

Abstract
Subject terms: Cancer genomics, Cancer genetics, Cancer genetics
TO THE EDITOR:
Genome profiling tests for multiple genes are essential in the treatment of leukemia, facilitating precise diagnosis, risk stratification, and identification of therapeutic targets. Targeted capture sequencing with a next-generation sequencer (NGS) is already in use for clinical cancer genome profiling tests [1]. However, several limitations remain to this approach, including the inferior capability for detecting structural variations (SVs) and copy number variations (CNVs), and the long turn-around time.
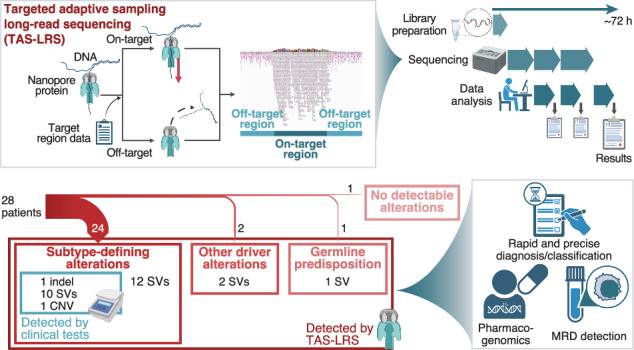
Nanopore sequencer, one of the long-read sequencers, is equipped with a computational targeted sequencing function called “adaptive sampling” (Supplemental Figure S1A) [2]. Targeted adaptive sampling long-read sequencing (TAS-LRS) can utilize a wide range of target regions without additional preparation such as capture-based enrichment. By taking advantage of long-read sequencing, TAS-LRS excels in SV detection and haplotype-aware variant calling, as well as the shorter time required for library preparation and sequencing than conventional short-read sequencing [3].
Except for previous studies on germline analysis in the oncology field [4, 5], no studies have examined the utility of TAS-LRS for cancer genome profiling. Because pediatric leukemia has a high frequency of SVs and CNVs directly related to diagnosis and classification [6, 7], rapid and comprehensive genome analysis is beneficial. Therefore, we evaluated the utility of TAS-LRS as a genome-profiling method in pediatric leukemia.
This study enrolled 28 consecutive children with leukemia (10 with acute myeloid leukemia [AML], 13 with B-cell acute lymphoblastic leukemia [B-ALL], and five with T-cell ALL [T-ALL]) in the University of Tokyo Hospital whose tumor and normal samples were available (Supplemental Table S1). TAS-LRS was performed using a GridION sequencer (Oxford Nanopore Technologies, OX, UK) for tumor/normal-paired samples. Target regions included 466 genes associated with hematologic malignancies (Supplemental Table S2). After read alignment to the GRCh38 using minimap2 v2.24 (ref. [8]), single-nucleotide variants (SNVs) were called using PEPPER-Margin-DeepVariant v0.8 (tumor-only analysis) [9] and ClairS (tumor/normal-paired analysis; github.com/HKU-BAL/ClairS), SVs using nanomonsv v0.5.0 (ref. [10]), and CNVs using CNVkit [11] (Supplemental Fig. S1B,C). Short-read whole genome sequencing (WGS) was also performed for 21 patients (Supplemental Methods).
As clinical tests, the patients underwent non-NGS-based genetic tests, including G-banding, DNA index analysis, and polymerase chain reaction-based screening for fusion genes and other genetic alterations (Supplemental Methods and Supplemental Tables S3 and S4). Genomic alterations consistent with subgroup classifications described in previous studies [6, 12] were designated as “genomic subtypes.”
Tumor/normal-paired TAS-LRS was successful in all 28 patients. The process from library preparation to result output took approximately 72 hours. For tumor samples, mean depth, N50, and mean base quality were 21.3× (16.1×–31.1×), 11 089 bp (8 423–12 654 bp), and 15.8 (14.3–19.1) in the median in the on-target regions, respectively (Supplemental Fig. S2A–C). These statistics for normal samples are also depicted in Supplemental Fig. S2D–F.
A total of 498 SNVs, 35 small indels, and 632 SVs were identified, among which 22 SNVs (4.4%), 15 small indels (42.9%), and 71 SVs (11.2%) were considered driver alterations (Supplemental Tables S5 and S6). Among the 12 patients whose genomic subtypes had been determined by clinical testing, TAS-LRS reconfirmed all of them. Among the other 16 patients, TAS-LRS could determine the genomic subtypes in 12. Thus, genomic subtypes were determined in 24 (85.7%) patients (Fig. 1). All of these newly identified variants were SVs. Other driver alterations that are not involved in genomic subtypes but are found recurrently in pediatric leukemia were detected in two [13, 14], and germline RUNX1 partial deletion in one (Supplemental Fig. S3A). In the other patient (AML4), driver alterations were detected neither by clinical testing nor TAS-LRS.
Fig. 1. Subtype-defining alterations and other important genome alterations detected by TAS-LRS and/or clinical tests.

AML, acute myeloid leukemia; B-ALL, B-cell acute lymphoblastic leukemia; T-ALL, T-cell acute lymphoblastic leukemia; TAS-LRS, targeted adaptive sampling long-read sequencing.
SV analyses could determine the accurate coordinates of the breakpoints even if they were located within intronic or intergenic regions. Therefore, TAS-LRS efficiently identified SVs, including cryptic fusions and large deletions overlooked by clinical tests (Supplemental Fig. S3). DUX4 rearrangements were identified in three patients. Besides the canonical SV detection approach, two additional approaches, single breakend SV detection approach and de novo assembly approach, were necessary to detect comprehensively these DUX4 rearrangements (Supplemental Methods and Supplemental Fig. S4).
CNV analyses identified chromosome-level CNVs by using data both in the on- and off-target regions, with a high sensitivity for the results of G-banding (Supplemental Table S7, Supplemental Figs. S5 and S6A). Focal CNVs were also identified, including those not accurately described in G-banding and those overlooked by SV analysis due to the large size (Supplemental Fig. S6B,C).
In addition to genome profiling, as mentioned above, the technical nature of the method could be used for other applications, including NUDT15 diplotyping and detecting fusion breakpoints for fusion-based minimal residual disease (MRD) assay (Supplemental Figures S7 and S8).
In the comparison of results between the tumor-only and tumor/normal-paired analyses, 93.6% of the SNVs, 88.6% of the small indels, and 50.5% of the SVs were called only by tumor-only analysis, and most were derived from germline variants or sequencing errors (Supplemental Tables S8–S10, Supplemental Fig. S9A). However, even in tumor-only analysis, the number of detected variants in each patient was not very high, with <45 for each mutation type (Supplemental Fig. S9B). Seven SNVs (31.8% of the driver SNVs) and 12 small indels (80.0% of the driver small indels) were overlooked by the tumor/normal-paired analysis (Supplemental Table S5). For example, NPM1 frameshift insertion in AML6 was detected in the tumor-only analysis but was excluded in the tumor/normal-paired analysis result because of its “low quality” classification by ClairS. Germline RUNX1 deletion in TALL4 was the only driver SV missed by tumor/normal-paired analysis (Supplemental Tables S5 and S10).
Lastly, to evaluate the variant detection capability of TAS-LRS, the results were compared with those of the short-read WGS. When variants detected by WGS were considered true positives, the precision and recall for TAS-LRS were 100% and 60.9% for the SNVs, 100% and 17.6% for the small indels, and 73.7% and 85.3% for the SVs, respectively (Fig. 2A,B, Supplemental Figure S10A,B, and Supplemental Tables S11–S14). SVs detected only by WGS had significantly lower variant allele frequencies (VAFs) than those detected by TAS-LRS (0.157 vs. 0.271 in the median, P < 0.001; Fig. 2C). A similar tendency in SNV was found (0.207 vs. 0.416 in the median, P = 0.083), whereas VAF was nearly the same between the indels detected by TAS-LRS and only by WGS (0.444 vs. 0.393 in the median, P = 0.509). Blast fraction did not differ between variants detected by TAS-LRS and only by WGS (Fig. 2D).
Fig. 2. Comparison of the results of TAS-LRS with those of short-read WGS.

A Number of SNVs and small indels detected by TAS-LRS and/or short-read WGS. The variants detected by both TAS-LRS and short-read WGS are annotated as “Common.” B Number of SVs detected by TAS-LRS and/or short-read WGS. “Common (as SV)” represents variants commonly detected by TAS-LRS as SVs and short-read WGS as SVs, and “Common (as CNV)” represents variants commonly detected by TAS-LRS as CNVs and short-read WGS as SVs. C Allele frequency of variants detected by TAS-LRS (including those commonly detected by short-read WGS) and those detected only by short-read WGS. Asterisks represent statistically significant differences (* P ≤ 0.001). NS, not significant. D Blast fraction of variants detected by TAS-LRS (including those commonly detected by short-read WGS) and those detected only by short-read WGS. Mann–Whitney U test was applied for the statistical analyses.
Some clinically important SNVs and small indels were missed by TAS-LRS, such as KRAS Gly13Asp and NPM1 frameshift indels (Supplemental Tables S11 and S12). Indeed, in AML4, the only patient whose driver alterations were not detected by TAS-LRS and clinical testing, NPM1 2-bp deletion out of the mutational hotspot (NM_002520:c.755_756del) was identified by WGS. As to SVs, it was noteworthy that 32 immunoglobulin gene rearrangements and seven T-cell receptor gene rearrangements were detected only by TAS-LRS. Upon verification using Integrative Genomics Viewer, most of these “TAS-LRS-only” SVs were also depicted in WGS results; however, they were likely to be excluded from the SV calling results because of the low mapping quality (Supplemental Figure S10C).
TAS-LRS demonstrated superior characteristics combining the selectivity of targeted capture sequencing and the comprehensiveness of WGS, enabling quick identification of multimodal types of gene variants, especially SVs including a fusion gene with a novel partner gene, cryptic translocations, and complex SVs. Because TAS-LRS outputs data not only from the on-target regions but also from the off-target regions, low-coverage read data in the off-target regions was useful to determine CNVs, from large ones at the chromosome level to small ones ranging over only several genes. In the current analysis method, SV and CNV analyses complemented each other and could comprehensively detect focal deletions.
Major genomic alterations should be detected in tumor-only analysis because peripheral blood as a control specimen may contain leukemia cells at the time of diagnosis, and waiting until remission would be time-consuming. Even with tumor-only analysis results, we could narrow down the candidates to a manageable count for individual verification by the appropriate filtering. Hence, in clinical applications, conducting analysis on only tumor samples at diagnosis and subsequently performing a paired analysis after achieving remission might be practical.
The major problem of TAS-LRS is the low accuracy of SNV and small indel calling which may lead to increased false negatives, as reported in previous studies [3, 15]. Further, TAS-LRS had considerably lower coverages with ~20× than those in WGS with ~120×, which could also contribute to missing variants, particularly in minor clones. Although this study did not show a significant effect of tumor purity on variant calling accuracy, the samples had a high tumor purity, no less than 67.6%. Therefore, some variants may be overlooked in patients with low tumor purity. However, these problems would be solved by improvements in devices, chemistry, and analysis tools [3, 15]. Also, long-read and short-read sequencing could complement each other, potentially enabling the construction of a more robust integrated analysis system.
In summary, TAS-LRS using nanopore sequencing was a promising method of genome profiling for pediatric leukemia with rapidity and efficacy, particularly advantageous for identifying SVs and CNVs. Thus, the application of TAS-LRS to genome profiling for pediatric leukemia would lead to the refinement of leukemia treatment practice.
Supplementary information
Acknowledgements
The supercomputing resource was provided by the Human Genome Center, the Institute of Medical Science, the University of Tokyo. Figures were created with BioRender.com. We also thank M. Matsumura and E. Mochizuki (the University of Tokyo, Tokyo, Japan) for their technical and instrumental support. This work was supported by the Japan Agency for Medical Research and Development (AMED) under grant numbers JP23ck0106876 and JP23ck0106870 (M. Kato) and by the Japan Leukemia Research Fund (S. Kato).
Author contributions
SK, ASO, and MK designed this study; SK, NT, TI, MH, MS, KW, and MK collected materials; SK, ASO, WN, MS, AO, KC, NT, YS, and MK analyzed data; SK made the figures and wrote the draft; ASO, WN, MS, AO, KC, NT, TI, MH, MS, KW, YS, and MK critically revised the manuscript; and all authors approved the final version of the manuscript and are accountable for all aspects of the work.
Data availability
The nanopore sequencing data obtained in this study are deposited in the National Bioscience Database Center (NBDC) Human Database and are available at the Japanese Genotype–phenotype Archive (JGA) with accession codes JGAS000692. The nonmatched control panel for SV analysis using nanomonsv was created using the 30 Nanopore sequencing data from Human Pangenome Reference Consortium, which is available at zenodo (zenodo.org/records/7017953).
Competing interests
The authors declare no competing interests.
Ethics
This study adhered to the Declaration of Helsinki and was approved by the ethics board of the University of Tokyo (approval number: 2022382). Written informed consent was obtained from their parents and/or legal guardians.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41408-024-01108-5.
References
- 1.Duncavage EJ, Bagg A, Hasserjian RP, DiNardo CD, Godley LA, Iacobucci I, et al. Genomic profiling for clinical decision making in myeloid neoplasms and acute leukemia. Blood. 2022;140:2228–47. 10.1182/blood.2022015853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Payne A, Holmes N, Clarke T, Munro R, Debebe BJ, Loose M. Readfish enables targeted nanopore sequencing of gigabase-sized genomes. Nat Biotechnol. 2020;39:442–50. 10.1038/s41587-020-00746-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y, Zhao Y, Bollas A, Wang Y, Au KF. Nanopore sequencing technology, bioinformatics and applications. Nat Biotechnol. 2021;39:1348–65. 10.1038/s41587-021-01108-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakamura W, Hirata M, Oda S, Chiba K, Okada A, Mateos RN, et al. Assessing the efficacy of target adaptive sampling long-read sequencing through hereditary cancer patient genomes. npj Genom Med. 2024;9:11. 10.1038/s41525-024-00394-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakamichi K, Stacey A, Mustafi D. Targeted long-read sequencing allows for rapid identification of pathogenic disease-causing variants in retinoblastoma. Ophthalmic Genet. 2022;43:762–70. 10.1080/13816810.2022.2141797 [DOI] [PubMed] [Google Scholar]
- 6.Brady SW, Roberts KG, Gu Z, Shi L, Pounds S, Pei D, et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet. 2022;54:1376–89. 10.1038/s41588-022-01159-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolouri H, Farrar JE, Triche T, Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. 2018;24:103–12. 10.1038/nm.4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H. New strategies to improve minimap2 alignment accuracy. Bioinformatics. 2021;37:4572–4. 10.1093/bioinformatics/btab705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shafin K, Pesout T, Chang P-C, Nattestad M, Kolesnikov A, Goel S, et al. Haplotype-aware variant calling with PEPPER-Margin-DeepVariant enables high accuracy in nanopore long-reads. Nat Methods. 2021;18:1322–32. 10.1038/s41592-021-01299-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shiraishi Y, Koya J, Chiba K, Okada A, Arai Y, Saito Y, et al. Precise characterization of somatic complex structural variations from tumor/control paired long-read sequencing data with nanomonsv. Nucleic Acids Res. 2023;51:e74. 10.1093/nar/gkad526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol. 2016;12:e1004873. 10.1371/journal.pcbi.1004873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Umeda M, Ma J, Westover T, Ni Y, Song G, Maciaszek JL, et al. A new genomic framework to categorize pediatric acute myeloid leukemia. Nat Genet. 2024;56:281–93. 10.1038/s41588-023-01640-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Rooij JDE, Beuling E, van den Heuvel-Eibrink MM, Obulkasim A, Baruchel A, Trka J, et al. Recurrent deletions of IKZF1 in pediatric acute myeloid leukemia. Haematologica. 2015;100:1151–9. 10.3324/haematol.2015.124321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Balducci E, Steimlé T, Smith C, Villarese P, Feroul M, Payet-Bornet D, et al. TREC mediated oncogenesis in human immature T lymphoid malignancies preferentially involves ZFP36L2. Mol Cancer. 2023;22:108. 10.1186/s12943-023-01794-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolmogorov M, Billingsley KJ, Mastoras M, Meredith M, Monlong J, Lorig-Roach R, et al. Scalable Nanopore sequencing of human genomes provides a comprehensive view of haplotype-resolved variation and methylation. Nat Methods. 2023;20:1483–92. 10.1038/s41592-023-01993-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The nanopore sequencing data obtained in this study are deposited in the National Bioscience Database Center (NBDC) Human Database and are available at the Japanese Genotype–phenotype Archive (JGA) with accession codes JGAS000692. The nonmatched control panel for SV analysis using nanomonsv was created using the 30 Nanopore sequencing data from Human Pangenome Reference Consortium, which is available at zenodo (zenodo.org/records/7017953).