

Abstract
Autoimmune encephalitis (AE) is a category of immune-mediated disorders of the central nervous system (CNS) affecting children and adults. It is characterized by the subacute onset of altered mentation, neurocognitive issues, refractory seizures/drug-resistant epilepsy, movement disorders, and/or autonomic dysfunction. AE is mediated by autoantibodies targeting specific surface components or intracytoplasmic antigens in the CNS, leading to functional or structural alterations. Multiple triggers that induce autoimmunity have been described, which are mainly parainfectious and paraneoplastic. The imaging features of AE often overlap with each other and with other common causes of encephalitis/encephalopathy (infections and toxic-metabolic etiologies). Limbic encephalitis is the most common imaging finding shared by most of these entities. Cortical, basal ganglia, diencephalon, and brainstem involvement may also be present. Cerebellar involvement is rare and is often a part of paraneoplastic degeneration. Owing to an improved understanding of AE, their incidence and detection have increased. Hence, in an appropriate setting, a high degree of suspicion is crucial when reporting clinical MRIs to ensure prompt treatment and better patient outcomes. In this review, we discuss the pathophysiology of AE and common etiologies encountered in clinical practice.
Keywords: Autoimmune encephalitis, Limbic encephalitis, Paraneoplastic syndrome
INTRODUCTION
Autoimmune encephalitis (AE) is a heterogeneous group of disorders mediated by antibodies targeting native epitopes in the central nervous system (CNS) [1]. With advanced detection techniques and an improved understanding of immunopathology, AE has been increasingly implicated in patients with intractable epilepsy, movement disorders, and psychiatric disturbances [2]. Autoimmune triggers usually include recent infections (typically viral but also bacterial, fungal, and parasitic) and tumor antigens (paraneoplastic syndromes), although some may be idiopathic [3].
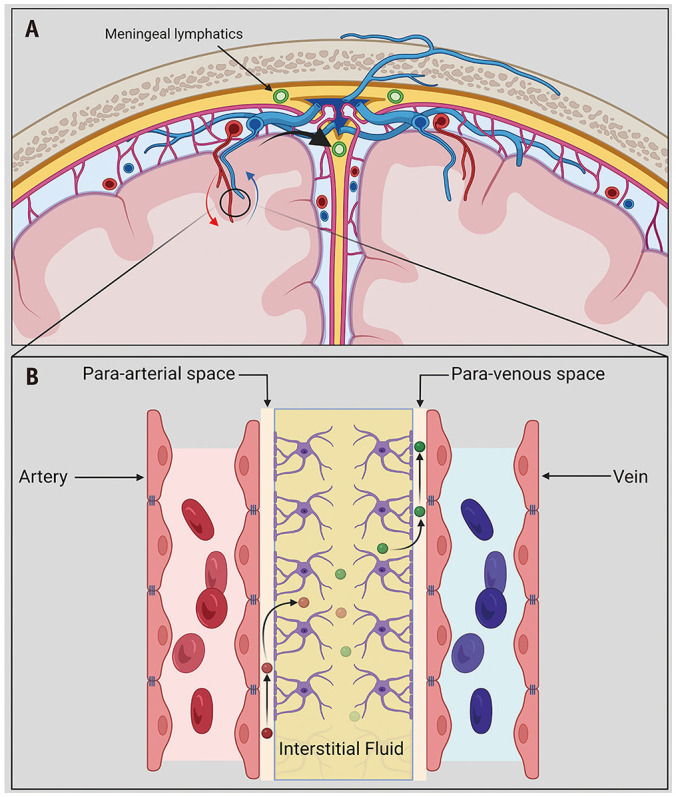
The route of CNS entry of autoantibodies is still unclear. The transfer of proteins and other substances into the CNS is tightly regulated by the blood-brain barrier (BBB), which is comprised of endothelial cells with tight junctions, basement membranes, and pericytes (Fig. 1) [4]. Physiological CNS immune surveillance is carried out by peripherally activated T-cells that can cross an intact BBB, a discovery that has challenged the previous misconception of the brain’s “immune privilege” [4]. However, inflammatory processes via the release of cytokines can breech the BBB and expose the brain’s self-antigens. Viral infections are a prototype of this phenomenon; they promote cross-reactive autoantibody production and facilitate entry across the BBB via pro-inflammatory cytokine release (e.g., interleukin [IL]-17) [1]. Another potential route of entry is the glymphatic system, a recently identified pathway that enables the flow of cerebrospinal fluid (CSF) through the periarteriolar and parenchymal extracellular spaces [1,5,6]. Its intended function is to transport essential nutrients (e.g., glucose) and waste products to and from the brain, respectively (Fig. 2) [7]. Another port of entry is the blood-CSF barrier (choroid plexus). However, evidence supporting this is lacking [8].
Fig. 1. Simplified illustration of the blood-brain barrier. Created using BioRender (BioRender.com).

Fig. 2. Illustration depicting the glymphatic and meningeal efflux pathway. A: Coronal cross section of the brain shows the location of the glymphatic pathway, surrounding the intraparenchymal vasculature. B: Simplified illustration of the glymphatic pathway. The cerebrospinal fluid provides nutrients (red) and other neuroactive substances to the central nervous system via para-arterial spaces pathway. It mixes with the interstitial fluid within the brain parenchyma, eventually draining waste material (green) across the para-venous clearance routes. The parasagittal meningeal lymphatic vessels within the dura mater contribute a major efflux pathway for downstream clearance of macromolecules (curved arrow in A). They also have a role in immunomodulation, facilitating trafficking of immune cells to the deep cervical nodal chain (not shown). Created using BioRender (BioRender.com).
Broadly, two major immune mechanisms can cause AE. The first is autoimmunity to synaptic surface components (receptors, ionic channels, and supporting proteins). The associated neuronal dysfunction is due to altered synaptic transmission secondary to cross-linking and receptor internalization (e.g., anti-N-methyl-D-aspartate receptor [NMDAR] encephalitis), functional disruption of ion channels (e.g., anti-voltage gated potassium channel [VGKC] encephalitis), or inhibition of neurotransmitter binding (e.g., anti-gamma aminobutyric acid-B [GABA-B] encephalitis). Autoantibodies targeting glial antigens (e.g., aquaporin 4 [AQP4], myelin oligodendrocyte glycoprotein [MOG]) cause CNS demyelination or perivascular inflammation, leading to neuronal loss through oligodendrocyte or astrocyte disruption [1]. These disorders have a favorable outcome if diagnosed early, because neuronal structure initially remains intact [2,9]. In the second group, antibodies induce cytotoxic T-cell-mediated damage. These include antibodies against intracytoplasmic antigens (e.g., Yo and CV2/collapsin response mediator protein 5 [CV2/CRMP5]) and nuclear onconeural antigens (e.g., Hu and Ri). The resulting structural damage culminates in neurodegeneration, despite aggressive treatment [2,9].
Microglia, the primary immune effector cells in the CNS, are thought to play a key role in the pathophysiology of AE. These cells are considered to be major stakeholders in the onset, maintenance, relapse, and progression of inflammation in the CNS. Under normal, physiological conditions, microglia are thought to be ‘resting,’ only to be activated upon tissue damage or inflammation. In the course of this activation, they release neurotrophic factors (e.g., nerve growth factor and brain-derived neurotrophic factor) as well as neurotoxic factors (nitric oxide) and proinflammatory cytokines (TNF-α, IL-1). Hence the brain pays a price for the microglia-medicated host defense (“bystander damage”). Studies on multiple sclerosis and experimental autoimmune encephalomyelitis have shown that microglia express major histocompatibility complexes (I and II) along with CD40 and CD80/86, which interact with, reprime, and reactivate T-cells at the lesion site, thereby exacerbating neuronal damage [10,11].
The diagnosis of AE requires a multidisciplinary approach, beginning with a thorough clinical examination. MRI is a crucial, often first-line diagnostic modality. Despite having a low yield and considering that most patients may have no imaging abnormalities in the acute phase, MRI remains valuable in excluding clinical mimics. MRI may reveal phenotypes common to a few AE-related disorders (e.g., limbic encephalitis [LE]) (Fig. 3). Abnormalities on MRI must instigate further workup for etiological triggers (e.g., evaluation of neoplasms causing paraneoplastic AE) (Fig. 4). Serum and CSF analyses constitute a confirmatory step in the diagnosis of AE and are often warranted based on MRI features and clinical suspicion [12]. The algorithmic approach is illustrated in Figure 5.
Fig. 3. Imaging features of limbic encephalitis in a 38-year-old patient with seronegative autoimmune encephalitis presenting with medically refractory focal seizures. A, B: Axial FLAIR images reveal confluent hyperintense signal abnormalities (arrows) involving both the temporal lobes, asymmetrically more on the right. C, D: Contrast enhanced T1 images show patchy areas of parenchymal enhancement (arrows) scattered within the areas of FLAIR hyperintense signal abnormality.

Fig. 4. Paraneoplastic syndrome in a 52-year-old patient presenting with expressive aphasia and speech apraxia. A: Axial FLAIR image reveals abnormal hyperintense, gyriform, cortical swelling involving the frontal lobes bilaterally (arrows), more on the left. B: Axial diffusion weighted image reveals mild facilitated diffusion in the frontal lobes (arrows) corresponding to the areas of FLAIR hyperintensity. C: Axial contrast-enhanced-T1 image reveals no definite enhancement within the affected frontal cortices. The possibility of AE was raised in addition to other differentials such as an atypical infective etiology. D: 68-Ga-DOTATATE PET, performed as part of the workup for AE, shows avid right paratracheal nodal disease (arrow) indicating somatostatin receptor positive neuroendocrine tumor in the mediastinum. E: Histopathological analysis (H&E stain) from the affected left frontal cortex reveals perivascular lymphocytic inflammation (arrow) and microglial nodules (circle). F: Histopathological analysis (H&E stain) from the affected left frontal cortex in another location reveals neuronophagia: neuron surrounded by microglia (circle). G: Immunocytochemical staining (CD8 stain) marked CD8+ T-cell infiltrates, with some directly apposed to neurons (circle), consistent with cytotoxic T-cell mediated neuronal attack. H: Neuronal nuclear protein (NeuN stain) stains obtained later in the course of the disease shows decreased neuronal population in the cortex (circle). The constellation of these pathological findings helped confirm the diagnosis of paraneoplastic AE. AE = autoimmune encephalitis.

Fig. 5. Algorithmic demonstration of the diagnostic approach in a patient with suspected AE. Modified from Rössling et al. Neuro Res Pract 2020;2:1 [12]. AE = autoimmune encephalitis, CSF = cerebrospinal fluid, PNS = paraneoplastic syndrome.

In this review, we discuss the common AE entities that affect children and adults. These disorders can be divided into two groups based on the location of the target: cell surface antigens or intracellular antigens [9].
Group I Antibodies: Antibodies Against Cell-Surface Antigens
Anti-NMDAR Encephalitis
The NMDAR is an ionotropic receptor that plays a role in neuroplasticity, behavior, and memory. Anti-NMDAR antibodies target the NMDAR (NR) 1 subunit of the receptor, causing reversible receptor internalization, for which tumors (especially teratomas) and post-viral states are common immunologic triggers [13]. However, almost 50% of cases have no identifiable triggers [14].
Anti-NMDAR encephalitis typically occurs in young patients (median age: 21 years; range: <1–85 years) [15]. Patients initially present with a viral-like prodrome (fever, malaise, headache), followed by psychiatric symptoms (anxiety, depression, psychosis) and temporal lobe dysfunction (amnesia, seizures), and eventually profound neurological deficits (autonomic dysfunction, dystonia, dyskinesia, and encephalopathy) [9].
In a study by Zhang et al. [16] (n = 53), brain MRI findings were divided into four groups: those with normal examinations (n = 28; 53%), only hippocampal lesions (n = 7; 13%), lesions sparing the hippocampus (n = 7; 13%), and mixed lesions (n = 11; 21%) (Figs. 6, 7). These lesions demonstrated T2/FLAIR hyperintensities and hypointense T1 signals (Figs. 6, 7) [16]. Leptomeningeal enhancement is rare [17,18].
Fig. 6. Anti-NMDAR encephalitis in a 12-year-old patient presenting with neuropsychiatric symptoms and movement disorder. A: Axial FLAIR image reveals bilateral asymmetric hyperintense signals involving the basal ganglia (right more than left; long arrows) and the right external capsule (short arrow). B: Axial FLAIR image on follow-up after intensive immunosuppressive therapy reveals complete resolution of the signal abnormality. NMDAR = N-methyl-D-aspartate receptor.

Fig. 7. Anti-NMDAR encephalitis in a 38-year-old patient presenting with neuropsychiatric symptoms. A, B: Presentation MRI. Axial (A) and coronal (B) FLAIR images reveal abnormal, gyriform hyperintense swelling along the superior and lateral aspects of the left temporal lobe (arrows). C, D: Follow up MRI. Axial FLAIR images reveal interim development of new areas of abnormal, hyperintense signals involving the cingulate gyri (arrows in C) and the left frontal lobe (arrow in D). NMDAR = N-methyl-D-aspartate receptor.

Anti-Voltage Gated Potassium Channel (VGKC) Encephalitis
The VGKC ensures neuronal return to the resting state following an action potential. Antibody-mediated disruption of this physiological process has been implicated in neuronal hyperexcitability disorders (Issacs’ syndrome [acquired neuromyotonia], Morvan’s syndrome [neuromyotonia, cognitive impairment, sleep disturbances, and dysautonomia], and LE) [19]. The antibodies target one of two VGKC cell surface antigens, leucine-rich glioma-inactivated protein 1 (LGI1) or contactin-associated protein 2 (CASPR2) [19].
Anti-LGI1 encephalitis shows a male predominance (50-70 years), with patients presenting with features of LE [20]. MRI may reveal unilateral or bilateral mesial temporal T2/FLAIR hyperintensities. Additionally, T2/FLAIR hyperintensities in the basal ganglia have also been reported in patients with anti-LG1 associated faciobrachial dystonic seizures. Interestingly, 18F-fluorodeoxyglucose (FDG)-PET demonstrates hypermetabolism in the mesial temporal lobes (MTL) and/or basal ganglia, even in the absence of MRI abnormalities [3]. Cerebellar involvement has rarely been described, particularly in children (Fig. 8) [21].
Fig. 8. Autoimmune cerebellitis in a 19-year-old patient with anti-VGKC encephalitis presenting with a one-month history of arm twitching (right > left). A, B: Axial FLAIR images reveal patchy, ill-defined hyperintensities involving the cerebellar hemispheres (arrows), more on the right. C, D: Contrast-enhanced-T1 images reveal questionable faint enhancement in the right cerebellar lesions (arrow in C) and subtle leptomeningeal enhancement along the right interfolial spaces (arrow in D). Anti-LGI1 encephalitis (a subgroup of anti-VGKC encephalitis) is known to present with cerebellar involvement. VGKC = voltage gated potassium channel.

Anti-CASPR2 antibodies cause LE and Morvan syndrome. Seizures and cognitive impairments are common. Additionally, almost 50% of patients develop peripheral nerve hyperexcitability (fasciculation, myokymia) [20]. In a retrospective study of anti-CASPR2 encephalitis (n = 38), 70% of the MRI studies did not reveal any abnormalities, while 24% showed bilateral T2/FLAIR hyperintensity in the MTL (Fig. 9). Brainstem involvement (n = 1) and cerebellar atrophy (n = 1) were also noted [22].
Fig. 9. Anti-CASPR2 encephalitis in a 64-year-old patient presenting with a one-year history of cognitive decline, mood and sleep disturbances, as well as gait difficulties. A-C: Axial 3D-FLAIR (A, B) and its coronal reconstruction (C) reveal abnormal FLAIR hyperintensity within the mesial temporal lobes on both sides (arrows in A), with asymmetric involvement of the right amygdala (arrows in B, C). There was no corresponding restricted diffusion or contrast enhancement noted (not shown). CASPR2 = contactin-associated protein 2.

Anti-GABA Encephalitis
Gamma-aminobutyric acid (GABA) is an inhibitory neurotransmitter. When GABA binds to specific receptors (i.e., GABA-AR and GABA-BR), it triggers the opening of chloride or potassium channels, causing a decrease in the transmembrane potential [3].
Antibodies against GABA-ARs cause a reduction in receptor density by internalization. Patients often present with epilepsia partialis continua, status epilepticus, anxiety and/or insomnia [3]. On MRI, confluent or patchy cerebral T2/FLAIR hyperintensities may be observed, with the latter associated with a better prognosis (Fig. 10). These areas of abnormal signals often involve the limbic system and frontal lobes [23].
Fig. 10. Anti-GABA-AR encephalitis in a 65-year-old patient initially presenting with focal motor seizures. A, B: MRI on presentation. Axial FLAIR (A) and CE-T1 (B) images reveal a focal patchy area of non-enhancing, hyperintense signal abnormality involving the posteromesial right frontal cortex and juxtacortical white matter (arrow). C, D: MRI on follow-up. Axial FLAIR (C) and CE-T1 (D) images reveals a new confluent, mildly expansile, non-enhancing area of hyperintense signal abnormality involving the right superior temporal gyrus (arrow). CE = contrast-enhanced.

GABA-BRs are concentrated in the hippocampus, thalamus, and cerebellum. GABA-BR antibodies directly block the receptor function. The patients present with seizures, movement disorders, memory loss, opsoclonus myoclonus, and cerebellar ataxia [3]. Anti-GABA-BR encephalitis is associated with small cell lung cancer (SCLC) and neuroendocrine tumors. On MRI, unilateral or bilateral T2/FLAIR hyperintensities involving MTLs may be observed, while 18F-FDG-PET may reveal hypermetabolism in the MTLs with diffuse cortical hypometabolism [3].
Neuromyelitis Optica (NMO)
NMO is an immunoglobulin G (IgG)-mediated disorder targeting AQP4, a transmembrane water channel located at the astrocytic foot processes [24]. AQP4 is abundantly expressed in the CNS (optic nerves, periventricular regions, hypothalamus, circumventricular organs, brainstem [area postrema], and spinal cord) [25]. NMO typically presents between 32–45 years of age, with pediatric presentation accounting for 3%–5% of cases [25]. Presentations include bilateral painless optic neuritis, transverse myelitis (motor, sensory, and sphincteric pathway involvement), area postrema syndrome (vomiting and hiccoughs), diencephalic syndrome (narcolepsy and altered temperature regulation), brainstem syndromes (cranial nerve palsies, ataxia, and weakness), encephalopathy, and seizures [24].
Confluent T2/FLAIR hyperintensities involving the periependymal and periaqueductal regions, the callososeptal interface, the diencephalon, and the brainstem are frequently observed on MRI. Approximately 9%–36% of lesions show contrast enhancement, a feature associated with higher relapse rates. Involvement of bilateral intracranial optic nerve segments is common, and T2 hyperintensity and avid enhancement (acute phase) or volume loss (chronic phase) are stereotypical features. Longitudinally extensive transverse myelitis (LETM) is the hallmark of spinal cord involvement, and T2 hyperintensity with lentiform areas of contrast enhancement is pathognomonic. Relatively “bright spotty” T2 hyperintensities may be seen intrinsic to the signal abnormality, representing necrotic and microcystic changes (Figs. 11, 12, 13) [25].
Fig. 11. Typical imaging features of a neuromyelitis optica in an 18-year-old patient presenting with altered mental status. A: Coronal CE T1 weighted image reveals patchy enhancement involving the hypothalamus (arrow) and the corpus striatum. B: Axial CE-T1FS shows long segment enhancement within the posterior aspect of the left optic nerve (arrow), relatively sparing the retrolaminar segment. C: Sagittal T2FS image of the spine reveals long segment T2 hyperintensity within the thoracic cord (arrow), sparing the conus. D: Axial T2FS image reveals diffuse involvement of the cord (i.e., involving both grey and white matter), with a punctate, “bright spotty” T2 hyperintense lesion (arrow) intrinsic to the area of signal abnormality. E: CE-T1 image through the thoracic cord reveals heterogeneous, biconvex enhancement within the mid-thoracic cord (arrow). CE = contrast-enhanced, FS = fat saturation.

Fig. 12. Imaging features of area postrema syndrome in a 15-year-old patient with neuromyelitis optica presenting with persistent vomiting. A, B: Axial FLAIR image (A) and axial diffusion weighted image (B) reveal a small area of hyperintense signal abnormality in the dorsal medulla, in the expected location of the area postrema (arrows).

Fig. 13. Atypical imaging features of longitudinally extensive transverse myelitis related to neuromyelitis optica in a 10-year-old patient presenting with lower limb weakness and gait ataxia. A, B: Sagittal (A) and axial (B) T2 images reveal a multicystic, mildly expansile lesion extensively involving the lower cervical and thoracic cord (arrows). C: Sagittal CE-T1 image reveals mild, nodular enhancement predominantly along the posterior aspect of the lesion (arrow). A glioma was initially considered, however active demyelination was confirmed on biopsy of the lesion and anti-aquaporin 4 immunoglobulin G was identified on the serum. D, E: Sagittal (D) and axial (E) T2 images obtained after 4 weeks of immunosuppressive therapy reveals marked reduction in the extent of the lesion and the associated cord expansion (arrows). CE-T1 image (not shown) was motion degraded; however, there was considerable reduction in the degree of enhancement. CE = contrast-enhanced.

Myelin Oligodendrocyte Glycoprotein Antibody–Associated Disease (MOGAD)
MOG is a minor component of myelin in the CNS. MOG-IgG binds to extracellular MOG domains located in the peripheral processes of oligodendrocytes. This binding subsequently activates the complement cascade, ultimately leading to demyelination [25]. Infectious prodromes frequently precede symptom onset in MOGAD (60% of the cases) [26]. Patients often present with monophasic or relapsing acute optic neuritis, transverse myelitis, encephalitis or rhombencephalitis [26]. Encephalopathy, fever, headache, seizures, ataxia, hemiparesis, and dysarthria have also been observed [27].
Less than 50% of adults have brain lesions on presentation MRI, seen as bilateral, ill-defined T2/FLAIR hyperintensities. Approximately 30% of patients show predominantly infratentorial involvement. Children may also have confluent brainstems and deep gray lesions. Involvement of the cerebellar peduncle is common in children. Cortical lesions, leptomeningeal enhancement, and cranial nerve involvement were rarely observed (Fig. 14). Bilateral, long-segment involvement of the anterior intraconal optic nerves is pathognomonic, commonly demonstrating edematous T2 hyperintense swelling with intrinsic enhancement, as well as enhancement of the perioptic sheaths and fat (Fig. 15). LETM and short-segment involvement of the spinal cord have been previously described. Spinal cord lesions demonstrate T2 hyperintensity involving both gray and white matter (>50% cord diameter). Illustrated contrast enhancement and conus involvement are characteristic of MOGAD [26].
Fig. 14. Imaging findings of myelin oligodendrocyte glycoprotein antibody–associated disease in a 6-year-old patient presenting with 2-week history of low-grade fever and headache. A, B: Coronal T2 (A) and axial FLAIR (B) images reveal asymmetric, hyperintense, gyriform swelling of the right mesial parieto-occipital lobes (arrows), whereas CE-T1 revealed no corresponding enhancement (not shown). C: CE-FLAIR image reveals mild leptomeningeal enhancement within the adjacent sulcal spaces (arrow). CE = contrast-enhanced.

Fig. 15. Imaging features of myelin oligodendrocyte glycoprotein antibody–associated disease-related optic neuritis in a 10-year-old patient presenting with acute loss of vision in the left eye. A: Axial T2FS image through the left orbit reveals longitudinally extensive hyperintense swelling of the left optic nerve along its entire intraconal length (long arrow). Note the faint peri-optic fat stranding in the retrobulbar region (short arrow). B: Contrast-enhanced-T1FS image reveals abnormal enhancement within the affected optic nerve along with ill-defined enhancement of the peri-optic fat (arrow) and marked enhancement of the peri-optic sheath posteriorly (dotted arrow). FS = fat saturation.

Group II Antibodies: Antibodies Against Intracellular Antigens
Anti-Ma2 Encephalitis
Ma1, Ma2, and Ma3 are homologous proteins involved in mRNA synthesis [28]. Ma1 and Ma2 are onconeuronal proteins [28,29]. Antibodies against Ma1 and Ma2 are associated with paraneoplastic cerebellar degeneration (PCD) and brainstem encephalitis in several primary cancers (lung and testicular tumors). Additionally, anti-Ma2 antibodies have also been associated with LE and diencephalic lesions in germ cell tumors of the testes [29,30,31].
Patients generally present with psychiatric disturbances, ataxia, or Parkinsonism (rigidity, tremor, and dystonic posturing) [31]. Patients may also present with narcolepsy or ophthalmoparesis due to diencephalic or upper brainstem involvement [31]. On MRI, lesions related to anti-Ma encephalitis often demonstrate hyperintense signals on T2/FLAIR sequences, with or without corresponding T1 hypointensity. Occasionally, lesions demonstrated contrast enhancement (Fig. 16). Lesions may show increased choline and acetate levels with reduced N-acetyl-aspartate levels on MR spectroscopy, as well as hypermetabolism on 18F-FDG-PET imaging [32].
Fig. 16. Anti-Ma2 encephalitis in a 20-year-old patient presenting with hyperphagia, hypersomnia, and endocrinopathy. A, B: Axial FLAIR (A) and coronal T2 (B) images reveal abnormal hyperintense signals centered within the hypothalamus (arrows). C: Contrast-enhanced-T1 reveals abnormal enhancement within the region of the median eminence (arrow).

Anti-GAD Encephalitis
Glutamic acid decarboxylase (GAD) catalyzes the conversion of glutamic acid into GABA, which is a potent inhibitory neurotransmitter. This enzyme is present in the GABAergic cytoplasm of neurons and secretory vesicles [33]. It is also expressed in multiple extra-CNS locations (pancreatic β-cells, liver, testes, fallopian tubes, kidney, adrenal glands). GAD has two isoforms (65 and 67 kDa) encoded on chromosomes 10 and 2, respectively [33].
Anti-GAD antibodies were first described in association with the stiff-person syndrome and subsequently in patients with type I diabetes mellitus [33]. It is also associated with refractory seizures, cerebellar degeneration, nystagmus, and other autoimmune disorders (psoriasis, celiac disease, autoimmune thyroiditis) [34]. Recently, anti-GAD antibodies have been implicated in paraneoplastic (SCLC and malignant thymoma) and non-paraneoplastic LE [34].
Patients with anti-GAD encephalitis present with seizures (97%) and cognitive impairment (66%). Other uncommon manifestations include psychiatric symptoms, dysautonomia, cerebellar dysfunction, and headaches [34]. In a retrospective study that included a cohort of 19 patients with confirmed anti-GAD65 encephalitis, Fredriksen et al. [35] found that parenchymal atrophy (47%) was the most common finding, followed by cortical/subcortical (37%) and hippocampal (26%) T2/FLAIR hyperintense signal abnormalities (Fig. 17). No lesions showed contrast enhancement [35].
Fig. 17. Anti-GAD65 encephalitis in a 20-year-old patient initially presenting with headache and episodes of bizarre behavior. A, B: MRI on presentation. Coronal (A) and axial (B) FLAIR images reveal unilateral hyperintense swelling involving the right hippocampal formation (arrows). There was no contrast enhancement noted on this study (not shown). C, D: MRI on follow-up. Axial FLAIR (C) reveals new hyperintense swelling of the left MTL, predominantly involving the left hippocampal formation (long arrow in C). Note the interim volume loss of the right MTL (short arrow). CE-T1 image (D) reveals a focal area of abnormal contrast enhancement along the left uncus (arrow). MTL = mesial temporal lobe.

Anti-Yo-Mediated PCD
PCD, related to progressive Purkinje cell loss, is an immune-mediated syndrome without structural, infectious, or toxic metabolic causes [36,37]. Although rare, they may be associated with SCLC, breast cancer, lymphoma, and gynecological malignancies [37]. Anti-Yo antineuronal antibodies (Purkinje cell cytoplasmic antibody type 1) are specific for PCD; however, multiple other antibodies have also been identified [37].
Anti-Yo-mediated PCD showed a female preponderance (>60 years old). Almost 75% and 15% of patients had an underlying gynecologic malignancy and breast cancer, respectively [37]. Symptomatology includes acute or subacute onset of ataxia, nystagmus, vertigo, diplopia, oscillopsia, stroke-like episodes, and cognitive deficits [37]. MR imaging may be normal in the acute phase or may demonstrate T2/FLAIR hyperintense cerebellar swelling. In the chronic phase, the signal abnormality resolves with residual cerebellar atrophy (Fig. 18). Additionally, 18F-FDG-PET may reveal cerebellar hypometabolism during the chronic phase [36].
Fig. 18. Anti-Yo-related paraneoplastic cerebellar degeneration in a 51-year-old patient, with high grade serous ovarian cancer, presenting with progressive truncal and appendicular ataxia along with impaired speech. A: Sagittal T1 image on presentation revealed no significant abnormality. B: Sagittal T1 image on follow up (after 2 years) reveals marked volume loss of the cerebellar vermis (arrow) with prominence of the interfolial spaces. C: Axial 3D-FLAIR-fat saturation reveals subtle, near symmetric hyperintense signal abnormality within the cerebellar hemispheres as well (arrows). An incidental left schwannoma along the left VII-VIII nerve complex (dotted arrow) was also noted.

Opsoclonus-Myoclonus-Ataxia Syndrome (OMAS)
Opsoclonus-myoclonus-ataxia syndrome (OMAS) is characterized by saccadic eye movements, cerebellar ataxia, and choreiform extremity movement [38]. It typically begins in the 2nd decade of life, with 50% of patients having an underlying peripheral neuroblastic neoplasm (i.e., neuroblastoma [most common], ganglioneuroblastoma, ganglioneuroma) [39]. Non-paraneoplastic, parainfectious OMAS (Mycoplasma, Streptococcus, Epstein-Barr virus, and adenovirus) have also been recorded, albeit less common [39]. Adult onset OMAS is rare and often paraneoplastic (lung, breast, ovarian carcinomas; ANNA2/anti-Ri antibodies) [1,40].
The exact pathophysiology of OMAS is unknown; however, its potential role in autoimmunity has been proposed [38]. At the neuronal level, the dysfunction of Purkinje cells or their connections with the fastigial nucleus has been proposed [38]. No specific OMAS biomarker has been identified; however, CSF studies may show B-cell expansion and oligoclonal bands [39].
Imaging plays an important role in detecting the underlying neoplastic associations. MR imaging of the brain is often normal during the acute phase; The cerebellar volume loss, sparing the supratentorial brain, develops over time (Fig. 19). Hayward et al. [41] found that atrophy was generally pan-cerebellar.
Fig. 19. Imaging features of opsoclonus-myoclonus-ataxia syndrome in a 3-year-old patient. A: Sagittal T1 image reveals marked volume loss of the vermis (arrow). B: Coronal T2 image also reveals volume loss of the cerebellar hemispheres, without signal abnormality. C: Coronal contrast-enhance-CT image of the abdomen reveals a neuroblastoma (arrow) involving the right adrenal gland.

CONCLUSION
Since the discovery of autoantibodies and their pathomechanistic effects, there has been a paradigm shift in the diagnostic and treatment considerations for patients presenting with subacute memory loss, altered mentation, seizures, movement disorders, and psychiatric symptoms. In recent decades, vast strides have been made in the identification of a myriad of potential autoimmune triggers and antibodies, enabling better treatment and clinical outcomes. Despite considerable imaging overlap with other entities, AE remains an important imaging consideration in an appropriate setting. This can initiate a more comprehensive evaluation of underlying conditions (e.g., neoplasms) and prompt accurate treatment.
Footnotes
Conflicts of Interest: The authors have no potential conflicts of interest to disclose.
- Conceptualization: Vivek Pai, Manohar Shroff.
- Supervision: Manohar Shroff.
- Writing—original draft: Vivek Pai.
- Writing—review & editing: Heejun Kang, Suradech Suthiphosuwan, Andrew Gao, Daniel Mandell, Manohar Shroff.
Funding Statement: None
References
- 1.Neaţu M, Jugurt A, Covaliu A, Davidescu EI, Popescu BO. Autoimmune encephalitis-a multifaceted pathology. Biomedicines. 2023;11:2176. doi: 10.3390/biomedicines11082176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel A, Meng Y, Najjar A, Lado F, Najjar S. Autoimmune encephalitis: a physician’s guide to the clinical spectrum diagnosis and management. Brain Sci. 2022;12:1130. doi: 10.3390/brainsci12091130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu S, Yu J, Wu Y, Peng J, Xie X, Zhang X, et al. Pathophysiology and clinical management of autoimmune encephalitis-associated seizures. Neuroimmunomodulation. 2022;29:282–295. doi: 10.1159/000524783. [DOI] [PubMed] [Google Scholar]
- 4.Ampie L, McGavern DB. Immunological defense of CNS barriers against infections. Immunity. 2022;55:781–799. doi: 10.1016/j.immuni.2022.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Louveau A, Plog BA, Antila S, Alitalo K, Nedergaard M, Kipnis J. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J Clin Invest. 2017;127:3210–3219. doi: 10.1172/JCI90603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rojas M, Restrepo-Jiménez P, Monsalve DM, Pacheco Y, Acosta-Ampudia Y, Ramírez-Santana C, et al. Molecular mimicry and autoimmunity. J Autoimmun. 2018;95:100–123. doi: 10.1016/j.jaut.2018.10.012. [DOI] [PubMed] [Google Scholar]
- 7.Jessen NA, Munk AS, Lundgaard I, Nedergaard M. The glymphatic system: a beginner’s guide. Neurochem Res. 2015;40:2583–2599. doi: 10.1007/s11064-015-1581-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Platt MP, Agalliu D, Cutforth T. Hello from the other side: how autoantibodies circumvent the blood-brain barrier in autoimmune encephalitis. Front Immunol. 2017;8:442. doi: 10.3389/fimmu.2017.00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelley BP, Patel SC, Marin HL, Corrigan JJ, Mitsias PD, Griffith B. Autoimmune encephalitis: pathophysiology and imaging review of an overlooked diagnosis. AJNR Am J Neuroradiol. 2017;38:1070–1078. doi: 10.3174/ajnr.A5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Könnecke H, Bechmann I. The role of microglia and matrix metalloproteinases involvement in neuroinflammation and gliomas. Clin Dev Immunol. 2013;2013:914104. doi: 10.1155/2013/914104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldmann T, Prinz M. Role of microglia in CNS autoimmunity. Clin Dev Immunol. 2013;2013:208093. doi: 10.1155/2013/208093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rössling R, Prüss H. SOP: antibody-associated autoimmune encephalitis. Neurol Res Pract. 2020;2:1. doi: 10.1186/s42466-019-0048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding H, Jian Z, Stary CM, Yi W, Xiong X. Molecular pathogenesis of anti-NMDAR encephalitis. Biomed Res Int. 2015;2015:643409. doi: 10.1155/2015/643409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang Q, Xie Y, Hu Z, Tang X. Anti-N-methyl-D-aspartate receptor encephalitis: a review of pathogenic mechanisms, treatment, prognosis. Brain Res. 2020;1727:146549. doi: 10.1016/j.brainres.2019.146549. [DOI] [PubMed] [Google Scholar]
- 15.Sun Y, Ren G, Ren J, Shan W, Han X, Lian Y, et al. The association between age and prognosis in patients under 45 years of age with anti-NMDA receptor encephalitis. Front Neurol. 2020;11:612632. doi: 10.3389/fneur.2020.612632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang T, Duan Y, Ye J, Xu W, Shu N, Wang C, et al. Brain MRI characteristics of patients with anti-N-methyl-D-aspartate receptor encephalitis and their associations with 2-year clinical outcome. AJNR Am J Neuroradiol. 2018;39:824–829. doi: 10.3174/ajnr.A5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Mater H. Pediatric inflammatory brain diseases: a diagnostic approach. Curr Opin Rheumatol. 2014;26:553–561. doi: 10.1097/BOR.0000000000000092. [DOI] [PubMed] [Google Scholar]
- 18.Budhram A, Britton JW, Liebo GB, Dubey D, Zekeridou A, Flanagan EP, et al. Use of diffusion-weighted imaging to distinguish seizure-related change from limbic encephalitis. J Neurol. 2020;267:3337–3342. doi: 10.1007/s00415-020-10007-1. [DOI] [PubMed] [Google Scholar]
- 19.Langille MM, Desai J. Encephalitis due to antibodies to voltage gated potassium channel (VGKC) with cerebellar involvement in a teenager. Ann Indian Acad Neurol. 2015;18:238–239. doi: 10.4103/0972-2327.150623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Sonderen A, Petit-Pedrol M, Dalmau J, Titulaer MJ. The value of LGI1, Caspr2 and voltage-gated potassium channel antibodies in encephalitis. Nat Rev Neurol. 2017;13:290–301. doi: 10.1038/nrneurol.2017.43. [DOI] [PubMed] [Google Scholar]
- 21.Weihua Z, Haitao R, Jie D, Changhong R, Ji Z, Anna Z, et al. Autoimmune cerebellar ataxia associated with anti-leucine-rich glioma-inactivated protein 1 antibodies: two pediatric cases. J Neuroimmunol. 2022;370:577918. doi: 10.1016/j.jneuroim.2022.577918. [DOI] [PubMed] [Google Scholar]
- 22.van Sonderen A, Ariño H, Petit-Pedrol M, Leypoldt F, Körtvélyessy P, Wandinger KP, et al. The clinical spectrum of Caspr2 antibody-associated disease. Neurology. 2016;87:521–528. doi: 10.1212/WNL.0000000000002917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deng B, Cai M, Qiu Y, Liu X, Yu H, Zhang X, et al. MRI characteristics of autoimmune encephalitis with autoantibodies to GABAA receptor: a case series. Neurol Neuroimmunol Neuroinflamm. 2022;9:e1158. doi: 10.1212/NXI.0000000000001158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clarke L, Arnett S, Lilley K, Liao J, Bhuta S, Broadley SA. Magnetic resonance imaging in neuromyelitis optica spectrum disorder. Clin Exp Immunol. 2021;206:251–265. doi: 10.1111/cei.13630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dutra BG, da Rocha AJ, Nunes RH, Maia ACM., Júnior Neuromyelitis optica spectrum disorders: spectrum of MR imaging findings and their differential diagnosis. Radiographics. 2018;38:169–193. doi: 10.1148/rg.2018170141. [DOI] [PubMed] [Google Scholar]
- 26.Shahriari M, Sotirchos ES, Newsome SD, Yousem DM. MOGAD: how it differs from and resembles other neuroinflammatory disorders. AJR Am J Roentgenol. 2021;216:1031–1039. doi: 10.2214/AJR.20.24061. [DOI] [PubMed] [Google Scholar]
- 27.Wegener-Panzer A, Cleaveland R, Wendel EM, Baumann M, Bertolini A, Häusler M, et al. Clinical and imaging features of children with autoimmune encephalitis and MOG antibodies. Neurol Neuroimmunol Neuroinflamm. 2020;7:e731. doi: 10.1212/NXI.0000000000000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sahashi K, Sakai K, Mano K, Hirose G. Anti-Ma2 antibody related paraneoplastic limbic/brain stem encephalitis associated with breast cancer expressing Ma1, Ma2, and Ma3 mRNAs. J Neurol Neurosurg Psychiatry. 2003;74:1332–1335. doi: 10.1136/jnnp.74.9.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenfeld MR, Eichen JG, Wade DF, Posner JB, Dalmau J. Molecular and clinical diversity in paraneoplastic immunity to Ma proteins. Ann Neurol. 2001;50:339–348. [PubMed] [Google Scholar]
- 30.Ortega Suero G, Sola-Valls N, Escudero D, Saiz A, Graus F. Anti-Ma and anti-Ma2-associated paraneoplastic neurological syndromes. Neurologia (Engl Ed) 2018;33:18–27. doi: 10.1016/j.nrl.2016.05.010. [DOI] [PubMed] [Google Scholar]
- 31.Neto ADP, de Aquino BCV, de Morais Brito PS, eSilva RDA, Neto MM, Barsottini OGP, et al. ANTI-MA2 encephalitis mimicking diencephalic demyelinating syndrome. Interdiscip Neurosurg. 2021;23:100980 [Google Scholar]
- 32.Co DO, Kwon JM. Autoimmune encephalitis: distinguishing features and specific therapies. Crit Care Clin. 2022;38:393–412. doi: 10.1016/j.ccc.2021.11.007. [DOI] [PubMed] [Google Scholar]
- 33.Vianello M, Tavolato B, Giometto B. Glutamic acid decarboxylase autoantibodies and neurological disorders. Neurol Sci. 2002;23:145–151. doi: 10.1007/s100720200055. [DOI] [PubMed] [Google Scholar]
- 34.Mansoor S, Murphy K. Anti-GAD-associated limbic encephalitis: an unusual clinical manifestation from northwest of Ireland. Egypt J Neurol Psychiatry Neurosurg. 2020;56:23 [Google Scholar]
- 35.Fredriksen JR, Carr CM, Koeller KK, Verdoorn JT, Gadoth A, Pittock SJ, et al. MRI findings in glutamic acid decarboxylase associated autoimmune epilepsy. Neuroradiology. 2018;60:239–245. doi: 10.1007/s00234-018-1976-6. [DOI] [PubMed] [Google Scholar]
- 36.Madhavan AA, Carr CM, Morris PP, Flanagan EP, Kotsenas AL, Hunt CH, et al. Imaging review of paraneoplastic neurologic syndromes. AJNR Am J Neuroradiol. 2020;41:2176–2187. doi: 10.3174/ajnr.A6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le May M, Dent S. Anti-Yo antibody-mediated paraneoplastic cerebellar degeneration associated with cognitive affective syndrome in a patient with breast cancer: a case report and literature review. Curr Oncol. 2018;25:e585–e591. doi: 10.3747/co.25.4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shah NS, Pathak J, Shah PC, Bharmal UF, Ansari MI. A rare case of opsoclonus myoclonus ataxia syndrome post viral illness. Cureus. 2023;15:e40396. doi: 10.7759/cureus.40396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rossor T, Yeh EA, Khakoo Y, Angelini P, Hemingway C, Irani SR, et al. Diagnosis and management of opsoclonus-myoclonus-ataxia syndrome in children: an international perspective. Neurol Neuroimmunol Neuroinflamm. 2022;9:e1153. doi: 10.1212/NXI.0000000000001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lino AMM, Spera RR, de Campos FPF, Freitas CHA, Garcia MRT, Lopes LDC, et al. Adult-onset opsoclonus-myoclonus-ataxia syndrome as a manifestation of Brazilian lyme disease-like syndrome: a case report and review of literature. Autops Case Rep. 2014;4:29–37. doi: 10.4322/acr.2014.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hayward K, Jeremy RJ, Jenkins S, Barkovich AJ, Gultekin SH, Kramer J, et al. Long-term neurobehavioral outcomes in children with neuroblastoma and opsoclonus-myoclonus-ataxia syndrome: relationship to MRI findings and anti-neuronal antibodies. J Pediatr. 2001;139:552–559. doi: 10.1067/mpd.2001.118200. [DOI] [PubMed] [Google Scholar]