

Abstract
Tumorigenesis arises from the dysfunction of cancer genes, leading to uncontrolled cell proliferation through various mechanisms. Establishing a complete cancer gene catalogue will make precision oncology possible. Although existing methods based on graph neural networks (GNN) are effective in identifying cancer genes, they fall short in effectively integrating data from multiple views and interpreting predictive outcomes. To address these shortcomings, an interpretable representation learning framework IMVRL-GCN is proposed to capture both shared and specific representations from multiview data, offering significant insights into the identification of cancer genes. Experimental results demonstrate that IMVRL-GCN outperforms state-of-the-art cancer gene identification methods and several baselines. Furthermore, IMVRL-GCN is employed to identify a total of 74 high-confidence novel cancer genes, and multiview data analysis highlights the pivotal roles of shared, mutation-specific, and structure-specific representations in discriminating distinctive cancer genes. Exploration of the mechanisms behind their discriminative capabilities suggests that shared representations are strongly associated with gene functions, while mutation-specific and structure-specific representations are linked to mutagenic propensity and functional synergy, respectively. Finally, our in-depth analyses of these candidates suggest potential insights for individualized treatments: afatinib could counteract many mutation-driven risks, and targeting interactions with cancer gene SRC is a reasonable strategy to mitigate interaction-induced risks for NR3C1, RXRA, HNF4A, and SP1.
Keywords: cancer gene identification, interpretable deep learning, multiview representation learning, graph neural network, precision oncology
Introduction
Cancer remains a leading global health threat, causing nearly 10 million deaths in 2020 [1]. Tumorigenesis involves gene function changes that provide a growth advantage to cells, thus identifying pivotal genes in the process as cancer genes [2]. Targeted therapies, such as vemurafenib for BRAF mutations [3] and IMAB362 for CLDN18.2 [4], have shown significant clinical success. However, tumor heterogeneity necessitates discovering new drivers to broaden targeted treatments.
Efforts to catalog known cancer genes (KCGs) include projects like the Network of Cancer Genes (NCG) [5] and the COSMIC Cancer Gene Census (CGC) [6]. Computational methods, leveraging genomic data, have accelerated this process. Methods like MuSiC [7], MutSigCV [8], deepDriver [9], and OncodriveCLUST [10] predict cancer genes through mutation analysis. Despite advances, relying solely on genomic data has limitations, ignoring mechanisms like epigenetic changes and protein interactions. Thus, the incorporation of diverse gene profiles is imperative.
Recent approaches predict cancer genes using complementary information from diverse gene profiles and interaction information from intricate protein–protein interaction (PPI) networks that finely control human traits. Methods like LOTUS [11] and ModulOmics [12] illustrate the value of multiview data and interaction data. However, they are not specifically designed for graph-structured data and cannot fully exploit resourceful interaction knowledge. Graph neural networks (GNNs) [13] are deep learning frameworks developed specifically for graph-structured data, enhancing characterization by leveraging multiview data and interaction knowledge. Recently, several GNN-based methods have been proposed for predicting cancer genes [14–16]. For example, EMOGI [14] integrates genomic, epigenomic, and transcriptomic features with PPI networks, demonstrating superior performance compared to methodologies lacking such integrative approaches. MTGCN [15] introduces an additional edge reconstruction task to utilize knowledge from unlabeled samples. MODIG [16] integrates multiple biological networks and utilizes multidimensional graph attention networks (GAT) to fuse multiple representations. These methods achieved elevated performances by considering multiview data and interaction knowledge simultaneously. Nevertheless, these methods straightforwardly integrated multiview data in a concatenated manner, thereby disregarding the significance of distinct views and failing to effectively leverage the consensus information inherent in multiple views. Another concern is the lack of interpretability and validation in the decision-making process for these deep learning methods, limiting the applicability and generalization of the results [17]. For cancer gene identification, obtaining a list of candidates without understanding the causal factors hinders further mechanism research and clinical therapy development.
Multiview representation learning is a promising approach for modeling multiview data. Existing approaches mainly adopt two strategies: fusion and alignment. The fusion strategy fuses features from multiple views into a condensed representation [18, 19], while the alignment strategy projects learned multiview representations into an aligned space [20–23]. Recently, a particular type, known as the shared-and-specific disentangled method [21, 22, 24], has attracted considerable interest. This method learns shared and specific representations from multiview data, demonstrating consensus and complementary effects. Research shows that the comprehensive consideration of consensus and complementary effects can augment downstream learning tasks’ performance [25, 26]. Moreover, these distinctly disentangled shared and specific representations make the model’s decision-making and prediction processes easier to understand.
In this work, we present IMVRL-GCN, an Interpretable Multi-View Representation Learning framework based on Graph Convolutional Network, to predict cancer genes using integrated multiview data and the PPI network. Compared to four self-implemented baselines and three state-of-the-art cancer gene identification models in different experiments, IMVRL-GCN demonstrated superior performance in both AUC and AUPR metrics. Ultimately, we compiled a list of 74 high-confidence candidate cancer genes (CCGs) by implementing IMVRL-GCN. To understand the model’s decisions, we applied a comprehensive analysis of the multiview representations, highlighting the importance of both shared and specific representations in novel predictions. Additionally, our in-depth analyses of these representations suggest potential therapeutic interventions. Overall, IMVRL-GCN presents a promising approach to complete the KCG catalogue and advance cancer treatment development.
Methods
Dataset collection
In this study, we employed Peng’s dataset [15] including multiview data (three-omics and one structural data) for cancer gene identification. The three-omics data includes genomic (somatic mutation), transcriptomic (gene expression), and epigenomic (DNA methylation) profiles. The structural data is the PPI network from the Carcinogenic Potency Database (CPDB version 34) [27]. The structural data not only serve as input to the GCNs but are also utilized for extracting structural features that can effectively improve the identification of cancer genes, as previously done by Peng et al. [15]. Our benchmark dataset comprises 796 positive samples from the Network of Cancer Genes (NCG version 6.0) [5] and DigSEE [28], and 2187 negative samples, obtained by excluding genes present in NCG [5], Kyoto Encyclopedia of Genes and Genomes(KEGG) cancer pathways [29], Online Mendelian Inheritance in Man (OMIM) database [30], MSigDB [31], and those correlated to cancer gene expression [32]. To investigate the influence of diverse PPI networks on model performance, we considered eight other PPI networks. The first PPI network was from the STRING database (version 11) [33], with a low-confidence interactions filtration threshold of 0.85. The remaining seven PPI networks were sourced from NDEx (version 2.5.1) [34]. We constructed corresponding datasets for these networks, and for ease of representation, we named the datasets according to the network’s name. The implementation is as follows: for each dataset, we utilized the same three-omics data as in Peng’s dataset, and for the structural data, we excluded genes that were deficient in multiview data and their connections.
Workflow overview of interpretable multiview representation learning framework based on graph convolutional network
Inspired by advances in multiview representation learning technology and accumulation of high-throughput biological data, we proposed IMVRL-GCN to improve the accuracy and interpretability of cancer gene identification. IMVRL-GCN comprises four modules: a multiview feature extractor to obtain gene multiview features, a shared representation learner to capture shared multiview information, a specific representation learner to learn view-specific information, and a cancer gene predictor to combine the shared information and specific information for cancer gene identification. The details of each module are outlined below and illustrated in Fig. 1.
Figure 1.

Overview of the IMVRL-GCN. The IMVRL-GCN is an end-to-end framework consisting of four modules, namely, the multiview feature extractor, the shared representation learner, the specific representation learner, and the cancer gene predictor. The shared and specific representation learners capture the consensus and complementary information from multiview features through their separate generators and discriminators, respectively. The cancer gene predictor yields the probability of a certain gene as a potential cancer gene based on the fusion of the consensus and complementary information.
The multiview feature extractor
Similar to MTGCN, we extracted multiview features from three-omics data and structural data. For genomic data, the gene mutation rate was defined as the average number of single-nucleotide variant (SNVs) across samples for a cancer type. For transcriptomic data, the differential expression rate per gene was calculated by averaging the log2 fold change between tumor and normal pairs. For epigenomic data, the differential DNA methylation rate per gene was characterized as the mean difference in the methylation signal values between tumor and normal pairs. These three-omics data were calculated for each of the 16 cancer types based on 8000 samples as the three-omics features [35]. For structural data, a network embedding algorithm (i.e. node2vec [36]) was performed on the PPI network from the CPDB database to obtain a feature of each gene, which can capture the topological structural information in the PPI network. Thus, we obtained four-view features for each gene. Given a set of genes, their multiview features can be expressed as 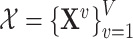 , where
, where 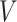 denotes the total number of views, which is equal to 4 for the four-view features in this study. For the
denotes the total number of views, which is equal to 4 for the four-view features in this study. For the 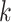 -th gene in the training set of
-th gene in the training set of 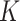 genes,
genes, 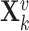 denotes its
denotes its 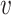 -th view feature,
-th view feature, 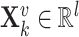 , with
, with 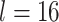 denoting the input gene feature dimension.
denoting the input gene feature dimension.
The shared representation learner
Shared representations should be consistent across multiview features to maintain their consensus impact on cancer gene identification. Specifically, we leveraged the GAN-based framework [22, 25] to render shared representations that present similar classification indicators for preserving their mutual influence on cancer gene identification. Given the 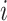 -th and the
-th and the 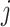 -th view features of the
-th view features of the 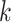 -th gene
-th gene 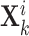 and
and 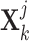 ,
, 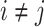 , we fed them into the well-designed generators
, we fed them into the well-designed generators 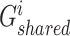 and
and 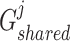 , respectively, which are multi-layer GCNs, where
, respectively, which are multi-layer GCNs, where 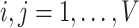 . After that, we obtained their shared representations
. After that, we obtained their shared representations 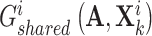 and
and 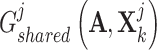 , where
, where 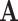 is the adjacent matrix of the PPI network, then employed a discriminator
is the adjacent matrix of the PPI network, then employed a discriminator 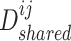 to distinguish the generated
to distinguish the generated 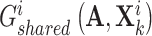 and
and 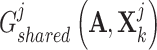 , resulting in the consensus score formulated as follows:
, resulting in the consensus score formulated as follows:
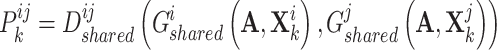 |
(1) |
where 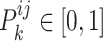 . In the training procedure, the designed generators and discriminator were assigned the opposite optimization objectives, with generators
. In the training procedure, the designed generators and discriminator were assigned the opposite optimization objectives, with generators 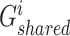 and
and 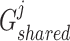 aiming at maximizing
aiming at maximizing 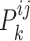 for capturing the consistent information between the
for capturing the consistent information between the 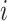 -th and the
-th and the 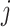 -th views and discriminator
-th views and discriminator 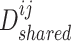 seeking to minimize
seeking to minimize 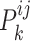 . That is, the generators
. That is, the generators 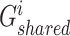 and
and 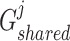 aim at deceiving discriminator
aim at deceiving discriminator 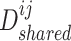 , resulting in the finally derived shared view representations
, resulting in the finally derived shared view representations 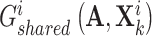 and
and 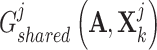 being as consistent as possible, contributing to the manifestation of consensus effects in cancer gene identification.
being as consistent as possible, contributing to the manifestation of consensus effects in cancer gene identification.
Therefore, the objective function of shared view representation learning can be written as:
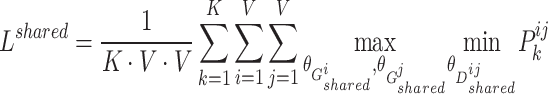 |
(2) |
where 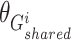 ,
, 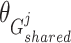 , and
, and 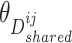 are the trainable parameters for generators
are the trainable parameters for generators 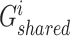 ,
, 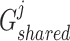 , and discriminator
, and discriminator 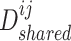 , respectively.
, respectively.
We then averaged all the shared view representations to get the final shared representations as 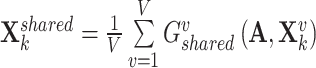 to retain the consistent contributions of its shared components.
to retain the consistent contributions of its shared components.
The specific representation learner
Specific view representations should own their respective distinctive classification indicators to preserve their individual influence on cancer gene identification. Given the 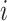 -th and the
-th and the 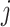 -th view representations
-th view representations 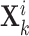 and
and 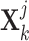 , we also first devised two separate multilayer GCNs
, we also first devised two separate multilayer GCNs 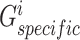 and
and 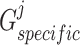 as generators and fed
as generators and fed 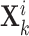 and
and 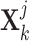 into
into 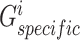 and
and 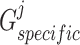 , respectively, to derive their specific view representations
, respectively, to derive their specific view representations 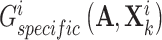 and
and 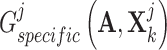 , and then exploited a discriminator
, and then exploited a discriminator 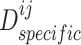 to discriminate the generated
to discriminate the generated 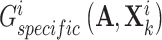 and
and 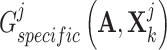 . Mathematically, the complementary score
. Mathematically, the complementary score 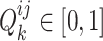 is defined as follows:
is defined as follows:
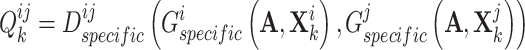 |
(3) |
Specifically, the generators 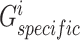 and
and 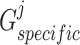 aim at minimizing
aim at minimizing 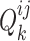 for capturing the distinctive information between the
for capturing the distinctive information between the 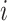 -th and the
-th and the 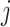 -th views and the discriminator
-th views and the discriminator 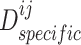 seeks to maximize
seeks to maximize 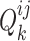 . In other words, the generated shared view representations
. In other words, the generated shared view representations 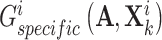 and
and 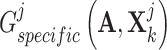 try to deceive discriminator
try to deceive discriminator 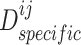 , resulting in the finally derived shared view representations
, resulting in the finally derived shared view representations 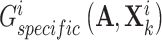 and
and 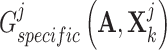 to be as unique as possible, contributing to the presentation of complementary effects in cancer gene identification. We concatenated specific view representations for preserving the unique characteristics of one gene:
to be as unique as possible, contributing to the presentation of complementary effects in cancer gene identification. We concatenated specific view representations for preserving the unique characteristics of one gene:
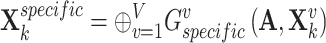 |
(4) |
where  denotes the concatenation operation.
denotes the concatenation operation.
Therefore, the objective function of shared view representation learning can be written as:
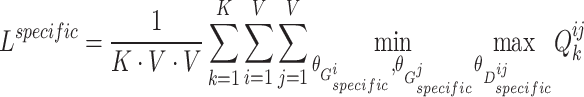 |
(5) |
Similar to the shared representation learner, 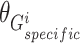 ,
, 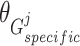 , and
, and 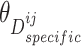 are the trainable parameters for generators
are the trainable parameters for generators 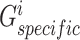 ,
, 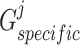 , and discriminator
, and discriminator 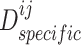 , respectively.
, respectively.
The cancer gene predictor
Shared and specific view representations were concatenated to generate the final view representations of the 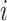 -th gene:
-th gene:
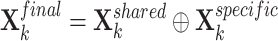 |
(6) |
Finally, we employed a classifier 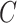 to obtain the cancer gene prediction scores
to obtain the cancer gene prediction scores 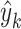 :
:
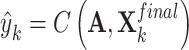 |
(7) |
where 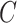 comprises a multilayer GCN followed by a fully connected layer, to map a given feature to a probability score.
comprises a multilayer GCN followed by a fully connected layer, to map a given feature to a probability score.
Hence, the loss of cancer gene identification is quantified using the binary cross-entropy:
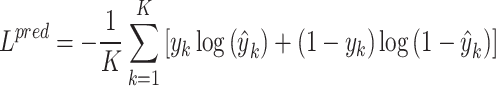 |
(8) |
where 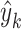 and
and 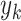 are the prediction score and the label (0 or 1) of gene
are the prediction score and the label (0 or 1) of gene 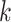 , respectively.
, respectively.
Our model was jointly trained in an end-to-end manner where both the representation loss and classification loss are back-propagated together. Therefore, the joint loss 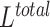 can be expressed as:
can be expressed as:
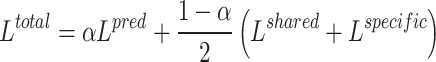 |
(9) |
where 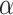 controls the weight of the cancer gene identification task.
controls the weight of the cancer gene identification task.
Results
Overview of interpretable multiview representation learning framework based on graph convolutional network
We introduced IMVRL-GCN, an end-to-end deep learning approach for predicting cancer genes utilizing genomic, transcriptomic, epigenomic, and structural data. As illustrated in Fig. 1, IMVRL-GCN comprises four modules: (i) multiview feature extractor, (ii) shared representation learner, (iii) specific representation learner, and (iv) cancer gene predictor. Initially, IMVRL-GCN extracts multiview features from the data using the multiview feature extractor. Subsequently, IMVRL-GCN discerns shared and specific representations from these features through dedicated learners. Finally, IMVRL-GCN combines these representations and feeds them into the cancer gene predictor to determine probabilities of identifying certain genes as cancer genes.
The multiview feature extractor implements strategies outlined by Peng et al. [15] for each view. The shared and specific representation learners utilize generative adversarial network–based (GAN) frameworks [22, 25] to disentangle interview and intraview information from the multiview features. These learners consist of four generators corresponding to the four-view features and one discriminator. To ensure structural consistency, the shared and specific learners are designed with identical architectures and share a common discriminator. However, the generators within each learner operate with distinct parameters to accommodate the generation of distinct representations. In order to obtain consistent representations, the discriminator in the shared representation learner instructs that shared outputs from the generators show similar classification indicators by maximizing their consensus scores for presenting their accordant contributions in cancer gene identification. The final shared representations are obtained by averaging the generators’ shared outputs. Conversely, the discriminator in the specific representation learner instructs that specific outputs from generators show respective unique classification indicators without sharing with others by minimizing their complementary scores for presenting their unique contributions to cancer gene identification. Finally, the integration of such two kinds of feature representations is fed into the cancer gene predictor, composed of graph convolutional layers and fully connected layers, to generate the cancer gene prediction score. More details of the IMVRL-GCN can be found in the Methods section.
Performance assessment of interpretable multiview representation learning framework based on graph convolutional network
To demonstrate IMVRL-GCN’s superiority, we compared it with four baselines (random forest (RF), GAT [37], GCN [38], Chebnet [39]), and three state-of-the-art cancer gene identification models (EMOGI [14], MODIG [16], MTGCN [15]). We used the same multiview features as IMVRL-GCN, concatenated as input for the baselines. RF sets the tree number to 1000, while GAT, GCN, and Chebnet each comprise three layers, with ReLU activation for the first two layers and sigmoid activation for the final layer. EMOGI, MODIG, and MTGCN were implemented using the same multiview features and were parameterized as recommended or adjusted for optimal performance.
Firstly, we conducted 10 five-fold cross-validations to evaluate IMVRL-GCN and compared models, using AUC and AUPR metrics, on the cross-validation set of Peng’s dataset (see Methods). As shown in Fig. 2A and B, IMVRL-GCN achieved an AUC of 0.9130 and an AUPR of 0.8372, outperforming all compared models. Among the compared models, RF outperformed EMOGI and MODIG, suggesting they may not efficiently integrate multiview data. IMVRL-GCN’s performance highlights its advanced multiview integration capability, capturing concise and valuable information from multiview data.
Figure 2.

IMVRL-GCN outperforms compared models and identifies 74 high-confidence CCGs. (A) ROC curve comparison with other models. (B) Precision–recall (PR) curve comparison with other models. (C) AUPR values for IMVRL-GCN and other models across different PPI networks. (D) Performance comparisons of IMVRL-GCN with other models on two independent cancer gene sets. (E) Multisource supporting evidence for the newly identified CCGs by IMVRL-GCN. (F) Venn diagram of the overlap between the CCGs identified by IMVRL-GCN and several compared models. (G) The supporting rate of different methods under List1 and List2. List1 contains convincing candidates in the independent cancer gene sets while List2 additionally compiles the results from CancerMine.
Further, we evaluated IMVRL-GCN and the compared models across different PPI networks and independent test sets to assess robustness. All models were retrained on these multiview datasets utilizing different PPI networks. Figure 2C illustrates that IMVRL-GCN performed best on CPDB and STRING datasets, demonstrating remarkable node characterization ability within complex networks, as detailed in Supplementary Table 1. Within sparse networks, performance dropped for IMVRL-GCN and state-of-the-art models due to their high reliance on network knowledge, yet IMVRL-GCN still excelled. We then trained these models using all training samples and tested them on the OncoKB and NCG + Bailey datasets separately. The OncoKB dataset comprises manually curated cancer genes with validated oncogenic effects from OncoKB [40], while the NCG + Bailey dataset comprises high-confidence cancer genes in publications from NCG [5] and those compiled using different computational tools by Bailey et al. [41]. Overlapping genes with training samples were removed for independence. Focusing on true-positive prediction performance, we used AUPR as the evaluation metric here. Figure 2D shows that IMVRL-GCN achieved the highest mean AUPR of 0.2088 on the two independent datasets, outperforming other models, which ranged from 0.1553 to 0.1994.
These studies confirm IMVRL-GCN’s superiority and robustness compared to state-of-the-art cancer gene identification models.
Ablation experiments
To investigate the importance of different components, we performed ablation experiments and designed the following seven variants of IMVRL-GCN:
‘IMVRL-GCN without genomic data’ (w/o GD) removes the genomic profiles.
‘IMVRL-GCN without transcriptomic data’ (w/o TD) removes the transcriptomic profiles.
‘IMVRL-GCN without epigenomic data’ (w/o ED) removes the epigenomic profiles.
‘IMVRL-GCN without structural data’ (w/o SD) removes the structural profiles.
‘IMVRL-GCN without shared representation learning module’ (w/o SH) removes the shared representation learning module.
‘IMVRL-GCN without specific representation learning module’ (w/o SP) removes the specific representation learning module.
‘IMVRL-GCN without representation learning module’ (w/o RL) integrates multiview features in a concatenated manner instead of representation learning module.
Table 1 shows the performance of IMVRL-GCN and its variants, evaluated by 10 five-fold cross-validations on Peng’s dataset. After removing view data, the mean AUC of variants (w/o SD, w/o GD, w/o TD, and w/o ED) ranged from 0.8786 to 0.9122, the mean AUPR from 0.7690 to 0.8359, and the mean MCC from 0.5384 to 0.6381, indicating the necessity of all view data. Performance dropped significantly without genomic and structural data, indicating the importance of genome-level aberration and functional interaction knowledge in cancer gene identification. The experiment on the variants (w/o RL, w/o SH, w/o SP) confirmed that the representation learning module is necessary and outperforms the direct concatenation of multiview features.
Table 1.
The results of IMVRL-GCN and variants in ablation experiments.
| Method | Description | AUC | AUPR | MCC |
|---|---|---|---|---|
| IMVRL-GCN (w/o GD) | IMVRL-GCN without genomic data | 0.8946 ± 0.0012 | 0.7937 ± 0.0034 | 0.5839 ± 0.0077 |
| IMVRL-GCN (w/o TD) | IMVRL-GCN without transcriptomic data | 0.9069 ± 0.0008 | 0.8281 ± 0.0018 | 0.6349 ± 0.0083 |
| IMVRL-GCN (w/o ED) | IMVRL-GCN without epigenomic data | 0.9122 ± 0.0010 | 0.8359 ± 0.0025 | 0.6381 ± 0.0050 |
| IMVRL-GCN (w/o SD) | IMVRL-GCN without structural data | 0.8786 ± 0.0013 | 0.7690 ± 0.0030 | 0.5384 ± 0.0073 |
| IMVRL-GCN (w/o SH) | IMVRL-GCN without shared representation learning module | 0.9103 ± 0.0012 | 0.8321 ± 0.0028 | 0.6334 ± 0.0068 |
| IMVRL-GCN (w/o SP) | IMVRL-GCN without specific representation learning module | 0.9105 ± 0.0005 | 0.8325 ± 0.0018 | 0.6349 ± 0.0067 |
| IMVRL-GCN (w/o RL) | IMVRL-GCN without representation learning module | 0.9110 ± 0.0012 | 0.8312 ± 0.0022 | 0.6211 ± 0.0087 |
| IMVRL-GCN | IMVRL-GCN | 0.9130 ± 0.0012 | 0.8372 ± 0.0024 | 0.6429 ± 0.0062 |
To further discuss how these data combinations affect model performance, we tested an additional six dual data combinations (GD + TD, GD + ED, GD + SD, TD + ED, TD + SD, ED + SD) (Supplementary Table 2). Among these combinations, GD + SD performed the best, closely followed by the TD + SD and ED + SD. This indicates that combining structural data with other data types significantly enhances model performance. The triple data combinations further improved model performance, particularly when transcriptomic data or epigenomic data were added to the GD + SD combination. However, in some cases, the inclusion of additional data types did not always result in significant performance improvements. For instance, the combination GD + TD + ED did not show an advantage over the combinations GD + TD or GD + ED. This suggests that there may be some degree of redundancy between transcriptomic data and epigenomic data.
Identification of 74 high-confidence novel candidate cancer genes
We trained IMVRL-GCN using all positive and negative samples from the benchmark dataset to predict the possibility of unlabeled genes being cancer genes. Setting a threshold of 0.99, we identified 74 high-confidence CCGs as specified in Supplementary Table 3 and verified their reliability as follows.
First, we compared the predicted CCGs with high-confidence candidates from OncoKB [40], NCG [5], and the study by Bailey et al. [41]. Figure 2E shows that 74.32% (55/74) of our CCGs were validated by at least one piece of evidence. For the remaining 19 CCGs, we used CancerMine [42], a text-mining-based, regularly updated cancer gene database, to avoid potential omissions from out-of-date resources. We noticed 14 of 19 candidates were supported by CancerMine, and ultimately 93.24% (69/74) of our CCGs had at least one piece of supporting evidence, confirming their potential role in tumorigenesis.
Next, we implemented the state-of-the-art models (MTGCN, EMOGI, and MODIG) and compared their respective top 74 predictions with our CCGs. Figure 2F shows that 74.32% (55/74) of our predictions overlapped with at least one of these models, and 18.92% (14/74) overlapped with two or more models. Our CCGs had the highest agreement with MTGCN, which performed second-best to IMVRL-GCN in cross-validation (Fig. 2A). We then calculated each model’s supporting rates against the aforementioned high-confidence cancer gene sets. For ease of representation, we refer to the candidates in OncoKB, NCG, and the study by Bailey et al. collectively as List 1, while List 2 additionally incorporates up-to-date candidates in CancerMine. Figure 2G shows that IMVRL-GCN had the highest supporting rate of 74.32%, surpassing other models (47.30%–67.57%). Consistently, for List 2, IMVRL-GCN exhibited an even higher supporting rate of 93.24%, outperforming other models (78.38%–91.89%).
In conclusion, the implementation of IMVRL-GCN led to the identification of 74 novel high-confidence CCGs, most supported by diverse evidence, validating their status as bona fide cancer genes.
Analysis of multiview representations interpreted their contributions to IMVRL-GCN’s predictions
In IMVRL-GCN, our disentangled shared and specific multiview representations are interpretable, aiding in understanding the model’s decisions. We conducted a comprehensive analysis of these representations to evaluate their contributions to the novel predictions. Specifically, we investigated the attribution scores of these representations for cancer gene identification using IntegratedGradients [43], a powerful axiomatic attribution method based on backpropagation (see Supplementary Methods).
As depicted in Fig. 3A, all types of multiview representations demonstrated a modest or even negative impact on the majority of the predictions. This is attributed to the fact that most predictions are noncancer-related genes, showing no discernible difference between tumor and normal samples. We then concentrated on the contributions of the 74 CCGs. In Fig. 3B, we observed that the distribution of shared, mutation-specific, and structure-specific representations displayed distinct peaks near the most important regions, denoted as Peak 1, Peak 2, and Peak 3, respectively, indicating that the corresponding representations are of great importance in the decision-making progress for a large portion of CCGs. Peak 1 included all CCGs, highlighting the vital roles of shared representations in all CCG predictions, with a median attribution score of 0.94, indicating that the consensus effect is a discerning indicator in cancer gene identification. Peak 2 and Peak 3 encompassed 52 and 15 CCGs, respectively, indicating the pivotal roles played by mutation-specific and structure-specific representations for these genes.
Figure 3.

Attribution score distribution of shared and specific representations on the predicted results for (A) all predicted genes. (B) 74 novel CCGs, each dot represents a CCG, and the arrow points to the peaks of prominent importance. Attribution scores have been subjected to min-max normalization.
The aforementioned analysis demonstrates the superior discrimination capabilities of shared, mutation-specific, and structure-specific representations in identifying cancer genes, and their respective meanings provided us with insights that elucidate the factors contributing to their discriminative capabilities. Shared representations denote consistent information across views, emphasizing the superior cancer gene discrimination capabilities inherent in the biological nature of these CCGs. Mutation-specific representations exhibit complementary information captured exclusively from the mutational view, suggesting that genomic aberrations in these 52 CCGs are noteworthy and may serve as the primary causes of their tumorigenesis. Structure-specific representations indicate the complementary information captured exclusively from the structural view, implying that these 15 CCGs may be involved in tumorigenesis through interactions in the PPI network.
Biological explanations for the most important multiview representations
In this section, we conducted in-depth studies on shared, mutation-specific, and structure-specific representations to validate the above hypotheses about their superior cancer gene discrimination capabilities and glean further insights for novel therapies.
Shared representations exhibit an association with gene functions
We clustered CCGs by their shared representations to examine if each cluster showed a common biological nature. As illustrated in Fig. 4A and Supplementary Figure 1, 74 CCGs are categorized into seven clusters using eigengap analysis, where a significant drop between consecutive eigenvalues indicates natural data separation [44].
Figure 4.

Analysis of shared representations reveals their connections to gene functions. (A) Spectral clustering results of 74 CCGs based on their shared representations. (B) Biological process terms shared by each cluster. The number of shared term values is averaged by the cluster size. (C) Consistent pattern between the shared representation similarities and the BP term similarities within Cluster 1. Each dot represents a gene–gene pair. The P values are calculated by the Spearman correlation analysis. (D) Hierarchical clustering of semantic similarities of genes in Cluster 1.
We used Gene Ontology (GO) annotations to examine common biological process (BP), cellular component (CC), and molecular function (MF) terms within each cluster, elucidating the relationship between shared representations and biological nature. Figure 4B highlights that Cluster 1 and 4 have recurrent BP terms, signifying functional commonalities among their genes. Genes in Cluster 1 are involved in axon guidance (GO:0007411) and the transmembrane receptor protein tyrosine kinase signaling pathway (GO:0007169), suggesting their crucial roles in regulating neuronal axon growth and signaling processes via tyrosine kinase activity. Genes in Cluster 4 participate in RNA polymerase II-mediated transcription (GO:0045944 and GO:0006357), essential for controlling gene expression during cell development. Notably, the shared pathways for Cluster 1 are intricately linked to tumorigenesis. Pancreatic cancer genomes undergo frequent somatic aberrations in axon guidance genes [45]. Mouse models further support the notion that disturbances in axon guidance signals can mitigate pancreatic ductal carcinoma progression [46]. Receptor tyrosine kinases (RTKs) are crucial for cell growth, motility, differentiation, and metabolism. Consequently, dysregulation of RTK signaling significantly contributes to many diseases, particularly cancer [47]. The correlation analysis between BP term semantic similarities and shared representation similarities in Cluster 1 showed a significant agreement, thereby substantiating the connection between gene biological function and shared representation (P = .026, Fig. 4C). Within Cluster 1, NOTCH3 and NEDD4 exhibit lower semantic similarity with other genes (Fig. 4D), likely due to their lack of RTK signaling pathway annotation. However, their close relationship with RTK signaling has been confirmed: NOTCH3 encodes a key receptor in NOTCH signaling, likely cross-regulates with RTK signaling [48], and NEDD4 enhances insulin-like growth factor (IGF, a member of the superfamily of RTKs) signaling by mediating IRS-2 and IGF1R ubiquitination [49, 50].
Regarding CC, an observed convergence in the cellular localization of genes within each cluster suggests similar function modes (Supplementary Figure 2A). Specifically, products of genes in Cluster 1 are predominantly located at the plasma membrane and cytosol, genes in Cluster 3 at the extracellular region, genes in Cluster 4 at the nucleoplasm and nucleus, genes in Cluster 5 at the cytoplasm, and genes in Cluster 7 at the cytosol. For MF in Supplementary Figure 2B, all clusters shared the GO:0005515 term, indicating their bindings to a protein but limited utility for cancer gene recognition. Apart from GO:0005515, terms such as GO:0046875 for Cluster 1 and GO:1990837, GO:0003700, GO:0000981, and GO:0000978 for Cluster 4 align with their respective shared BP terms.
In conclusion, shared representations play a significant role in recognizing cancer genes, possibly stemming from their association with gene functions. Moreover, these representations offer a novel perspective for studying gene functions, where deep learning models autonomously assimilate consistent information from multiview data and infer unknown gene functions based on those sharing similar consistent information.
Mutation-specific representations mirror the susceptibility of candidate cancer genes to genomic aberrations
Firstly, to validate the primary causative roles of the mutational information in tumorigenesis for the 52 CCGs in Peak 2 (Fig. 3B), we cross-referenced them with the CCGD database [51], a mouse model-based mutation-driven cancer genes database, and found that ~63% (33/52) are included. We calculated the mutation rate of all CCGs in the pan-cancer patient cohort [52] and observed a significantly higher rate of CCGs in Peak 2 compared to others (Fig. 5A). The gene mutation burden, defined as the number of mutations per mega-base (MB) in the coding region, eliminating the influence of gene length, was also higher for CCGs in Peak 2, indicating their susceptibility to mutations and potential carcinogenesis risk (Fig. 5B).
Figure 5.

Analysis of mutation-specific representations indicates their connections to gene mutagenic propensity. (A) Different genetic alteration rates between CCGs in Peak 2 and other CCGs. The P values are calculated using unpaired Student’s t-tests. ***P value <.001. (B) Different mutation burden between CCGs in Peak 2 and other CCGs. The P values are calculated using unpaired Student’s t-tests. ***P value <.001. (C) Univariate Cox regression analysis identifies 12 risk factors associated with poor overall survival in the pan-cancer cohort. The P values are calculated by the log-rank test. (D) Waterfall chart showing the genomic alteration distribution of 12 risk factors in the pan-cancer cohort. (E) Risk factors display a notable difference in expression between the altered type and wild type. The P values are calculated using an unpaired Wilcoxon rank-sum test. *P value <.05, **P value <.01, ***P value <.001. (F) Responses to several drugs are correlated with risk factor expression levels across diverse cancer cell lines. The P values are calculated using two-side Spearman’s correlation test.
Subsequently, we conducted a univariate Cox regression analysis to assess these CCGs as prognostic biomarkers. As depicted in Fig. 5C, we identified 12 potential tumor prognostic biomarkers. Genes like MACF1 in cholangiocarcinoma [53]; SPTA1 in hepatocellular carcinoma [54]; HMCN1 and TTN in hepatocellular carcinoma [55]; and OBSCN in highly aggressive tumors like glioblastoma, melanoma, and pancreatic carcinoma [56] are known mutational drivers. Furthermore, we observed that the copy numbers of these genes frequently altered consistent with point mutations, collectively impacting 4%–24% of patients in the pan-cancer cohort (Fig. 5D), suggesting a broader at-risk population and a need for effective therapeutic interventions.
Finally, we screened existing drugs for potential therapeutics against these risk genes (see Supplementary Methods). Among the 12 risk genes, 6 exhibited significant expression changes due to genomic alterations, indicating potential concurrent function dysregulation (Fig. 5E). Leveraging the Genomics of Drug Sensitivity in Cancer (GDSC) dataset [57], we identified drugs sensitive to the expression of these risk genes (Fig. 5F). The screening revealed sensitivity of 40, 204, 112, 93, 228, and 1 drugs to RYR1, SYNE1, ANK2, HMCN1, TTN, and OBSCN, respectively, with 19 drugs exhibiting sensitivity to at least four risk factors (Supplementary Table 4). Notably, Afatinib, an inhibitor of the ErbB tyrosine kinases used for nonsmall cell lung cancer, demonstrated sensitivity to five of the six risk genes, suggesting its potential therapeutic utility against these risk genes.
Structure-specific representations indicate the hub node status of candidate cancer genes in the protein–protein interaction network
We analyzed the 15 CCGs in Peak 3 (Fig. 3B) for their roles in biological mechanisms. Figure 6A shows that these genes are enriched in cellular response processes, such as to organic cyclic compound, hormone stimulus, and growth factor stimulus, indicating they are crucial hubs in signal transduction. Further evidence supported that these CCGs have a higher number of interactions (Fig. 6B), highlighting their central roles in the PPI network.
Figure 6.

Analysis of structure-specific representations indicates their connections to the central role of genes in functional synergy. (A) Bubble chart of enriched biological processes for genes in Peak 3. The circle color and size represent the significance of the difference and hit count, respectively. (B) Different cancer gene interaction numbers between CCGs in Peak 3 and other CCGs. The P values are calculated using unpaired Student’s t-tests. **P value <.01. (C) Extraction of PPI subnetwork components containing genes in Peak 3 and their top five contributory neighbors. The circles represent genes in Peak 3, and the triangles represent their important neighbors. The triangle size represents the node degree, and the color indicates the neighbor type. (D, E) The mRNA alteration frequency of TFs HNF4A in (D) and SP1 in (E) in pan-cancer cohort.
To pinpoint causal interactions for these structure-driven CCGs, we utilized IntegratedGradients to compute the link importance by accumulating gradients along the path between the baseline (all-zero graph) and the state of the graph. Consequently, we extracted these CCGs and the subnetwork containing their causal interactions (Fig. 6C). These interactions implicated several KCGs, such as IL6ST, which encodes a signal transducer, interacts with nonreceptor tyrosine kinase LYN, and adapter proteins GRB2 and SHC1, crucial contributors to signal transduction. These interactions indicated that LYN, GRB2, and SHC1 are key participants in IL6ST-induced adverse consequences. Notably, another KCG, SRC, implicated in the most interactions, encoding a steroid receptor coactivator, not only interacts with SHC1 as a nonreceptor tyrosine kinase to impact signal transduction, but also with four transcription factors (TFs) including NR3C1, RXRA, HNF4A, and SP1, boosting transcription by participating in all gene expression substeps [58].
Previous research highlighted the potential oncogenic roles of HNF4A and SP1. HNF4A is involved in the AMPK-HNF4A-WNT signaling cascade, a novel targetable oncogenic mechanism in gastric cancer, while SP1 serves as a central TF regulating pathway crucial to tumorigenesis [59, 60]. Their expression patterns across cancer types revealed distinct profiles. HNF4A is highly expressed in a few cancer types including colorectal, hepatobiliary, and esophagogastric cancers (Fig. 6D), indicating that HNF4A encodes a functionally specific TF. In contrast, SP1 has variable expression across cancers, with low expression in glioma and high expression in pancreatic and esophagogastric cancers (Fig. 6E), indicating that SP1 encodes a pleiotropic TF. Therefore, enhancing or blocking their interactions with SRC based on cancer type may be a promising therapeutic strategy for SRC-induced cancers.
Discussion
The traditional view of cancer as primarily a genetic mutation-driven disease has evolved to encompass various biological mechanisms. This includes epigenetic changes and TF-mediated modifications. Despite advances, our understanding of KCGs remains limited, hindering the development of effective treatments. Computational methods, particularly GNN-based approaches, have improved KCG identification by handling relational data well. However, existing methods fail to integrate multiview data effectively, as they overlook the distinct value of each perspective and neglect to leverage the consensus information from multiple views. Additionally, these methods lack interpretability. Although some can determine the importance of different views, identifying causal factors remains challenging due to the complex associations across views. This complicates understanding prediction processes and hinders further exploration of mechanisms and clinical applications of novel targets.
To address these issues, we developed IMVRL-GCN. This framework skillfully extracts shared and specific multiview representations for each gene, not only synthesizing this information to enhance cancer gene identification but also facilitating comprehension of the model’s decisions as such disentangled representations are explicit and easy to understand. IMVRL-GCN outperforms state-of-the-art models and baselines, identifying 74 CCGs with high-confidence evidence. Furthermore, we explain each CCG prediction by identifying crucial view representations, highlighting the importance of shared, mutation-specific, and structure-specific representations for many CCGs. This insight underlines the importance of the multiview representation learning-based approach in cancer research.
We observed that genes with similar shared representations showed high semantic similarity, suggesting a link between consensus effects and biological functions. Clustering the 74 CCGs based on shared representations revealed that genes in Cluster 1 are implicated in carcinogenesis via the RTK signaling pathway, whereas Cluster 4 influences carcinogenesis by impacting RNA polymerase II-mediated transcription. Our conclusions stemmed from examining the correlation between shared representations and three gene function descriptors, i.e. BP, CC, and MF. However, it is noteworthy that these descriptors may not fully encapsulate the complexity of gene function. Therefore, developing more precise descriptions of biological functions, such as semantic embeddings based on gene function descriptions, could offer additional evidence linking shared representations and gene function. In summary, the importance of consensus effects in cancer gene identification may stem from their ability to glean valuable biological insights. This revelation also inspires innovative approaches to studying candidates with unknown functions, i.e. one can speculate on their roles by summarizing and comparing the functions of genes with similar shared representations.
Prominent mutational and structural complementary effects emphasize the importance of their respective perspectives. Our analysis identified 52 CCGs with prominent mutational effects, noting their high mutation burdens. Among these, 15 prognostic biomarkers were recognized, many of which are known mutational drivers, indicating that we can effectively reproduce their causal molecular factors. In terms of treatment, Afatinib demonstrated widespread sensitivity to these mutation-driven CCGs, underscoring its therapeutic promise. For the 15 CCGs with prominent structural effects, we identified their extensive interactions within the PPI network. Prioritizing the most important interactions, we observed that several causal interactions implicated well-known cancer genes, particularly SRC, whose interactions resulted in the highest number of predicted outcomes for those CCGs. Specifically, SRC protein boosts gene transcription by binding with the TFs NR3C1, RXRA, HNF4A, and SP1, whose expression patterns were investigated across various cancers to warn potential risks.
Our proposed IMVRL-GCN is an extensible framework, which can not only accommodate various view profiles, including chromosome conformation, proteome, metabolome, etc., but also extend different gene association profiles, such as coexpression network, to construct a multidimensional network. Our method heralds a promising paradigm for multiview integration in pan-cancer research, and in the future, we could refine it for cancer type–specific research using transfer learning methods. By leveraging the knowledge acquired at the pan-cancer level, we can identify more reliable cancer type–specific cancer genes and therapeutic targets. Considering the advantages of IMVRL-GCN in multiview data integration and interpretability, we believe it can be applied to other research areas in the future.
Key Points
Innovative cancer gene identification with GCN and shared-and-specific disentangled representation learning.
Disentangled shared and specific view representations from multiview data are crucial for accurate cancer gene identification.
Assessing the importance of interpretable disentangled representations helps to understand the model’s decisions.
Significantly outperforms state-of-the-art methods and demonstrates robustness and generalization across diverse datasets.
Analyzing the compiled 74 high-confidence candidates aids in comprehending their molecular mechanisms and directing personalized treatments.
Supplementary Material
Contributor Information
Jianye Yang, College of Informatics, Huazhong Agricultural University, Wuhan 430070, China.
Haitao Fu, College of Informatics, Huazhong Agricultural University, Wuhan 430070, China; School of Artificial Intelligence, Hubei University, Wuhan 430070, China.
Feiyang Xue, College of Informatics, Huazhong Agricultural University, Wuhan 430070, China.
Menglu Li, College of Informatics, Huazhong Agricultural University, Wuhan 430070, China.
Yuyang Wu, College of Informatics, Huazhong Agricultural University, Wuhan 430070, China.
Zhanhui Yu, College of Informatics, Huazhong Agricultural University, Wuhan 430070, China.
Haohui Luo, College of Informatics, Huazhong Agricultural University, Wuhan 430070, China.
Jing Gong, College of Informatics, Huazhong Agricultural University, Wuhan 430070, China; College of Biomedicine and Health, Huazhong Agricultural University, Wuhan 430062, China.
Xiaohui Niu, College of Informatics, Huazhong Agricultural University, Wuhan 430070, China.
Wen Zhang, College of Informatics, Huazhong Agricultural University, Wuhan 430070, China.
Funding
This work was supported by the National Key Research and Development Program of China (2021YFF0703703), National Science and Technology Innovation 2030 Major Program of China (2023ZD0404702), National Natural Science Foundation of China (62072206, 61772381), Fundamental Research Funds for the Central Universities (2662021JC008, 2662022JC004), Natural Science Foundation of Hubei Province (2021CFB404), and Huazhong Agricultural University Scientific & Technological Self-innovation Foundation (2662024SZ006).
Data availability
Datasets and source code are publicly available at https://github.com/YJY-98/IMVRL-GCN.
References
- 1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71:209–49. [DOI] [PubMed] [Google Scholar]
- 2. Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science 2013;339:1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Al-Batran S-E, Schuler MH, Zvirbule Z, et al. FAST: an international, multicenter, randomized, phase II trial of epirubicin, oxaliplatin, and capecitabine (EOX) with or without IMAB362, a first-in-class anti-CLDN18.2 antibody, as first-line therapy in patients with advanced CLDN18.2+ gastric and gastroesophageal junction (GEJ) adenocarcinoma. J Clin Oncol 2016;34:LBA4001. [Google Scholar]
- 5. Repana D, Nulsen J, Dressler L, et al. The network of cancer genes (NCG): a comprehensive catalogue of known and candidate cancer genes from cancer sequencing screens. Genome Biol 2019;20:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sondka Z, Bamford S, Cole CG, et al. The COSMIC cancer gene census: describing genetic dysfunction across all human cancers. Nat Rev Cancer 2018;18:696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dees ND, Zhang Q, Kandoth C, et al. MuSiC: identifying mutational significance in cancer genomes. Genome Res 2012;22:1589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Luo P, Ding Y, Lei X, et al. deepDriver: predicting cancer driver genes based on somatic mutations using deep convolutional neural networks. Front Genet 2019;10:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tamborero D, Gonzalez-Perez A, Lopez-Bigas N. Oncodrive CLUST: exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 2013;29:2238–44. [DOI] [PubMed] [Google Scholar]
- 11. Collier O, Stoven V, Vert JP. LOTUS: a single- and multitask machine learning algorithm for the prediction of cancer driver genes. PLoS Comput Biol 2019;15:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Silverbush D, Cristea S, Yanovich-Arad G, et al. Simultaneous integration of multi-omics data improves the identification of cancer driver modules. Cell Syst 2019;8:456–466.e455. [DOI] [PubMed] [Google Scholar]
- 13. Scarselli F, Gori M, Tsoi AC, et al. The graph neural network model. IEEE Trans Neural Netw 2009;20:61–80. [DOI] [PubMed] [Google Scholar]
- 14. Schulte-Sasse R, Budach S, Hnisz D, et al. Integration of multiomics data with graph convolutional networks to identify new cancer genes and their associated molecular mechanisms. Nat Mach Intell 2021;3:513–26. [Google Scholar]
- 15. Peng W, Tang Q, Dai W, et al. Improving cancer driver gene identification using multi-task learning on graph convolutional network. Brief Bioinform 2022;23:bbab432. [DOI] [PubMed] [Google Scholar]
- 16. Zhao WY, Gu X, Chen SQ, et al. MODIG: integrating multi-omics and multi-dimensional gene network for cancer driver gene identification based on graph attention network model. Bioinformatics 2022;38:4901–7. [DOI] [PubMed] [Google Scholar]
- 17. Kaur D, Uslu S, Rittichier KJ, et al. Trustworthy artificial intelligence: a review. ACM Comput Surv 2022;55:1–38. [Google Scholar]
- 18. Chen N, Zhu J, Sun F, et al. Large-margin predictive latent subspace learning for multiview data analysis. IEEE Trans Pattern Anal Mach Intell 2012;34:2365–78. [DOI] [PubMed] [Google Scholar]
- 19. Li J, Zhang B, Lu G, et al. Generative multi-view and multi-feature learning for classification. Inf Fusion 2019;45:215–26. [Google Scholar]
- 20. Chen X, Chen S, Xue H, et al. A unified dimensionality reduction framework for semi-paired and semi-supervised multi-view data. Pattern Recognit 2012;45:2005–18. [Google Scholar]
- 21. Wang W, Arora R, Livescu K, et al. On Deep Multi-View Representation Learning. In: Francis B, David B. (eds) Proceedings of the 32nd International Conference on International Conference on Machine Learning - Volume 37. Lille, France: PMLR, 2015, 1083–92. [Google Scholar]
- 22. Xu J, Han J, Nie F. Multi-View Feature Learning with Discriminative Regularization. In: Sierra C. (ed) Proceedings of the 26th International Joint Conference on Artificial Intelligence. Melbourne, Australia: AAAI Press, 2017, 3161–7. [Google Scholar]
- 23. Jing X-Y, Hu R-M, Zhu Y-P, et al. Intra-view and inter-view supervised correlation analysis for multi-view feature learning. Proc AAAI Conf Artif Intell 2014;28:1882–9. [Google Scholar]
- 24. Zhu J, Liu Y, Zhang Y, et al. Multi-attribute discriminative representation learning for prediction of adverse drug-drug interaction. IEEE Trans Pattern Anal Mach Intell 2022;44:10129–44. [DOI] [PubMed] [Google Scholar]
- 25. Jia X, Jing XY, Zhu X, et al. Semi-supervised multi-view deep discriminant representation learning. IEEE Trans Pattern Anal Mach Intell 2021;43:2496–509. [DOI] [PubMed] [Google Scholar]
- 26. Li Y, Yang M, Zhang Z. A survey of multi-view representation learning. IEEE Trans Knowl Data Eng 2019;31:1863–83. [Google Scholar]
- 27. Herwig R, Hardt C, Lienhard M, et al. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat Protoc 2016;11:1889–907. [DOI] [PubMed] [Google Scholar]
- 28. Kim J, So S, Lee HJ, et al. DigSee: disease gene search engine with evidence sentences (version cancer). Nucleic Acids Res 2013;41:W510–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000;28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McKusick VA. Mendelian inheritance in man and its online version, OMIM. Am J Hum Genet 2007;80:588–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liberzon A, Birger C, Thorvaldsdottir H, et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Szklarczyk D, Gable AL, Nastou KC, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res 2021;49:10800–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pillich RT, Chen J, Churas C, et al. NDEx: accessing network models and streamlining network biology workflows. Curr Protoc 2021;1:e258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, et al. The Cancer genome atlas Pan-Cancer analysis project. Nat Genet 2013;45:1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grover A, Leskovec J. node2vec: scalable feature learning for networks. KDD 2016;2016:855–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Veličković P, Cucurull G, Casanova A, et al. Graph Attention Networks. In: Bengio Y, LeCun Y. (eds) International Conference on Learning Representations. Vancouver, Canada: OpenReview.net, 2018, Poster.
- 38. Kipf TN, Welling M. Semi-Supervised Classification with Graph Convolutional Networks. In: Bengio Y, LeCun Y. (eds) International Conference on Learning Representations. Toulon, France: OpenReview.net, 2017, Poster.
- 39. Defferrard M, Bresson X, Vandergheynst P. Convolutional neural networks on graphs with Fast localized spectral filtering. Adv Neural Inf Process Syst (Nips 2016) 2016;29:3844–52. [Google Scholar]
- 40. Chakravarty D, Gao JJ, Phillips S, et al. OncoKB: a precision oncology Knowledge Base. JCO Precis Oncol 2017;1:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bailey MH, Tokheim C, Porta-Pardo E, et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018;174:1034–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lever J, Zhao EY, Grewal J, et al. CancerMine: a literature-mined resource for drivers, oncogenes and tumor suppressors in cancer. Nat Methods 2019;16:505–7. [DOI] [PubMed] [Google Scholar]
- 43. Sundararajan M, Taly A, Yan Q. Axiomatic Attribution for Deep Networks. In: Jebara T. (ed) Proceedings of the 34th International Conference on Machine Learning. Sydney, Australia: JMLR.org, 2017, 3319–28. [Google Scholar]
- 44. Lapuschkin S, Waldchen S, Binder A, et al. Unmasking clever Hans predictors and assessing what machines really learn. Nat Commun 2019;10:1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012;491:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jurcak NR, Rucki AA, Muth S, et al. Axon guidance molecules promote perineural invasion and metastasis of orthotopic pancreatic tumors in mice. Gastroenterology 2019;157:838–850.e836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Du Z, Lovly CM. Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer 2018;17:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hurlbut GD, Kankel MW, Artavanis-Tsakonas S. Nodal points and complexity of Notch-Ras signal integration. Proc Natl Acad Sci U S A 2009;106:2218–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fukushima T, Yoshihara H, Furuta H, et al. Nedd4-induced monoubiquitination of IRS-2 enhances IGF signalling and mitogenic activity. Nat Commun 2015;6:6780. [DOI] [PubMed] [Google Scholar]
- 50. Vecchione A, Marchese A, Henry P, et al. The Grb10/Nedd4 complex regulates ligand-induced ubiquitination and stability of the insulin-like growth factor I receptor. Mol Cell Biol 2003;23:3363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Abbott KL, Nyre ET, Abrahante J, et al. The candidate cancer gene database: a database of cancer driver genes from forward genetic screens in mice. Nucleic Acids Res 2015;43:D844–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Consortium TITP-CAoWG . Pan-cancer analysis of whole genomes. Nature 2023;614:E39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Y, Ma Z, Li C, et al. The genomic landscape of cholangiocarcinoma reveals the disruption of post-transcriptional modifiers. Nat Commun 2022;13:3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Candia J, Bayarsaikhan E, Tandon M, et al. The genomic landscape of Mongolian hepatocellular carcinoma. Nat Commun 2020;11:4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen P, Chen D, Bu D, et al. Dominant neoantigen verification in hepatocellular carcinoma by a single-plasmid system coexpressing patient HLA and antigen. J Immunother Cancer 2023;11:e006334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Balakrishnan A, Bleeker FE, Lamba S, et al. Novel somatic and germline mutations in cancer candidate genes in glioblastoma, melanoma, and pancreatic carcinoma. Cancer Res 2007;67:3545–50. [DOI] [PubMed] [Google Scholar]
- 57. Yang W, Soares J, Greninger P, et al. Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 2013;41:D955–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. York B, O'Malley BW. Steroid receptor coactivator (SRC) family: masters of systems biology. J Biol Chem 2010;285:38743–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huang C, Xie K. Crosstalk of Sp1 and Stat3 signaling in pancreatic cancer pathogenesis. Cytokine Growth Factor Rev 2012;23:25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chang HR, Nam S, Kook MC, et al. HNF4alpha is a therapeutic target that links AMPK to WNT signalling in early-stage gastric cancer. Gut 2016;65:19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets and source code are publicly available at https://github.com/YJY-98/IMVRL-GCN.