2 mm, detailed image with 20x magnification = 200 μm). Statistical analysis was performed using Student’s t-test and the one-way ANOVA with Tukey correction for multiple comparison. The p values are indicated in respective graphs. All data are presented as mean ± standard deviation.
TC-RK mice demonstrated a significantly decreased lifespan, marked by severe symptoms including accelerated breathing, diminished activity, and pronounced hepato- and splenomegaly, as shown by enlarged organ size and weight (Fig. 1c, d; Supplementary Fig. 1a, b). Unexpectedly, flow cytometric analysis of terminally ill TC-RK mice revealed low levels of CD19+CD5+ cells in both peripheral blood and spleen (Fig. 1e), suggesting that CLL cells might not directly contribute to the observed morbidity and mortality. Intriguingly, these mice showed a marked increase in GFP-expressing, CD19-negative (GFPhiCD19neg) cells in the bone marrow, spleen, and liver, indicating a deviation from the expected CLL phenotype (Fig. 1f–g). Further characterization of this population revealed high CD138 expression likely representing terminally differentiated plasma cells (Fig. 1h–j). This assertion is further corroborated by their increase in cell size, low levels of surface immunoglobulins, IgM and IgD, and the detection of intracellular expression of IRF4 and BLIMP1, critical transcription factors for plasma cell differentiation (Fig. 1k, Supplementary Fig. 1c) [22–25]. Immunohistochemical analysis demonstrated that the bone marrow from TC-RK mice is characterized by focal plasma cell regions that disrupt normal hematopoiesis, closely mirroring the infiltration patterns observed in human myeloma (Fig. 1l). However, while normal plasma cells are typically confined to the bone marrow, their malignant counterparts in TC-RK mice infiltrated the spleen and liver, in some cases, the peripheral blood, mimicking the pathology of plasma cell leukemia in humans, which is associated with a poor prognosis [26, 27]. These results suggest that the pathology in TC-RK mice might closely resemble aggressive MM, with a significant expansion of plasma cells across various organs.
TC-RK-induced disease reflects clinical features of human multiple myeloma
To further investigate the potential characteristics of multiple myeloma in our mouse model, we performed serum protein electrophoresis to check for immunoglobulin secretion by plasma cells in TC-RK mice, a diagnostic and monitoring tool for plasma cell malignancies in clinical settings [28]. Remarkably, we observed distinct M-spikes exclusively in the serum of TC-RK mice, indicating the presence of monoclonal immunoglobulins (Fig. 2a). Further analysis of immunoglobulin isotypes revealed that the expanded plasma cell clones in TC-RK mice predominantly secreted IgM, IgG2b, or IgA (Fig. 2b), suggesting that both pre-germinal center and class-switched B-cells contribute to the clonally expanded plasma cell pool.
Fig. 2. TC-RK induced disease mirrors features of human multiple myeloma.

a Representative serum electrophoresis of immunoglobulins from TC-RK and control mice. b Representative quantification of immunoglobulin isotypes in plasma samples of five diseased TC-RK mice by flow cytometry-based multiplex immunoassay. c Red blood cell count (RBC) of diseased TC-RK mice and indicated controls (n = 3–6 per genotype). d Representative images of H&E staining from bone marrow of five-month-old RK and TC-RK mice (scale bars: overview = 600 μm, detailed images: 60 μm) e Representative images of H&E staining from kidneys of diseased TC-RK mice and indicated diseased or aged control mice (scale bars: 100 μm). f Kaplan-Meier OS analysis of Rag2ko mice (n = 13) transplanted with TC-RK splenocytes derived from four different sick donor mice compared to Rag2ko control mice. g Spleen weight of Rag2ko mice (n = 8) transplanted with TC-RK splenocytes at end point and aged-matched Rag2ko mice (n = 4). h Percentages of GFPhiCD19neg cells and GFP+CD19+ cells of viable cells from BM, SP, PB and LI isolated from diseased Rag2ko mice (n = 9–10 per organ) transplanted with TC-RK splenocytes derived from three different donor mice. Statistical analysis was performed using Student’s t-test and one-way ANOVA with Tukey correction for multiple comparison. P values are indicated in respective graphs and all data are presented as mean ± standard deviation.
We also assessed for symptoms typical of human myeloma in TC-RK mice, such as anemia, bone lesions, and kidney failure. At the onset of disease symptoms, TC-RK mice exhibited significantly lower red blood cell counts compared to controls (Fig. 2c). Bone histology in ill TC-RK mice revealed myeloma-like changes, including osteolytic lesions adjacent to multifocal plasma cell infiltrations and altered spongiotic architecture in the myeloma-infiltrated regions, as evaluated by a certified pathologist, confirming the induction of a myeloma-like disease in these mice (Fig. 2d). Kidney analysis showed severe renal disease characterized by tubular degeneration, intratubular protein aggregates, and the presence of intravascular tumor cells (Fig. 2e). Conversely, control and TC mice displayed normal kidney histology, while RK mice showed signs of mild to moderate protein accumulation, possibly mediated by autoimmune immunoglobulin aggregates [9]. Spleen architecture was also disrupted by the severe expansion of CD138/IRF4+ plasma cells (Supplementary Fig. 1d). To also test the disease-inducing potential of TC-RK-derived plasma cells, we injected TC-RK-derived splenocytes into Rag2KO mice, which are devoid of both B and T-cells, resulting in lethal disease in all recipients (Fig. 2f). These mice showed symptoms requiring euthanasia 5 to 10 weeks post transplantation and organ analysis revealed a significant increase in GFPhiCD19neg plasma cells in the bone marrow, spleen, and liver (Fig. 2g, h). Plasma cells were detectable in the blood only in cases where the donor mouse exhibited a leukemic phenotype. Bone marrow derived plasma cells were equally efficient in promoting disease burden (Supplementary Fig. 1e, f).
These findings collectively demonstrate that TC-RK mice exhibit a clonal plasma cell expansion highly similar to human myeloma with full penetrance by 7-months of age. This malignancy impacts bone niches, leading to anemia, osteolytic changes, and significant renal damage. Importantly, this disease is transplantable from affected to unaffected mice, underscoring its value as a model for studying the characteristics of multiple myeloma.
B2 B-cells are the cell-of-origin of TC-RK driven multiple myeloma
Observing CLL-like B1-lymphocyte expansion in younger TC-RK mice prior to the overt onset of myeloma, we next aimed to explore the relationship between CLL and plasma cell populations. Given that myeloma typically originates from CD5-negative, post-germinal center B2-cells, whereas murine CLL is believed to arise from the B1-lineage that express CD5 [12], our goal was to examine the impact of the B1-cell compartment on myeloma development. We investigated this by deleting CD19 through homozygous CD19Cre/Cre expression (CD19KO mice) [29], which substantially reduces the B1-cell population [30]. We first verified that CD19 deletion halted the development of both CLL and B1-cells in TC-RKCD19KO mice in the spleen and the blood, while the B2-cell development was less affected, still comprising approximately 20% of peripheral blood cells (Fig. 3a, b). Despite the lack of B1/CLL cell expansion in CD19KO mice, expression of TC-RK still resulted in significant plasma cell expansion and transformation. These effects were comparable to those observed in heterozygous CD19-expressing TC-RK mice (Fig. 3c, d). The malignant phenotype of the plasma cells derived from CD19KO TC-RK mice was further validated by their successful transplantation into Rag2KO mice, leading to rapid disease onset (Fig. 3e, f). This highlights that TC-RK specifically facilitates myeloma development within the B2-cell lineage, irrespective of CLL formation and the B1-lineage. This finding was further corroborated by transplanting sorted B2 (CD19+CD5neg) bone marrow cells from young TC-RK mice into Rag2KO mice. Again, these TC-RK-derived B2-cells progressed into myeloma in the absence of B1-cells and CLL (Fig. 3g), confirming the myeloma development is driven in B2-cells by TC-RK, independent of B1-cell involvement.
Fig. 3. Origin of myeloma cells in TC-RK driven mouse models.

a Representative flow cytometric analysis of CD5 and CD19 expression on B220+ splenocytes from mice with indicated genotypes. b Percentage of B220+CD5+ and B220+CD5neg cells of total viable lymphocytes in the peripheral blood from animals with indicated genotypes (n = 2–6 per genotype) determined by flow cytometry. Pooled data from three different experiments. c Kaplan-Meier OS analysis of TC-RKCD19KO (n = 4) mice compared to TC-RK (n = 13) and RKCD19KO (n = 3) and CD19KO (n = 5) mice. d Percentages of CD138+B220low and B220+CD138neg cells of viable cells from BM, SP and LI isolated from diseased TC-RKCD19KO mice (n = 3). e Kaplan-Meier OS analysis of Rag2ko mice (n = 6) transplanted with TC-RKCD19KO splenocytes derived from two different sick donor mice compared to Rag2ko control mice. f Percentages of CD138+B220neg cells and B220+CD138neg cells of viable cells from BM, SP and LI isolated from diseased Rag2ko mice (n = 6) transplanted with TC-RKCD19KO splenocytes derived from two different donor mice. g Percentages of GFPhiCD19neg and GFP+CD19+ cells of viable splenocytes isolated from Rag2ko mice (n = 3) after transplantation of sorted CD19+CD5neg BM cells from three different 3-month-old TC-RK donor mice. h Graphical summary of B2-cells giving rise to MM cells and B1-cells giving rise to CLL cells in TC-RK mice. Graphic was generated with BioRender.com. Statistical analysis was performed using Student’s t-test and one-way ANOVA with Tukey correction for multiple comparison. P values are indicated in respective graphs and all data are presented as mean ± standard deviation.
Therefore, our analysis reveals that TC-RK uniquely fosters myeloma originating from B2-cells, separate from the CLL phenotype (Fig. 3h). This underscore specific B2-cell-intrinsic mechanisms as pivotal for myeloma genesis and this delineation between the pathways leading to CLL and myeloma highlights the complexity of oncogenic events driving B-cell malignancies.
Mechanistic insights into the role of RANK and TCL1 in driving myeloma formation
To further elucidate myeloma development in the TC-RK mouse model, we examined the effects of transgene expression on B-cell differentiation and maturation in young mice. At six weeks, RANK and TCL1 transgenes did not significantly impact B-cell precursor development in the bone marrow (Supplementary Fig. 2a). However, significant increases in marginal zone B-cells and B1-cells were observed in the spleens of RK and TC-RK mice compared to TC and wt controls (Supplementary Fig. 2b, c). Notably, RK and TC-RK mice also showed a higher proportion of plasma cells in the spleen, though not in the bone marrow, than their TC and wt counterparts (Fig. 4a), suggesting that RANK transgene expression alone might promote splenic plasma cell differentiation. We therefore sought to assess the intrinsic potential for plasma cell differentiation of B-cells in vitro. Naive splenic B-cells (excluding B1-cells) from TC-RK and control mice were stimulated ex vivo (Fig. 4b). At low LPS concentrations in the absence of IL4, insufficient to induce plasma cell differentiation in wt and TC mice [31], B-cells from RK and TC-RK mice successfully differentiated into plasma cells and secreted higher levels of IgM, IgG2b, IgG1, and IgA (Fig. 4c, d; Supplementary Fig. 2d). This suggests that RK expression may mediate a non-canonical pathway to plasma cell differentiation and isotype switching, reducing the requirement for IL-4 signaling. The differentiation potential in RK-expressing B-cells was less pronounced following optimal combined LPS and IL-4 stimulation (Supplementary Fig. 2e, f). We also tested whether suboptimal levels of T-cell-dependent stimuli for plasma cell differentiation could cooperate with active RANK signaling, hypothesizing that active RANK signaling reduces the requirement for BCR/CD40 co-stimulation to initiate a comprehensive B-cell activation program for plasma cell generation. However, neither suboptimal BCR stimulation nor anti-CD40 stimulation alone induced meaningful plasma cell differentiation in RK-expressing B-cells (Supplementary Fig. 2g, h). This suggests that low levels of LPS, but not BCR or CD40 signaling, are sufficient to cooperate with RK signaling to trigger plasma cell differentiation in TC-RK mice.
Fig. 4. Active RANK signaling drives a plasma cell differentiation program via Blimp1 expression.

a Percentages of CD138+B220low plasma cells of viable cells from SP (left) and BM (right) of six-week-old TC-RK (n = 5) and control mice (n = 4–5 per genotype). Pooled data from three different experiments. b Experimental set up of in vitro differentiation of naive B-cells using low concentrations of LPS (LPSlow, 100 ng/mL). B-cell stage and immunoglobulins in supernatants were analyzed by flow cytometry four days after stimulation. Graphic was created using BioRender.com. c Percentages of CD138+B220low plasma cells after in vitro differentiation of naive B-cells derived from animals with indicated genotype (n = 4–5 per genotype) with LPSlow were determined by flow cytometry after four days. Biological replicates were pooled from two individual experiments. d Quantification of immunoglobulin isotypes in supernatants from in vitro differentiated naive B-cells, derived from animals with indicated genotype (n = 3–5 per genotype) and stimulated for four days with LPSlow, by flow cytometry-based multiplex immunoassay. Pooled data from two independent experiments. e Experimental set up of in vitro differentiation of naive B-cells derived from TC-RK mice using recombinant RANKL in presence or absence of LPSlow for four days. f Total TC-RK-derived B-cell count (n = 4) per well after in vitro stimulation with RANKL and LPSlow as indicated. Biological replicates were pooled from two individual experiments. g TC-RK-derived CD138+B220low plasma cell count (n = 4) per well after in vitro differentiation using RANKL and LPSlow as indicated. Biological replicates were pooled from two individual experiments. h Geometric mean (geo. mean) of BLIMP1 from living cells after in vitro differentiation of TC-RK-derived B-cells (n = 4) using RANKL and LPSlow as indicated. Biological replicates were pooled from two individual experiments. i Macroscopic appearance of representative spleens (left) and dot plot graph (right) depicts SP weight in gram (g) of 20-week-old TC-RK (n = 10) and aged-matched control mice (n = 7–9 per genotype). j Percentages of CD138+B220low plasma cells of viable cells from SP (left) and BM (right) from 20-week-old TC-RK (n = 6–7) and aged-matched control mice (n = 6–10 per genotype). Pooled data from more than three experiments. Statistical analysis was performed using Student’s t-test and one-way ANOVA with Tukey correction for multiple comparison. P values are indicated in respective graphs. All data are presented as mean ± standard deviation.
As TLR-mediated signaling seems critical to mediate the full differentiation to plasma cells in RK-expressing B-cells, we next analyzed the effects of recombinant murine RANKL on the viability of LPS-stimulated, naive TC-RK-derived B-cells and found a significant boost in B-cell population viability, with a notable, though not statistically significant, increase in plasma cell numbers (Fig. 4e–g). The presence of RANKL also elevated BLIMP1 protein levels [32], crucial for terminal B-cell differentiation, irrespective of LPS presence (Fig. 4h), emphasizing the essential role of RANK in promoting B-cell viability and differentiation into the plasma cell lineage, thus facilitating cellular transformation in aged mice. To gain further insight into signals regulated by RANK activation and how RANK signaling converges with BCR stimulation, we used RANK-overexpressing murine BAL17 lymphoma B-cell lines to dissect pathway activation following acute RANK and BCR signaling. We found that RANKL stimulation activated the JNK and NF-κB pathways in RANK-overexpressing BAL17 cell lines, while BCR activation led to rapid SRC/PLCγ2/AKT/ERK pathway activation. Simultaneous activation of RANK and BCR signaling resulted in the activation of both pathways but did not show significant enhancement in our model (Supplementary Fig. 3a, b). This suggests that dual RANK/BCR stimulation cooperates but do not potentiate differential signaling events early after activation, eventually driving plasma cell differentiation in our model.
To further track plasma cell expansion in vivo, we analyzed five-month-old mice. At this age, TC-RK mice displayed significantly enlarged spleens and increased plasma cell populations in both the spleen and bone marrow, the latter being the primary expansion site for malignant myeloma cells (Fig. 4i, j). Together with our in vitro analysis, this suggests that active RANK signaling is vital for initiating plasma cell differentiation, while the concurrent expression of TCL1 markedly intensifies this effect in vivo, leading to rapid and extensive plasma cell expansion within the bone marrow niche. This prompted us to analyze specific alterations within the bone marrow that contribute to this progression, thereby deepening our understanding of the mechanisms driving myeloma pathogenesis.
Transcriptional alterations within the bone marrow niche upon TC-RK-mediated myeloma progression
To elucidate the changes within the bone marrow niche that facilitate myeloma cell proliferation and potentially evade immune responses, we performed single-cell RNA sequencing on bone marrow-derived cells from five-month-old TC-RK and RK mice. Our analysis revealed a pronounced shift in cell population dynamics in the bone marrow of TC-RK mice, with a marked increase in plasma cells identified by Sdc1 (CD138), Slamf7, and Tnfrsf17 (BCMA) expression and loss of Cd19, at expense of nearly all other immune cell types (Fig. 5a–c). Further detailed analysis within the B-cell/plasma cell population showed a diminished presence of B-cells marked by Cd19 and Pax5 expression in TC-RK mice, in stark contrast to the prevalent CD138/Sdc1+ plasma cells, also characterized by the expression of genes associated with myeloma, such as Slamf7, Tnfrsf17 (BCMA)—a target for CAR-T-cell therapy—along with Cd74, Mzb1, Enpp1, and the autophagy gene Sqstm1 (p62) (Fig. 5d). Furthermore, we detected high expression of the BLIMP1-encoding Prdm1 gene and other genes associated with plasma cells, such as Jchain and Xbp1 within the expanded plasma cell cluster (Fig. 5e). Additional B-cell (Pax5, Ebf1) and plasma cell markers (Mzb1, Ptprclow) as well as plasma cell scores are depicted in Supplementary Fig. 4a, b. Examination of the T-cell compartment exposed an increase in markers linked to T-cell activation/exhaustion (Icos, Tnfrsf9, Pdcd1, Tigit, Tox) and cytotoxicity (Ifng, Gzmb, Gzmk; Fig. 5f), with gene set enrichment analysis reinforcing the presence of an exhausted, effector phenotype among cytotoxic T-cells (Fig. 5g; scores depicted in Supplementary Fig. 4c). Therefore, our single-cell transcriptional profiling not only highlights a substantial expansion of the plasma cell lineage, enriched with myeloma-associated gene expressions within the bone marrow niche, but also underscores the presence of a significant activated and exhausted T-cell phenotype, reflecting potential roles in myeloma progression.
Fig. 5. Landscape of the bone marrow microenvironment in TC-RK mice.

a Uniform manifold approximation and projection (UMAP) visualizations of unsupervised clustering analysis of all cells that passed quality filtering. Cells are colored according to their genotype in the upper row and according to their Sdc1, Cd19, Tnfrsf17 and Slamf7 expression respectively below. b UMAP visualization of annotated clusters (left) and frequency of clusters (right) in RK (pooled from 3 mice) and TC-RK (pooled from 4 mice) derived BM cells. Plasma cells are highlighted with a green circle in the UMAP. c Differential abundance (DA) analysis for changes in cluster abundance (left) and log-fold change for each annotated cluster (right). Plasma cell cluster is highlighted by a green circle (left) or a green arrow (right). d Gene expression dot plot depicts the percent and average expression of B-cell markers within the B and plasma cell subcluster. e UMAP visualization of unsupervised clustering analysis of B-cells and plasma cells (defined by Sdc1 and Slamf7 expression). Cells are color coded according to their genotype (left) or according to Sdc1, Jchain, Prdm1 and Xpb1 expression respectively (right). f Gene expression profile visualize the percent and average expression of T-cell markers within the T-cell cluster. g Gene set enrichment analysis (GSEA) plot for CD8 T-cell exhaustion enriched T-cells from TC-RK mice (top) and GSEA plot of naive CD8 T-cells enriched in T-cells of RK mice (bottom).
Comparative analysis of TC-RK induced with myeloma of other murine and human origin
To determine whether our new myeloma model shares additional features to previously described models, we next compared the transcriptional profile of (pre-)malignant plasma cells of TC-RK mice with those of RK mice at five months age using bulk RNA sequencing analysis and subsequently performed gene set enrichment analysis (Fig. 6a). This revealed upregulation of several pathways, most of which were previously identified in both newly developed murine myeloma models and in human myeloma [33]. These include upregulation of E2F transcription factor-related gene expression and a MYC signature, pathways that have recently been identified as responsive to targeted therapies in myeloma [33]. Notably, we also observed an enhanced unfolded protein response (UPR) pathway, presumably triggered by the substantial immunoglobulin production in TC-RK plasma cells. A detailed cross-comparison with other myeloma models [33] showed pronounced parallels (Fig. 6b, Supplementary Fig. 4a), most notably with the model induced by KRASG12D in conjunction with BCL2 overexpression, which exhibited the highest similarity (p = 1.8 × 10−246; odds ratio (OR) = 3.7). Furthermore, when comparing the transcriptomic profiles of TC-RK induced myeloma with human myeloma cases, there was a notable correspondence, particularly in the upregulation of myeloma-associated genes such as Birc5, Ezh2, Prc1, and Foxm1 (p = 1.2 × 10−5; OR = 1.7), and in the downregulation of genes like Cd19, Ctsh, and Tmsb4x (p = 1.7 × 10−4; OR = 1.6; Supplementary Table 1) [34, 35].
Fig. 6. Comparative analysis between TC-RK induced myeloma and both other murine MM mouse models and human disease.

a GSEA analysis showing hallmarks enriched in splenic plasma cells of TC-RK mice compared to plasma cells of RK mice. NES, normalized enrichment score. b Venn diagram depicts the overlap of deregulated genes in myeloma cells from Kras-BIγ1 mice [33] and TC-RK mice. Fisher’s exact test was applied and both p value and odd ratio are depicted. Illustrations were generated using BioRender.com. c Representative copy number variation patterns of myeloma cells from TC-RK mice (n = 7). d Representative immunohistochemistry staining of primary patient biopsies confirmed RANK expression in myeloma cells. Illustrations were generated using BioRender.com. e Treatment schedule of xenograft model with anti-RANKL antibody prior and after injection of L363 in NSG mice (top) and Kaplan-Meier survival analysis (bottom) is shown for NSG mice received anti-RANKL (n = 3) or vehicle (n = 4) therapy prior and after L363 transplantation (Tx L363). Statistical significance with corresponding p values calculated by log-rank (Mantel-Cox) test is depicted in the graph. Graph represents one out of three independent experiments.
Using whole exome sequencing, we discovered that a substantial proportion of the mutated genes in TC-RK myeloma cells, specifically 44% (72 out of 164), have previously been identified in human myeloma datasets [36] (p = 1.7 × 10−6; OR = 2.1), underscoring a notable genetic similarity between our model and human myeloma. Additionally, 23% of the mutations in our study (p = 3.1 × 10−10; OR = 3.8) were found in genes that are recurrently mutated in human myeloma (defined as a mutation present in at least 2 patients [36]) including the BCL6 interacting factor Dnah9 [37, 38] and the immunomodulating factor Irf2bp2 (Supplementary Table 2) [39, 40]. These findings suggest a significant overlap in the mutational landscape between our model and the genetic alterations observed in human myeloma, reinforcing the relevance of our model in the context of human disease. We also explored copy number variations (CNV) in our dataset, revealing two mutually exclusive CNV patterns previously identified in murine myeloma models [33, 41]. Specifically, we observed recurrent gains in chromosomes 7 and 17 in five out of seven TC-RK-derived myelomas [33]. The remaining two mice demonstrated a loss of chromosome 5 (Fig. 6c), consistent with other reports in the myeloma literature [33, 41]. While the precise impact of these chromosomal changes warrants further investigation, their presence in our model and others confirms overlapping disease evolutions.
Given the parallels between our TC-RK model and human myeloma, we explored the potential role of RANK signaling in human myeloma. Analysis of bone marrow immunohistochemistry from myeloma patients revealed stable RANK expression in most cases (Fig. 6d; Quantification in Supplementary Fig. 4b). We tested the functional significance of RANK signaling by treating L363-xenotransplanted mice, finding that early RANKL blockade significantly slowed myeloma progression and extended disease-free survival in this aggressive model (Fig. 6e). These findings underscore a potential relevance of B-cell intrinsic RANK in human myeloma and clarify the previously suggested anti-myeloma effects of denosumab, which targets RANKL in the treatment of bone disease [42].
Our findings collectively demonstrate that active RANK signalling in combination with TCL1 initially promote CLL expansion which is predominated by myeloma formation that accurately mirrors key aspects of the human disease. Characterized by a rapid onset of myeloma within seven months and complete penetrance, this model stands as a significant advancement in simulating critical disease features. Its efficiency and reliability offer an invaluable tool for further exploring immunosurveillance processes and possible resistance mechanisms in myeloma, underscoring its potential to significantly impact future research and therapeutic strategies.
Discussion
In this study, we introduce a novel mouse model that reveals an unexpected phenotype of initial CLL expansion followed by aggressive myeloma formation through constitutive RANK signaling and TCL1 overexpression. This model accurately reflects the phenotypic, transcriptional, and genomic hallmarks of other murine myeloma models [33], while overcoming limitations of previous models that exhibited delayed onset and inconsistent disease development [43–45]. Our model not only provides insights into the complex consequences of lineage-specific oncogene expression but also serves as a valuable tool for precise monitoring of immunosurveillance dynamics and evaluating potential therapeutic interventions. Furthermore, our data suggest a significant contribution of RANK signaling to the pathology of human multiple myeloma, underscoring the potential for targeted therapies in this context.
We demonstrate that oncogenes driving B-cell lymphoma, specifically TCL1 and the mutant RANKK240E, synergize to initially drive CLL progression, yet invariably shift towards a myeloma phenotype originating from the B2-cell lineage. The interplay between specific oncogenes and the cellular context in which they are expressed plays a critical role in the pathogenesis of malignancies. Our findings underscore that the manifestation of disease can vary significantly depending on the cell type harbouring the oncogene and the presence of additional oncogenic events. For instance, in our study, the expression of TCL1 and active RANK in B2-cells specifically led to the development of multiple myeloma, independent of the CLL-like phenotype observed with similar oncogene expression in B1-cells.
Mechanistically, we discovered that low LPS levels are sufficient to induce plasma cell differentiation in RK- and TC-RK-derived B2-cells, but not wild type B-cells. In contrast, BCR signaling alone was insufficient to drive RK-expressing B-cells into the plasma cell lineage.
LPS activates B-cells via TLR4 in a polyclonal manner and induces plasma cell differentiation in vitro. In contrast, anti-BCR stimulation in vitro leads to B-cell activation and proliferation but does not promote differentiation of wild-type B-cells into plasma cells. We chose these stimuli to test whether RK- or TC-RK expression amplifies plasma cell differentiation signals or replaces them. Since we did not observe plasma cell differentiation upon BCR activation in TC-RK expressing B-cells, we conclude that TC-RK expression lowers the threshold for signals needed to commit to the plasma cell fate but is not sufficient to replace these signals. Additionally, we stimulated CD40 and assessed plasma cell differentiation. In vivo, B-cells are exposed to CD40L during immune responses to thymus-dependent antigens, inducing pro-survival signaling. In vitro, CD40 stimulation alone induces low levels of plasma cell differentiation, but the signal is substantially weaker than LPS stimulation. For comparison, the frequency of plasma cells in wild-type B-cells stimulated with suboptimal LPS levels was around 5%, but only 1% with CD40 stimulation. TC-RK expressing B-cells showed a trend towards increased plasma cell differentiation compared to B-cells expressing TC alone; however, overall plasma cell differentiation remained low. This suggests that while TC-RK signaling can enhance plasma cell differentiation upon various stimulations, the strength of the differentiation signal is crucial for the final outcome.
Consistent with our functional assays, we found that RANK signaling does not alter the quality of BCR signaling but provides the cells with additional pro-survival signals. To study RANK induced signaling we have transduced BAL17 B lymphoma cells with the wildtype and constitutively active form of RANK (RANKwt and RANKK240E, respectively). We chose BAL17 cells since they do not express RANK and BAL17 cells transduced with an empty vector can thus serve as a negative control for RANK-mediated signaling. Treatment with RANKL induced phosphorylation of the NF-κB pathway signaling molecule IKKα/β as well as JNK phosphorylation in both RANKwt- or RANKK240E- expressing cells but not BAL17 cells transduced with an empty vector. Activation of NF-κB is critical for driving human myeloma progression [14]. In mouse models, active IKKβ in combination with pro-survival signals has been found to drive multiple myeloma [33], highlighting the importance of NF-κB activation in driving the disease. Active JNK phosphorylates nuclear transcription factors such as c-Jun and ATF2, leading to the transcription of target genes that protect myeloma cells from apoptosis [46]. Additionally, JNK activation by BLyS, another TNFR superfamily member, creates a positive feedback loop that enhances cell survival and proliferation in myeloma-derived plasma cells [47]. In summary RANK signaling increases critical downstream effectors with known relevance in driving myeloma formation.
Importantly we found, the phosphorylation of IKK and JNK to be higher in cells expressing RANKK240E rather than RANKwt. This suggests that while wildtype RANK can provide B-cells with pro-survival signaling, the hyperactive form is more efficient in doing so. Consistent with our signaling studies, our functional experiments indicate that wild-type RANK provides multiple myeloma cells with survival signals, as blocking RANK signaling in multiple myeloma xenotransplantation models reduces tumor burden. To test whether RANK activation modulates BCR dependent signaling, we stimulated the cells with an anti-BCR antibody alone or in combination with RANKL treatment. Early events of BCR activation such as SRC/PLCγ/AKT and ERK phosphorylation were not altered by RANKwt- or RANKK240E expression, suggesting no direct regulation of BCR signaling via RANK.
Therefore, our study demonstrates that RANK hyperactivation increases pro-survival signaling via NF-κB and JNK and amplifies signals inducing plasma cell differentiation. Previous studies have shown that RANK is not required for the activation, proliferation, and differentiation of normal B-cells [48]. Our findings reveal that RANK hyperactivation boosts plasma cell differentiation in vitro. This suggests that while RANK is not essential, it can enhance plasma cell differentiation. In the context of malignant transformation, RANK hyperactivation alone can drive CLL development in a mouse model [9]. However, multiple myeloma only develops in combination with additional pro-survival signals, such as those provided by TCL1. Our signaling studies indicate that RK and TC expression alone is insufficient to induce plasma cell development without differentiation signals. Thus, we hypothesize that RANK lowers the threshold for both survival and plasma cell differentiation. B1-cells are known to exhibit increased survival properties compared to B2-cells, making RANK expression alone sufficient to drive their malignant transformation into CLL. In contrast, transforming B2-cells may require additional survival signals, provided by TCL1 in our model. A key question remains why TC-RK induces multiple myeloma rather than DLBCL, where RANKK240E expression was discovered [5]. Our studies demonstrate that Blimp1 expression and plasma cell differentiation are enhanced by RANKK240E upon plasma cell differentiation stimuli. Future research should investigate whether low levels of TLR ligand exposure, e.g., from gut microbiota, initiate multiple myeloma development in this mouse model. Additionally, it would be interesting to assess whether inhibiting plasma cell differentiation by deleting Blimp1 in our mouse model would result in DLBCL rather than multiple myeloma development.
Interestingly, the transition to the severe myeloma phenotype is marked by a striking absence of CLL cells, suggesting competition for growth and/or survival limiting factors such as nutrients [49] or cytokines like BAFF, potentially facilitated through BCMA expression and its shedding [50], a hypothesis that warrants further exploration. This observation suggests a complex interplay of cellular and molecular factors influencing disease progression and highlights not only the cell-specific actions of oncogenes but also suggests that the co-occurrence of other molecular abnormalities may amplify or modify the oncogenic potential of a given gene. This complexity is crucial for understanding the full spectrum of oncogene-driven pathologies and indicates that targeted therapies must consider not just the oncogenic drivers but also the cellular and molecular context of their expression.
Emerging evidence, from our study and beyond, reinforces the potential of the RANKL-blocking antibody denosumab, not only for its bone-protective properties in late-stage myeloma, but also for its potential anti-myeloma effects. Recent clinical studies demonstrate that denosumab therapy is associated with significantly better progression-free survival rates over those receiving zoledronic acid, which is similar effective in preventing bone loss [17]. Our findings, alongside other preclinical research [51–53], highlight the importance of B-cell intrinsic RANK signaling in the progression of myeloma and support denosumab’s capability to interfere in these processes. This insight is particularly relevant for the ongoing denosumab clinical trial in smoldering myeloma (Eudra-CT 2018-000924-32) and future applications of combination treatments. Notably, our model also highlights high expression of the scaffolding protein SQSTM1/p62, a factor promoting RANK signalling [54] but also conferring resistance to proteasome inhibitors in myeloma [55, 56]. Further insight into the regulation of p62 and RANK signaling may unveil novel strategies to counteract drug resistance in myeloma.
In conclusion, our novel mouse model, combining constitutive RANK signaling with TCL1 expression in B-cells, not only promotes CLL formation but also mirrors aggressive forms of human myeloma and thereby for the first time highlights a possible role of this signaling pathway in a cell-intrinsic manner in MM development or propagation. The bone marrow niche is known to play a pro-tumorigenic role in multiple myeloma and our data highlight a new signaling node amenable to medical intervention. Our findings pave the way for further research into the mechanisms of myeloma development and treatment, with the potential to significantly improve patient outcomes.
Material and methods
For Material and Methods please refer to Supplementary Data.
Supplementary information
Acknowledgements
This work was funded by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) Sachbeihilfe Project-ID 516923543 and FOR5560 Project-ID 505372148 to MB and Project BA 2851/6-1 and BA 2851/7-1 to FB, the German Cancer Aid (Deutsche Krebshilfe, Max Eder Grant to MB, Project ID: 70114720); and by the Wilhelm Sander Foundation (Project ID 2018.111.1 to MB). We thank Marc Schmidt-Supprian for helpful discussions and the biobank tissue (IBioTUM tissue) and the Comparative Experimental Pathology (CEP) of the School of Medicine and Health, TUM for excellent technical support.
Author contributions
Lisa Pfeuffer: Data acquisition, methodology, analysis, manuscript preparation, conceptualization; Viola Siegert: Data acquisition; Julia Frede: Data methodology, analysis, Leonie Rieger: Data acquisition, methodology, analysis; Riccardo Trozzo & Niklas Andrade-Kraetzig: Data analysis; Sandra Ring: Data acquisition, methodology; Shamim Sarhadi & Christof Winter: Data methodology, analysis; Nicole Beck & Stefan Niedermeier & Mar Abril-Gil: Data acquisition; Mohamed Elbahloul: Data acquisition and methodology; Marianne Remke: Data acquisition, methodology, analysis; Katja Steiger: Data acquisition, methodology, analysis, Ruth Eichner: conceptualization; Julia Jellusova: conceptualization; Roland Rad: Data methodology; Florian Bassermann: provided guidance and helpful discussion; Jürgen Ruland: provided guidance and helpful discussion; Maike Buchner: Conceptualization, project administration, funding acquisition, and manuscript preparation. All authors discussed the results and contributed to the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Data availability
For original data, please contact maike.buchner@tum.de.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41408-024-01123-6.
References
- 1.LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112:1570–80. 10.1182/blood-2008-02-078071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–30. 10.1016/j.ccr.2007.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gasparini C, Celeghini C, Monasta L, Zauli G. NF-kappaB pathways in hematological malignancies. Cell Mol Life Sci. 2014;71:2083–102. 10.1007/s00018-013-1545-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim KH, Yang Y, Staudt LM. Pathogenetic importance and therapeutic implications of NF-kappaB in lymphoid malignancies. Immunol Rev. 2012;246:359–78. 10.1111/j.1600-065X.2012.01105.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459:717–21. 10.1038/nature07968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Secchiero P, Corallini F, Barbarotto E, Melloni E, di Iasio MG, Tiribelli M, et al. Role of the RANKL/RANK system in the induction of interleukin-8 (IL-8) in B chronic lymphocytic leukemia (B-CLL) cells. J Cell Physiol. 2006;207:158–64. 10.1002/jcp.20547 [DOI] [PubMed] [Google Scholar]
- 7.Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–24. 10.1101/gad.13.18.2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cowan AJ, Allen C, Barac A, Basaleem H, Bensenor I, Curado MP, et al. Global Burden of Multiple Myeloma: A Systematic Analysis for the Global Burden of Disease Study 2016. JAMA Oncol. 2018;4:1221–7. 10.1001/jamaoncol.2018.2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alankus B, Ecker V, Vahl N, Braun M, Weichert W, Macher-Goppinger S, et al. Pathological RANK signaling in B cells drives autoimmunity and chronic lymphocytic leukemia. J Exp Med. 2021;218:e20200517. 10.1084/jem.20200517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seifert M, Sellmann L, Bloehdorn J, Wein F, Stilgenbauer S, Durig J, et al. Cellular origin and pathophysiology of chronic lymphocytic leukemia. J Exp Med. 2012;209:2183–98. 10.1084/jem.20120833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barwick BG, Gupta VA, Vertino PM, Boise LH. Cell of Origin and Genetic Alterations in the Pathogenesis of Multiple Myeloma. Front Immunol. 2019;10:1121. 10.3389/fimmu.2019.01121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayakawa K, Formica AM, Brill-Dashoff J, Shinton SA, Ichikawa D, Zhou Y, et al. Early generated B1 B cells with restricted BCRs become chronic lymphocytic leukemia with continued c-Myc and low Bmf expression. J Exp Med. 2016;213:3007–24. 10.1084/jem.20160712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hideshima T, Anderson KC. Signaling Pathway Mediating Myeloma Cell Growth and Survival. Cancers (Basel). 2021;13:216. 10.3390/cancers13020216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demchenko YN, Kuehl WM. A critical role for the NFkB pathway in multiple myeloma. Oncotarget. 2010;1:59–68. 10.18632/oncotarget.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fragioudaki M, Tsirakis G, Pappa CA, Aristeidou I, Tsioutis C, Alegakis A, et al. Serum BAFF levels are related to angiogenesis and prognosis in patients with multiple myeloma. Leuk Res. 2012;36:1004–8. 10.1016/j.leukres.2012.03.012 [DOI] [PubMed] [Google Scholar]
- 16.Raje NS, Bhatta S, Terpos E. Role of the RANK/RANKL Pathway in Multiple Myeloma. Clin Cancer Res. 2019;25:12–20. 10.1158/1078-0432.CCR-18-1537 [DOI] [PubMed] [Google Scholar]
- 17.Terpos E, Raje N, Croucher P, Garcia-Sanz R, Leleu X, Pasteiner W, et al. Denosumab compared with zoledronic acid on PFS in multiple myeloma: exploratory results of an international phase 3 study. Blood Adv. 2021;5:725–36. 10.1182/bloodadvances.2020002378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl J Med. 2016;374:311–22. 10.1056/NEJMoa1513257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woyach JA, Johnson AJ, Byrd JC. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood. 2012;120:1175–84. 10.1182/blood-2012-02-362624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chauhan D, Singh AV, Brahmandam M, Carrasco R, Bandi M, Hideshima T, et al. Functional interaction of plasmacytoid dendritic cells with multiple myeloma cells: a therapeutic target. Cancer Cell. 2009;16:309–23. 10.1016/j.ccr.2009.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bichi R, Shinton SA, Martin ES, Koval A, Calin GA, Cesari R, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci USA. 2002;99:6955–60. 10.1073/pnas.102181599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. 2003;19:607–20. 10.1016/S1074-7613(03)00267-X [DOI] [PubMed] [Google Scholar]
- 23.Angelin-Duclos C, Cattoretti G, Lin KI, Calame K. Commitment of B lymphocytes to a plasma cell fate is associated with Blimp-1 expression in vivo. J Immunol. 2000;165:5462–71. 10.4049/jimmunol.165.10.5462 [DOI] [PubMed] [Google Scholar]
- 24.Falini B, Fizzotti M, Pucciarini A, Bigerna B, Marafioti T, Gambacorta M, et al. A monoclonal antibody (MUM1p) detects expression of the MUM1/IRF4 protein in a subset of germinal center B cells, plasma cells, and activated T cells. Blood. 2000;95:2084–92. 10.1182/blood.V95.6.2084 [DOI] [PubMed] [Google Scholar]
- 25.Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol. 2006;7:773–82. 10.1038/ni1357 [DOI] [PubMed] [Google Scholar]
- 26.Perez-Soler R, Esteban R, Allende E, Tornos Salomo C, Julia A, Guardia J. Liver involvement in multiple myeloma. Am J Hematol. 1985;20:25–9. 10.1002/ajh.2830200105 [DOI] [PubMed] [Google Scholar]
- 27.Gundesen MT, Lund T, Moeller HEH, Abildgaard N. Plasma Cell Leukemia: Definition, Presentation, and Treatment. Curr Oncol Rep. 2019;21:8. 10.1007/s11912-019-0754-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Longsworth LG, Shedlovsky T, Macinnes DA. Electrophoretic Patterns of Normal and Pathological Human Blood Serum and Plasma. J Exp Med. 1939;70:399–413. 10.1084/jem.70.4.399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997;25:1317–8. 10.1093/nar/25.6.1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376:352–5. 10.1038/376352a0 [DOI] [PubMed] [Google Scholar]
- 31.Ozcan E, Garibyan L, Lee JJ, Bram RJ, Lam KP, Geha RS. Transmembrane activator, calcium modulator, and cyclophilin ligand interactor drives plasma cell differentiation in LPS-activated B cells. J Allergy Clin Immunol. 2009;123:1277–86. e5 10.1016/j.jaci.2009.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyauchi Y, Ninomiya K, Miyamoto H, Sakamoto A, Iwasaki R, Hoshi H, et al. The Blimp1-Bcl6 axis is critical to regulate osteoclast differentiation and bone homeostasis. J Exp Med. 2010;207:751–62. 10.1084/jem.20091957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larrayoz M, Garcia-Barchino MJ, Celay J, Etxebeste A, Jimenez M, Perez C, et al. Preclinical models for prediction of immunotherapy outcomes and immune evasion mechanisms in genetically heterogeneous multiple myeloma. Nat Med. 2023;29:632–45. 10.1038/s41591-022-02178-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boiarsky R, Haradhvala NJ, Alberge JB, Sklavenitis-Pistofidis R, Mouhieddine TH, Zavidij O, et al. Single cell characterization of myeloma and its precursor conditions reveals transcriptional signatures of early tumorigenesis. Nat Commun. 2022;13:7040. 10.1038/s41467-022-33944-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davies FE, Dring AM, Li C, Rawstron AC, Shammas MA, O’Connor SM, et al. Insights into the multistep transformation of MGUS to myeloma using microarray expression analysis. Blood. 2003;102:4504–11. 10.1182/blood-2003-01-0016 [DOI] [PubMed] [Google Scholar]
- 36.Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25:91–101. 10.1016/j.ccr.2013.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bolli N, Biancon G, Moarii M, Gimondi S, Li Y, de Philippis C, et al. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia. 2018;32:2604–16. 10.1038/s41375-018-0037-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hideshima T, Mitsiades C, Ikeda H, Chauhan D, Raje N, Gorgun G, et al. A proto-oncogene BCL6 is up-regulated in the bone marrow microenvironment in multiple myeloma cells. Blood. 2010;115:3772–5. 10.1182/blood-2010-02-270082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pastor TP, Peixoto BC, Viola JPB. The Transcriptional Co-factor IRF2BP2: A New Player in Tumor Development and Microenvironment. Front Cell Dev Biol. 2021;9:655307. 10.3389/fcell.2021.655307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blotta S, Tassone P, Prabhala RH, Tagliaferri P, Cervi D, Amin S, et al. Identification of novel antigens with induced immune response in monoclonal gammopathy of undetermined significance. Blood. 2009;114:3276–84. 10.1182/blood-2009-04-219436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Croucher DC, Richards LM, Tsofack SP, Waller D, Li Z, Wei EN, et al. Longitudinal single-cell analysis of a myeloma mouse model identifies subclonal molecular programs associated with progression. Nat Commun. 2021;12:6322. 10.1038/s41467-021-26598-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borcherding N, Bormann NL, Kraus G. scRepertoire: An R-based toolkit for single-cell immune receptor analysis. F1000Res. 2020;9:47. 10.12688/f1000research.22139.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell. 2008;13:167–80. 10.1016/j.ccr.2008.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morito N, Yoh K, Maeda A, Nakano T, Fujita A, Kusakabe M, et al. A novel transgenic mouse model of the human multiple myeloma chromosomal translocation t(14;16)(q32;q23). Cancer Res. 2011;71:339–48. 10.1158/0008-5472.CAN-10-1057 [DOI] [PubMed] [Google Scholar]
- 45.Boylan KL, Gosse MA, Staggs SE, Janz S, Grindle S, Kansas GS, et al. A transgenic mouse model of plasma cell malignancy shows phenotypic, cytogenetic, and gene expression heterogeneity similar to human multiple myeloma. Cancer Res. 2007;67:4069–78. 10.1158/0008-5472.CAN-06-3699 [DOI] [PubMed] [Google Scholar]
- 46.Podar K, Raab MS, Tonon G, Sattler M, Barila D, Zhang J, et al. Up-regulation of c-Jun inhibits proliferation and induces apoptosis via caspase-triggered c-Abl cleavage in human multiple myeloma. Cancer Res. 2007;67:1680–8. 10.1158/0008-5472.CAN-06-1863 [DOI] [PubMed] [Google Scholar]
- 47.Xu G, Shen XJ, Pu J, Chu SP, Wang XD, Wu XH, et al. BLyS expression and JNK activation may form a feedback loop to promote survival and proliferation of multiple myeloma cells. Cytokine. 2012;60:505–13. 10.1016/j.cyto.2012.06.317 [DOI] [PubMed] [Google Scholar]
- 48.Perlot T, Penninger JM. Development and function of murine B cells lacking RANK. J Immunol. 2012;188:1201–5. 10.4049/jimmunol.1102063 [DOI] [PubMed] [Google Scholar]
- 49.Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162:1229–41. 10.1016/j.cell.2015.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanchez E, Smith EJ, Yashar MA, Patil S, Li M, Porter AL, et al. The Role of B-Cell Maturation Antigen in the Biology and Management of, and as a Potential Therapeutic Target in, Multiple Myeloma. Target Oncol. 2018;13:39–47. 10.1007/s11523-017-0538-x [DOI] [PubMed] [Google Scholar]
- 51.Sordillo EM, Pearse RN. RANK-Fc: a therapeutic antagonist for RANK-L in myeloma. Cancer. 2003;97:802–12. 10.1002/cncr.11134 [DOI] [PubMed] [Google Scholar]
- 52.Terpos E, Szydlo R, Apperley JF, Hatjiharissi E, Politou M, Meletis J, et al. Soluble receptor activator of nuclear factor kappaB ligand-osteoprotegerin ratio predicts survival in multiple myeloma: proposal for a novel prognostic index. Blood. 2003;102:1064–9. 10.1182/blood-2003-02-0380 [DOI] [PubMed] [Google Scholar]
- 53.Pearse RN, Sordillo EM, Yaccoby S, Wong BR, Liau DF, Colman N, et al. Multiple myeloma disrupts the TRANCE/ osteoprotegerin cytokine axis to trigger bone destruction and promote tumor progression. Proc Natl Acad Sci USA. 2001;98:11581–6. 10.1073/pnas.201394498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duran A, Serrano M, Leitges M, Flores JM, Picard S, Brown JP, et al. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev Cell. 2004;6:303–9. 10.1016/S1534-5807(03)00403-9 [DOI] [PubMed] [Google Scholar]
- 55.Milan E, Perini T, Resnati M, Orfanelli U, Oliva L, Raimondi A, et al. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy. 2015;11:1161–78. 10.1080/15548627.2015.1052928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Riz I, Hawley TS, Hawley RG. KLF4-SQSTM1/p62-associated prosurvival autophagy contributes to carfilzomib resistance in multiple myeloma models. Oncotarget. 2015;6:14814–31. 10.18632/oncotarget.4530 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For original data, please contact maike.buchner@tum.de.