

Abstract
Testosterone (T) and 17β-estradiol (E2) are produced in male and female humans and are potent metabolic regulators in both sexes. When E2 and T production stops or decreases during aging, metabolic dysfunction develops and promotes degenerative metabolic and vascular disease. Here, we discuss the shared benefits afforded by E2 and T for metabolic function human females and males. In females, E2 is central to bone and vascular health, subcutaneous adipose tissue distribution, skeletal muscle insulin sensitivity, antiinflammatory immune function, and mitochondrial health. However, T also plays a role in female skeletal, vascular, and metabolic health. In males, T’s conversion to E2 is fundamental to bone and vascular health, as well as prevention of excess visceral adiposity and the promotion of insulin sensitivity via activation of the estrogen receptors. However, T and its metabolite dihydrotestosterone also prevent excess visceral adiposity and promote skeletal muscle growth and insulin sensitivity via activation of the androgen receptor. In conclusion, T and E2 are produced in both sexes at sex-specific concentrations and provide similar and potent metabolic benefits. Optimizing levels of both hormones may be beneficial to protect patients from cardiometabolic disease and frailty during aging, which requires further study.
Introduction
Testosterone (T) and 17β-estradiol (E2) are considered male and female sex hormones, respectively, because they are secreted by gonads in the circulation at sex-specific concentrations and are involved in sexual differentiation and reproduction. E2, however, is not exclusively a female hormone since, for example, it is essential for erection and libido in male individuals (1). Likewise, T is not exclusively a male hormone, as it is essential for libido in female individuals (2). Most importantly, E2 and T are central to metabolic homeostasis of most cells and in both sexes. When E2 and T production stops or decreases during aging, metabolic dysfunction develops and promotes degenerative metabolic and vascular disease. Understanding the sex-specific and shared benefits of E2 and T in metabolic function in both sexes is critical to medicine and healthy aging. Here, we analyze sex differences and similarities in E2 and T benefits for metabolic homeostasis in male and female humans, including glucose and lipid metabolism, bone, vascular, adipose, muscle, and immune functions, and the prevention of metabolic dysfunction leading to cardiometabolic disease. We use the terms male and female to describe the biological sex of human subjects through the paper and we specify when animal studies are discussed. For details on mechanisms of E2 and T’s actions, we will refer to recent and landmark reviews.
Origin of T and E2 in both sexes
In males, all T is produced by Leydig cells of the testis. T behaves as a hormone by binding the androgen receptor (AR), and also behaves as a prohormone that is converted in peripheral tissues to E2 or dihydrotestosterone (DHT), a pure AR agonist that cannot be converted to E2. In males, most E2 (80%) is formed via aromatization of circulating T in the periphery. The testes directly produce approximately 20% of circulating E2 (3) (Figure 1A). Circulating concentrations of E2 in males are half of those of females and are essential to metabolic homeostasis, as we will discuss. In females of reproductive age, the granulosa cells of the ovaries produce E2, the major circulating estrogen (Figure 1B). After menopause, estrone (E1) becomes the major circulating estrogen (4). E1 is produced by aromatization from the adrenal androgen androstenedione in adipose tissue (5) (Figure 1C). E1 is a weak estrogen and should be considered a reservoir of the more potent E2 in postmenopausal females. E2 is produced locally in extra-ovarian tissues and acts locally as a paracrine and intracrine factor (Figure 1). In females, T is the most abundant circulating active sex steroid throughout the life span (Figure 2). In females of reproductive age, T is produced by the ovary (25%), the adrenal gland (25%), and in peripheral tissues (50%), following conversion from circulating androstenedione (equally produced by the ovary and the adrenal gland) (6–9) (Figure 1B). After natural menopause, ovarian T production decreases slowly. T is mainly produced by the ovaries (50%) and via peripheral conversion from androstenedione (40%) mainly of adrenal origin (6–9). Direct adrenal production of T is minor (around 10%) (Figure 1C). Although T is ten times less abundant in the blood of females than males, in females across the life span, circulating T is 5–50 times more abundant than E2 (Figure 2), the implications of which we will discuss below.
Figure 1. Origin of T and E2 in males and females.

(A) In males, all T is produced by Leydig cells of the testis. Most E2 (80%) is formed via aromatization of circulating T in the periphery. The testes directly produce approximately 20% of circulating E2. (B) In females of reproductive age, the granulosa cells of the ovaries produce E2, the major circulating estrogen. T is produced by the ovary (25%), the adrenal gland (25%), and in peripheral tissues (50%) following conversion from circulating androstenedione (A4, an androgen that is equally produced by the ovary and the adrenal gland). (C) After menopause, estrone (E1) becomes the major circulating estrogen and is produced by aromatization from A4 (mainly produced by the adrenal gland) in adipose tissue. E1 serves as a reservoir of E2. T is mainly produced by the ovaries (50%) and peripheral conversion of A4 (40%). 17β-HSD, 17β-hydroxysteroid dehydrogenase.
Figure 2. T and E2 concentrations in males and females.

(A) Circulating T and E2 in males and females over the life span. (B) Ratio of T to E2 in males and females. Data in both panels derived from the CDC’s NHANES sex steroids data from 2013–2014 and 2015–2016 databases using sex steroids data from 2013–2014 and 2015–2016 for 7201 males and 7561 females (156, 157). In these data, total hormone (free and protein-bound) was measured using isotope dilution liquid chromatography–tandem mass spectrometry (ID-LC-MS/MS). We binned data from participants ages 6 years and up into decades and plotted as 95% confidence intervals (shown as lighter shading around averaged line). Data outside of the reported range of values were excluded (E2: 2.117 to 1220 pg/mL and T: 4.1 to 15,500 pg/mL).
E2 promotes metabolic homeostasis in females
In females of reproductive age, E2 is instrumental to skeletal, vascular, and energy homeostasis. The central role of E2 in maintenance of bone metabolism, the detrimental effect of postmenopausal E2 deficiency on osteopenia and osteoporosis, and their prevention by estrogen therapy in postmenopausal females is evidence-based medicine (10, 11) and will not be discussed here.
E2 promotes female vascular function and health.
Females with early E2 deficiency because of surgical oophorectomy (12, 13), premature ovarian insufficiency (14), or early menopause (15) are at increased risk of cardiovascular disease (CVD) and mortality compared with females who experience natural menopause. As we will discuss below, the vascular protection provided by E2 extends to males through T conversion. E2 protects arteries by promoting vasodilation, either through stimulation of nitric oxide (NO) production in endothelial cells or direct effects on dilatory mechanisms within vascular smooth muscle. Brachial artery flow–mediated dilation (FMD) is NO mediated (16) and is considered the gold standard for assessing macrovascular endothelial health because it is a strong predictor of future CVD (17). E2 increases FMD at puberty in females (18) and maintains greater FMD in reproductive-aged females versus males (19), while E2 deficiency after menopause reduces FMD (20). This ability of E2 to improve vascular tone is integral for its protection against high blood pressure, supported by the increased incidence of hypertension after surgical or early menopause (21). In rodent models that display male predominance in hypertension, ovariectomy in females increased blood pressure to the level of male rodents (22). The association of menopausal hormone therapy with hypertension is observed only with oral estrogens, especially conjugated equine estrogens (CEEs) and oral estrogen in combination with synthetic progestogens, not progesterone, highlighting the importance of differentiating endogenous versus synthetic hormones as well as route of administration (23, 24).
The second mechanism for the vascular protection provided by E2 relates to its ability to prevent detrimental remodeling, including fibrosis, stiffening, and calcification. Pulse wave velocity is a clinical measure of arterial stiffness and a strong predictor of cardiovascular events (25). Supporting the importance of E2 in protecting from arterial stiffness, females exhibited lower arterial stiffness than males only between puberty and menopause (26). Stiffness significantly increased in females at menopause (27), and in fact females developed higher arterial stiffness than age-matched males despite similar blood pressure (28). Thus, E2 deficiency amplifies arterial stiffness in a female-specific manner.
The third mechanism of E2 vascular protection involves its ability to lower atherogenic lipids (discussed in the corresponding section) and to decrease systemic inflammation. Females display a more robust immune response to infection and vaccination than males, but are more susceptible to autoimmune diseases (29). E2 reduces proinflammatory cytokines through direct immunomodulatory actions on immune cells (30). Atherosclerosis is a chronic inflammatory disease, characterized by elevated lipids and macrophage infiltration into the vascular wall, and mouse models show that E2 is atheroprotective, especially in the early stages of lesion formation (31).
Why the vascular benefits provided by endogenous E2 and demonstrated in females with early E2 deficiency do not always translate to protection by exogenous menopausal estrogen therapy is a subject of ongoing debate. Several hypotheses have been proposed, the first of which is that endogenous E2 prevents or slows the progression of CVD, but does not reverse established vascular damage. If E2 is not restored early, then irreversible damage develops that cannot be reversed. This theory underlies the “timing hypothesis,” which postulates that E2 therapy started at the time of menopause in a woman with healthy arteries prevents the development of CVD, but beyond a certain point, the age- and E2 deficiency–related damage renders the effects of E2 less beneficial and potentially harmful (32). In support of this, a meta-analysis of 19 randomized controlled trials of over 40,000 postmenopausal women concluded that women who initiate estrogen therapy within 10 years of menopause show a 50% reduction in cardiovascular mortality and myocardial infarction (MI) (33). The mechanism for this early protection could be that actions mediated by ERα, but not ERβ, are protective, but prolonged E2 deficiency decreases the vascular ERα/ERβ ratio (34). In addition, the increased CVD observed in older postmenopausal women was related to the use of CEE therapy, not E2 (35). CEE contains mostly E1, a poor ERα agonist, along with several equine estrogens that exhibit greater affinity for ERβ (36) and are inferior to E2 with regard to NO production (37). Thus, CEE is likely to exhibit different vascular actions than E2. In summary, current evidence indicates that endogenous E2 prevents damage in a healthy vascular system following short-term E2 deficiency, but does not protect vessels exposed to prolonged E2 deficiency (Figure 3).
Figure 3. Cardiometabolic effect of E2 and T in females.
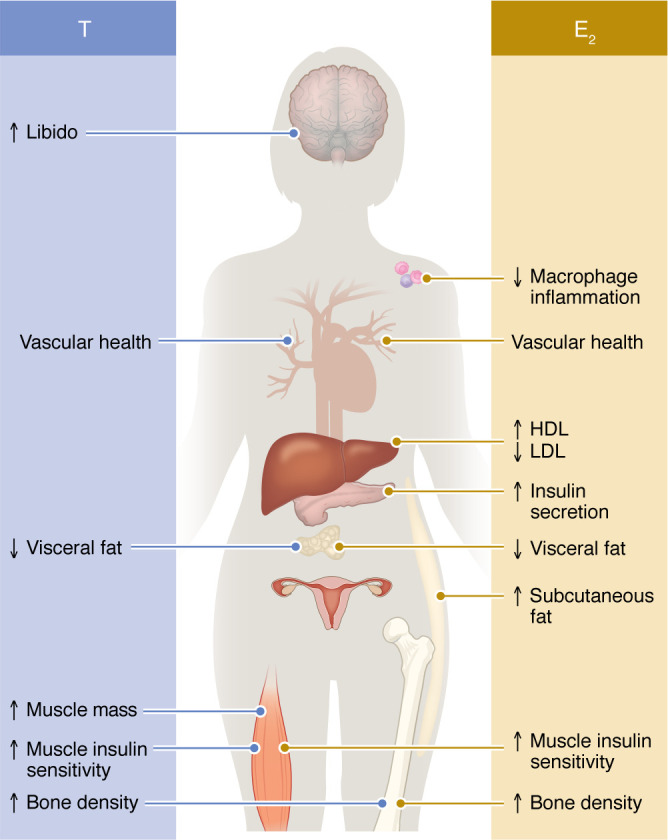
E2’s effects on immune, vascular, lipid, islet, adipose, muscle, and bone biology are represented on the right, while T’s effects on vascular, adipose, muscle, and bone biology are represented on the left.
E2 promotes subcutaneous lipid storage in females.
A major evolutionary function of E2 is to facilitate postprandial lipid storage in subcutaneous adipose tissue (SCAT) to prepare for pregnancy (38). Thus, premenopausal females carry more SCAT than males because higher circulating concentrations of E2 in females favors SCAT expansion and inhibits visceral adipose tissue (VAT) development. The best evidence is found in transgender individuals assigned male sex at birth who were treated with high doses of estrogens (in the presence of antiandrogens) as gender-affirming therapy. These individuals accumulated preferential SCAT in the leg and gynoid region, which increased hip circumference (39). After menopause, E2 deficiency leads to VAT accumulation, but it is reduced by estrogen therapy (40). As we will discuss below, T’s conversion to E2 is also instrumental in preventing VAT accumulation in males. In females, endogenous E2 also promoted lipid oxidation in skeletal muscle during fasting and exercise, but inhibited hepatic lipid oxidation during the fed and resting periods, which promoted energy storage in SCAT (38). Estrogens taken orally also increased hepatic de novo lipogenesis and triglyceride synthesis for export into very-low-density lipoproteins (VLDLs) that can be taken up by the expanded SCAT to promote lipid storage (41). After menopause, E2 deficiency decreases lipid oxidation and leads to disinhibition of VAT accumulation. In summary, E2 promotes lipid oxidation in fasting and SCAT expansion to promote lipid storage in fed and resting states while inhibiting VAT development, which produces the female gynoid phenotype.
E2 promotes glucose and lipid homeostasis in females.
E2 is an antidiabetic hormone; thus, deficiency increases the risk of new-onset type 2 diabetes (T2D) (42). In postmenopausal women, estrogen therapy reduces the incidence of new-onset T2D and improved glycemia in women with diabetes. In postmenopausal women with T2D, estrogen therapy reduced fasting glucose and insulin as well as HbA1c (a marker of chronic hyperglycemia), and decreased the homeostatic model assessment for insulin resistance (HOMA-IR) index to a greater extent than in postmenopausal women without diabetes (40, 43). Estrogens administered orally produced a greater decrease in diabetes risk than the transdermal route (44). The stronger effect of oral estrogens on blood glucose results from first-pass liver metabolism, which better suppresses hepatic glucose output (45). The beneficial effects of endogenous E2 can be inferred from studies using animal models suggesting that E2 enhances insulin sensitivity via ERα in liver and skeletal muscle (42, 45) and protects muscle mitochondrial function, which is essential to female insulin sensitivity (46) (see also section below).
Endogenous and exogenous estrogens also protect β cell function and insulin secretion, as shown in preclinical and clinical studies (42, 47–51). This effect is less apparent clinically because the hyperbolic relationship between insulin sensitivity and β cell function (i.e., disposition index) produces a dynamic compensation of the E2-induced improvement in insulin sensitivity by reducing insulin secretion. Endogenous E2 and exogenous estrogens produce beneficial effects on cholesterol and inflammatory markers. Women experience an increase in low-density lipoprotein (LDL) cholesterol during perimenopause (52), and estrogen therapy is protective. In meta-analyses, estrogens reduce the ratio of LDL/high-density lipoprotein (HDL) cholesterol, lipoprotein (a), and the inflammatory markers E-selectin and plasminogen activator inhibitor-1 (40). In summary, E2 is critical in females for maintaining glucose and lipid homeostasis, which is reproduced by estrogen therapy and, as discussed below in “T promotes metabolic homeostasis in males,” is also true in males.
E2 promotes mitochondrial fitness in females.
E2 allows women to transmit the fittest mitochondria to prevent the transmission of inherited disease (38). In female rodents, E2 promotes higher mitochondrial antioxidant enzyme activity, decreases reactive oxygen species production, and reduces damage to mitochondria DNA (mtDNA) compared with male rodents. In addition, E2 via nuclear ERα and ERβ activates a transcriptional cascade culminating in the expression of mitochondrial respiratory chain complexes (53, 54). E2 acting on mitochondrial ERα or ERβ also maintains mitochondrial dynamics and promotes mitochondrial fusion while attenuating fission (46, 55). In summary, E2 promotes female mitochondrial quality with higher respiratory capacities, biogenesis, and resistance to oxidative stress (56). Figure 3 summarizes the effects of E2 on female metabolic homeostasis.
The importance of T in female metabolic homeostasis
T production favors healthy body composition in females.
Supraphysiological levels of T in women such as those achieved during polycystic ovarian syndrome are associated with insulin resistance, visceral obesity, and T2D, demonstrating the metabolic impact of T in females (57). However, although clinical trials have focused of the effects of T supplementation in postmenopausal women with regard to libido and well being, the physiological impact of T in female metabolic homeostasis has not been explored. This lack of knowledge is surprising since, as discussed above, in females, T is always more abundant than E2 (Figure 2). In addition, studies have documented wide AR expression across female human tissues (58) and strong AR genomic localization in female rat tissues despite low levels of AR protein compared with male rats (59). An example that illustrates the physiological role of T in female metabolism is its conversion to active steroids in pancreatic islet β cells. Female mouse and human β cells are equipped with the enzymes aromatase and 5α-reductase (5α-R) to convert circulating T to E2 and DHT, respectively (60). Intracrine conversion of T to E2 or DHT by these enzymes was observed in female human islets, and this enhanced insulin secretion (60). In androgen-deficient women (as a result of hypopituitarism, oophorectomy, or natural menopause), T treatment that produced concentrations in the female physiological range increased lean mass (bone density and muscle mass) and decreased fat mass (61–67), improved insulin resistance (62, 68), and decreased inflammation (69, 70). T even improved aerobic capacity, muscle performance, and effort tolerance in postmenopausal females with advanced chronic heart failure (62). It is unknown to what extent the effect of T on fat mass and insulin sensitivity in females is mediated via aromatization to E2. However, the effect on muscle mass is likely mediated via T or DHT acting on AR, as discussed for males below. In addition, in postmenopausal females, T enhanced the effect of E2 in increasing bone mineral density, suggesting that T acting on AR is also important for maintenance of female bone strength (66, 67). Indeed, female mice lacking AR display reduced trabecular bone mass (71).
Physiological T production protects female vascular health.
Hyperandrogenism in women of reproductive age has been associated with subclinical markers of atherosclerotic CVD, such as arterial stiffness, carotid intima media thickness, coronary artery calcification, endothelial dysfunction, and CVD (72). The administration of high-dose T was also associated with atherosclerosis in postmenopausal women (73). In contrast, low endogenous T in women has been prospectively associated with increased all-cause mortality and incident CVD independent of other risk factors (74). Thus, a physiological window of T seems necessary for female vascular health. Indeed, throughout the female life span, higher T concentrations within the physiological range have been associated with lower carotid intimal-medial thickness (75). Conversely, lower T concentrations were associated with carotid atherosclerosis (76, 77) and coronary artery disease (CAD) (78) in females. The mechanisms by which T promotes vascular health in females may involve a reduction in CV risk factors; apolipoprotein CIII (apoCIII) impairs the metabolism of VLDL and LDL, increasing triglycerides, and thus is a strong predictor of CAD (79). In women with surgical menopause, T added to estrogens reduced the apoCIII concentration selectively in VLDL and LDL compared with estrogens alone, which was expected to improve CAD risk (70). Addition of T to oral E2 counteracts the E2-induced rise in the inflammatory marker C-reactive protein (CRP) in postmenopausal females (69). T also promoted arterial vasodilation in postmenopausal females who were already using estrogen therapy (80), suggesting the existence of a synergism between E2 and T in control of blood pressure. Foam cell formation is an early event in atherosclerosis due to the uptake of LDL by macrophages in the arterial wall (81). Female mice are protected from atherosclerosis compared with males, which is believed to be due to E2 (31). DHT caused a dose-dependent and AR-mediated increase in macrophage cholesterol loading and atherosclerosis-related genes in cultured human male, but not female, macrophages (82, 83). T decreased atherosclerosis in female mice generated on an atherosclerosis-prone apoE-deficient background, but increased atherosclerosis in apoE-deficient male mice (84). In addition, apoE-deficient female mice lacking AR developed diet-induced obesity, dyslipidemia, and atherosclerosis (85). In summary, the physiological importance of T in female metabolic homeostasis is underestimated and may involve beneficial effects on body composition, vascular health, and prevention of atherosclerosis. Figure 3 summarizes T’s actions in female biology.
T promotes metabolic homeostasis in males
In males, T is a hormone that binds the AR and a prohormone that provides a circulating reservoir of E2 and DHT. T deficiency in males leads to sexual dysfunction, depressed mood, anemia, osteoporosis, metabolic syndrome and T2D, and CVD. In the following section, we discuss the effect of T on metabolic homeostasis separated into the effects induced by E2 versus T/DHT.
T-to-E2 conversion maintains bone mass in males.
T’s conversion to E2 by aromatase is instrumental to both normal bone development and preservation of healthy bone metabolism during aging. Support for the importance of E2 in T’s action comes from studies in young males with inactivating mutations of either ERα or aromatase who exhibit abnormal bone growth and development as well as early osteoporosis (86, 87). Furthermore, T treatment of males rendered hypogonadal using gonadotropin-releasing hormone (GnRh) agonists improved bone mineral density, but this effect was abolished with simultaneous administration of an aromatase inhibitor, which blocks T’s conversion to E2 (1). In aging men, E2 is the dominant sex steroid preventing bone resorption, whereas both E2 and T are important in increasing bone formation (88). It is estimated that in males, E2 accounts for approximately 70% of the maintenance of bone mass, with T contributing 30%.
T is an anti-obesity hormone in males.
T deficiency promotes VAT accumulation, and the development of metabolic syndrome in males (reviewed in ref. 89).
T’s aromatization to E2 prevents visceral adiposity in male individuals. Orchiectomized male rodents treated with T or E2 remained lean, while those treated with DHT, which cannot be converted to E2, developed obesity (90). Similarly, in human males rendered hypogonadal using GnRh agonists, T replacement prevented VAT accumulation, an effect that was blocked in the presence of an aromatase inhibitor (1). In addition, human and rodent studies confirmed that inactivating mutations of aromatase increase VAT in males (87, 91). The mechanism by which T’s conversion to E2 prevents VAT in male individuals likely involves an inhibition of adipocytes and adipose progenitors as well as the promotion of lipid oxidation, as described in female individuals.
T has anti-obesity properties mediated via AR actions. In males with genetic androgen resistance (linked to CAG-repeat polymorphisms in the AR gene that decrease AR-mediated gene transcription), a low number of CAG repeats (which increases AR action) was associated with low adiposity and plasma insulin, demonstrating that intact AR action is necessary to prevent VAT accumulation (92). Second, male mice lacking AR developed late-onset visceral obesity and insulin resistance (93, 94). These effects of T on VAT are likely mediated via AR in skeletal muscle, as overexpression of AR selectively in muscle cells of male rats increased muscle mass, which elevated metabolic rate and reduced adipose tissue mass (95). In contrast, male adipocyte-specific AR-deficient mice exhibited no increase in VAT, demonstrating that direct AR action in adipocytes is not necessary for the control of VAT mass (96). In summary, in male individuals, T prevents VAT accumulation via E2’s action on ERα in muscle and adipose (like in females) as well as T/DHT’s action on AR in skeletal muscle.
T prevents T2D in males.
Androgen deprivation therapy (ADT), the standard of treatment of prostate cancer, produces severe T deficiency and is a severe risk factor for developing T2D in males (97, 98). Moderate T deficiency also predisposes to T2D, while T replacement therapy (TRT) prevents or reverses T2D in T-deficient men (99). The antidiabetic effects of T are mediated via a decrease in VAT (described above), an increase in skeletal muscle mass and glycolytic capacity (both of which increase insulin sensitivity), and improved β cell function, as we describe below.
T improves insulin sensitivity via conversion to E2 and DHT, or via the effect of T itself. T promotes insulin sensitivity in skeletal muscle at least partially via an increase in peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC1α), which stimulates mitochondrial biogenesis and skeletal muscle oxidative fibers, and is a molecular marker of muscle insulin sensitivity. A decrease in PGC1α in skeletal muscle was associated with insulin resistance in males (100). Similarly, men with low T exhibited low PGC1α expression in muscle (101). T’s effect on PGC1α is likely to be E2 mediated, as E2 treatment of males increases PGC1α in muscle (102). T’s improvement of insulin sensitivity also requires conversion to DHT. Dual inhibition of the T-to-DHT–converting enzymes 5α-R1 and -R2, but not inhibition of 5α-R2 alone, produced peripheral insulin resistance (103), which is associated with hepatic lipid accumulation in males (104). This suggests that T’s conversion to DHT via 5α-R1 is necessary for insulin sensitivity. T also promotes insulin sensitivity by increasing muscle mass. Surprisingly, the inhibition of T’s conversion to DHT by 5α-R inhibitors had no effect on the ability of T to increase muscle mass and strength (105), indicating that in this context, T directly binds AR and does not require conversion to DHT to promote muscle growth. T also promotes carbohydrate utilization, glycolysis, and glycogen synthesis in skeletal muscle (106, 107), which enhances insulin sensitivity via AR (106). Overexpression of AR in skeletal muscle of male mice produced hypertrophy of glycolytic muscle fibers and increased glucose metabolism (95). Activation of AR also increased glycolysis in male pancreatic islet β cells (108). In contrast, E2 treatment of males (which also decreases T) enhanced lipid oxidation, decreased carbohydrate oxidation during exercise (109) and in cultured male myotubes (110), and increased skeletal muscle expression of medium chain acyl-CoA dehydrogenase, a marker of lipid oxidation (102). Note that individuals assigned male sex at birth who were treated with estrogens (and androgen depletion) as gender-affirming therapy developed insulin resistance (111), suggesting that in males, E2 improves insulin sensitivity in the presence of intact AR action. In summary, in males, T promotes insulin sensitivity with mixed actions of E2 on ERα (insulin sensitivity), DHT on AR (insulin sensitivity), and T on AR (muscle mass).
T’s conversion to DHT enhances insulin secretion in male individuals. Human and rodent male β cells express 5α-R1, which is necessary to convert T to DHT and enhances glucose-stimulated insulin secretion in cultured islets (60). Male mice lacking AR in β cells (βARKO mice) developed β cell failure, leading to inadequate compensation for insulin resistance and hyperglycemia (112). βARKO islets displayed dysregulated genes involved in inflammation and insulin secretion (113). Thus, in the absence of AR in β cells, T cannot maintain normoglycemia, demonstrating the importance of the β cell AR pool to glucose homeostasis in the male. The mechanism involves DHT activation of AR, which amplifies the insulinotropic action of glucagon-like peptide 1 (GLP-1) via its receptor in human β cells, thus enhancing the hypoglycemic and anabolic actions of insulin (108, 112, 114).
T’s conversion to E2 is also important to β cell protection in males. First, male human β cells express aromatase, which is necessary to convert T to E2 and enhances insulin secretion (60). Indeed, in male mice, T’s conversion to E2 via aromatase was necessary to prevent β cell damage from the toxin streptozotocin (115). Second, in multiple male animal models of T2D or β cell failure, E2 protected male islets in vivo from diabetic injuries such as glucolipotoxicity or ER stress (47, 49, 51, 115), suggesting that T’s conversion to E2 is necessary to protect β cell function in males.
Endogenous T promotes cardiovascular health in males.
Endogenous T directly protects the male cardiovascular system. T is a potent vasodilator that acutely increases coronary blood flow (116) and exerts beneficial effects on blood pressure (117). Observational studies demonstrate a direct association between low serum T concentrations and increased risk of CVD in males (118, 119). A meta-analysis of 70 studies concluded that patients with CVD exhibit lower T concentrations (120). Similarly, GnRh agonists, which suppress T production, promote vascular damage (121, 122). Accordingly, a retrospective examination of over 83,000 hypogonadal males showed that normalization of T levels by TRT decreased all-cause mortality, risk of MI, and stroke (123). Moreover, in men with T deficiency and high risk of CVD, the TRAVERSE trial using transdermal T confirmed that TRT does not increase the incidence of major adverse cardiac events (124), providing reassurance about the cardiovascular safety of TRT (125). In summary, despite controversy about T’s effects on CVD, endogenous T prevents CVD and accordingly low T predisposes to CVD. In hypogonadal men, TRT is safe regarding CVD.
Endogenous T promotes cardiovascular health in males via conversion to E2, as demonstrated by the development of endothelial dysfunction and CAD in a young male with absence of functional ERα (126, 127). In middle-aged healthy males, circulating concentrations of E2, not T, are positively associated with FMD (128), while a reduction in plasma E2, through aromatase inhibition, decreases FMD (129). This effect is likely mediated via NO production, as in females. However, the beneficial effect of E2 in males seems to occur within a tight physiological window and in the presence of physiological T concentrations. The early Coronary Drug Project, designed to evaluate the ability of high doses of oral CEE to prevent CAD in males with prior MIs, was discontinued because of increased incidence of MI (130). Similarly, high-dose diethylstilbestrol, a synthetic estrogen, increased the incidence of atherothrombotic disease in males (131), and high-dose ethinyl estradiol, a potent synthetic estrogen used for contraception, increased CVD risk when used as a gender-affirming therapy in transgender individuals assigned male sex at birth (132). However, lower doses of CEE, ethinyl estradiol, or E2 for shorter duration in transgender individuals on gender-affirming therapy improved vascular function (133), enhanced endothelial function and arterial reactivity (134), and promoted endothelium-dependent vasodilation in the microcirculation (135). In older hypogonadal males, E2 enhanced endothelium-mediated vasorelaxation, attenuated vasoconstriction, and reduced blood pressure (136). Estradiol also induced male human coronary relaxation in vitro (137). Studies using genetically modified mice confirmed that the beneficial effects of E2 on vasodilation in male mice, as in female mice, are mediated by ERα (138). Taken together, these data demonstrate that E2 at physiological doses is beneficial for male vascular health.
The T/E2 ratio seems to be a critical parameter for optimal male CVD protection. In the general male population, the T/E2 ratio (both in pg/mL) ranges between 150 and 200 (Figure 2D). In males with existing atherosclerotic disease, a low T/E2 ratio (<120) was associated with increased systemic inflammation and inflammatory plaques, as well as an increased risk of future major adverse cardiovascular events compared with males with a higher T/E2 ratio (>160) (139). In older males, low T and high E2 levels (which decrease the T/E2 ratio) were also associated with an unhealthy artery wall on ultrasound (140, 141). In these studies, the low T/E2 reflected low T with higher E2 concentrations, but still in the physiological range. Thus, it is possible that higher E2 production in the face of low T reflects an endogenous compensatory increase in aromatase activity to lower E2 output in tissue and developing atherosclerosis. The importance of the T/E2 ratio and the stoichiometry of T and E2’s actions may explain why data in male or transgender patients receiving gender-affirming therapy with high-dose estrogens, which suppress T, display increased CVD risk (130–132). However, in transgender individuals receiving gender-affirming therapies, psychosocial stressors may also be implicated in CVD risk (142).
T supplementation decreases HDL in hypogonadal men (143), but produces no change in cholesterol efflux capacity (CEC) of serum HDL, a more reliable CAD risk predictor (144). This decrease in HDL is likely mediated via AR and reproduced by a selective AR modulator (145). In contrast, T is likely to improve atherogenic lipids via conversion to E2, as men with aromatase mutations exhibit low HDL, high LDL, and increased triglycerides, which are corrected by E2 treatment (87, 146). In fact, in males, oral E2 increased HDL (136) and decreased LDL (147), as it does in females. Oral E2 also decreased triglyceride and homocysteine (147). In summary, in males, T promotes vascular protection via conversion to E2, likely by increasing NO and promoting a less atherogenic lipid profile. Consequently, low T, which is associated with low E2, predisposes to CVD. Figure 4 summarizes T’s actions in males.
Figure 4. Cardiometabolic effect of T and E2 in males.
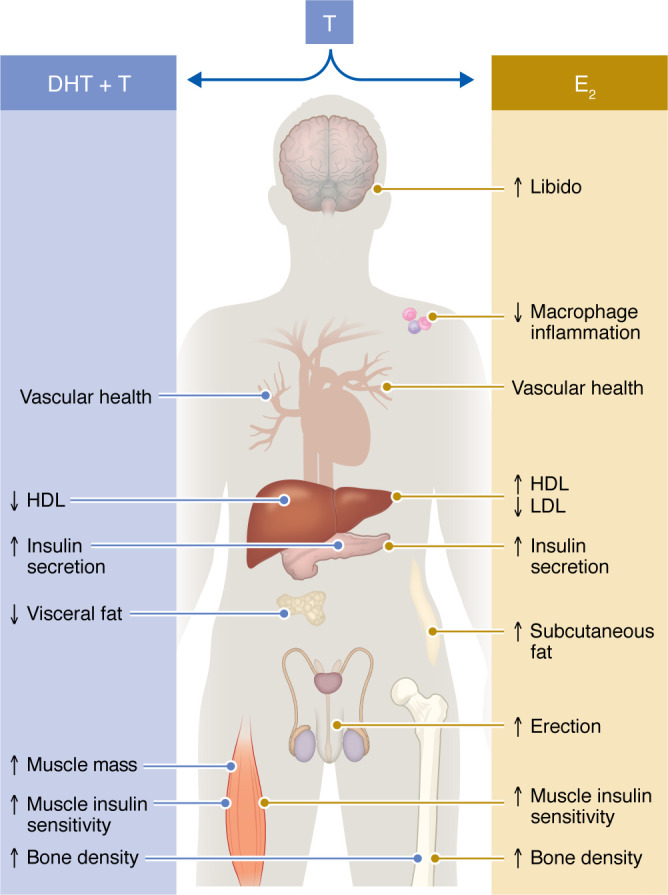
In males, T is converted to E2 and DHT. T’s effects that are mediated via conversion to E2 on immune, vascular, lipid, islet, adipose, muscle, and bone biology as well as sexual function are represented on the right, while T’s effects mediated via direct action or conversion to DHT on vascular, lipid, islet adipose, muscle, and bone biology are represented on the left. DHT, dihydrotestosterone.
Conclusions and clinical implications
T and E2 are produced in both sexes at sex-specific concentrations and share similar and potent metabolic functions. The loss of E2 after menopause in females and the decrease in T in aging males both produce metabolic dysfunction and are serious health threats leading to cardiometabolic disease and frailty. The reason that these important metabolic mediators are not prescribed more often relates to myths about the danger of hormones. In particular, there are persistent misconceptions about the risks of estrogen-based therapies in females (148–154). Apart from the purported risk of breast cancer, which has been attributed to synthetic progestins, confusion about the risks of estrogens lies in the too often ignored biological difference between synthetic hormones like CEE, which is associated with CVD, and endogenous and bioidentical E2, which is not associated with negative CVD outcomes. In the case of males and T, myths about risk of prostate cancer and CVD along with its cultural associations with illegally enhancing athletic performance and toxic masculinity has created resistance to consider aging as a treatable condition of T deficiency (155).
It is not known what the role of T in female metabolism is. Is it mediated via T or DHT acting on AR, as animal studies suggest, or is T an additional reservoir for local E2 synthesis in tissues? Clinical trials assessing the effect of T supplementation in postmenopausal women to achieve serum concentrations in the upper limit of female physiology should be considered to ascertain its ability to improve muscle and metabolic function along with its beneficial effects on libido.
Anecdotally, male patients on TRT often enquire about their E2 levels due to fear of “too much female hormone.” Mens’ health clinics even prescribe aromatase inhibitors to suppress E2 production while raising T concentrations. However, we discussed the essential role of T’s conversion to E2 in male bone and vascular health, as well as glucose and lipid homeostasis (not to mention libido and erectile function). Thus, it is our view that E2 should not be suppressed in men, and in fact clinical trials of E2 supplementation should be considered in some men on TRT to decrease LDL cholesterol and improve endothelial function.
Finally, current laboratory measurements of serum T and E2 levels (total or free) poorly reflect tissue and cellular T and E2 concentrations, catabolism, and elimination. Novel assays that provide accurate measures of cellular T and E2 outputs will be informative in clinical studies and are desperately needed.
Acknowledgments
This work was supported by NIH grants DK074970 (to FMJ), HL133619 (to SHL), P20GM152305 (to FMJ and SHL), and US Department of Veterans Affairs Merit Award BX005812 (to FMJ). Figure 2 was developed from material that is freely available in the CDC’s NHANES databases. Reference to this material does not constitute its endorsement or recommendation by the US government, Department of Health and Human Services or the CDC.
Version 1. 09/03/2024
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2024, Mauvais-Jarvis et al. This is an open access article published under the terms of the Creative Commons Attribution 4.0 International License.
Reference information: J Clin Invest. 2024;134(17):e180073. https://doi.org/10.1172/JCI180073.
Contributor Information
Franck Mauvais-Jarvis, Email: fmauvais@tulane.edu.
Sarah H. Lindsey, Email: lindsey@tulane.edu.
References
- 1.Finkelstein JS, et al. Gonadal steroids and body composition, strength, and sexual function in men. N Engl J Med. 2013;369(11):1011–1022. doi: 10.1056/NEJMoa1206168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davis SR, et al. Efficacy and safety of a testosterone patch for the treatment of hypoactive sexual desire disorder in surgically menopausal women: a randomized, placebo-controlled trial. Menopause. 2006;13(3):387–396. doi: 10.1097/01.gme.0000179049.08371.c7. [DOI] [PubMed] [Google Scholar]
- 3.Vermeulen A, et al. Estradiol in elderly men. Aging Male. 2002;5(2):98–102. doi: 10.1080/tam.5.2.98.102. [DOI] [PubMed] [Google Scholar]
- 4.Vermeulen A, Verdonck L. Factors affecting sex hormone levels in postmenopausal women. J Steroid Biochem. 1979;11(1c):899–904. doi: 10.1016/0022-4731(79)90027-X. [DOI] [PubMed] [Google Scholar]
- 5.Forney JP, et al. Aromatization of androstenedione to estrone by human adipose tissue in vitro. Correlation with adipose tissue mass, age, and endometrial neoplasia. J Clin Endocrinol Metab. 1981;53(1):192–199. doi: 10.1210/jcem-53-1-192. [DOI] [PubMed] [Google Scholar]
- 6.Burger HG. Androgen production in women. Fertil Steril. 2002;77 Suppl 4:S3–S5. doi: 10.1016/S0015-0282(02)03008-X. [DOI] [PubMed] [Google Scholar]
- 7.Longcope C. Adrenal and gonadal androgen secretion in normal females. Clin Endocrinol Metab. 1986;15(2):213–228. doi: 10.1016/S0300-595X(86)80021-4. [DOI] [PubMed] [Google Scholar]
- 8.Judd HL, Fournet N. Changes of ovarian hormonal function with aging. Exp Gerontol. 1994;29(3–4):285–298. doi: 10.1016/0531-5565(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 9.Adashi EY. The climacteric ovary as a functional gonadotropin-driven androgen-producing gland. Fertil Steril. 1994;62(1):20–27. doi: 10.1016/S0015-0282(16)56810-1. [DOI] [PubMed] [Google Scholar]
- 10.Manson JE, et al. Menopausal hormone therapy and health outcomes during the intervention and extended poststopping phases of the Women’s Health Initiative randomized trials. JAMA. 2013;310(13):1353–1368. doi: 10.1001/jama.2013.278040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lufkin EG, et al. Treatment of postmenopausal osteoporosis with transdermal estrogen. Ann Intern Med. 1992;117(1):1–9. doi: 10.7326/0003-4819-117-1-1. [DOI] [PubMed] [Google Scholar]
- 12.Rocca WA, et al. Bilateral oophorectomy and accelerated aging: cause or effect? J Gerontol A Biol Sci Med Sci. 2017;72(9):1213–1217. doi: 10.1093/gerona/glx026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mytton J, et al. Removal of all ovarian tissue versus conserving ovarian tissue at time of hysterectomy in premenopausal patients with benign disease: study using routine data and data linkage. BMJ. 2017;356:j372. doi: 10.1136/bmj.j372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roeters van Lennep JE, et al. Cardiovascular disease risk in women with premature ovarian insufficiency: A systematic review and meta-analysis. Eur J Prev Cardiol. 2016;23(2):178–186. doi: 10.1177/2047487314556004. [DOI] [PubMed] [Google Scholar]
- 15.Muka T, et al. Association of age at onset of menopause and time since onset of menopause with cardiovascular outcomes, intermediate vascular traits, and all-cause mortality: a systematic review and meta-analysis. JAMA Cardiol. 2016;1(7):767–776. doi: 10.1001/jamacardio.2016.2415. [DOI] [PubMed] [Google Scholar]
- 16.Green DJ, et al. Is flow-mediated dilation nitric oxide mediated?: A meta-analysis. Hypertension. 2014;63(2):376–382. doi: 10.1161/HYPERTENSIONAHA.113.02044. [DOI] [PubMed] [Google Scholar]
- 17.Matsuzawa Y, et al. Prognostic value of flow-mediated vasodilation in brachial artery and fingertip artery for cardiovascular events: a systematic review and meta-analysis. J Am Heart Assoc. 2015;4(11):e002270. doi: 10.1161/JAHA.115.002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hopkins ND, et al. Age and sex relationship with flow-mediated dilation in healthy children and adolescents. J Appl Physiol (1985) 2015;119(8):926–933. doi: 10.1152/japplphysiol.01113.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skaug EA, et al. Age and gender differences of endothelial function in 4739 healthy adults: the HUNT3 Fitness Study. Eur J Prev Cardiol. 2013;20(4):531–540. doi: 10.1177/2047487312444234. [DOI] [PubMed] [Google Scholar]
- 20.Moreau KL, et al. Endothelial function is impaired across the stages of the menopause transition in healthy women. J Clin Endocrinol Metab. 2012;97(12):4692–4700. doi: 10.1210/jc.2012-2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samargandy S, et al. Trajectories of blood pressure in midlife women: does menopause matter? Circ Res. 2022;130(3):312–322. doi: 10.1161/CIRCRESAHA.121.319424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chappell MC, et al. Estrogen and salt sensitivity in the female mRen(2). Lewis rat. Am J Physiol Regul Integr Comp Physiol. 2006;291(5):R1557–R1563. doi: 10.1152/ajpregu.00051.2006. [DOI] [PubMed] [Google Scholar]
- 23.Madika AL, et al. Menopausal hormone therapy and risk of incident hypertension: role of the route of estrogen administration and progestogens in the E3N cohort. Menopause. 2021;28(11):1204–1208. doi: 10.1097/GME.0000000000001839. [DOI] [PubMed] [Google Scholar]
- 24.Kalenga CZ, et al. Association between the route of administration and formulation of estrogen therapy and hypertension risk in postmenopausal women: a prospective population-based study. Hypertension. 2023;80(7):1463–1473. doi: 10.1161/HYPERTENSIONAHA.122.19938. [DOI] [PubMed] [Google Scholar]
- 25.Blacher J, et al. Aortic pulse wave velocity as a marker of cardiovascular risk in hypertensive patients. Hypertension. 1999;33(5):1111–1117. doi: 10.1161/01.HYP.33.5.1111. [DOI] [PubMed] [Google Scholar]
- 26.Laogun AA, Gosling RG. In vivo arterial compliance in man. Clin Phys Physiol Meas. 1982;3(3):201–212. doi: 10.1088/0143-0815/3/3/004. [DOI] [PubMed] [Google Scholar]
- 27.Khan ZA, et al. Serial studies in subclinical atherosclerosis during menopausal transition (from the Study of Women’s Health Across the Nation) Am J Cardiol. 2018;122(7):1161–1168. doi: 10.1016/j.amjcard.2018.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waddell TK, et al. Women exhibit a greater age-related increase in proximal aortic stiffness than men. J Hypertens. 2001;19(12):2205–2212. doi: 10.1097/00004872-200112000-00014. [DOI] [PubMed] [Google Scholar]
- 29.Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. 2016;16(10):626–638. doi: 10.1038/nri.2016.90. [DOI] [PubMed] [Google Scholar]
- 30.Mauvais-Jarvis F, et al. Estradiol, progesterone, immunomodulation, and COVID-19 outcomes. Endocrinology. 2020;161(9):bqaa127. doi: 10.1210/endocr/bqaa127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cann JA, et al. Timing of estrogen replacement influences atherosclerosis progression and plaque leukocyte populations in ApoE-/- mice. Atherosclerosis. 2008;201(1):43–52. doi: 10.1016/j.atherosclerosis.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clarkson TB, et al. Timing hypothesis for postmenopausal hormone therapy: its origin, current status, and future. Menopause. 2013;20(3):342–353. doi: 10.1097/gme.0b013e3182843aad. [DOI] [PubMed] [Google Scholar]
- 33.Boardman HM, et al. Hormone therapy for preventing cardiovascular disease in post-menopausal women. Cochrane Database Syst Rev. 2015;2015(3):CD002229. doi: 10.1002/14651858.CD002229.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.SenthilKumar G, et al. Estrogen and the vascular endothelium: the unanswered questions. Endocrinology. 2023;164(6):bqad079. doi: 10.1210/endocr/bqad079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rossouw JE, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288(3):321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 36.Bhavnani BR. Estrogens and menopause: pharmacology of conjugated equine estrogens and their potential role in the prevention of neurodegenerative diseases such as Alzheimer’s. J Steroid Biochem Mol Biol. 2003;85(2-5):473–482. doi: 10.1016/S0960-0760(03)00220-6. [DOI] [PubMed] [Google Scholar]
- 37.Novensa L, et al. Equine estrogens impair nitric oxide production and endothelial nitric oxide synthase transcription in human endothelial cells compared with the natural 17{beta}-estradiol. Hypertension. 2010;56(3):405–411. doi: 10.1161/HYPERTENSIONAHA.110.151969. [DOI] [PubMed] [Google Scholar]
- 38.Mauvais-Jarvis F. Sex differences in energy metabolism: natural selection, mechanisms and consequences. Nat Rev Nephrol. 2024;20(1):56–69. doi: 10.1038/s41581-023-00781-2. [DOI] [PubMed] [Google Scholar]
- 39.Klaver M, et al. Changes in regional body fat, lean body mass and body shape in trans persons using cross-sex hormonal therapy: results from a multicenter prospective study. Eur J Endocrinol. 2018;178(2):163–171. doi: 10.1530/EJE-17-0496. [DOI] [PubMed] [Google Scholar]
- 40.Salpeter SR, et al. Meta-analysis: effect of hormone-replacement therapy on components of the metabolic syndrome in postmenopausal women. Diabetes Obes Metab. 2006;8(5):538–554. doi: 10.1111/j.1463-1326.2005.00545.x. [DOI] [PubMed] [Google Scholar]
- 41.Marlatt KL, et al. Effect of conjugated estrogens and bazedoxifene on glucose, energy and lipid metabolism in obese postmenopausal women. Eur J Endocrinol. 2020;183(4):439–452. doi: 10.1530/EJE-20-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mauvais-Jarvis F, et al. Menopausal hormone therapy and type 2 diabetes prevention: evidence, mechanisms, and clinical implications. Endocr Rev. 2017;38(3):173–188. doi: 10.1210/er.2016-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Speksnijder EM, et al. Effect of postmenopausal hormone therapy on glucose regulation in women with type 1 or type 2 diabetes: a systematic review and meta-analysis. Diabetes Care. 2023;46(10):1866–1875. doi: 10.2337/dc23-0451. [DOI] [PubMed] [Google Scholar]
- 44.de Lauzon-Guillain B, et al. Menopausal hormone therapy and new-onset diabetes in the French Etude Epidemiologique de Femmes de la Mutuelle Générale de l’Education Nationale (E3N) cohort. Diabetologia. 2009;52(10):2092–2100. doi: 10.1007/s00125-009-1456-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mauvais-Jarvis F, et al. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev. 2013;34(3):309–338. doi: 10.1210/er.2012-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ribas V, et al. Skeletal muscle action of estrogen receptor α is critical for the maintenance of mitochondrial function and metabolic homeostasis in females. Sci Transl Med. 2016;8(334):334ra54. doi: 10.1126/scitranslmed.aad3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tiano JP, Mauvais-Jarvis F. Importance of oestrogen receptors to preserve functional β-cell mass in diabetes. Nat Rev Endocrinol. 2012;8(6):342–351. doi: 10.1038/nrendo.2011.242. [DOI] [PubMed] [Google Scholar]
- 48.Lovre D, et al. Conjugated estrogens and bazedoxifene improve β cell function in obese menopausal women. J Endocr Soc. 2019;3(8):1583–1594. doi: 10.1210/js.2019-00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tiano JP, et al. Estrogen receptor activation reduces lipid synthesis in pancreatic islets and prevents β cell failure in rodent models of type 2 diabetes. J Clin Invest. 2011;121(8):3331–3342. doi: 10.1172/JCI44564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wong WP, et al. Extranuclear estrogen receptor-{alpha} stimulates NeuroD1 binding to the insulin promoter and favors insulin synthesis. Proc Natl Acad Sci U S A. 2010;107(29):13057–13062. doi: 10.1073/pnas.0914501107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu B, et al. Estrogens promote misfolded proinsulin degradation to protect insulin production and delay diabetes. Cell Rep. 2018;24(1):181–196. doi: 10.1016/j.celrep.2018.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.El Khoudary SR, et al. Low-density lipoprotein subclasses over the menopausal transition and risk of coronary calcification and carotid atherosclerosis: the SWAN Heart and HDL ancillary studies. Menopause. 2023;30(10):1006–1013. doi: 10.1097/GME.0000000000002245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kobayashi A, et al. Mechanisms underlying the regulation of mitochondrial respiratory chain complexes by nuclear steroid receptors. Int J Mol Sci. 2020;21(18):6683. doi: 10.3390/ijms21186683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klinge CM. Estrogenic control of mitochondrial function. Redox Biol. 2020;31:101435. doi: 10.1016/j.redox.2020.101435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beikoghli Kalkhoran S, Kararigas G. Oestrogenic regulation of mitochondrial dynamics. Int J Mol Sci. 2022;23(3):1118. doi: 10.3390/ijms23031118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ventura-Clapier R, et al. Mitochondria: a central target for sex differences in pathologies. Clin Sci (Lond) 2017;131(9):803–822. doi: 10.1042/CS20160485. [DOI] [PubMed] [Google Scholar]
- 57.Ehrmann DA, et al. Prevalence and predictors of the metabolic syndrome in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91(1):48–53. doi: 10.1210/jc.2005-1329. [DOI] [PubMed] [Google Scholar]
- 58.Uhlen M, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 59.Nash C, et al. Genome-wide analysis of androgen receptor binding and transcriptomic analysis in mesenchymal subsets during prostate development. Dis Model Mech. 2019;12(7):dmm039297. doi: 10.1242/dmm.039297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu W, et al. Intracrine testosterone activation in human pancreatic β-cells stimulates insulin secretion. Diabetes. 2020;69(11):2392–2399. doi: 10.2337/db20-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller KK, et al. Effects of testosterone replacement in androgen-deficient women with hypopituitarism: a randomized, double-blind, placebo-controlled study. J Clin Endocrinol Metab. 2006;91(5):1683–1690. doi: 10.1210/jc.2005-2596. [DOI] [PubMed] [Google Scholar]
- 62.Iellamo F, et al. Testosterone therapy in women with chronic heart failure: a pilot double-blind, randomized, placebo-controlled study. J Am Coll Cardiol. 2010;56(16):1310–1316. doi: 10.1016/j.jacc.2010.03.090. [DOI] [PubMed] [Google Scholar]
- 63.Zang H, et al. Effects of treatment with testosterone alone or in combination with estrogen on insulin sensitivity in postmenopausal women. Fertil Steril. 2006;86(1):136–144. doi: 10.1016/j.fertnstert.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 64.Huang G, et al. Testosterone dose-response relationships in hysterectomized women with or without oophorectomy: effects on sexual function, body composition, muscle performance and physical function in a randomized trial. Menopause. 2014;21(6):612–623. doi: 10.1097/GME.0000000000000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tapper J, et al. The effects of testosterone administration on muscle areas of the trunk and pelvic floor in hysterectomized women with low testosterone levels: proof-of-concept study. Menopause. 2019;26(12):1405–1414. doi: 10.1097/GME.0000000000001410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Davis SR, et al. Testosterone enhances estradiol’s effects on postmenopausal bone density and sexuality. Maturitas. 1995;21(3):227–236. doi: 10.1016/0378-5122(94)00898-H. [DOI] [PubMed] [Google Scholar]
- 67.Miller BE, et al. Sublingual administration of micronized estradiol and progesterone, with and without micronized testosterone: effect on biochemical markers of bone metabolism and bone mineral density. Menopause. 2000;7(5):318–326. doi: 10.1097/00042192-200007050-00006. [DOI] [PubMed] [Google Scholar]
- 68.Miller KK, et al. Effects of testosterone therapy on cardiovascular risk markers in androgen-deficient women with hypopituitarism. J Clin Endocrinol Metab. 2007;92(7):2474–2479. doi: 10.1210/jc.2007-0195. [DOI] [PubMed] [Google Scholar]
- 69.Kocoska-Maras L, et al. Testosterone addition to estrogen therapy - effects on inflammatory markers for cardiovascular disease. Gynecol Endocrinol. 2009;25(12):823–827. doi: 10.3109/09513590903056134. [DOI] [PubMed] [Google Scholar]
- 70.Chiuve SE, et al. Effect of the combination of methyltestosterone and esterified estrogens compared with esterified estrogens alone on apolipoprotein CIII and other apolipoproteins in very low density, low density, and high density lipoproteins in surgically postmenopausal women. J Clin Endocrinol Metab. 2004;89(5):2207–2213. doi: 10.1210/jc.2003-031564. [DOI] [PubMed] [Google Scholar]
- 71.Määttä JA, et al. Inactivation of the androgen receptor in bone-forming cells leads to trabecular bone loss in adult female mice. Bonekey Rep. 2013;2:440. doi: 10.1038/bonekey.2013.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hirschberg AL. Hyperandrogenism and cardiometabolic risk in pre- and postmenopausal women-what is the evidence? J Clin Endocrinol Metab. 2023;109(5):1202–1213. doi: 10.1210/clinem/dgad590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hak AE, et al. High-dose testosterone is associated with atherosclerosis in postmenopausal women. Maturitas. 2007;56(2):153–160. doi: 10.1016/j.maturitas.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 74.Sievers C, et al. Low testosterone levels predict all-cause mortality and cardiovascular events in women: a prospective cohort study in German primary care patients. Eur J Endocrinol. 2010;163(4):699–708. doi: 10.1530/EJE-10-0307. [DOI] [PubMed] [Google Scholar]
- 75.Bernini GP, et al. Endogenous androgens and carotid intimal-medial thickness in women. J Clin Endocrinol Metab. 1999;84(6):2008–2012. doi: 10.1210/jcem.84.6.5824. [DOI] [PubMed] [Google Scholar]
- 76.Golden SH, et al. Endogenous postmenopausal hormones and carotid atherosclerosis: a case-control study of the atherosclerosis risk in communities cohort. Am J Epidemiol. 2002;155(5):437–445. doi: 10.1093/aje/155.5.437. [DOI] [PubMed] [Google Scholar]
- 77.Montalcini T, et al. Role of endogenous androgens on carotid atherosclerosis in non-obese postmenopausal women. Nutr Metab Cardiovasc Dis. 2007;17(10):705–711. doi: 10.1016/j.numecd.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 78.Kaczmarek A, et al. The association of lower testosterone level with coronary artery disease in postmenopausal women. Int J Cardiol. 2003;87(1):53–57. doi: 10.1016/S0167-5273(02)00203-6. [DOI] [PubMed] [Google Scholar]
- 79.Mendivil CO, et al. Low-density lipoproteins containing apolipoprotein C-III and the risk of coronary heart disease. Circulation. 2011;124(19):2065–2072. doi: 10.1161/CIRCULATIONAHA.111.056986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Worboys S, et al. Evidence that parenteral testosterone therapy may improve endothelium-dependent and -independent vasodilation in postmenopausal women already receiving estrogen. J Clin Endocrinol Metab. 2001;86(1):158–161. doi: 10.1210/jcem.86.1.7103. [DOI] [PubMed] [Google Scholar]
- 81.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362(6423):801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 82.McCrohon JA, et al. Androgen receptor expression is greater in macrophages from male than from female donors. A sex difference with implications for atherogenesis. Circulation. 2000;101(3):224–226. doi: 10.1161/01.CIR.101.3.224. [DOI] [PubMed] [Google Scholar]
- 83.Ng MK, et al. Androgens up-regulate atherosclerosis-related genes in macrophages from males but not females: molecular insights into gender differences in atherosclerosis. J Am Coll Cardiol. 2003;42(7):1306–1313. doi: 10.1016/j.jacc.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 84.von Dehn G, et al. Atherosclerosis in apolipoprotein E-deficient mice is decreased by the suppression of endogenous sex hormones. Horm Metab Res. 2001;33(2):110–114. doi: 10.1055/s-2001-12405. [DOI] [PubMed] [Google Scholar]
- 85.Fagman JB, et al. The androgen receptor confers protection against diet-induced atherosclerosis, obesity, and dyslipidemia in female mice. FASEB J. 2015;29(4):1540–1550. doi: 10.1096/fj.14-259234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Smith EP, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. 1994;331(16):1056–1061. doi: 10.1056/NEJM199410203311604. [DOI] [PubMed] [Google Scholar]
- 87.Carani C, et al. Effect of testosterone and estradiol in a man with aromatase deficiency. N Engl J Med. 1997;337(2):91–95. doi: 10.1056/NEJM199707103370204. [DOI] [PubMed] [Google Scholar]
- 88.Falahati-Nini A, et al. Relative contributions of testosterone and estrogen in regulating bone resorption and formation in normal elderly men. J Clin Invest. 2000;106(12):1553–1560. doi: 10.1172/JCI10942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Navarro G, et al. The role of androgens in metabolism, obesity, and diabetes in males and females. Obesity (Silver Spring) 2015;23(4):713–719. doi: 10.1002/oby.21033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Moverare-Skrtic S, et al. Dihydrotestosterone treatment results in obesity and altered lipid metabolism in orchidectomized mice. Obesity (Silver Spring) 2006;14(4):662–672. doi: 10.1038/oby.2006.75. [DOI] [PubMed] [Google Scholar]
- 91.Jones ME, et al. Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci U S A. 2000;97(23):12735–12740. doi: 10.1073/pnas.97.23.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zitzmann M, et al. The CAG repeat polymorphism in the androgen receptor gene modulates body fat mass and serum concentrations of leptin and insulin in men. Diabetologia. 2003;46(1):31–39. doi: 10.1007/s00125-002-0980-9. [DOI] [PubMed] [Google Scholar]
- 93.Fan W, et al. Androgen receptor null male mice develop late-onset obesity caused by decreased energy expenditure and lipolytic activity but show normal insulin sensitivity with high adiponectin secretion. Diabetes. 2005;54(4):1000–1008. doi: 10.2337/diabetes.54.4.1000. [DOI] [PubMed] [Google Scholar]
- 94.Lin HY, et al. Insulin and leptin resistance with hyperleptinemia in mice lacking androgen receptor. Diabetes. 2005;54(6):1717–1725. doi: 10.2337/diabetes.54.6.1717. [DOI] [PubMed] [Google Scholar]
- 95.Fernando SM, et al. Myocyte androgen receptors increase metabolic rate and improve body composition by reducing fat mass. Endocrinology. 2010;151(7):3125–3132. doi: 10.1210/en.2010-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yu IC, et al. Hyperleptinemia without obesity in male mice lacking androgen receptor in adipose tissue. Endocrinology. 2008;149(5):2361–2368. doi: 10.1210/en.2007-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Keating NL, et al. Diabetes and cardiovascular disease during androgen deprivation therapy for prostate cancer. J Clin Oncol. 2006;24(27):4448–4456. doi: 10.1200/JCO.2006.06.2497. [DOI] [PubMed] [Google Scholar]
- 98.Keating NL, et al. Diabetes and cardiovascular disease during androgen deprivation therapy: observational study of veterans with prostate cancer. J Natl Cancer Inst. 2010;102(1):39–46. doi: 10.1093/jnci/djp404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wittert G, et al. Testosterone treatment to prevent or revert type 2 diabetes in men enrolled in a lifestyle programme (T4DM): a randomised, double-blind, placebo-controlled, 2-year, phase 3b trial. Lancet Diabetes Endocrinol. 2021;9(1):32–45. doi: 10.1016/S2213-8587(20)30367-3. [DOI] [PubMed] [Google Scholar]
- 100.Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 101.Pitteloud N, et al. Relationship between testosterone levels, insulin sensitivity, and mitochondrial function in men. Diabetes Care. 2005;28(7):1636–1642. doi: 10.2337/diacare.28.7.1636. [DOI] [PubMed] [Google Scholar]
- 102.Maher AC, et al. Men supplemented with 17beta-estradiol have increased beta-oxidation capacity in skeletal muscle. Physiol Genomics. 2010;42(3):342–347. doi: 10.1152/physiolgenomics.00016.2010. [DOI] [PubMed] [Google Scholar]
- 103.Upreti R, et al. 5α-reductase type 1 modulates insulin sensitivity in men. J Clin Endocrinol Metab. 2014;99(8):E1397–E1406. doi: 10.1210/jc.2014-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hazlehurst JM, et al. Dual-5α-reductase inhibition promotes hepatic lipid accumulation in man. J Clin Endocrinol Metab. 2016;101(1):103–113. doi: 10.1210/jc.2015-2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bhasin S, et al. Effect of testosterone supplementation with and without a dual 5α-reductase inhibitor on fat-free mass in men with suppressed testosterone production: a randomized controlled trial. JAMA. 2012;307(9):931–939. doi: 10.1001/jama.2012.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ghaibour K, et al. Androgen receptor coordinates muscle metabolic and contractile functions. J Cachexia Sarcopenia Muscle. 2023;14(4):1707–1720. doi: 10.1002/jcsm.13251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Haren MT, et al. Testosterone modulates gene expression pathways regulating nutrient accumulation, glucose metabolism and protein turnover in mouse skeletal muscle. Int J Androl. 2011;34(1):55–68. doi: 10.1111/j.1365-2605.2010.01061.x. [DOI] [PubMed] [Google Scholar]
- 108.Xu W, et al. Architecture of androgen receptor pathways amplifying glucagon-like peptide-1 insulinotropic action in male pancreatic β cells. Cell Rep. 2023;42(5):112529. doi: 10.1016/j.celrep.2023.112529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hamadeh MJ, et al. Estrogen supplementation reduces whole body leucine and carbohydrate oxidation and increases lipid oxidation in men during endurance exercise. J Clin Endocrinol Metab. 2005;90(6):3592–3599. doi: 10.1210/jc.2004-1743. [DOI] [PubMed] [Google Scholar]
- 110.Salehzadeh F, et al. Testosterone or 17{beta}-estradiol exposure reveals sex-specific effects on glucose and lipid metabolism in human myotubes. J Endocrinol. 2011;210(2):219–229. doi: 10.1530/JOE-10-0497. [DOI] [PubMed] [Google Scholar]
- 111.Deischinger C, et al. Effects of gender-affirming hormone therapy on cardiovascular risk factors focusing on glucose metabolism in an Austrian transgender cohort. Int J Transgend Health. 2023;24(4):499–509. doi: 10.1080/26895269.2022.2123425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Navarro G, et al. Extranuclear actions of the androgen receptor enhance glucose-stimulated insulin secretion in the male. Cell Metab. 2016;23(5):837–851. doi: 10.1016/j.cmet.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xu W, et al. Intracrine testosterone activation in human pancreatic β cells stimulates insulin secretion. Diabetes. 2020;69(11):2392–2399. doi: 10.2337/db20-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Xu W, et al. Emerging role of testosterone in pancreatic β-cell function and insulin secretion [posted on January 1, 2019]. J Endocrinol. [DOI] [PMC free article] [PubMed]
- 115.Le May C, et al. Estrogens protect pancreatic beta-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc Natl Acad Sci U S A. 2006;103(24):9232–9237. doi: 10.1073/pnas.0602956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Webb CM, et al. Effects of testosterone on coronary vasomotor regulation in men with coronary heart disease. Circulation. 1999;100(16):1690–1696. doi: 10.1161/01.CIR.100.16.1690. [DOI] [PubMed] [Google Scholar]
- 117.Zitzmann M, Nieschlag E. Androgen receptor gene CAG repeat length and body mass index modulate the safety of long-term intramuscular testosterone undecanoate therapy in hypogonadal men. J Clin Endocrinol Metab. 2007;92(10):3844–3853. doi: 10.1210/jc.2007-0620. [DOI] [PubMed] [Google Scholar]
- 118.Yeap BB, et al. Lower testosterone levels predict incident stroke and transient ischemic attack in older men. J Clin Endocrinol Metab. 2009;94(7):2353–2359. doi: 10.1210/jc.2008-2416. [DOI] [PubMed] [Google Scholar]
- 119.Soisson V, et al. A J-shaped association between plasma testosterone and risk of ischemic arterial event in elderly men: the French 3C cohort study. Maturitas. 2013;75(3):282–288. doi: 10.1016/j.maturitas.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 120.Araujo AB, et al. Clinical review: Endogenous testosterone and mortality in men: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2011;96(10):3007–3019. doi: 10.1210/jc.2011-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dockery F, et al. Testosterone suppression in men with prostate cancer leads to an increase in arterial stiffness and hyperinsulinaemia. Clin Sci (Lond) 2003;104(2):195–201. doi: 10.1042/cs1040195. [DOI] [PubMed] [Google Scholar]
- 122.Smith JC, et al. The effects of induced hypogonadism on arterial stiffness, body composition, and metabolic parameters in males with prostate cancer. J Clin Endocrinol Metab. 2001;86(9):4261–4267. doi: 10.1210/jcem.86.9.7851. [DOI] [PubMed] [Google Scholar]
- 123.Sharma R, et al. Normalization of testosterone level is associated with reduced incidence of myocardial infarction and mortality in men. Eur Heart J. 2015;36(40):2706–2715. doi: 10.1093/eurheartj/ehv346. [DOI] [PubMed] [Google Scholar]
- 124.Lincoff AM, et al. Cardiovascular safety of testosterone-replacement therapy. N Engl J Med. 2023;389(2):107–117. doi: 10.1056/NEJMoa2215025. [DOI] [PubMed] [Google Scholar]
- 125.Lim GB. Testosterone-replacement therapy does not increase cardiac events in men with hypogonadism. Nat Rev Cardiol. 2023;20(9):581. doi: 10.1038/s41569-023-00909-8. [DOI] [PubMed] [Google Scholar]
- 126.Sudhir K, et al. Endothelial dysfunction in a man with disruptive mutation in oestrogen-receptor gene. Lancet. 1997;349(9059):1146–1147. doi: 10.1016/S0140-6736(05)63022-X. [DOI] [PubMed] [Google Scholar]
- 127.Sudhir K, et al. Premature coronary artery disease associated with a disruptive mutation in the estrogen receptor gene in a man. Circulation. 1997;96(10):3774–3777. doi: 10.1161/01.CIR.96.10.3774. [DOI] [PubMed] [Google Scholar]
- 128.Saltiki K, et al. Endogenous estrogen levels are associated with endothelial function in males independently of lipid levels. Endocrine. 2010;37(2):329–335. doi: 10.1007/s12020-010-9307-7. [DOI] [PubMed] [Google Scholar]
- 129.Lew R, et al. Endogenous estrogens influence endothelial function in young men. Circ Res. 2003;93(11):1127–1133. doi: 10.1161/01.RES.0000103633.57225.BC. [DOI] [PubMed] [Google Scholar]
- 130.The Coronary Drug Project. The Coronary Drug Project. Findings leading to discontinuation of the 2.5-mg day estrogen group. The coronary Drug Project Research Group. JAMA. 1973;226(6):652–657. doi: 10.1001/jama.1973.03230060030009. [DOI] [PubMed] [Google Scholar]
- 131.Byar DP, Corle DK. Hormone therapy for prostate cancer: results of the Veterans Administration Cooperative Urological Research Group studies. NCI Monogr. 1988;(7):165–170. [PubMed] [Google Scholar]
- 132.Gooren LJ, et al. Cardiovascular disease in transsexual persons treated with cross-sex hormones: reversal of the traditional sex difference in cardiovascular disease pattern. Eur J Endocrinol. 2014;170(6):809–819. doi: 10.1530/EJE-14-0011. [DOI] [PubMed] [Google Scholar]
- 133.New G, et al. Long-term estrogen therapy improves vascular function in male to female transsexuals. J Am Coll Cardiol. 1997;29(7):1437–1444. doi: 10.1016/S0735-1097(97)00080-6. [DOI] [PubMed] [Google Scholar]
- 134.McCrohon JA, et al. Arterial reactivity is enhanced in genetic males taking high dose estrogens. J Am Coll Cardiol. 1997;29(7):1432–1436. doi: 10.1016/S0735-1097(97)00063-6. [DOI] [PubMed] [Google Scholar]
- 135.New G, et al. Long-term oestrogen therapy is associated with improved endothelium-dependent vasodilation in the forearm resistance circulation of biological males. Clin Exp Pharmacol Physiol. 2000;27(1-2):25–33. doi: 10.1046/j.1440-1681.2000.03195.x. [DOI] [PubMed] [Google Scholar]
- 136.Komesaroff PA, et al. Low-dose estrogen supplementation improves vascular function in hypogonadal men. Hypertension. 2001;38(5):1011–1016. doi: 10.1161/hy1101.095006. [DOI] [PubMed] [Google Scholar]
- 137.Chester AH, et al. Oestrogen relaxes human epicardial coronary arteries through non-endothelium-dependent mechanisms. Coron Artery Dis. 1995;6(5):417–422. doi: 10.1097/00019501-199505000-00009. [DOI] [PubMed] [Google Scholar]
- 138.Rubanyi GM, et al. Vascular estrogen receptors and endothelium-derived nitric oxide production in the mouse aorta. Gender difference and effect of estrogen receptor gene disruption. J Clin Invest. 1997;99(10):2429–2437. doi: 10.1172/JCI119426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.van Koeverden ID, et al. Testosterone to oestradiol ratio reflects systemic and plaque inflammation and predicts future cardiovascular events in men with severe atherosclerosis. Cardiovasc Res. 2019;115(2):453–462. doi: 10.1093/cvr/cvy188. [DOI] [PubMed] [Google Scholar]
- 140.Naessen T, et al. Higher endogenous estrogen levels in 70-year-old women and men: an endogenous response to counteract developing atherosclerosis? Menopause. 2012;19(12):1322–1328. doi: 10.1097/gme.0b013e31825ea8c1. [DOI] [PubMed] [Google Scholar]
- 141.Naessen T, et al. Endogenous steroids measured by high-specificity liquid chromatography-tandem mass spectrometry and prevalent cardiovascular disease in 70-year-old men and women. J Clin Endocrinol Metab. 2010;95(4):1889–1897. doi: 10.1210/jc.2009-1722. [DOI] [PubMed] [Google Scholar]
- 142.Streed CG, Jr, et al. Assessing and addressing cardiovascular health in people who are transgender and gender diverse: a scientific statement from the American Heart Association. Circulation. 2021;144(6):e136. doi: 10.1161/CIR.0000000000001003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Whitsel EA, et al. Intramuscular testosterone esters and plasma lipids in hypogonadal men: a meta-analysis. Am J Med. 2001;111(4):261–269. doi: 10.1016/S0002-9343(01)00833-6. [DOI] [PubMed] [Google Scholar]
- 144.Rohatgi A, et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371(25):2383–2393. doi: 10.1056/NEJMoa1409065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Guo W, et al. Effect of selective androgen receptor modulator on cholesterol efflux capacity, size, and subspecies of HDL particles. J Endocr Soc. 2022;6(8):bvac099. doi: 10.1210/jendso/bvac099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bilezikian JP, et al. Increased bone mass as a result of estrogen therapy in a man with aromatase deficiency. N Engl J Med. 1998;339(9):599–603. doi: 10.1056/NEJM199808273390905. [DOI] [PubMed] [Google Scholar]
- 147.Giri S, et al. Oral estrogen improves serum lipids, homocysteine and fibrinolysis in elderly men. Atherosclerosis. 1998;137(2):359–366. doi: 10.1016/S0021-9150(98)00022-7. [DOI] [PubMed] [Google Scholar]
- 148.Huang AJ, Grady D. Menopausal hormone therapy for prevention of chronic conditions: when is enough, enough? JAMA. 2022;328(17):1712–1713. doi: 10.1001/jama.2022.19098. [DOI] [PubMed] [Google Scholar]
- 149.Pickar JH. Independent conclusions? Climacteric. 2023;26(2):73–74. doi: 10.1080/13697137.2022.2159168. [DOI] [PubMed] [Google Scholar]
- 150.Glynne S, et al. Hormone therapy for the prevention of chronic conditions in postmenopausal persons. JAMA. 2023;329(11):940–941. doi: 10.1001/jama.2023.0180. [DOI] [PubMed] [Google Scholar]
- 151.Chlebowski RT, Aragaki AK. Hormone therapy for the prevention of chronic conditions in postmenopausal persons. JAMA. 2023;329(11):942. doi: 10.1001/jama.2023.0186. [DOI] [PubMed] [Google Scholar]
- 152.Nachtigall M, et al. Hormone therapy for the prevention of chronic conditions in postmenopausal persons. JAMA. 2023;329(11):940. doi: 10.1001/jama.2023.0183. [DOI] [PubMed] [Google Scholar]
- 153.Gartlehner G, Kahwati L. Hormone therapy for the prevention of chronic conditions in postmenopausal persons-reply. JAMA. 2023;329(11):943. doi: 10.1001/jama.2023.0192. [DOI] [PubMed] [Google Scholar]
- 154.Anderson GL. Hormone therapy for the prevention of chronic conditions in postmenopausal persons. JAMA. 2023;329(11):941. doi: 10.1001/jama.2023.0189. [DOI] [PubMed] [Google Scholar]
- 155.Morgentaler A, et al. Recognizing the true value of testosterone therapy in health care. Androg Clin Res Ther. 2022;3(1):217–223. doi: 10.1089/andro.2022.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Centers for Disease Control and Prevention. Laboratory Data - Continuous NHANES. https://wwwn.cdc.gov/Nchs/Nhanes/Search/DataPage.aspx?Component=Laboratory&Cycle=2013-4 Updated June 2024. Accessed July 16, 2024.
- 157. Centers for Disease Control and Prevention (CDC), and National Center for Health Statistics (NCHS). Hyattsville, MD: U.S. Department of Health and Human Services; 2015-16: https://wwwn.cdc.gov/Nchs/Nhanes/Search/DataPage.aspx?Component=Laboratory&Cycle=2015-6.