

Abstract
Background/Aim
The application of next-generation sequencing (NGS) technology in the genetic investigation of hereditary cancer is important for clinical surveillance, therapeutic approach, and reducing the risk of developing new malignancies. The aim of the study was to explore genetic predisposition in individuals referred for hereditary cancer.
Materials and Methods
A total of 8,261 individuals were referred for multigene genetic testing, during the period 2020-2023, in the laboratory, and underwent multigene genetic testing using NGS. Among the examined individuals, 56.17% were diagnosed with breast cancer, 6.77% with ovarian cancer, 2.88% with colorectal cancer, 1.91% with prostate cancer, 6.43% were healthy with a significant family history of cancer, while 3.06% had a different type of cancer and 0.21% had not provided any information. Additionally, in 85 women with breast cancer we performed whole exome sequencing analysis.
Results
20% of the examined individuals carried a pathogenic variant. Specifically, 54.8% of the patients had a pathogenic variant in a clinically significant gene (BRCA1, BRCA2, PALB2, RAD51C, PMS2, CDKN2A, MLH1, MSH2, TP53, MSH6, APC, RAD51D, PTEN, RET, CDH1, MEN1, and VHL). Among the different types of pathogenic variants detected, a significant percentage (6.52%) represented copy number variation (CNV). With WES analysis, the following findings were detected: CTC1: c.880C>T, p.(Gln294*); MLH3: c.405del, p.(Asp136Metfs*2), PPM1D: c.1426_1430del, p.(Glu476Leufs*3), and SDHB: c.395A>G, p.(His132Arg).
Conclusion
Comprehensive multigene genetic testing is necessary for appropriate clinical management of pathogenic variants’ carriers. Additionally, the information obtained is important for determining the risk of malignancy development in family members of the examined individuals.
Keywords: Hereditary cancer, next generation sequencing, whole exome sequencing
Hereditary cancers arise due to specific alterations in genes critical for controlling cell growth, division, and repair. These genetic variants are typically found in germ cells, and they can be transmitted from one generation to the next (1). Hereditary cancer syndromes often adhere to Mendelian inheritance patterns, including autosomal dominant, autosomal recessive, or X-linked inheritance, wherein autosomal dominant conditions necessitate only one copy of the mutated gene from either parent to heighten the risk of cancer (2). Several genes contribute to hereditary cancers. For instance, pathogenic/likely pathogenic variants (PVs/LPVs) in BRCA1 and BRCA2 are linked to an increased risk of breast, ovarian, and other cancers (3), while TP53 PVs/LPVs are associated with Li-Fraumeni syndrome, elevating the risk of various cancer types (4). PVs/LPVs in APC, MLH1, MSH2, MSH6, and PMS2 relate to Lynch syndrome, leading to an elevated risk of colorectal and other cancers (5).
Penetrance indicates the likelihood that an individual with a specific gene variant will develop cancer. In some instances, the presence of a PVs/LPVs does not assure cancer development, as other factors can influence penetrance. Hereditary cancers often manifest at an earlier age compared to sporadic cases of the same cancer, a characteristic feature distinguishing them (6). The risk of hereditary cancers within families can have a cascade effect. Upon identifying a specific gene PV/LPV, other family members may undergo genetic testing to assess their own risk (7).
Genetic testing involves examining an individual’s DNA to identify specific PVs/LPVs associated with hereditary cancer syndromes. Prior to and following genetic testing, genetic counseling is recommended to furnish individuals and families with information about the implications of the test results and guidance on managing cancer risk (8). Understanding the genetic foundation of hereditary cancers is essential for formulating targeted prevention and early detection strategies. While hereditary cancers constitute a relatively small proportion of all cancer cases, the identification of high-risk individuals facilitates proactive management and personalized medical care (9).
Next-generation sequencing (NGS) approach has transformed the domain of genetics and has been pivotal in uncovering and comprehending hereditary cancer. NGS stands out as a high-throughput sequencing technology that expeditiously and cost-effectively analyzes extensive DNA segments (10). It permits the simultaneous sequencing of multiple genes or even entire genomes, delivering a holistic perspective on an individual’s genetic composition. In contrast to labor-intensive and time-consuming traditional genetic testing methods like Sanger sequencing, NGS allows the concurrent analysis of numerous genes linked to hereditary cancers in a single test, emerging as a potent tool for all-encompassing screening (11).
NGS finds application in panel testing, where a predetermined set of genes associated with specific hereditary cancer syndromes is concurrently examined. Hereditary cancer panels typically encompass genes, such as BRCA1, BRCA2, TP53, APC, and MLH1, among others, contingent upon the suspected syndrome (12). Whole exome sequencing (WES) targets the sequencing of gene protein-coding regions (exons), where disease-causing variants are commonly located. On the other hand, whole genome sequencing (WGS) scrutinizes the entire genome, offering a comprehensive overview of both coding and non-coding regions (13).
NGS simplifies the identification of PVs/LPVs within genes linked to hereditary cancers. Uncovering these variants aids in evaluating an individual’s cancer risk and formulating personalized management strategies. The high-throughput nature of NGS enhances diagnostic efficacy compared to conventional sequencing methods, allowing for the detection of rare or novel mutations that might escape targeted approaches (14).
Despite its advantages, NGS introduces challenges, including grappling with variants of uncertain significance (VUS) and the imperative need for meticulous variant interpretation. Genetic counseling is paramount in both pre- and post-testing phases to facilitate individuals in comprehending the implications of the results (15). Moreover, NGS has played a pivotal role in large-scale genomic research, contributing to the identification of novel genes associated with hereditary cancers and deepening our understanding of the genetic underpinnings of these conditions (16).
The aim of this study was to investigate the genetic predisposition in individuals referred for hereditary cancer using NGS technology. Moreover, genetic testing was conducted on a sample of 85 women diagnosed with breast cancer, employing the advanced technology WES. This comprehensive analysis sheds light on the diverse genetic landscapes that contribute to breast cancer susceptibility, offering valuable insights for further research and potential clinical implications.
Materials and Methods
During the period from 2020 to 2023, a total of 8,261 individuals were referred to at Genekor’s laboratory, with a focus on genetic analysis using NGS. Before undergoing molecular genetic testing, everyone participated in an informed consent process, signifying their understanding and agreement to have their genetic information analyzed. Importantly, they also granted explicit permission for the anonymized utilization of their data in scientific research endeavors and potential publication of research findings. To provide a comprehensive context for the genetic analysis, additional information was gathered from these individuals. Clinicians and healthcare providers collaborating in the study facilitated the collection of these vital data, ensuring that each patient’s unique medical and family background was thoroughly documented.
In all 8,261 individuals we performed NGS analysis using a customized gene panel including 52 genes. Moreover, we analyzed using WES 85 breast cancer patients. Of these patients, a significant subgroup of 47 individuals exhibited no observable PVs/LPVs in a comprehensive panel of 52 genes known to be associated with susceptibility to hereditary cancer. A distinct group of 20 participants displayed PVs/LPVs in moderate genes. Furthermore, 18 individuals exhibited PVs/LPVs within low-penetrance genes associated with breast cancer (Figure 1).
Figure 1.

Schematic representation of the workflow used in this study. PV: Pathogenic variant; LPV: likely pathogenic variant; NGS: next-generation sequencing; WES: whole exome sequencing.
According to the manufacturer’s protocol, genomic DNA extraction from peripheral blood samples was performed utilizing the MagCore® Genomic DNA Whole Blood Kit (RBC Bioscience, New Taipei City, Taipei, Taiwan, ROC). This extraction process ensured the retrieval of high-quality DNA suitable for downstream genetic analysis. For the comprehensive analysis of hereditary cancer predisposition, a solution-based capture technique was employed, targeting 52 genes implicated in such predisposition (17). This capture method utilized the Roche NimbleGen SeqCap EZ Choice kit (Pleasanton, CA, USA), along with WES facilitated by the KAPA HyperExome kit (Pleasanton, CA, USA). The sample preparation adhered meticulously to the instructions provided in the SeqCap EZ Choice Library User’s Guide by Roche NimbleGen, ensuring consistency and accuracy throughout the process. Briefly, the assay creates a library using a solution-based capture method to enrich targeted genomic DNA regions. Initially, each sample required 150 ng of double-stranded DNA, which was enzymatically fragmented. To maintain the stability of this reaction, an EDTA neutralizing solution was applied before fragmentation. The fragmented DNA was then processed through end-repair, A-tailing, and ligation with paired-end indexed adapters. Finally, the library was amplified using ligation-mediated PCR and hybridized overnight with custom probes. Library preparation concluded with post-capture LM-PCR, following the manufacturer’s protocol. Sequencing of the prepared samples was carried out utilizing DNBG400 technology provided by MGI Tech Co., Ltd (Beishan Industrial Zone, Shenzhen, PR China). The generated sequencing data underwent analysis using the SeqNext version 4.4.0 software suite developed by JSI Medical Systems GmbH (Ettenheim, Germany). This software facilitated the identification and interpretation of sequence alterations within the context of clinically relevant transcripts.
Bioinformatics tools were instrumental in the identification of various genetic variations, including single nucleotide variations (SNVs), insertions, deletions (indels), and copy number variations (CNVs). Following variant identification, thorough annotation and prioritization processes were implemented, with a specific emphasis on discerning potentially pathogenic alterations. This meticulous approach ensured the comprehensive evaluation of genetic variants, ultimately contributing to the identification of clinically relevant findings relevant to hereditary cancer predisposition.
Results
Demographics. The demographic characteristics of individuals, clinical history related to cancer diagnosis, and a thorough family history assessment are shown in Table I. The breakdown of individuals based on their countries of origin was as follows: 5,733 (69.4%) from Greece, 991 (12%) from Romania, 816 (9.8%) from Turkey, and 721 from other countries (8.7%). The median age at testing for individuals in our cohort was 46.8 years, with ages ranging from 18 to 82 years. In terms of sex distribution, the majority of individuals tested were female, constituting 89.7% (7,407 out of 8,261) of the cohort. Conversely, males comprised only 10.3% (1,854 out of 8,261) of the individuals referred for testing. A notable finding was that a significant proportion of individuals, specifically 86.4% (5,697 out of 6,591 for whom family history data was available), had a family history of cancer. Within this subgroup, 42% (2,396 out of 5,697) had a family history of the same type of cancer, indicating potential hereditary factors contributing to the prevalence of certain cancer types within families.
Table I. Demographic characteristics of the individuals.

Out of the examined individuals, 4,641 (76.6%) were diagnosed with breast cancer, 560 (6.7%) with ovarian cancer, 238 with colorectal cancer (3.9%), 210 (3.5%) with pancreatic cancer, and 158 with prostate cancer (2.6%). Additionally, 532 were referred as healthy individuals with a significant family history of cancer, while 253 (4.2%) had a different type of cancer, and 1,670 (20.2%) had not provided any information at the time of genetic testing.
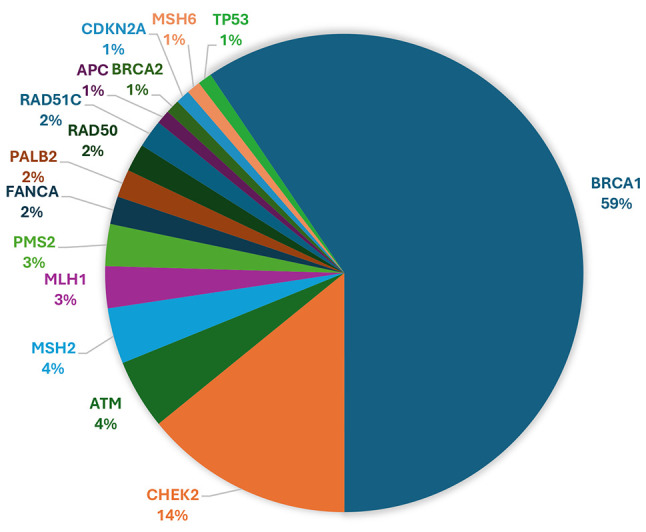
Heredigene results. Approximately 20% (1,656 out of 8,261) of the individuals studied showed a LP/P variant (Figure 2). More precisely, within this group, 54.8% (908 out of 1,656) of the patients exhibited a LP/P in a gene of clinical significance. The breakdown of these variants in specific genes is as follows: BRCA1 (23.13%), BRCA2 (15.16%), PALB2 (4.95%), RAD51C (1.87%), PMS2 (1.57%), CDKN2A (1.45%), MLH1 (1.45%), MSH2 (1.03%), TP53 (1.03%), MSH6 (0.97%), APC (0.85%), RAD51D (0.48%), PTEN (0.36%), RET (0.24%), CDH1 (0.18%), MEN1 (0.06%), and VHL (0.06%) (Figure 3). Notably, among the identified PVs, a significant proportion (6.52%, 108 out of 1,656) were CNVs (Figure 4).
Figure 2.
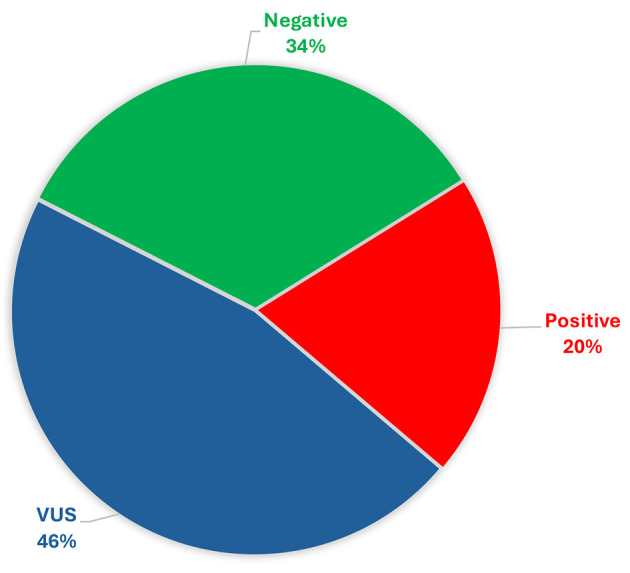
Results from the panel testing conducted on 8,216 individuals. Positive outcomes pertain to instances where a pathogenic variant/likely pathogenic variant was detected. VUS: Variant of unknown significance.
Figure 3.

Percentage of pathogenic/likely pathogenic findings identified in each gene.
Figure 4.
Percentage of pathogenic/likely pathogenic copy number variation findings identified in each gene.
Heredigene results by cancer type. Among the patients diagnosed with various types of cancer, including breast cancer, ovarian cancer, colorectal cancer, pancreatic cancer, and prostate cancer, specific PVs/LPVs have been identified in a subset of cases. Specifically, out of 4,641 patients with breast cancer, 20% were found to have a PV/LPV. For ovarian cancer patients (totaling 560), this assumption rose to 27%, while 21% of the 238 colorectal cancer patients were found to carry PVs/LPVs. In the case of pancreatic cancer, affecting 210 individuals, 27% were found to have identifiable PVs. Lastly, among the 158 patients with prostate cancer, 17% were identified as having PVs/LPVs. The contribution of genes that identify PVs in each cancer type is shown in Figure 5A-E.
Figure 5.


Results of panel testing for individuals diagnosed with A) breast, B) colorectal, C) pancreatic, D) ovarian, and E) prostate cancer.
Double heterozygosity. Within the cohort of positive individuals, comprising 95 out of 1,656 cases (5.73%), it was observed that some individuals carry two PVs concurrently (Figure 6). One notable case involves a patient diagnosed with breast cancer at the age of 27, who was found to harbor two PVs within the BRCA1 gene. These variants are identified as c.1287_1291del AGACT, p.(Asp430Thrfs*4), and c.1300_1307dupAGTG ATCC, p.(His437Valfs*7). Moreover, two patients were identified as carrying one PV in BRCA1 and another in the BRCA2 gene. In one instance, a patient diagnosed with breast cancer at 37 years old exhibited the pathogenic variant c.5467G>A, p.(Ala1823Thr) in BRCA1, alongside the variant c.2490_2491insT, p.(Val831Cysfs*2) in BRCA2. In another case, a patient diagnosed with ovarian cancer at 38 years old was found to harbor the pathogenic variant c.3607C>T, p.(Arg1203), in BRCA1, and the pathogenic variant c.9371A>T, p.(Asn3124Ile), in the BRCA2 gene.
Figure 6.

Configurations of genes in instances where there are two pathogenic variants/likely pathogenic variants (double heterozygotes).
CHEK2 variants. Among the genetic variants in the CHEK2 gene, missense variants are the most prevalent (59%). These mutations entail a single nucleotide change, leading to the substitution of one amino acid with another in the associated protein (Figure 7). One particular variant in the CHEK2 gene, denoted as c.470T>C, results in the amino acid alteration p.(Ile157Thr) (Table II).
Figure 7.
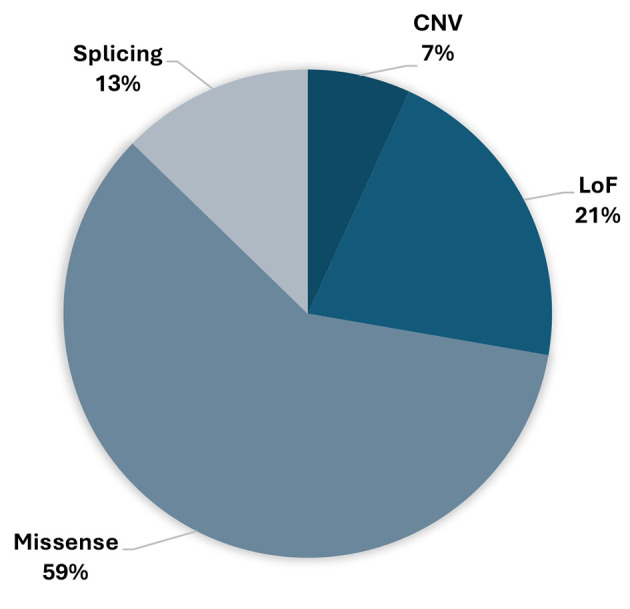
The involvement of different types of variants detected in the CHEK2 gene within this study.
Table II. List of pathogenic and likely pathogenic variants (PVs/LPVs) in the CHEK2 gene.

CNV: Copy number variation; LoF: loss of function.
TP53 variants. Within the examined group, 17 individuals were identified to carry PVs within the TP53 gene. Remarkably, most of these variants are present as missense variants, signifying changes where a single nucleotide substitution leads to the substitution of one amino acid with another within the TP53 protein sequence (Table III).
Table III. List of pathogenic and likely pathogenic variants (PVs/LPVs) in the TP53 gene.

LoF: Loss of function.
Variants in homologous recombination (HR) genes. In the group of 1,656 individuals who yielded positive test results, a significant segment, precisely 1,310 individuals, were discovered to possess a PV in one or more genes linked to HR repair (Figure 8). These HR genes are pivotal in the precise restoration of double-stranded DNA breaks, safeguarding the stability and completeness of the genome.
Figure 8.

The occurrence rate of pathogenic/likely pathogenic (P/LP) variants in genes associated with homologous recombination (HR).
WES results. The results of the study involve WES conducted on a total of 85 samples. During the analysis, the data underwent alignment, and variant annotation using JSI, leading to the identification of a substantial number of variants – 29,511 to be precise. These variants were further scrutinized, focusing on 588 genes associated with cancer, employing the guidelines set by the American College of Medical Genetics and Genomics (ACMG). This refined the pool of variants to 3,615, narrowing down the focus to those with potential relevance to cancer. From this subset, a more targeted investigation was carried out, resulting in the identification of four candidate variants that stood out as potential indicators of interest. Using WES analysis, the following findings were identified: CTC1:c.880C>T, p.(Gln294*); MLH3: c.405del, p.(Asp136Metfs*2), PPM1D: c.1426_1430del, p.(Glu476Leufs*3), and SDHB: c.395A>G, p.(His132Arg) (Figure 9, Table IV).
Figure 9.
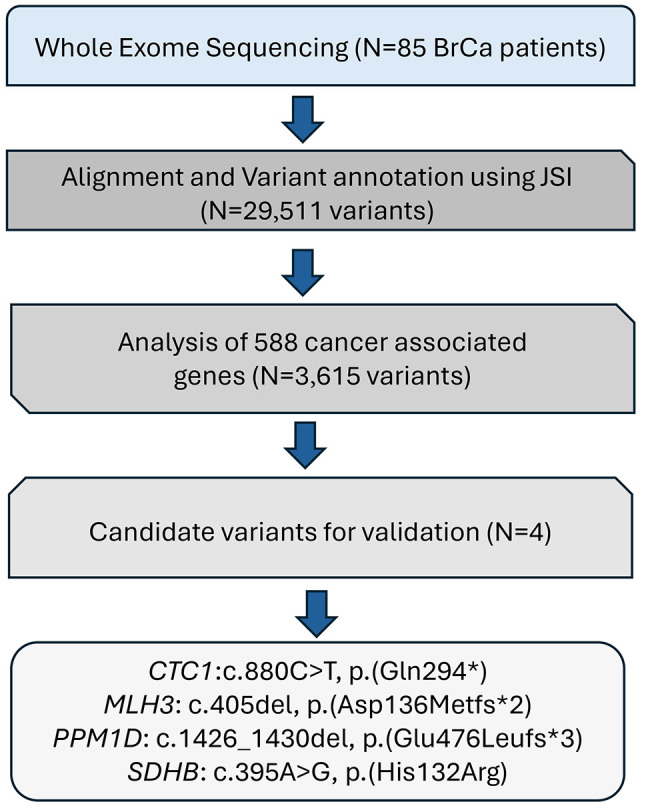
Workflow for prioritizing genes based on genetic information.
Table IV. Genetic variants observed in breast cancer patients through whole exome sequencing (WES).

Discussion
The current study utilized advanced genetic analysis techniques, employing NGS technology to detect P/PL variants associated with hereditary cancer. This cutting-edge technique enabled the simultaneous examination of multiple genes, allowing for a comprehensive assessment of genetic predispositions to various forms of cancer. NGS technology revolutionizes genetic analysis by facilitating high-throughput sequencing of DNA samples, thereby enabling the rapid and cost-effective analysis of vast genomic regions (18). By examining a panel of genes associated with various cancer types, the study sought to capture a broad spectrum of potential genetic risk factors. By harnessing the power of NGS technology and multi-gene analysis, the study aimed to enhance our understanding of the genetic underpinnings of cancer susceptibility, ultimately paving the way for improved risk assessment, early detection, and personalized management strategies for individuals at heightened risk of hereditary cancer.
This study encompassed a comprehensive cohort of 8,261 individuals who were consecutively referred to our laboratory for the analysis of genes associated with hereditary cancer predisposition. This sizable and consecutive inclusion of individuals underscores the breadth and depth of the study’s scope, reflecting a significant sample size representative of those seeking genetic testing for hereditary cancer concerns. The inclusion of such a large and consecutive cohort speaks to the substantial demand for genetic testing in this field and highlights the importance of addressing hereditary factors in cancer risk assessment and management.
Upon variant analysis, findings unveiled the presence of at least one PV or LPV in a notable 20% of the studied cohort. This observation underscores the significance of genetic alterations in the context of hereditary cancer predisposition, suggesting a substantial proportion of individuals harboring genetic mutations with potential clinical implications. These PVs/LPVs represent alterations in genes known to confer an increased risk of developing cancer. Their identification within the study cohort highlights the importance of genetic testing in uncovering potential genetic predispositions and underscores the utility of such analyses in clinical practice. The 20% prevalence of PVs/LPVs underscores the importance of genetic screening and counseling in individuals with suspected hereditary cancer predisposition. Identifying these variants allows for targeted risk assessment, surveillance, and potentially preventive measures to mitigate the risk of cancer development in affected individuals and their families.
Among the subset of individuals harboring P/LP variants, a noteworthy proportion, comprising 54.8% (908 out of 1,656), were found to carry these variants within genes recognized for their clinical significance. This finding underscores the importance of identifying PVs/LPVs within genes known to play a pivotal role in hereditary cancer predisposition. Delving deeper into the breakdown of PVs within clinically significant genes, several notable patterns emerged. BRCA1 and BRCA2, well-established tumor suppressor genes associated with hereditary breast and ovarian cancer syndrome, accounted for the largest proportions, with BRCA1 contributing to 23.13% and BRCA2 to 15.16% of the variants detected. These findings align with the established role of BRCA1 and BRCA2 variants in predisposing individuals to breast, ovarian, and other cancers (19). Additionally, other genes implicated in hereditary cancer syndromes exhibited varying frequencies of PVs within the cohort. Notable examples include PALB2, RAD51C, and PMS2, each contributing to 4.95%, 1.87%, and 1.57% of the variants, respectively, further highlighting the diverse genetic landscape of hereditary cancer predisposition. Moreover, the detection of PVs in genes, such as CDKN2A, MLH1, MSH2, TP53, MSH6, APC, RAD51D, PTEN, RET, CDH1, MEN1, and VHL underscores the importance of comprehensive genetic testing in uncovering potential risk factors across a spectrum of cancer types and syndromes (20,21).
Furthermore, a significant and noteworthy discovery among the identified PVs was the occurrence of CNV in a substantial subset of cases, accounting for 6.52% (108 out of 1,656) of the individuals studied. This observation sheds light on the diverse spectrum of genetic alterations contributing to hereditary cancer predisposition, highlighting the importance of comprehensive genetic analysis techniques capable of detecting various types of genomic aberrations (22). CNVs represent a class of genetic mutations characterized by duplications, deletions, or rearrangements of large segments of DNA, which can have profound implications for gene function and disease susceptibility. In the context of hereditary cancer predisposition, CNVs can encompass alterations in critical tumor suppressor genes or oncogenes, thereby significantly impacting an individual’s risk of developing cancer. These findings emphasize the necessity of utilizing advanced genomic technologies capable of detecting CNVs alongside SNVs and small insertions/deletions (indels) for a comprehensive assessment of genetic risk factors (23). Understanding the prevalence and significance of CNVs in hereditary cancer predisposition has important clinical implications, as it can inform risk assessment, screening, and management strategies for individuals at heightened risk of developing cancer. By integrating CNV analysis into genetic testing protocols, healthcare providers can provide more tailored and personalized care for individuals with suspected hereditary cancer predisposition, ultimately improving patient outcomes and reducing the burden of cancer within affected families (24,25).
Within the cohort of individuals who tested positive for PVs/LPVs, an intriguing subset emerged, comprising 5.73% (95 out of 1,656) who were identified as carrying not just one, but two P/LP variants. This discovery underscores the complexity and potential severity of genetic predispositions observed in certain cases, suggesting a multifactorial contribution to cancer susceptibility within this subset of individuals (26). Finding individuals with dual PVs/LPVs raises several compelling questions about the interplay between these genetic alterations and their cumulative effect on cancer risk. It suggests the presence of synergistic or additive effects, where the combination of multiple PVs may confer an even higher risk of cancer development compared to having a single variant alone (27). Furthermore, identifying individuals with two PVs/LPVs highlights the importance of thorough genetic analysis and counseling. It underscores the need for healthcare providers to consider the cumulative impact of multiple genetic variants when assessing cancer risk and developing personalized management strategies for affected individuals.
The CHEK2 gene produces a protein called checkpoint kinase 2, which is essential for DNA damage response and cell cycle regulation. Variants in this gene are linked to an increased risk of various cancers, including breast cancer (28). It is crucial to recognize that the prevalence of CHEK2 PVs in breast cancer patients can differ among populations and regions. In European studies, these variants have been found in 1-5% of breast cancer patients, indicating that CHEK2 mutations could contribute significantly to hereditary breast cancer cases in these populations. In contrast, the prevalence of CHEK2 PVs seems to be lower in other populations, such as Asians or Africans (29).
The clinical importance of CHEK2 gene variants can be influenced by the specific mutation and its effect on protein function. Some variants might cause a moderate risk increase, while others could be linked to a higher risk (30). Frameshift variants result from insertions or deletions of nucleotides that disrupt the reading frame of a gene. In the context of the CHEK2 gene, frameshift variants can lead to a truncated or altered protein product. These variants can introduce premature stop codons, resulting in a non-functional or partially functional protein. Frameshift variants in the CHEK2 gene have been linked to an increased risk of breast, prostate, and other cancers (31). Missense variants are single nucleotide changes that lead to the substitution of one amino acid for another in the protein sequence. In the CHEK2 gene, missense mutations can affect the protein’s structure and function by altering its three-dimensional conformation or enzymatic activity. Depending on the location and nature of the amino acid change, missense variants can either increase or decrease the risk of cancer. Some missense variants may result in a hyperactive protein, disrupting normal cellular processes. Others may lead to a non-functional protein, compromising the cell’s ability to respond to DNA damage and maintain genomic stability (32). In the spectrum of genetic variants observed within the CHEK2 gene, a prominent pattern emerges, with missense variants standing out as the most prevalent, constituting a significant 59% of the identified P/LP variants.
Li-Fraumeni syndrome (LFS) is a rare hereditary cancer predisposition syndrome characterized by a significantly increased risk of developing a wide range of cancers at a young age. LFS is primarily caused by germline variants in the TP53 gene, which encodes the tumor suppressor protein p53 (33). TP53 gene plays a crucial role in regulating cell growth, DNA repair, and apoptosis, making it a key guardian of genomic stability (34). Individuals with LFS have a high lifetime risk of developing various types of cancers, including soft tissue sarcomas, osteosarcomas, breast cancer, brain tumors, leukemia, colorectal cancer, melanoma, other rare cancers (35). The hallmark of LFS is the early onset of multiple primary cancers across different organ systems, often occurring before the age of 45. The pattern of cancer inheritance in LFS follows an autosomal dominant pattern, meaning that a person with a mutation in one copy of the TP53 gene has a 50% chance of passing the mutation on to each of their offspring (36).
Like Li-Fraumeni Syndrome (LFL) refers to families who exhibit some, but not all, of the classic features of LFS. These families typically have a history of early-onset cancers, often involving multiple generations, suggestive of a hereditary predisposition to cancer. However, they may not meet the strict diagnostic criteria for LFS due to a lack of specific cancer types or the presence of additional genetic or environmental factors influencing cancer risk (37). The genetic basis of LFL is heterogeneous, and variants in genes other than TP53 may contribute to the observed cancer predisposition in these families. For example, variants in genes, such as CHEK2, BRCA1, BRCA2, and others have been implicated in familial cancer syndromes with overlapping features with LFS (38). Management of individuals with LFS or LFL typically involves regular cancer screening and surveillance starting at an early age to detect tumors at their earliest, most treatable stages. Genetic counseling and testing are essential for at-risk individuals and their families to assess their cancer risk, inform medical management decisions, and facilitate personalized cancer screening and prevention strategies. Additionally, lifestyle modifications, such as avoiding tobacco use, maintaining a healthy weight, and limiting exposure to environmental carcinogens, may help reduce cancer risk in these individuals (39).
In this study, the most common variants observed were missense variants, accounting for 88.2% of alterations within the TP53 gene. Missense variants in the TP53 gene are particularly significant due to the critical role TP53 plays in regulating cell division and preventing the proliferation of cells with damaged DNA. As a tumor suppressor gene, TP53 is instrumental in orchestrating cellular responses to stressors such as DNA damage, thereby safeguarding genomic integrity and inhibiting the development of cancer (40).
The identification of P/LP variants in HR genes among these individuals has significant implications for their cancer risk and treatment options. Disruption of HR pathway genes can impair the cell’s ability to repair DNA damage effectively, leading to genomic instability and an increased predisposition to cancer development (41,42). In our study, 79.1% of individuals possess P/LP variants in genes associated with HR. Understanding the prevalence of P/LP in HR genes within this cohort underscores the importance of genetic testing and counseling in identifying individuals at increased risk of hereditary cancer syndromes associated with HR pathway dysfunction.
WES is a breakthrough in genomics, enabling scientists and healthcare professionals to deeply analyze an individual’s genetic makeup. This cutting-edge method primarily concentrates on meticulously examining the exome, the DNA segment responsible for protein coding. Interestingly, this portion constitutes a small percentage, around 1-2%, of the entire genome (43). Despite its small size compared to the vast genome, the exome holds great significance in genetic research. This compact genetic database is where most mutations causing diseases are found, making it an invaluable focus for understanding the origins and mechanisms of various disorders (44). A crucial application of WES lies in the field of oncology, particularly in deciphering the intricate relationships of genetic alterations in cancer development. WES has been instrumental in unraveling the complex network surrounding cancer progression. Breast cancer, a multifaceted disease influenced by a combination of genetic and environmental factors, serves as an example. Within the realm of genetics, specific gene variants, especially those in key genes, significantly contribute to heightened breast cancer risk. To fully grasp the intricate dance between genetic predispositions and environmental influences, it is vital to understand the complexity of breast cancer. This comprehensive approach is essential for developing well-rounded strategies for prevention, early detection, and tailored treatment plans to combat this widespread and impactful disease (45).
This study detected the following genetic variations in 85 female breast cancer patients through WES: CTC1:c.880C>T, p.(Gln294*); MLH3: c.405del, p.(Asp136Metfs*2); PPM1D: c.1426_1430del, p.(Glu476Leufs*3); and SDHB: c.395A>G, p.(His132Arg).
The CTC1 gene, part of the CST complex, is vital for maintaining telomere stability at chromosome ends. Telomeres protect genetic material during DNA replication and shield chromosomes from degradation (46). The variant c.880C>T, p.(Gln294*) leads to a premature stop codon, forming a shortened protein. This can be detrimental as it disrupts normal protein function. In telomere maintenance, such variants may hinder CTC1’s interaction with the CST complex or its role in telomere length regulation (47). Consequently, telomeres may destabilize and shorten faster, causing cellular issues, genome instability, and a heightened risk of genetic disorders.
The MLH3 gene is a vital component of the DNA mismatch repair (MMR) pathway, crucial for maintaining genome stability and integrity. MMR corrects errors during DNA replication to ensure accurate genetic information (48). The variant c.405del, p.(Asp136Metfs*2) involves a nucleotide deletion at position 405, causing a frameshift mutation. This alters the protein-building instructions, disrupting the normal amino acid sequence. The variant results in a premature stop codon, producing a truncated MLH3 protein. Variants like c.405del, p.(Asp136Metfs*2), are often pathogenic or deleterious, affecting MLH3’s normal function in the MMR pathway. This impairment may lead to a higher accumulation of genetic mutations and an increased risk of certain cancers, such as those associated with Lynch syndrome (49).
The gene protein phosphatase Mg2+/Mn2+ dependent 1D (PPM1D), also known as Wip1, plays a vital role in regulating cellular responses to stress and DNA damage. It encodes a protein that acts as a phosphatase, which means it removes phosphate groups from other proteins. This action can modulate the activity of various proteins involved in critical cellular processes (50). The variant c.1426_1430del, p.(Glu476Leufs*3) in the PPM1D gene involves the deletion of five nucleotides starting from position 1426. This deletion results in a frameshift mutation that leads to the creation of a premature stop codon, which results in a truncated or abnormally shortened PPM1D protein. This disrupts the normal function of the PPM1D protein, which can have significant implications for cellular responses to stress and DNA damage. This disruption may lead to an increased susceptibility to genetic instability and the development of various types of cancer. Notably, certain variants in the PPM1D gene have been associated with a predisposition to a range of cancers, including breast cancer and ovarian cancer (51).
The gene succinate dehydrogenase complex iron sulfur subunit B (SDHB) is a crucial component of the mitochondrial enzyme complex known as succinate dehydrogenase (SDH) or complex II. This enzyme complex plays a pivotal role in both the citric acid cycle (Krebs cycle) and the electron transport chain within the mitochondria. It is involved in the oxidation of succinate to fumarate, a critical step in energy production through oxidative phosphorylation (52). The variant c.395A>G, p.(His132Arg) in the SDHB gene involves a change in a single nucleotide at position 395. Variants in genes like SDHB are of particular interest in the context of hereditary paraganglioma and pheochromocytoma syndromes, which are genetic conditions associated with the development of certain neuroendocrine tumors.
In international literature, several studies have identified potential new genes linked to breast cancer through WES (53,54). However, it is crucial to conduct additional analysis to confirm these associations. Contemporary research endeavors are prioritizing the analysis of entire exomes and, more recently, whole genomes, rather than solely focusing on specific candidate genes. This approach is driving the exploration of novel connections between genes and diseases. By examining the entirety of the genome, which includes non-coding regions, studies utilizing whole-genome sequencing hold significant potential for identifying previously unrecognized genetic variants that play a role in disease susceptibility. Furthermore, recent advancements in technology aimed at detecting structural variations within the genome are starting to reveal genetic variants that may have been missed by conventional short-read sequencing techniques.
Conclusion
This study marks a significant milestone as the most comprehensive research undertaken by a single clinical diagnostic facility in Greece, encompassing the examination of over 10,000 individuals for hereditary cancer utilizing NGS technology since 2015 (55). The insights gleaned from this investigation hold transformative potential for healthcare providers, enabling them to customize patient care strategies based on the intricate nuances of genetic makeup rather than solely relying on traditional personal and family medical histories. Notably, the identification of PVs across multiple genes can offer invaluable insights into the complex array of tumor types observed within certain families, shedding light on potential genetic predispositions, and informing targeted treatment approaches.
However, while these initial findings are promising, further research endeavors are imperative to consolidate and expand upon these discoveries. Future studies, encompassing a larger cohort of families and employing advanced WES techniques, are warranted to validate and build upon the initial findings, ultimately paving the way for more precise and personalized approaches to cancer management and prevention.
Conflicts of Interest
The Authors have no conflicts of interest to declare in relation to this study.
Authors’ Contributions
All Authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Nikolaos Tsoulos, Konstantinos Agiannitopoulos, Kevisa Potska, Anastasia Katseli, Christina Ntogka, Georgia Pepe, Dimitra Bouzarelou, Athanasios Papathanasiou, Dimitrios Grigoriadis, Georgios N. Tsaousis, Helen Gogas, Theodore Troupis, Konstantinos Papazisis, Ioannis Natsiopoulos, Vassileios Venizelos, Kyriakos Amarantidis, Stylianos Giassas, Christos Papadimitriou, Elena Fountzilas, Maroulio Stathoulopoulou, Anna Koumarianou, Grigorios Xepapadakis, Alexandru Blidaru, Daniela Zob, Oana Voinea, Mustafa Özdoğan, Mahmut Çerkez Ergören, Alinta Hegmane, Eirini Papadopoulou, George Nasioulas and Christos Markopoulos. The first draft of the article was written by Nikolaos Tsoulos and Konstantinos Agiannitopoulos and all Authors commented on previous versions of the article. All Authors read and approved the final article.
References
- 1.Garutti M, Foffano L, Mazzeo R, Michelotti A, Da Ros L, Viel A, Miolo G, Zambelli A, Puglisi F. Hereditary cancer syndromes: a comprehensive review with a visual tool. Genes (Basel) 2023;14(5):1025. doi: 10.3390/genes14051025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Imyanitov EN, Kuligina ES, Sokolenko AP, Suspitsin EN, Yanus GA, Iyevleva AG, Ivantsov AO, Aleksakhina SN. Hereditary cancer syndromes. World J Clin Oncol. 2023;14(2):40–68. doi: 10.5306/wjco.v14.i2.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pozzar RA, Seven M. Interventions to support decision making in people considering germline genetic testing for BRCA 1/2 pathogenic and likely pathogenic variants: A scoping review. J Genet Couns. 2024;33(2):392–401. doi: 10.1002/jgc4.1738. [DOI] [PubMed] [Google Scholar]
- 4.Rocca V, Blandino G, D’Antona L, Iuliano R, Di Agostino S. Li-Fraumeni syndrome: Mutation of TP53 is a biomarker of hereditary predisposition to tumor: new insights and advances in the treatment. Cancers (Basel) 2022;14(15):3664. doi: 10.3390/cancers14153664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dal Buono A, Puccini A, Franchellucci G, Airoldi M, Bartolini M, Bianchi P, Santoro A, Repici A, Hassan C. Lynch syndrome: from multidisciplinary management to precision prevention. Cancers (Basel) 2024;16(5):849. doi: 10.3390/cancers16050849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mighton C, Lerner-Ellis JP. Principles of molecular testing for hereditary cancer. Genes Chromosomes Cancer. 2022;61(6):356–381. doi: 10.1002/gcc.23048. [DOI] [PubMed] [Google Scholar]
- 7.Levine R, Kahn RM, Perez L, Brewer J, Ratner S, Li X, Yeoshoua E, Frey MK. Cascade genetic testing for hereditary cancer syndromes: a review of barriers and breakthroughs. Fam Cancer. 2024;23(2):111–120. doi: 10.1007/s10689-024-00373-4. [DOI] [PubMed] [Google Scholar]
- 8.Charron M, Kaiser B, Dauge A, Gallois H, Lapointe J, Dorval M, Nabi H, Joly Y. Integrating hereditary breast and ovarian cancer genetic counselling and testing into mainstream clinical practice: Legal and ethical challenges. Crit Rev Oncol Hematol. 2022;178:103797. doi: 10.1016/j.critrevonc.2022.103797. [DOI] [PubMed] [Google Scholar]
- 9.Ueki A, Yoshida R, Kosaka T, Matsubayashi H. Clinical risk management of breast, ovarian, pancreatic, and prostatic cancers for BRCA1/2 variant carriers in Japan. J Hum Genet. 2023;68(8):517–526. doi: 10.1038/s10038-023-01153-1. [DOI] [PubMed] [Google Scholar]
- 10.Price KS, Svenson A, King E, Ready K, Lazarin GA. Inherited cancer in the age of next-generation sequencing. Biol Res Nurs. 2018;20(2):192–204. doi: 10.1177/1099800417750746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamps R, Brandão RD, Bosch BJ, Paulussen AD, Xanthoulea S, Blok MJ, Romano A. Next-generation sequencing in oncology: Genetic diagnosis, risk prediction and cancer classification. Int J Mol Sci. 2017;18(2):308. doi: 10.3390/ijms18020308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pei XM, Yeung MHY, Wong ANN, Tsang HF, Yu ACS, Yim AKY, Wong SCC. Targeted sequencing approach and its clinical applications for the molecular diagnosis of human diseases. Cells. 2023;12(3):493. doi: 10.3390/cells12030493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imyanitov E, Sokolenko A. Integrative genomic tests in clinical oncology. Int J Mol Sci. 2022;23(21):13129. doi: 10.3390/ijms232113129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serratì S, De Summa S, Pilato B, Petriella D, Lacalamita R, Tommasi S, Pinto R. Next-generation sequencing: advances and applications in cancer diagnosis. Onco Targets Ther. 2016;9:7355–7365. doi: 10.2147/OTT.S99807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monteiro AN, Bouwman P, Kousholt AN, Eccles DM, Millot GA, Masson JY, Schmidt MK, Sharan SK, Scully R, Wiesmüller L, Couch F, Vreeswijk MPG. Variants of uncertain clinical significance in hereditary breast and ovarian cancer genes: best practices in functional analysis for clinical annotation. J Med Genet. 2020;57(8):509–518. doi: 10.1136/jmedgenet-2019-106368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davidson AL, Dressel U, Norris S, Canson DM, Glubb DM, Fortuno C, Hollway GE, Parsons MT, Vidgen ME, Holmes O, Koufariotis LT, Lakis V, Leonard C, Wood S, Xu Q, McCart Reed AE, Pickett HA, Al-Shinnag MK, Austin RL, Burke J, Cops EJ, Nichols CB, Goodwin A, Harris MT, Higgins MJ, Ip EL, Kiraly-Borri C, Lau C, Mansour JL, Millward MW, Monnik MJ, Pachter NS, Ragunathan A, Susman RD, Townshend SL, Trainer AH, Troth SL, Tucker KM, Wallis MJ, Walsh M, Williams RA, Winship IM, Newell F, Tudini E, Pearson JV, Poplawski NK, Mar Fan HG, James PA, Spurdle AB, Waddell N, Ward RL. The clinical utility and costs of whole-genome sequencing to detect cancer susceptibility variants-a multi-site prospective cohort study. Genome Med. 2023;15(1):74. doi: 10.1186/s13073-023-01223-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agiannitopoulos K, Potska K, Katseli A, Ntogka C, Tsaousis GN, Pepe G, Bouzarelou D, Tsoulos N, Papathanasiou A, Ziogas D, Venizelos V, Markopoulos C, Iosifidou R, Karageorgopoulou S, Giassas S, Natsiopoulos I, Papazisis K, Vasilaki-Antonatou M, Psyrri A, Koumarianou A, Matthaios D, Zairi E, Blidaru A, Banu E, Jinga DC, Laçin Ş, Özdoğan M, Papadopoulou E, Nasioulas G. Only 32.3% of breast cancer families with pathogenic variants in cancer genes utilized cascade genetic testing. Cancers (Basel) 2023;15(21):5218. doi: 10.3390/cancers15215218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones JC, Golafshar MA, Coston TW, Rao R, Wysokinska E, Johnson E, Esplin ED, Nussbaum RL, Heald B, Klint M, Barrus K, Uson PL Jr, Nguyen CC, Colon-Otero G, Bekaii-Saab TS, Dronca R, Kunze KL, Samadder NJ. Universal genetic testing vs. guideline-directed testing for hereditary cancer syndromes among traditionally underrepresented patients in a community oncology program. Cureus. 2023;15(4):e37428. doi: 10.7759/cureus.37428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valentini V, Bucalo A, Conti G, Celli L, Porzio V, Capalbo C, Silvestri V, Ottini L. Gender-specific genetic predisposition to breast cancer: BRCA genes and beyond. Cancers (Basel) 2024;16(3):579. doi: 10.3390/cancers16030579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrish N, Snowsill T, Dodman S, Medina-Lara A. Preferences for genetic testing to predict the risk of developing hereditary cancer: a systematic review of discrete choice experiments. Med Decis Making. 2024;44(3):252–268. doi: 10.1177/0272989X241227425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamio T, Kamio H, Aoki T, Ondo Y, Uchiyama T, Yamamoto-Shimojima K, Watanabe M, Okamoto T, Kanno H, Yamamoto T. Molecular profiles of breast cancer in a single institution. Anticancer Res. 2020;40(8):4567–4570. doi: 10.21873/anticanres.14462. [DOI] [PubMed] [Google Scholar]
- 22.Agiannitopoulos K, Pepe G, Tsaousis GN, Potska K, Bouzarelou D, Katseli A, Ntogka C, Meintani A, Tsoulos N, Giassas S, Venizelos V, Markopoulos C, Iosifidou R, Karageorgopoulou S, Christodoulou C, Natsiopoulos I, Papazisis K, Vasilaki-Antonatou M, Kabletsas E, Psyrri A, Ziogas D, Lalla E, Koumarianou A, Anastasakou K, Papadimitriou C, Ozmen V, Tansan S, Kaban K, Ozatli T, Eniu DT, Chiorean A, Blidaru A, Rinsma M, Papadopoulou E, Nasioulas G. Copy number variations (CNVs) account for 10.8% of pathogenic variants in patients referred for hereditary cancer testing. Cancer Genomics Proteomics. 2023;20(5):448–455. doi: 10.21873/cgp.20396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quinodoz M, Kaminska K, Cancellieri F, Han JH, Peter VG, Celik E, Janeschitz-Kriegl L, Schärer N, Hauenstein D, György B, Calzetti G, Hahaut V, Custódio S, Sousa AC, Wada Y, Murakami Y, Fernández AA, Hernández CR, Minguez P, Ayuso C, Nishiguchi KM, Santos C, Santos LC, Tran VH, Vaclavik V, Scholl HPN, Rivolta C. Detection of elusive DNA copy-number variations in hereditary disease and cancer through the use of noncoding and off-target sequencing reads. Am J Hum Genet. 2024;111(4):701–713. doi: 10.1016/j.ajhg.2024.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agaoglu NB, Unal B, Akgun Dogan O, Zolfagharian P, Sharifli P, Karakurt A, Can Senay B, Kizilboga T, Yildiz J, Dinler Doganay G, Doganay L. Determining the accuracy of next generation sequencing based copy number variation analysis in Hereditary Breast and Ovarian Cancer. Expert Rev Mol Diagn. 2022;22(2):239–246. doi: 10.1080/14737159.2022.2048373. [DOI] [PubMed] [Google Scholar]
- 25.Ueda T, Tsubamoto H, Takimoto Y, Isono-Taniguchi R, Narita S, Nakagawa K, Wakimoto Y, Nishimura Y, Muroi Y, Nagahashi M, Hirota S, Sawai H, Shibahara H. Comprehensive genomic profiling detects hereditary cancers and confers survival advantage in patients with gynaecological cancers. Anticancer Res. 2023;43(5):2091–2101. doi: 10.21873/anticanres.16370. [DOI] [PubMed] [Google Scholar]
- 26.Madar L, Majoros V, Szűcs Z, Nagy O, Babicz T, Butz H, Patócs A, Balogh I, Koczok K. Double heterozygosity for rare deleterious variants in the BRCA1 and BRCA2 genes in a Hungarian patient with breast cancer. Int J Mol Sci. 2023;24(20):15334. doi: 10.3390/ijms242015334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Megid TBC, Barros-Filho MC, Pisani JP, Achatz MI. Double heterozygous pathogenic variants prevalence in a cohort of patients with hereditary breast cancer. Front Oncol. 2022;12:873395. doi: 10.3389/fonc.2022.873395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boonen RA, Vreeswijk MP, van Attikum H. CHEK2 variants: linking functional impact to cancer risk. Trends Cancer. 2022;8(9):759–770. doi: 10.1016/j.trecan.2022.04.009. [DOI] [PubMed] [Google Scholar]
- 29.Pavlovica K, Irmejs A, Noukas M, Palover M, Kals M, Tonisson N, Metspalu A, Gronwald J, Lubinski J, Murmane D, Kalnina A, Loza P, Maksimenko J, Trofimovics G, Subatniece S, Daneberga Z, Miklasevics E, Gardovskis J. Spectrum and frequency of CHEK2 variants in breast cancer affected and general population in the Baltic states region, initial results and literature review. Eur J Med Genet. 2022;65(5):104477. doi: 10.1016/j.ejmg.2022.104477. [DOI] [PubMed] [Google Scholar]
- 30.LaDuca H, Polley EC, Yussuf A, Hoang L, Gutierrez S, Hart SN, Yadav S, Hu C, Na J, Goldgar DE, Fulk K, Smith LP, Horton C, Profato J, Pesaran T, Gau CL, Pronold M, Davis BT, Chao EC, Couch FJ, Dolinsky JS. A clinical guide to hereditary cancer panel testing: evaluation of gene-specific cancer associations and sensitivity of genetic testing criteria in a cohort of 165,000 high-risk patients. Genet Med. 2020;22(2):407–415. doi: 10.1038/s41436-019-0633-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kleiblova P, Stolarova L, Krizova K, Lhota F, Hojny J, Zemankova P, Havranek O, Vocka M, Cerna M, Lhotova K, Borecka M, Janatova M, Soukupova J, Sevcik J, Zimovjanova M, Kotlas J, Panczak A, Vesela K, Cervenkova J, Schneiderova M, Burocziova M, Burdova K, Stranecky V, Foretova L, Machackova E, Tavandzis S, Kmoch S, Macurek L, Kleibl Z. Identification of deleterious germline CHEK2 mutations and their association with breast and ovarian cancer. Int J Cancer. 2019;145(7):1782–1797. doi: 10.1002/ijc.32385. [DOI] [PubMed] [Google Scholar]
- 32.Mundt E, Mabey B, Rainville I, Ricker C, Singh N, Gardiner A, Manley S, Slavin T Jr. Breast and colorectal cancer risks among over 6,000 CHEK2 pathogenic variant carriers: A comparison of missense versus truncating variants. Cancer Genet. 2023;278-279:84–90. doi: 10.1016/j.cancergen.2023.10.002. [DOI] [PubMed] [Google Scholar]
- 33.Guha T, Malkin D. Inherited TP53 mutations and the Li-Fraumeni syndrome. Cold Spring Harb Perspect Med. 2017;7(4):a026187. doi: 10.1101/cshperspect.a026187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu KL, Su F, Yang JR, Xiao RW, Wu RY, Cao MY, Ling XL, Zhang T. TP53 to mediate immune escape in tumor microenvironment: an overview of the research progress. Mol Biol Rep. 2024;51(1):205. doi: 10.1007/s11033-023-09097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mansur MB, Greaves M. Convergent TP53 loss and evolvability in cancer. BMC Ecol Evol. 2023;23(1):54. doi: 10.1186/s12862-023-02146-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abdel-Razeq H. Surgical options for patients with early-stage breast cancer and pathogenic germline variants: an oncologist perspectives. Front Oncol. 2023;13:1265197. doi: 10.3389/fonc.2023.1265197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giacomazzi CR, Giacomazzi J, Netto CB, Santos-silva P, Selistre SG, Maia AL, Oliveira VZD, Camey SA, Goldim JR, Ashton-prolla P. Pediatric cancer and Li-Fraumeni/Li-Fraumeni-like syndromes: a review for the pediatrician. Rev Assoc Med Bras (1992) 2015;61(3):282–289. doi: 10.1590/1806-9282.61.03.282. [DOI] [PubMed] [Google Scholar]
- 38.Vogel WH. Li-Fraumeni syndrome. J Adv Pract Oncol. 2017;8(7):742–746. doi: 10.6004/jadpro.2017.8.7.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Consul N, Amini B, Ibarra-Rovira JJ, Blair KJ, Moseley TW, Taher A, Shah KB, Elsayes KM. Li-Fraumeni syndrome and whole-body MRI screening: Screening guidelines, imaging features, and impact on patient management. AJR Am J Roentgenol. 2021;216(1):252–263. doi: 10.2214/AJR.20.23008. [DOI] [PubMed] [Google Scholar]
- 40.Kennedy MC, Lowe SW. Mutant p53: it’s not all one and the same. Cell Death Differ. 2022;29(5):983–987. doi: 10.1038/s41418-022-00989-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collet L, Hanvic B, Turinetto M, Treilleux I, Chopin N, Le Saux O, Ray-Coquard I. BRCA1/2 alterations and reversion mutations in the area of PARP inhibitors in high grade ovarian cancer: state of the art and forthcoming challenges. Front Oncol. 2024;14:1354427. doi: 10.3389/fonc.2024.1354427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Voutsadakis IA, Stravodimou A. Homologous recombination defects and mutations in DNA damage response (DDR) genes besides BRCA1 and BRCA2 as breast cancer biomarkers for PARP inhibitors and other DDR targeting therapies. Anticancer Res. 2023;43(3):967–981. doi: 10.21873/anticanres.16241. [DOI] [PubMed] [Google Scholar]
- 43.Udupa P, Ghosh DK. Implementation of exome sequencing to identify rare genetic diseases. Methods Mol Biol. 2024;2719:79–98. doi: 10.1007/978-1-0716-3461-5_5. [DOI] [PubMed] [Google Scholar]
- 44.Hartley T, Gillespie MK, Graham ID, Hayeems RZ, Li S, Sampson M, Boycott KM, Potter BK. Exome and genome sequencing for rare genetic disease diagnosis: A scoping review and critical appraisal of clinical guidance documents produced by genetics professional organizations. Genet Med. 2023;25(11):100948. doi: 10.1016/j.gim.2023.100948. [DOI] [PubMed] [Google Scholar]
- 45.Zelli V, Compagnoni C, Cannita K, Capelli R, Capalbo C, Di Vito Nolfi M, Alesse E, Zazzeroni F, Tessitore A. Applications of next generation sequencing to the analysis of familial breast/ovarian cancer. High Throughput. 2020;9(1):1. doi: 10.3390/ht9010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lim CJ, Cech TR. Shaping human telomeres: from shelterin and CST complexes to telomeric chromatin organization. Nat Rev Mol Cell Biol. 2021;22(4):283–298. doi: 10.1038/s41580-021-00328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nasheuer HP, Onwubiko NO. Lagging strand initiation processes in DNA replication of eukaryotes-strings of highly coordinated reactions governed by multiprotein complexes. Genes (Basel) 2023;14(5):1012. doi: 10.3390/genes14051012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muro Y, Sugiura K, Mimori T, Akiyama M. DNA mismatch repair enzymes: Genetic defects and autoimmunity. Clin Chim Acta. 2015;442:102–109. doi: 10.1016/j.cca.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 49.Valle L, de Voer RM, Goldberg Y, Sjursen W, Försti A, Ruiz-Ponte C, Caldés T, Garré P, Olsen MF, Nordling M, Castellvi-Bel S, Hemminki K. Update on genetic predisposition to colorectal cancer and polyposis. Mol Aspects Med. 2019;69:10–26. doi: 10.1016/j.mam.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 50.Husby S, Hjermind Justesen E, Grønbæk K. Protein phosphatase, Mg2+/Mn2+-dependent 1D (PPM1D) mutations in haematological cancer. Br J Haematol. 2021;192(4):697–705. doi: 10.1111/bjh.17120. [DOI] [PubMed] [Google Scholar]
- 51.Vodolazhsky DI, Mayakovskaya AV, Kubyshkin AV, Aliev KA, Fomochkina II. Clinical significance of gene polymorphisms for hereditary predisposition to breast and ovarian cancer (review of literature) Klin Lab Diagn. 2021;66(12):760–767. doi: 10.51620/0869-2084-2021-66-12-760-767. [DOI] [PubMed] [Google Scholar]
- 52.Liu C, Zhou D, Yang K, Xu N, Peng J, Zhu Z. Research progress on the pathogenesis of the SDHB mutation and related diseases. Biomed Pharmacother. 2023;167:115500. doi: 10.1016/j.biopha.2023.115500. [DOI] [PubMed] [Google Scholar]
- 53.Glentis S, Dimopoulos AC, Rouskas K, Ntritsos G, Evangelou E, Narod SA, Mes-Masson AM, Foulkes WD, Rivera B, Tonin PN, Ragoussis J, Dimas AS. Exome sequencing in BRCA1- and BRCA2-negative Greek families identifies MDM1 and NBEAL1 as candidate risk genes for hereditary breast cancer. Front Genet. 2019;10:1005. doi: 10.3389/fgene.2019.01005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee NY, Hum M, Amali AA, Lim WK, Wong M, Myint MK, Tay RJ, Ong PY, Samol J, Lim CW, Ang P, Tan MH, Lee SC, Lee ASG. Whole-exome sequencing of BRCA-negative breast cancer patients and case-control analyses identify variants associated with breast cancer susceptibility. Hum Genomics. 2022;16(1):61. doi: 10.1186/s40246-022-00435-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tsaousis GN, Papadopoulou E, Apessos A, Agiannitopoulos K, Pepe G, Kampouri S, Diamantopoulos N, Floros T, Iosifidou R, Katopodi O, Koumarianou A, Markopoulos C, Papazisis K, Venizelos V, Xanthakis I, Xepapadakis G, Banu E, Eniu DT, Negru S, Stanculeanu DL, Ungureanu A, Ozmen V, Tansan S, Tekinel M, Yalcin S, Nasioulas G. Analysis of hereditary cancer syndromes by using a panel of genes: novel and multiple pathogenic mutations. BMC Cancer. 2019;19(1):535. doi: 10.1186/s12885-019-5756-4. [DOI] [PMC free article] [PubMed] [Google Scholar]