

Abstract
Benvitimod has been successfully used in the treatment of psoriasis and atopic dermatitis (AD). However, the mechanism remains to be clarified. We aim to assess the effects of benvitimod on MC903-induced dermatitis in mice and to investigate the effects of benvitimod on filaggrin (FLG), involucrin (IVL), and loricrin (LOR) expressions and possible mechanism. MC903-induced mouse AD model was used to evaluate the effects of benvitimod. Filaggrin, involucrin, and loricrin protein and mRNA expressions in lesions of mice dermatitis were measured by Western blot and quantitative real-time PCR. In vitro, normal human epidermal keratinocytes (NHEKs) were cultured and benvitimod was used to treat NHEKs primed with IL-4 and IL-13. Then AHR and OVOL1 in NHEKs were knocked down to evaluate the role of AHR and OVOL1 in the effects of benvitimod. Topical treatment of benvitimod repaired skin barrier and alleviated skin inflammation in mouse AD model. This effect was inhibited by pretreatment with an AHR antagonist. Benvitimod upregulated the filaggrin, involucrin, and loricrin expressions in lesions of mouse AD model. In addition, benvitimod upregulated the filaggrin, involucrin, and loricrin expressions in NHEKs. Knockdown of AHR or OVO-like (OVOL)1 abrogated the upregulation of filaggrin, involucrin, and loricrin induced by benvitimod. Benvitimod attenuated MC903-induced mouse dermatitis and upregulated filaggrin, involucrin, and loricrin expressions via AHR-OVOL1 axis.
Graphical Abstract
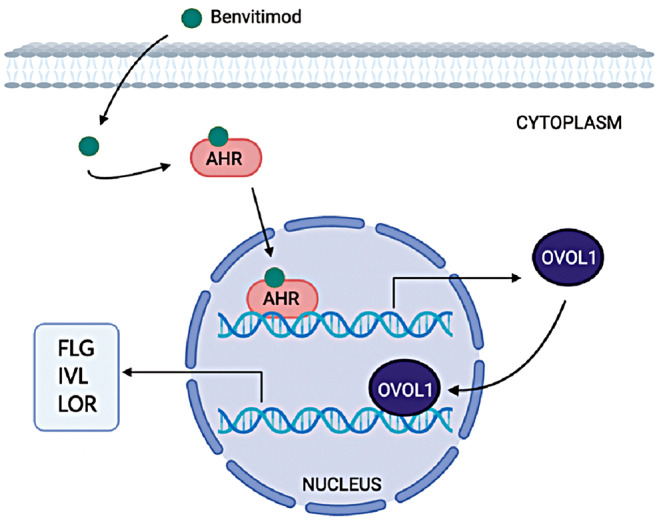
Possible pathway of benvitimod upregulating FLG, IVL and LOR expression. Benvitimod activates AHR, then AHR translocates into the nucleus and upregulates OVOL1 expression. OVOL1 further translocates into the nucleus and upregulates the FLG, IVL and LOR expression
Supplementary Information
The online version contains supplementary material available at 10.1007/s00403-024-03268-7.
Keywords: Aryl hydrocarbon receptor, Benvitimod, Filaggrin, Loricrin, Involucrin and OVOL1
Introduction
Atopic dermatitis (AD) is a chronic, inflammatory skin disease characterized by recurrent eczematous lesions and severe pruritus that significantly impact patient’s quality of life [1–3]. The pathogenesis of AD mainly involves genetic predisposition, epidermal barrier impairment and immune dysregulation [2, 4]. Loss-of-function mutations in the filaggrin (FLG) gene and reduced levels of FLG contribute to skin barrier impairment [5–7]. Besides FLG, involucrin (IVL) and loricrin (LOR) also play crucial roles in maintaining the skin barrier [7, 8]. The immune dysregulation in AD is predominantly driven by Th2 inflammation, with IL-4 and IL-13 being the main cytokines [1, 9, 10]. Previous studies have reported that FLG, IVL, and LOR can be downregulated by IL-4 and IL-13, leading to skin barrier dysfunction and the development of AD [8, 11]. Conversely, epidermal keratinocytes in barrier-disrupted skin promote type 2 immune deviation [11, 12]. Therefore, breaking this vicious cycle by repairing the barrier may be a crucial breakthrough in treating AD.
Benvitimod (BVM, also known as tapinarof) is an aryl hydrocarbon receptor (AHR)-modulator which has been reported to be effective for AD [13, 14]. However, the mechanism by which benvitimod is effective for AD is far less recognized. It has been reported that benvitimod could upregulate FLG, LOR, and IVL expressions in vitro [15], indicating that benvitimod may improve AD by repairing skin barriers. Moreover, benvitimod has been proven to activate the AHR, thereby stimulating the expression of CYP1A1 and CYP1B1 in cultured primary human keratinocytes [15, 16]. AHR is a cytosolic ligand-activated transcription factor that senses diverse endogenous and exogenous stimuli and is involved in multiple biological processes [17, 18]. AHR is widely expressed in skin and was thought to be involved in skin barrier function regulation [19, 20]. However, the impact of benvitimod on the skin barrier of AD in vivo and the potential involvement of AHR in benvitimod’s effect on barrier repair remain unclear.
OVO-like (OVOL) proteins are transcribed from ubiquitously conserved genes encoding a C2H2 zinc finger transcription factor, which are susceptibility genes for AD [21, 22]. It has been reported that OVOL1 is regulated by AHR [23, 24]. Additionally, other AHR ligands, such as 6-formylindolo(3,2-b) carbazole (FICZ) and Rhodiola crenulata root extract, have been reported to upregulate FLG and LOR via the AHR-OVOL1 pathway [23, 24]. Nevertheless, different ligands trigger the interaction of AHR with different transcriptional partners, thereby inducing diverse biological responses [19, 25]. Further investigation is needed to determine whether benvitimod regulates OVOL1 via AHR and if OVOL1 is involved in the modulation of barrier function by benvitimod.
In the present study, an MC903-induced mouse model of AD was used to evaluate the effects of benvitimod on AD. Additionally, the AHR antagonist CH223191 was added to investigate the role of AHR in the effects of benvitimod. Furthermore, AHR and OVOL1 were knocked down to explore their roles in benvitimod’s effects on the expressions of FLG, LOR, and IVL in cultured normal human epidermal keratinocytes (NHEKs).
Results
Benvitimod improved MC903-induced mouse dermatitis
Consecutive application of MC903 induced significant AD-like dermatitis in mice, characterized by erythema, swelling, scaling, and epidermal thickening. Topical treatment with benvitimod for 14 days significantly improved the dermatitis (Fig. 1A). The effects of benvitimod could be blocked by AHR antagonist CH223191. As shown in Fig. 1A, redness, scales, and ear swelling were more prominent in BVM + CH (benvitimod + CH223191) group than in BVM (benvitimod) group. Histopathology demonstrated that benvitimod treatment inhibited epidermal thickening, spongiosis, and dermal inflammatory cell infiltration, whereas these abnormalities were not inhibited in BVM + CH group, even the inflammation was more severe in the murine skin of BVM + CH group than that of AD group (Fig. 1B). The dermatitis score was considerably reduced in BVM group compared with AD group, while they were higher in the BVM + CH group (Fig. 1C). The frequency of scratching was also significantly decreased after topical treatment with benvitimod and this effect was inhibited in the BVM + CH group. (Fig. 1D). Benvitimod improved the ear thickness significantly compared with AD group and this effect was also blocked by CH223191 (Fig. 1E).
Fig. 1.

Effects of topical benvitimod on MC903-induced mouse dermatitis. MC903 was applied on dorsal ears of the BALB/c mice for 10 consecutive days to establish mouse AD model. Then the mice were divided into 4 groups and treated with benvitimod (BVM) or benvitimod + CH223191 (BVM + CH) for 14 days. (A) Gross appearance of mice ears in different treatment groups. (B) Histopathology of the mice ears in different treatment groups. (HE staining, ×100, scale bar = 100 μm) (C) Scoring of dermatitis. (D) Frequency of scratching. (E) Ear thickness (µm). n = 6; ****P < 0.0001 (NC: naïve control group, AD: atopic dermatitis group, BVM: benvitimod treatment group, BVM + CH: AHR inhibition group)
Benvitimod inhibited downregulation of FLG, IVL and LOR expressions in MC903-induced mouse dermatitis via AHR pathway
In animal experiments, benvitimod treatment induced expression of AHR target genes CYP1A1 and CYP1B1 and this could be blocked by CH223191(Figs. 2A, B). The marked downregulation of LOR mRNA was observed in the MC903-induced dermatitis, whereas the mRNA levels of FLG and IVL were not decreased by benvitimod significantly compared to the NC group (Fig. 2C-E). FLG and IVL mRNA levels were upregulated by benvitimod significantly compared with AD group, however, benvitimod showed weak effects on LOR mRNA expression (Fig. 2C-E). Meanwhile, pretreatment with CH223191 inhibited the upregulation of FLG, IVL, and LOR mRNA levels induced by benvitimod (Fig. 2C-E).
Fig. 2.

Effects of benvitimod on FLG, IVL and LOR expressions in mouse dermatitis. The mouse ear dermatitis was treated with benvitimod (BVM) or benvitimod + CH223191 (BVM + CH) for 14 days. CYP1A1, CYP1B1, FLG, IVL and LOR mRNA expressions were measured by RT-qPCR. (A) CYP1A1 mRNA expression. (B) CYP1B1 mRNA expression. (C) FLG mRNA expression. (D) IVL mRNA expression. (E) LOR mRNA expression. (F) CYP1A1, CYP1B1, FLG, IVL, LOR and OVOL1 protein levels measured by Western blotting. Data were representative of repeated six experiments. n = 6; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001
The downregulation of FLG, IVL and LOR protein levels induced by MC903 treatment were recovered by benvitimod, and the effects of benvitimod were blocked by AHR antagonist CH223191 (Fig. 2F).
Benvitimod inhibited IL-4/IL-13-induced downregulation of FLG, IVL and LOR via AHR pathway in NHEKs
Significant downregulation of FLG, IVL and LOR mRNA levels and protein levels were observed when NHEKs were treated with IL-4 and IL-13 (Fig. 3A-D). Additionally, IL-4 and IL-13 downregulated the levels of FLG, IVL and LOR mRNA in a time-dependent manner (Fig. 3A-C). The mRNA levels of FLG, IVL and LOR were markedly downregulated by IL-4 and IL-13 at 1 ng/mL (Fig. 3E-G). Western blotting results showed that IL-4 and IL-13 significantly downregulated the expression of LOR at 1 ng/mL and significantly downregulated the expression of FLG and IVL at 10 ng/mL (Fig. 3H).
Fig. 3.

Benvitimod inhibited IL-4/IL-13-induced downregulation of FLG, IVL and LOR in NHEKs. (A-D) Normal human epidermal keratinocytes (NHEKs) were treated with IL-4 (10 ng/ml) and IL-13 (10 ng/ml) for indicated period of time. (A-C) FLG, IVL and LOR mRNA expressions were assessed by RT-qPCR. (D) FLG, IVL and LOR protein levels were measured by Western blotting (E-H) NHEKs were treated with IL-4 and IL-13 at indicated concentrations for 24 h. (E-G) FLG, IVL and LOR mRNA expressions were measured by RT-qPCR. (H) FLG, IVL and LOR protein levels were measured by Western blotting. (I-K) NHEKs were treated with IL-4 and IL-13 (10 ng/mL) in presence or absence of benvitimod (BVM) (0.1, 1, and 10 µM) for 24 h, and FLG, IVL and LOR mRNA expressions were assessed by RT-qPCR. n = 3; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001
Benvitimod recovered the mRNA levels of FLG and LOR dose-dependently, however the upregulation of IVL mRNA level induced by benvitimod was not dose-dependent (Fig. 3I-K). Benvitimod also upregulated the FLG, IVL and LOR mRNA levels significantly when NHEKs were not pretreated with IL-4 and IL-13 (Fig. 3I-K).
Transfection of AHR siRNA into NHEKs induced downregulation of the AHR protein (Figure S1). The FLG, IVL and LOR mRNA levels were not affected by benvitimod in AHR-knockdown NHEKs (Fig. 4A–C). In addition, transfection of AHR siRNA also inhibited benvitimod-induced upregulation of FLG, LOR and IVL protein levels (Fig. 4E). Moreover, the OVOL1 expression was upregulated by benvitimod and this effect was abrogated in AHR-knockdown NHEKs (Fig. 4D, E), similar to findings in animal experiments (Fig. 2F). Meanwhile, there was an obvious decrease of OVOL1 expression after silencing AHR in NHEKs (Fig. 4D, E).
Fig. 4.

Effects of benvitimod on FLG, LOR, and IVL expressions. (A-E) NHEKs transfected with siRNA against AHR (si-AHR) were treated with IL-4 (10 ng/mL) and IL-13 (10 ng/mL) for 24 h in the presence or absence of benvitimod (BVM, 1 µM). (A-D) FLG, IVL, LOR and OVOL1 mRNA expressions measured by RT-qPCR. (E) FLG, IVL, LOR, and OVOL1 protein levels measured by Western blotting. Data were representative of three experiments. n = 3; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001
Benvitimod-induced FLG, IVL and LOR upregulation is OVOL1-dependent
To determine if OVOL1 was involved in the upregulation of FLG, IVL and LOR induced by benvitimod, we transfected NHEKs with OVOL1-specific siRNA. The mRNA levels of FLG, IVL and LOR were upregulated by benvitimod in NHEKs treated by control siRNA (Fig. 5A-C). Upon OVOL1 knockdown (Figure S1), benvitimod-induced upregulation of LOR and IVL mRNA expressions were canceled completely, and upregulation of FLG mRNA expression was inhibited partially (Fig. 5A-C). Benvitimod could reverse the decreased expression of FLG, IVL and LOR mRNA induced by IL-4 and IL-13 in NHEKs treated by control siRNA, and this effect was inhibited after knockdown of OVOL1 (Fig. 5A-C). Benvitimod-induced upregulation of FLG, IVL and LOR protein levels was also abrogated in NHEKs transfected with OVOL1 siRNA compared to NHEKs transfected with control siRNA (Fig. 5D). These results suggested the upregulation of FLG, IVL and LOR expressions induced by benvitimod is OVOL1-dependent.
Fig. 5.

Benvitimod-induced FLG, IVL and LOR upregulation is OVOL1-dependent. (A-D) NHEKs transfected with si-OVOL1 were treated with IL-4 (10 ng/mL) and IL-13 (10 ng/mL) for 24 h in presence or absence of benvitimod (BVM, 1 µM). (A-C) FLG, IVL and LOR mRNA expressions measured by RT-qPCR. (D) FLG, IVL and LOR protein levels measured by Western blotting. Data were representative of three experiments. n = 3; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001
Discussion
Atopic dermatitis (AD) is a highly prevalent inflammatory skin disease caused by skin barrier dysfunction and chronic immune activation, primarily involving Th2 and Th17 inflammation [2, 3]. In MC903-induced dermatitis, a Th2 immune response is triggered and FLG, IVL, and LOR is downregulated [26, 27]. The therapeutic effects of benvitimod on MC903-induced dermatitis was firstly evaluated in mice in our present study. In this study, we demonstrated that benvitimod could alleviate symptoms such as scaling and itching caused by MC903, and it can improve epidermal thickening, spongiotic edema, and dermal inflammatory cell infiltration. Previous studies have shown that another AHR ligand, IAID, can improve dermatitis caused by MC903 through AHR [28], suggesting that benvitimod, as an AHR ligand, may also exert its effects through AHR. Our research indicated that the AHR antagonist CH223191 can block the therapeutic effect of benvitimod on MC903-induced dermatitis, further indicating that benvitimod may act through AHR.
Previous studies have shown that benvitimod can activate AHR and upregulate the expressions of FLG, IVL, and LOR in NHEKs [15]. However, there has been no relevant research on whether benvitimod can exert these effects in vivo. In our study, we found that benvitimod can restore the downregulation of FLG, IVL, and LOR in MC903-induced AD-like dermatitis in mice, which may be mediated by AHR. Our study further confirmed this conclusion in vivo and highlighted the important role of AHR in this process. In addition, the expression of FLG, IVL, and LOR in keratinocytes is also regulated by various cells and factors in the skin [11, 29, 30]. For example, Th2 cytokines IL-4 and IL-13 have been reported to downregulate the expression of FLG, IVL, and LOR in vitro [8, 11]. Our research also validated this conclusion. However, IL-4 and IL-13 could not significantly downregulate the expression of FLG, IVL, and LOR in NHEKs in the control siRNA group, which is inconsistent with the results in Fig. 3. This discrepancy may be due to the different sensitivities of NHEKs to IL-4 and IL-13 and the required stimulation time after control siRNA was added. Notably, in our study, the mRNA level of FLG was not significantly downregulated by MC903 in the AD group compared to the NC group, which is somewhat inconsistent with the protein level. A previous study demonstrated that FLG, IVL, and LOR were downregulated after MC903 was used for 6 days [31]. In the MC903 model, MC903 stimulates keratinocytes to produce TSLP [26], which further stimulates CD4 + T cells to produce IL-4 and IL-13 [2, 32]. These cytokines can downregulate the expressions of FLG, IVL, and LOR in keratinocytes [1, 30]. Additionally, skin treated with MC903 shows increased dermal inflammatory infiltrates consisting of eosinophils and CD3+, CD11c+, GR-1+, and mast cells, as well as increased levels of IL-5, IL-31, IL-10, IL-8, IFN-γ, and TNF-β [26, 33]. The immune status at different time points varies, affecting the regulation of barrier-related factors. The inability of IL-4 and IL-13 to significantly downregulate the expression of FLG, IVL, and LOR in NHEKs in the control siRNA group may be due to the different durations of MC903 stimulation and the different time points of detection, leading to varying expression levels of barrier-related factors.
AHR has been reported to be involved in the regulation of keratinocyte differentiation and barrier function [19, 25]. Upon activation, AHR translocates from the cytoplasm to the nucleus, forming AHR/ARNT dimers that further activate the AHR-dependent expression of CYP1A1 and genes encoded in the epidermal differentiation complex (EDC) region [34]. Different exogenous and endogenous AHR agonists induce diverse downstream effects [19, 34], making the specific role of AHR activation in keratinocyte differentiation controversial. A recent study showed barrier impairment in the epidermis of keratinocytes in AHR-knockout mice, and AHR-deficient mice had higher interindividual differences in their microbiome [20]. Consistently, severe functional defects in epidermal barrier have been observed in mice with targeted ablation of ARNT (aryl hydrocarbon receptor nuclear translocator) in keratinocytes [35]. It has been reported that the AHR ligand coal tar induces epidermal differentiation and stimulates filaggrin expression in keratinocytes by activating AHR signaling [7]. Additionally, previous studies revealed that AHR ligands such as FICZ and glyteer can activate AHR and upregulate FLG expression [24]. Jennifer A. Loertscher et al. demonstrated that exposure of mouse fetuses to the AHR ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in utero results in alterations in keratinocyte terminal differentiation [36]. Our research demonstrated that benvitimod can upregulate the expression of FLG, LOR, and IVL in vivo and in NHEKs, which may be mediated by AHR. Our study indicated that the binding of benvitimod to AHR has beneficial effects on epidermal differentiation and barrier function overall. In addition to the direct effect of benvitimod on FLG, LOR, and IVL through AHR, the influence of benvitimod on STAT6 may also have an indirect effect [8, 37]. Previous studies have shown that IL-4 and IL-13 can downregulate FLG, LOR, and IVL through STAT6 [37]. Our previous research indicated that benvitimod inhibits the phosphorylation of STAT6 in keratinocytes [37]. Therefore, it is possible that benvitimod reverses the IL-4 and IL-13 induced downregulation of FLG, IVL, and LOR by inhibiting the phosphorylation of STAT6.
OVO-like (OVOL) proteins are transcribed from ubiquitously conserved genes encoding a C2H2 zinc finger transcription factor [21]. OVOL1 has been identified as a susceptibility gene for AD and has been linked to AHR signaling in the skin barrier [21, 22]. OVOL1 knockout mice showed increased skin permeability to blue dye [21], suggesting that OVOL1 plays an important role in skin barrier functions. Additionally, activation of AHR induced by FICZ upregulated OVOL1 expression, leading to the translocation of cytoplasmic OVOL1 into the nucleus and subsequent upregulation of FLG expression [24]. Another study revealed that Rhodiola crenulata root extract induced FLG and LOR upregulation in an AHR-OVOL1-dependent manner [23]. It has been known that different ligands trigger the interaction of AHR with different transcriptional partners, thereby inducing different biological responses [34]. Our study demonstrated that OVOL1 expression is downregulated after AHR knockdown, suggesting that AHR may influence the baseline expression of OVOL1 in NHEKs. Furthermore, our research showed that after AHR knockdown, benvitimod’s ability to upregulate OVOL1 was abolished, indicating that benvitimod may upregulate OVOL1 expression through AHR. Additionally, after OVOL1 knockdown, benvitimod’s upregulation of FLG, LOR, and IVL was also abolished, indicating that OVOL1 plays a significant role in the upregulation of these proteins by benvitimod. However, the specific mechanism by which OVOL1 is crucial for skin barrier function in atopic dermatitis still requires further investigation.
We acknowledge several limitations in our study. Benvitimod was found to upregulate FLG, IVL, and LOR expressions in MC903-induced dermatitis of mice, possibly mediated by AHR. However, various immune cells in the skin that express AHR, such as CD4 + T cells and ILCs [19, 34], may also be affected by the AHR ligand benvitimod [15], indicating the need for further research into the role of benvitimod on other immune cells in the skin. Additionally, we have only studied the impact of the AHR pathway activated by benvitimod on FLG, LOR, and IVL in keratinocytes. Benvitimod may also affect FLG, LOR, and IVL through other pathways [11, 29, 38]. For example, a previous study has shown that benvitimod can induce the secretion of IL-24, leading to the downregulation of FLG and LOR expression [39]. Therefore, the impact of benvitimod on FLG, LOR, and IVL through other pathways requires further research.
In conclusion, this study suggests that benvitimod could improve MC903-induced dermatitis and upregulate the expressions of FLG, IVL, and LOR in mice. Furthermore, benvitimod was found to upregulate FLG, IVL and LOR expressions in cultured NHEKs, possibly mediated by AHR-OVOL1 axis. Therefore, benvitimod may help treat AD by breaking the vicious cycle of barrier dysfunction and inflammation through skin barrier repair. Further research is needed to elucidate the specific mechanisms of benvitimod in treating AD.
Materials and methods
Animal model and treatments
BALB/c mice (8 to 12 weeks old) were purchased from HFK Bioscience Co. Ltd. (Beijing, China). Mice were maintained in a 12-hour light/dark cycle under specific pathogen-free condition. Animal experiments were performed in compliance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Ethics Committee of Peking University People’s Hospital, Beijing, China (Permit number: 2020PHE083).
Mouse AD model was established by topical application of 2 nmol MC903 (dissolved in ethanol; Selleck) on dorsal ears for 10 consecutive days. Mice in naïve control group received 20 µl ethanol on the dorsal ears. The dermatitis was confirmed by clinical manifestation and histopathology.
After the establishment of animal model, mice were divided into naïve control group, atopic dermatitis group, benvitimod treatment group and AHR inhibition group. Mice in atopic dermatitis group received vehicle on the dorsal ears. Mice in benvitimod treatment group and AHR inhibition group received topical 1% benvitimod (Guangdong Zhonghao Pharma Ltd. China) once a day for 14 days. Mice in AHR inhibition group were treated with 20 µL CH223191 (10 mmol/L, Selleck) 30 min before benvitimod application. On days 14, the ear inflammation was evaluated by dermatitis scoring, with erythema/hemorrhage (0–4), scale/dryness (0–4), edema (0–4), and excoriation/erosion (0–4). The ear full thickness was measured and scratching frequency per 10 min was also recorded. Then all mice were sacrificed. Ear tissues were collected for RNA or protein extraction, and for histopathology.
Keratinocyte culture
Human skin specimens were obtained from circumcision. After digestion with Dispase (Sigma-Aldrich, St. Louis, MO) at 4 °C for 16–20 h, the epidermis was peeled off and disaggregated with trypsin (0.25 mg/mL) at 37 °C for 5 min. NHEKs were cultured in Keratinocyte Medium (ScienCell, USA) at 37 °C with 5% CO2. The culture medium was replaced every 2 days. When NHEKs reached 70–90% confluence, they were disaggregated with trypsin/0.01% ethylenediaminetetraacetic acid and subcultured. The fourth to sixth passage NHEKs were used for experiments.
In each experiment, NHEKs (1 × 106 per well) were seeded in 6-well culture plates, allowing to attach for 24 h before experiments.
This study was approved by the Ethics Committee of the Peking University People’s Hospital (Permit number: 2020PHB353-01) and conducted in accordance with the Declaration of Helsinki principles. All subjects provided written informed consent.
Transfection of siRNAs against AHR and OVOL1
Small interfering RNAs (siRNAs) against AHR or OVOL1, and scrambled siRNA that did not specifically degrade RNA (control siRNA) were purchased from Tsingke (China). AHR and OVOL1 siRNAs or the control siRNA were transfected into cells with Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. After incubation for 24 h, siRNA-transfected NHEKs were used for experiments. SiRNA sequences against AHR or OVOL1 for transient silencing were 5ʹ-GGAACACCUACAUCUAGAA-3ʹ and 5ʹ-AGUGUCACAACGACGUCAA-3ʹ.
Histopathology
Mice ear tissues were fixed in 4% phosphate-buffered formalin solution, embedded in paraffin, cut into 5-µm-thick sections, and stained with hematoxylin and eosin.
Quantitative real-time PCR
Total RNA was extracted from skin tissues and NHEKs using a tissue RNA purification kit (EZBioscience, USA) and performed according to the manufacturer’s protocol. EZscript reverse transcription mix (EZBioscience) was used to synthesize cDNA. mRNA levels were measured with the 7500 Fast Real Time PCR Detection System (Applied Biosystems, CA, USA) and SYBR Green qPCR master mix (EZBioscience). Fold changes in expression were normalized to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA level. Relative expression was calculated using the comparative threshold cycle (Ct) and 2−ΔΔCt method.
Primer sequences are shown in Table S1.
Western blotting
Total protein was extracted from mouse skin tissues and cultured NHEKs using tissue protein extraction kit (Solarbio, Beijing, China). The protein concentrations were measured with a BCA Protein Assay Kit (Beyotime, Jiangsu, China). Equal amounts of protein were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred onto a polyvinylidene difluoride membrane. After blocking with 5% dry skim milk, the membrane was incubated overnight at 4 °C with primary antibodies (Primary antibodies are listed in Table S2). Anti-rabbit IgG (1:2,000, Cell Signaling Technology) or anti-mouse IgG (1:2,000, Cell Signaling Technology) were used as secondary antibodies. Specific bands were observed using the ChemiDoc Touch Imaging System (Bio-Rad) with standard chemiluminescence (Beyotime). Results were analyzed with ImageJ software (National Institutes of Health, MD, USA) and normalized to GAPDH.
Statistical analysis
The statistical significance of differences between groups was examined by Student’s unpaired two-tailed t-test (for two groups) or one-way ANOVA (multiple groups) using GraphPad PRISM 9.0 software (GraphPad Software, La Jolla, CA, USA). A p-value less than 0.05 was considered significant.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (82003336, 82273524). The authors thank volunteers for providing their skin specimens and thank Guangdong Zhonghao Pharma Ltd for providing benvitimod cream and vehicle. The authors also thank staff of Animal Laboratory of Peking University People’s Hospital for running and advice in the animal studies.
Abbreviations
- AD
Atopic dermatitis
- AHR
Aryl hydrocarbon receptor
- ARNT
Aryl hydrocarbon receptor nuclear translocator
- BVM
Benvitimod
- CH
- CYP1A1
Cytochrome P450 family subfamily A member 1
- CYP1B1
Cytochrome P450 family subfamily B member 1
- FLG
Filaggrin
- IVL
Involucrin
- LOR
Loricrin
- NHEKs
Normal human epidermal keratinocytes
- OVOL
OVO-like
- TSLP
Thymic stromal lymphopoietin
- RT-qPCR
Real-time quantitative PCR
- siRNAs
Small interfering RNAs
Author contributions
The manuscript was prepared by Qiuyu Jia and reviewed by Xiaojie Wang, Ping Liu, Houmin Li, and Jianzhong Zhang. Conceptualization and design of experiments was conducted by Ping Liu, Qiuyu Jia, Xiaojie Wang, Houmin Li, and Jianzhong Zhang. Experiments were performed by Qiuyu Jia, Xiaojie Wang, Jian Hu, and Jun Jia. Data analysis was performed by Qiuyu Jia. All authors read and approved the final manuscript.
Data availability
No datasets were generated or analysed during the current study.
Declarations
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Qiuyu Jia and Ping Liu contributed equally to this work.
Contributor Information
Jianzhong Zhang, Email: rmzjz@126.com.
Houmin Li, Email: lhoumin@sina.cn.
References
- 1.Schuler CFt et al (2023) Novel insights into atopic dermatitis. J Allergy Clin Immunol 151(5):1145–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sroka-Tomaszewska J, Trzeciak M (2021) Molecular mechanisms of atopic Dermatitis Pathogenesis. Int J Mol Sci, 22(8) [DOI] [PMC free article] [PubMed]
- 3.Stander S (2021) Atopic dermatitis. N Engl J Med 384(12):1136–1143 [DOI] [PubMed] [Google Scholar]
- 4.Li H et al (2021) Update on the Pathogenesis and therapy of atopic dermatitis. Clin Rev Allergy Immunol 61(3):324–338 [DOI] [PubMed] [Google Scholar]
- 5.Dale BA, Resing KA, Lonsdale-Eccles JD (1985) Filaggrin: a keratin filament associated protein. Ann N Y Acad Sci 455:330–342 [DOI] [PubMed] [Google Scholar]
- 6.Harding CR, Scott IR (1983) Histidine-rich proteins (filaggrins): structural and functional heterogeneity during epidermal differentiation. J Mol Biol 170(3):651–673 [DOI] [PubMed] [Google Scholar]
- 7.van den Bogaard EH et al (2013) Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J Clin Invest 123(2):917–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim BE et al (2008) Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin Immunol 126(3):332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esaki H et al (2016) Early-onset pediatric atopic dermatitis is T(H)2 but also T(H)17 polarized in skin. J Allergy Clin Immunol 138(6):1639–1651 [DOI] [PubMed] [Google Scholar]
- 10.Salimi M et al (2013) A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med 210(13):2939–2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furue M (2020) Regulation of skin barrier function via competition between AHR Axis versus IL-13/IL-4JAKSTAT6/STAT3 Axis: pathogenic and therapeutic implications in atopic dermatitis. J Clin Med, 9(11) [DOI] [PMC free article] [PubMed]
- 12.Imai Y et al (2013) Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc Natl Acad Sci U S A 110(34):13921–13926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peppers J et al (2019) A phase 2, randomized dose-finding study of tapinarof (GSK2894512 cream) for the treatment of atopic dermatitis. J Am Acad Dermatol 80(1):89–98e3 [DOI] [PubMed] [Google Scholar]
- 14.Paller AS et al (2021) Efficacy and patient-reported outcomes from a phase 2b, randomized clinical trial of tapinarof cream for the treatment of adolescents and adults with atopic dermatitis. J Am Acad Dermatol 84(3):632–638 [DOI] [PubMed] [Google Scholar]
- 15.Smith SH et al (2017) Tapinarof is a natural AhR agonist that resolves skin inflammation in mice and humans. J Invest Dermatol 137(10):2110–2119 [DOI] [PubMed] [Google Scholar]
- 16.Tsuji G et al (2022) Natural compounds Tapinarof and Galactomyces Ferment Filtrate Downregulate IL-33 expression via the AHR/IL-37 Axis in Human keratinocytes. Front Immunol 13:745997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stockinger B et al (2014) The aryl hydrocarbon receptor: multitasking in the immune system. Annu Rev Immunol 32:403–432 [DOI] [PubMed] [Google Scholar]
- 18.Fernandez-Gallego N, Sanchez-Madrid F, Cibrian D (2021) Role of AHR Ligands in skin homeostasis and cutaneous inflammation. Cells, 10(11) [DOI] [PMC free article] [PubMed]
- 19.Napolitano M, Patruno C (2018) Aryl hydrocarbon receptor (AhR) a possible target for the treatment of skin disease. Med Hypotheses 116:96–100 [DOI] [PubMed] [Google Scholar]
- 20.Haas K et al (2016) Aryl Hydrocarbon receptor in Keratinocytes is essential for murine skin Barrier Integrity. J Invest Dermatol 136(11):2260–2269 [DOI] [PubMed] [Google Scholar]
- 21.Sun P et al (2021) OVOL1 regulates Psoriasis-Like skin inflammation and Epidermal Hyperplasia. J Invest Dermatol 141(6):1542–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsuji G et al (2018) The role of the OVOL1-OVOL2 axis in normal and diseased human skin. J Dermatol Sci 90(3):227–231 [DOI] [PubMed] [Google Scholar]
- 23.Hashimoto-Hachiya A et al (2018) Upregulation of FLG, LOR, and IVL expression by Rhodiola Crenulata Root Extract via Aryl Hydrocarbon receptor: Differential involvement of OVOL1. Int J Mol Sci, 19(6) [DOI] [PMC free article] [PubMed]
- 24.Tsuji G et al (2017) Aryl hydrocarbon receptor activation restores filaggrin expression via OVOL1 in atopic dermatitis. Cell Death Dis 8(7):e2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Furue M, Hashimoto-Hachiya A, Tsuji G (2019) Aryl Hydrocarbon Receptor in atopic dermatitis and psoriasis. Int J Mol Sci, 20(21) [DOI] [PMC free article] [PubMed]
- 26.Moosbrugger-Martinz V, Schmuth M, Dubrac S (2017) A mouse model for atopic dermatitis using topical application of vitamin D3 or of its Analog MC903. Methods Mol Biol 1559:91–106 [DOI] [PubMed] [Google Scholar]
- 27.Li M et al (2006) Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc Natl Acad Sci U S A 103(31):11736–11741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu J et al (2019) A tryptophan metabolite of the skin microbiota attenuates inflammation in patients with atopic dermatitis through the aryl hydrocarbon receptor. J Allergy Clin Immunol 143(6):2108–2119e12 [DOI] [PubMed] [Google Scholar]
- 29.Dai X et al (2022) Nuclear IL-33 plays an important role in IL-31Mediated downregulation of FLG, Keratin 1, and Keratin 10 by Regulating Signal Transducer and activator of transcription 3 activation in human keratinocytes. J Invest Dermatol 142(1):136–144e3 [DOI] [PubMed] [Google Scholar]
- 30.Furue K et al (2019) The IL-13-OVOL1-FLG axis in atopic dermatitis. Immunology 158(4):281–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nasanbat B et al (2023) Kaempferol therapy improved MC903 induced-atopic dermatitis in a mouse by suppressing TSLP, oxidative stress, and type 2 inflammation. J Dermatol Sci 111(3):93–100 [DOI] [PubMed] [Google Scholar]
- 32.Hvid M et al (2011) IL-25 in atopic dermatitis: a possible link between inflammation and skin barrier dysfunction? J Invest Dermatol 131(1):150–157 [DOI] [PubMed] [Google Scholar]
- 33.Kim BS et al (2013) TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med 5(170):170ra16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rothhammer V, Quintana FJ (2019) The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol 19(3):184–197 [DOI] [PubMed] [Google Scholar]
- 35.Geng S et al (2006) Targeted ablation of Arnt in mouse epidermis results in profound defects in desquamation and epidermal barrier function. J Cell Sci 119(Pt 23):4901–4912 [DOI] [PubMed] [Google Scholar]
- 36.Loertscher JA et al (2002) In utero exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin causes accelerated terminal differentiation in fetal mouse skin Toxicol Sci, 68(2): pp. 465 – 72 [DOI] [PubMed]
- 37.Wang X et al (2024) Benvitimod inhibits IL-4- and IL-13-Induced tight Junction impairment by activating AHR/ARNT pathway and inhibiting STAT6 phosphorylation in human keratinocytes. J Invest Dermatol 144(3):509–519e7 [DOI] [PubMed] [Google Scholar]
- 38.Yang G et al (2020) Skin barrier abnormalities and Immune Dysfunction in atopic dermatitis. Int J Mol Sci, 21(8) [DOI] [PMC free article] [PubMed]
- 39.Vu YH et al (2020) IL-24 negatively regulates keratinocyte differentiation Induced by Tapinarof, an aryl hydrocarbon receptor modulator: implication in the treatment of atopic dermatitis. Int J Mol Sci, 21(24) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analysed during the current study.