

Abstract
The patient was a 54-year-old woman with familial hypercholesterolemia and remarkable Achilles tendon thickening. At 20 years old, the patient had a total cholesterol level of approximately 300 mg/dL. She started receiving rosuvastatin (5 mg/day) for low-density lipoprotein cholesterol (LDL-C) 235 mg/dL at 42 years old, which was increased to 10 mg/day at 54 years old, decreasing her serum LDL-C level to approximately 90 mg/dL. The serum Lp(a) level was 9 mg/dL. A computed tomography coronary angiogram showed no significant stenosis. Next-generation sequencing revealed a frameshift variant in LDL receptor (LDLR) (heterozygous) and a missense variant in proprotein convertase subtilisin/kaxin type 9 (PCSK9) (heterozygous). Continued statin therapy, in addition to low Lp(a) and female sex, can help prevent cardiovascular disease.
Keywords: heterozygous familial hypercholesterolemia, a protein-truncating variant in the LDLR, Lp(a), computerized tomography (CT) coronary angiogram, PCSK9 variant
Introduction
Familial hypercholesterolemia (FH) is characterized by high levels of low-density lipoprotein cholesterol (LDL-C), skin and tendon xanthomas, and premature coronary artery disease (CAD) (1), with a prevalence of 1 heterozygous FH (HeFH) patient per 200-500 individuals in the general population (2,3). LDL receptor (LDLR), proprotein convertase subtilisin/kexin type 9 (PCSK9), and apolipoprotein B gene variants have been reported in patients in Japan (4-6).
Recently, the impact of positive clinical signs (xanthoma and/or family history) and a positive FH mutation status on the risk of coronary artery disease (CAD) over and above that predicted by LDL-C levels alone has been emphasized (7). The presence of clinical signs of FH and a positive FH variant status additively increased the risk of CAD. A separate study also showed that pathogenic variants, especially protein-truncating variants (PTVs), are significantly associated with poor outcomes in patients with FH (8).
We recently encountered a 54-year-old woman with FH without coronary artery stenosis who underwent multidetector-row computed tomography (MDCT) angiography and carotid artery thickening by carotid artery ultrasonography despite having a PTV in the LDLR and marked Achilles tendon xanthomas. She was also found to have a rare variant of PCSK9.
We herein report the detailed clinical profile of this patient with FH and describe potential factors that might have contributed to the slow progression of coronary artery stenosis despite her having a PTV in the LDLR and marked Achilles tendon xanthomas.
Case Report
A 54-year-old Japanese woman was diagnosed with heterozygous FH. At 20 years old, a blood test had revealed serum total cholesterol (TC) levels in the 300 mg/dL range. At 42 years old, she had been started on 5 mg/day of rosuvastatin by her previous physician for TC 335 mg/dL and LDL-C 235 mg/dL.
At 43 years old, she developed Achilles tendon rupture when her LDL-C level was approximately 140 mg/dL. At 44 years old (Fig. 1), she was referred to our hospital for the further investigation of dyslipidemia. A total of 500 mg/day of probucol was then added, and the serum LDL-C level remained at approximately 100 mg/dL. At 45 years old, she was diagnosed with breast cancer, underwent hormone therapy, and subsequently experienced menopause.
Figure 1.

Changes in serum LDL cholesterol levels of the patient during her 10-year clinical course.
At 49 years old, ezetimibe 10 mg was added. At 54 years old, the rosuvastatin dose was increased to 10 mg, and serum LDL-C levels were in the 80-90 mg/dL range. After 1 month of washing out the lowering drugs, her serum LDL-C and Lp(a) levels were 246 mg/dL and 9 mg/dL, respectively (Table 1). She did not have an abnormal liver function or kidney function (Table 2).
Table 1.
Clinical Profile of the Patient’s Family.
| Proband | First daughter | Second daughter | Son | |||||
|---|---|---|---|---|---|---|---|---|
| Age, y | 54 | 19 | 15 | 15 | ||||
| Body mass index , kg/m2 | 20.7 | 20.8 | 19.7 | 16.5 | ||||
| Achilles tendon thickening | yes | no | no | yes | ||||
| Total cholesterol, mg/dL | 325 | 197 | 187 | 269 | ||||
| Triglycerides, mg/dL | 50 | 77 | 117 | 52 | ||||
| LDL cholesterol, mg/dL | 246 | 104 | 113 | 203 | ||||
| HDL cholesterol, mg/dL | 64 | 75 | 58 | 55 | ||||
| Lp(a), mg/dL | 9 | ND | 3 | 3 | ||||
| LDL receptor gene | p.Val827SerfsTer102 | ND | Wild type | p.Val827SerfsTer102 | ||||
| PCSK 9 gene | p.Ser668Arg | ND | p.Ser668Arg | p.Ser668Arg | ||||
| p.Val644Ile |
ND: not done, Second daughter and Son were fraternal twins.
Table 2.
Laboratory Data for This Patient after One Month Discontinuation of Medication.
| WBC, 103 | 3,600 | T-Bil, mg/dL | 0.7 | TC, mg/dL | 325 | |||||
| Hb, g/dL | 12.6 | TP, g/dL | 8.2 | TG, mg/dL | 50 | |||||
| Plt, 104 | 14.3 | ALB, g/dL | 5.0 | HDL-C, mg/dL | 64 | |||||
| AST, U/L | 21 | Uric acid, mg/dL | 4.6 | LDL-C, mg/dL | 246 | |||||
| ALT, U/L | 11 | BUN, mg/dL | 13 | ApoA1, mg/dL | 133 | |||||
| LDH, U/L | 218 | Cre, mg/dL | 0.64 | ApoA2, mg/dL | 26.7 | |||||
| ALP, U/L | 76 | GFR,mL/min/1.73m2 | 70.8 | ApoB , mg/dL | 139 | |||||
| γGTP, U/L | 17 | Na, mmol/L | 138 | ApoC2, mg/dL | 2.9 | |||||
| CK, U/L | 181 | K, mmol/L | 4.3 | ApoC3, mg/dL | 7 | |||||
| Blood glucose, mg/dL | 87 | Cl, mmol/L | 105 | ApoE, mg/dL | 6.5 | |||||
| HbAc, % | 5.7 | U-protein | - | Lp(a), mg/dL | 9 |
On a physical examination, she had marked Achilles tendon thickening, increasing from (right/left) 19.9/20.4 mm at 44 years old to 15.6/17.9 mm at 54 years old, but she showed no corneal ring or xanthelasma. She had a family history of dyslipidemia in her mother and sister, based on a medical interview. She had three children living with her, and we conducted blood tests of these children for cascade screening. We found that 1 of her 3 children (15-year-old son), had a high serum LDL-C level (203 mg/dL) with Achilles tendon thickening (right/left: 7.4/8.2 mm), while the other 2 were normolipidemic (the 17-year-old older sister and the 15-year-old fraternal female twin of the hyperlipidemic son). Both her son and his fraternal twin showed low serum Lp(a) levels (Table 1), suggesting that there might be some genetic background for low Lp(a) levels in this family, although the detailed mechanism was unclear.
We conducted next-generation sequencing of the genes from the proband and her children (Table 1). Details of the sequencing analysis are described in our previous report (9). In brief, genomic DNA isolated from peripheral white blood cells was pooled, selected for size, ligated to sequencing adapters, and amplified to enrich for targets that were sequenced using the KAPA DNA Library Preparation. A custom NimbleGen in-solution DNA capture library (Roche NimbleGen Inc., Madison, WI, USA) was designed to capture all coding exons in 21 dyslipidemia-related Mendelian genes (ABCA1, ABCG5, ABCG8, ANGPTL3, APOA1, APOB, APOC2, APOC3, APOA5, APOE, CETP, GPIHBP1, LCAT, LDLR, LDLRAP1, LIPG, LMF1, LPL, MTTP, PCSK9, and SAR1B).
The mother was found to have a frameshift variant in LDLR (heterozygous), (NM_000527.5):c.2478del (p.Val827SerfsTer102), and a missense variant in PCSK9 (heterozygous), (NM_174936.4):c.2004C>A (p.Ser668Arg), revealing not only the same variants in LDLR and PCSK9 but also a missense variant in PCSK9 (heterozygous), (NM_174936.4):c.1930G>A (p.Val644Ile). His fraternal normolipidemic twin was found to have the PCSK9 (heterozygous) variant (NM_174936.4):c.2004C>A (p.Ser668Arg).
We performed 64-slice mechanical MDCT angiography (10). Three-dimensional volume-rendered images acquired with 64-slice coronary computed tomography angiography for the right (a) and left (b) coronary arteries showed no stenosis in either the right or left coronary arteries (Fig. 2). The multi-planar recontraction image also showed that there was no significant coronary artery stenosis in the right coronary artery (Fig. 3a), left anterior descending artery (Fig. 3b), or circumflex artery (Fig. 3c). We also conducted carotid ultrasonography to evaluate the carotid artery and found no intima-media complex thickening or plaque (Fig. 4).
Figure 2.
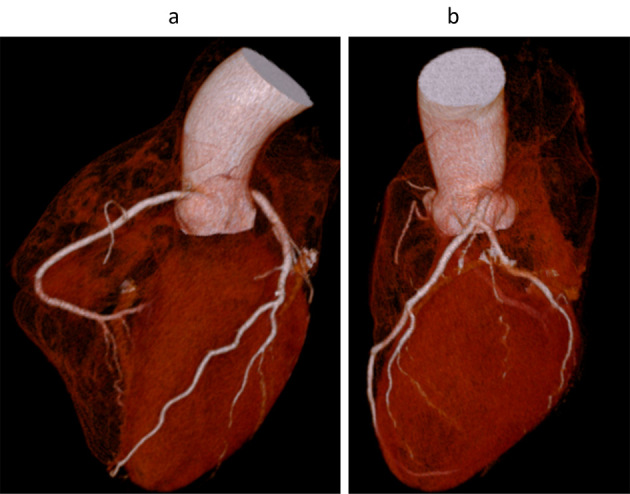
Three-dimensional volume rendering images acquired with 64-slice coronary computed tomography angiography of the right (a) and left (b) coronary arteries of the patient. There was no obvious coronary artery stenosis in either the right or the left coronary arteries.
Figure 3.

Multiplanar reconstruction of the right coronary artery (a), left anterior descending artery (b), and circumflex artery (c).
Figure 4.

Intima-media thickness (IMT) of the right (a) and left (b) common carotid arteries determined using B-mode ultrasound. The average IMT of the right (a) and left (b) common carotid arteries was 0.43 and 0.46 mm, respectively.
The genetic analyses were approved by the Ethics Committees of Chiba University and Kanazawa University. Written informed consent for the genetic analysis was obtained from each patient. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and the 1975 Declaration of Helsinki, as revised in 2008. Informed consent for the genetic analysis was obtained from each patient.
Discussion
The present patient with FH was found to have heterozygous variants in not only LDLR (p.Val827SerfsTer102) but also PCSK9 (p.Ser668Arg); the former is an established pathogenic variant causing amino acid truncation in LDLR protein and is reported to be associated with severe and poor outcomes of FH (8), while the clinical significance of the latter has not yet been established, although a previous study (4) suggested that this variant may be clinically insignificant.
Positive clinical signs of FH, such as Achilles tendon xanthomata and the FH variant status, are important contributors to the development of CAD in FH patients (7). Previous studies have shown that Achilles tendon thickening is associated with CAD in patients with FH (11-13). The current FH patient had marked Achilles tendon xanthomata and the LDLR truncated variant, both of which are expected to contribute to marked CAD development. However, in reality, the patient showed little development of cardiovascular disease, either on coronary artery MDCT or carotid artery ultrasonography. It is highly likely that continued statin treatment helped prevent the development of coronary heart disease in this patient.
Another factor possibly associated with the lack of development of cardiovascular disease in this patient may have been her low serum Lp(a) concentration, as Lp(a) has been shown to be an important contributor to the development of atherosclerosis, even in FH (14,15). Elevations in Lp(a) frequently accompany high levels of LDL in patients with FH, and both FH and elevated Lp(a) levels independently correlate with premature-onset CAD. In addition, it has been shown that CAD develops much later in women than in men (16). Indeed, most women were shown to be free of CAD in their 50s in a previous study (16). In addition, our patient's serum HDL-C and apo A1 levels may have been protective factors against the development of coronary artery disease.
The PCSK9 p.Ser668Arg variant, found in the proband, the second daughter, and the son, is not likely pathogenic because the second daughter had no elevation in serum LDL-C. This variant was reported by Hori et al. (4) as a variant of uncertain significance with an allele frequency of 0.001. Furthermore, given that the serum LDL-C level with LDLR variant alone is reported to be around 250 mg/dL (17), similar to the proband's serum LDL-C value of 246 mg/dL, this PCSK9 variant would have little if any effect on serum LDL-C levels.
Interestingly, the patient's 15-year-old son with FH had a heterozygous pVal644Ile variant in PCSK9 in addition to the same genetic variants as the proband (Table 2). This variant was reported by Hori et al. (4) and may not be related to high LDL-C levels. Pitavastatin (2 mg) treatment for 3 months produced a considerable reduction in the serum LDL-C level (203 to 117 mg/dL). He had already begun to have Achilles tendon thickening according to the new criteria for the diagnosis of FH in Japan (18).
The earlier the age at which treatment is initiated for FH, the better the prognosis. Nordesgaard et al. (3) suggested that, in cases of heterozygous FH, starting treatment with a mild statin at 12 years old was more effective at delaying the onset of coronary artery disease than starting treatment with a strong statin as an adult. In addition, the importance of cascade screening has been emphasized (19), since the identification of patients with FH by child-family cascade screening at a younger age appeared to result in a better prognosis (20). In the proband's family, two of her three children were fraternal twins (a 15-year-old boy and girl), and dyslipidemia was observed in the male twin. Next-generation sequencing revealed that he carried the same heterozygous variants in LDLR and PCSK9 as the proband but also a heterozygous pVal644Ile variant in PCSK9. We initiated treatment with pitavastatin for this 15-year-old boy.
In conclusion, we found a heterozygous LDLR variant (p.Val827SerfsTer102) and a heterozygous PCSK9 variant (p.Ser668Arg) in a middle-aged Japanese woman with FH. We suggest that continued statin therapy besides low Lp(a) and female sex may be associated with the prevention of atherosclerotic cardiovascular disease, even in cases with a PTV in the LDLR and considerable Achilles tendon thickening. Cascade screening allowed her 15-year-old son with genetically determined FH to start receiving statin treatment promptly.
Author's disclosure of potential Conflicts of Interest (COI).
Koutaro Yokote: Lecture fees, Astellas, AstraZeneca, Daiichi Sankyo, Kowa, Mitsubishi Tanabe, MSD, Ono, Sumitomo, Takeda, Nippon Boehr inger Ingelheim, Novartis, Novo Nordisk, Pfizer Japan and Taisho; Research funding, Abott Japan, Astellas, Bayer Yakuhin, Daiichi Sankyo, Kowa, Mitsubishi Tanabe, Takeda, MSD, Nippon Boehringer Ingelheim, Novo Nordisk, Ono, Shionogi, Sumitomo, Taisho and Teijin; Consultant, AstraZeneca, Bayer Yakuhin, Eli Lilly Japan, Kowa, Mitsubishi Tanabe, Nippon Boehringer Ingelheim, Novartis, Novo Nordisk and Pfizer Japan.
References
- 1. Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: The Metabolic and Molecular Bases of Inherited Disease. 8th. Scriver CR, Beaudet AL, Sly WS, Valle D, Eds. McGraw-Hill, Inc., New York, 2001: 2863-2913. [Google Scholar]
- 2. Mabuchi H, Nohara A, Noguchi T, et al. Molecular genetic epidemiology of homozygous familial hypercholesterolemia in the Hokuriku district of Japan. Atherosclerosis 214: 404-407, 2011. [DOI] [PubMed] [Google Scholar]
- 3. Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J 34: 3478-3490a, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hori M, Ohta N, Takahashi A, et al. Impact of LDLR and PCSK9 pathogenic variants in Japanese heterozygous familial hypercholesterolemia patients. Atherosclerosis 289: 101-108, 2019. [DOI] [PubMed] [Google Scholar]
- 5. Hori M, Takahashi A, Son C, Ogura M, Harada-Shiba M. The first Japanese cases of familial hypercholesterolemia due to a known pathogenic APOB gene variant, c.10580G>A: p.(Arg3527Gln). J Clin Lipidol 14: 482-486, 2020. [DOI] [PubMed] [Google Scholar]
- 6. Hori M, Takahashi A, Hosoda K, Ogura M, Harada-Shiba M. A low-frequency APOB p.(Pro955Ser) variant contributes to the severity of/variability in familial hypercholesterolemia. J Clin Endocrinol Metab 108: 422-432, 2023. [DOI] [PubMed] [Google Scholar]
- 7. Tada H, Kawashiri MA, Nohara A, Inazu A, Mabuchi H, Yamagishi M. Impact of clinical signs and genetic diagnosis of familial hypercholesterolaemia on the prevalence of coronary artery disease in patients with severe hypercholesterolemia. Eur Heart J 38: 1573-1579, 2017. [DOI] [PubMed] [Google Scholar]
- 8. Tada H, Kojima N, Yamagami K, et al. Effects of different types of pathogenic variants on phenotypes of familial hypercholesterolemia. Front Genet 13: 872056, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tada H, Kawashiri MA, Nomura A, et al. Oligogenic familial hypercholesterolemia, LDL cholesterol, and coronary artery disease. J Clin Lipidol 12: 1436-1444, 2018. [DOI] [PubMed] [Google Scholar]
- 10. Minamizuka T, Kobayashi J, Tada H, Koshizaka M, Maezawa Y, Yokote K. Homozygous familial lipoprotein lipase deficiency without obvious coronary artery stenosis. Clin Biochem 108: 42-45, 2022. [DOI] [PubMed] [Google Scholar]
- 11. Ogura M, Hori M, Harada-Shiba M. Association between cholesterol efflux capacity and atherosclerotic cardiovascular disease in patients with familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 36: 181-188, 2016. [DOI] [PubMed] [Google Scholar]
- 12. Hopkins PN, Stephenson S, Wu LL, Riley WA, Xin Y, Hunt SC. Evaluation of coronary risk factors in patients with heterozygous familial hypercholesterolemia. Am J Cardiol 87: 547-553, 2001. [DOI] [PubMed] [Google Scholar]
- 13. Oosterveer DM, Versmissen J, Yazdanpanah M, Hamza TH, Sijbrands EJ. Differences in characteristics and risk of cardiovascular disease in familial hypercholesterolemia patients with and without tendon xanthomas: a systematic review and meta-analysis. Atherosclerosis 207: 311-317, 2009. [DOI] [PubMed] [Google Scholar]
- 14. Alonso R, Andres E, Mata N, et al. Lipoprotein(a) levels in familial hypercholesterolemia: an important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J Am Coll Cardiol 63: 1982-1989, 2014. [DOI] [PubMed] [Google Scholar]
- 15. Allard MD, Saeedi R, Yousefi M, Frohlich J. Risk stratification of patients with familial hypercholesterolemia in a multi-ethnic cohort. Lipids Health Dis 13: 65, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harada-Shiba M, Sugisawa T, Makino H, et al. Impact of statin treatment on the clinical fate of heterozygous familial hypercholesterolemia. J Atheroscler Thromb 17: 667-674, 2010. [DOI] [PubMed] [Google Scholar]
- 17. Ohta N, Hori M, Takahashi A, et al. Proprotein convertase subtilisin/kexin 9V4I variant with LDLR mutations modifies the phenotype of familial hypercholesterolemia. J Clin Lipidol 10: 547-555.e5, 2016. [DOI] [PubMed] [Google Scholar]
- 18. Tada H, Hori M, Matsuki K, et al. Achilles tendon thickness assessed by X-ray predicting a pathogenic mutation in familial hypercholesterolemia gene. J Atheroscler Thromb 29: 816-824, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tada H, Okada H, Nomura A, et al. Prognostic impact of cascade screening for familial hypercholesterolemia on cardiovascular events. J Clin Lipidol 15: 358-365, 2021. [DOI] [PubMed] [Google Scholar]
- 20. Luirink IK, Wiegman A, Kusters DM, et al. 20-year follow-up of statins in children with familial hypercholesterolemia. N Engl J Med 381: 1547-1556, 2019. [DOI] [PubMed] [Google Scholar]