

Abstract
Ca2+ is a highly abundant ion involved in numerous biological processes, particularly in multicellular eukaryotic organisms where it exerts many of these functions through interactions with Ca2+ binding proteins. The laminin N-terminal (LN) domain is found in members of the laminin and netrin protein families where it plays a critical role in the function of these proteins. The LN domain of laminins and netrins is a Ca2+ binding domain and in many cases requires Ca2+ to perform its biological function. Here, we conduct a detailed examination of the molecular basis of the LN domain Ca2+ interaction combining structural, computational, bioinformatics, and biophysical techniques. By combining computational and bioinformatic techniques with x-ray crystallography we explore the molecular basis of the LN domain Ca2+ interaction and identify a conserved sequence present in Ca2+ binding LN domains. These findings enable a sequence-based prediction of LN domain Ca2+ binding ability. We use thermal shift assays and isothermal titration calorimetry to explore the biophysical properties of the LN domain Ca2+ interaction. We show that the netrin-1 LN domain exhibits a high affinity and specificity for Ca2+, which structurally stabilizes the LN domain. This study elucidates the molecular foundation of the LN domain Ca2+ binding interaction and provides a detailed functional characterization of this essential interaction, advancing our understanding of protein-Ca2+ dynamics within the context of the LN domain.
Graphical abstract
Significance
The Ca2+ binding laminin N-terminal (LN) domain is a protein domain found in members of the netrin and laminin protein families. The LN domain enables fundamental biological processes such as neurogenesis and formation of the cell basement membrane to occur. While it is known that LN domain Ca2+ binding is required for these biological process to occur, the biophysical understanding of the LN domain-Ca2+ interaction is limited. In this work we demonstrate the molecular basis of the LN domain Ca2+ interaction and characterize its biophysical properties. This work greatly expands the understanding of the role Ca2+ has in enabling the function of this important protein domain.
Introduction
Ca2+ is a critical element in biology, where it serves a variety of structural, functional, and regulatory roles. Intracellular Ca2+ is a highly versatile second messenger playing roles in various processes from muscle contraction to signal transduction (1,2). In the extracellular space, Ca2+ is also known to play a critical role in diverse cellular processes such as cell signaling, adhesion, and blood coagulation (3,4,5,6,7,8). In all cases, Ca2+ exerts these effects through interactions with Ca2+ binding proteins. The extracellular matrix contains many Ca2+ binding proteins, some of which require Ca2+ for fixed roles such as structural stabilization, while others require Ca2+ for dynamic roles such as cofactors in catalysis and receptor binding (9,10,11,12,13).
Laminin and related netrin protein families are extracellular Ca2+ binding glycoproteins that serve structural, functional, and regulatory roles (14,15,16). Laminins are a major component of cell basement membranes, where they polymerize into a structural network of heterotrimers composed of the N-terminal short arms of α, β, and γ laminin subunits. Laminin polymerization occurs through Ca2+-dependent interactions between the laminin N-terminal (LN) domains of the α, β, and γ subunits (17). The laminin subunit-γ (Lam-γ) LN domain is responsible for this Ca2+-dependent polymerization and is the only laminin subunit LN domain known to bind Ca2+ (18). Members of the netrin family arose from the laminin family by gene duplication, domain shuffling, and domain loss and, like the laminins, they contain the LN domain (19,20). The netrin family is primarily involved in development of the nervous system through axon guidance and neurite outgrowth (21,22,23,24,25,26). Like Lam-γ, the LN domains of netrin-1, netrin-4, netrin-G1, and netrin-G2 are involved in receptor binding and bind Ca2+ in a similar manner to the Lam-γ LN domain (27,28,29,30).
While the role of LN domain Ca2+ binding has been studied from a structural and biochemical perspective for some laminins, the role of Ca2+ with respect to the netrin LN domain has been under-researched, despite the availability of structural information for members of the netrin family. In this work we use netrin-1 (Net-1) as a model protein due to the broad availability of structural and functional information. By combining molecular dynamics (MD) simulations and bioinformatics analyses with x-ray crystallography we identify the molecular basis of the LN domain Ca2+ interaction, including the requirement of an uncommon bidentate coordination of Ca2+ by a conserved threonine. The structural analysis is combined with a biophysical characterization of the Net-1 LN domain Ca2+ binding interaction. Using isothermal titration calorimetry (ITC) and a differential scanning fluorimetry (DSF)-based thermal shift assay (TSA) we explore the affinity, stoichiometry, and specificity of the Net-1-Ca2+ interaction that demonstrates the role of Ca2+ in the stabilization of Net-1 and Lam-γ. Altogether, this study expands the structural and functional understanding of the role of Ca2+ in an important extracellular protein domain.
Methods
Protein expression and purification
Mus musculus netrin-1 consisting of domains LN to LE3 (NCBI reference sequence: NP_032770, amino acids 24–457) used for crystallography was cloned, expressed, and purified as published previously (31). Gallus netrin-1 domains LN to LE3 (NCBI reference sequence: NP_990750, amino acids 26–458), and laminin subunit-γ1 (NCBI reference sequence: XP_040561204.1, amino acids 27–391) used for biophysical studies was cloned, expressed, and purified using previously described methods (32,33).
Net-1-Sm3+ complex crystallization
Net-1 domains LN to LE3 from Mus musculus were crystallized in a hanging drop vapor diffusion experiment at 293 K in 0.1 M HEPES (pH 7.7), 2.8 M NaCl, and 0.2 M glycine at a concentration of 10.0 mg/mL. Crystals appeared after 1 month and grew to a final size of 0.1–0.5 mm after 3 months. Crystals were soaked with samarium(III) acetate hydrate (Hampton Research, Aliso Viejo, California) and diffracted on the X06SA beamline at the Swiss Light Source with a wavelength of 1.0349 Å in 1.0° wedges at 100 K. The diffraction images were processed with XDS and the CCP4 package to final P3221 space group (34,35). Phases were calculated in Phaser using a Net-1 search model (PDB: 4OVE) (31,36). The structure was built and refined crystallographically using the Coot suite and Phenix software packages (37,38). The crystallographic data table (Table S1) is available in the supporting material. The netrin-1-Sm3+ complex crystallographic data set and model (PDB: 8SNP) are available on the RCSB Protein Data Bank (https://www.rcsb.org/).
MD simulations of apo- and holo-Net-1
MD simulations for Net-1 with and without a Ca2+ ion were performed using the GROMACS MD simulation package (39) with the CHARMM36 force field parameters (40) and an SPC/E water model (41). For both trajectories, Na+ and Cl− ions were added to counter the protein charges to maintain a neutral charge in the overall system. Both systems were subjected to 50,000 steps of steepest decent energy minimization, heated from 0 to 310 K for 100 ps in a canonical ensemble (NVT ensemble), and then equilibrated in an isothermal and isobaric ensemble (NPT ensemble) under a constant pressure of 1.0 bar for 100 ps. The energetically minimized and stable systems were then placed on a 100 ns trajectory with time integration steps of 2 fs. The MD trajectory used a particle mesh Ewald method with a short-range columbic interaction force cut-off at 12 Å and was temperature equilibrated at 310 K. The completed trajectory was post processed, which included centering of the protein throughout the simulation and fixing rotational and translational trajectories. Protein stability as part of the overall system was confirmed by a backbone root mean-square deviation and radius of gyration analysis. Further analysis included a molecular mechanics Poisson-Boltzmann surface area (MMPBSA) interaction analysis of the Net-1-Ca2+ complex. The MMPBSA method used the GROMACS and APBS packages to calculate internal energies and decomposed interaction energies between the protein and calcium ligand with the gmx_MMPBSA toolkit (42). Interacting residues were cut off by a physical distance of 6 Å from the calcium ligand within the entire trajectory, after which interaction energies were calculated across the 100 ns trajectory at 100 ps time steps, averaged, and reported with standard deviation. This method was repeated for Net-1 mutants (D110/T118) to generate altered calcium binding loop trajectories. Finally, structural examinations and the generation of a movie covering 10 ns of the trajectory were performed using PyMOL 2.4.1 (Schrödinger, New York, New York). These included removing solvent molecules and averaging residue trajectories to minimize high-frequency vibrations. MD simulation run parameters are listed in the supporting material (Table S2) and additional supporting data are available on the Open Science Framework (10.17605/OSF.IO/C9VQX).
Net-1-Ca2+ binding region bioinformatics
The sequence of netrin-1 from Mus musculus lacking the C-terminal domain (NP_032770, amino acids 24–457) was compared with other species’ sequences through a database similarity search using BLAST (43,44). This generated a multiple sequence alignment file that was further supplemented through the HHblits method and filtered in GREMLIN for overall residue coverage and gaps (45). The filtered multiple sequence alignment file contained 831 netrin-1-related sequences from various species with a minimum sequence coverage of 75% on the target sequence. Furthermore, individual sequences of the Ca2+ binding loops across laminin family proteins were compared based on gene identity (NTN1, NTN3, NTN4, NTNG1, NTNG2, LAMA5, LAMB1, and LAMC1). For each gene, 150 sequences from varying species were aligned using Clustal Omega (46). Lastly, the multiple sequence alignments were analyzed in WebLogo3 for residue position probabilities to produce sequence logos (47).
Preparation of apo-protein and Ca2+ free buffer
Chelex-100 molecular biology grade resin, 200–400 mesh (Bio-Rad Laboratories, Hercules, California), packed into a 1 cm diameter by 11 cm length cylindrical chromatography column was used to remove Ca2+ from proteins and buffers. The column was equilibrated in a two-step process, starting with 0.5 M HEPES (pH 7.3) until the eluent reached a pH of 7.3, next the column was equilibrated with 20 mM HEPES (pH 7.3) and 150 mM NaCl until the eluent reached a stable conductivity. One milliliter of protein at a concentration of 1 mg/mL was injected on the column and the flow rate was set to 0.1 mL/min. Fractions containing protein were pooled and concentrated, fractions not containing protein were used as buffer in ITC and TSA experiments.
DSF-based TSAs
All DSF-based TSA experiments measuring intrinsic protein fluorescence were performed on a Prometheus Panta (NanoTemper Technologies, Munich, Germany), using standard Prometheus capillaries (PR-C002), in Ca2+ free 20 mM HEPES (pH 7.3) and 150 mM NaCl buffer. Protein melts were performed from 25°C until complete unfolding of the protein, using a temperature gradient of 1°C/min. Protein melting was measured by monitoring the change in 350/330 nm fluorescence ratio. Data analysis and Tm fitting was done with the Panta analysis software, version 1.2. To assess the complete removal of Ca2+ from proteins and to measure the Tm of the apo-protein, the protein melt before and after Ca2+ removal was recorded. To verify the complete removal of Ca2+ from the Chelex-100-treated protein, between 1.6 and 50 mM EDTA was added to the treated protein and the resulting Tm was recorded. To assess the Tm of the holo-protein under physiological Ca2+ concentrations, 1.4 mM CaCl2 was added to the apo-protein. To detect binding of divalent cations, the Tm of 10 μM apo-Net-1 incubated with 1 and 10 mM of the chloride salt (except for Pb2+, for which the acetate salt was used) of each cation was measured. Cations that showed a measurable dose-dependent increase in Tm were tested further for binding affinity. To measure cation binding affinity by TSA, the Tm of 29 μM apo Net-1 or apo Lam-γ was incubated with a 1/3 dilution series of the chloride salt of the divalent cation ranging from 5.3 mM to 10 nM. Data fitting and analysis was performed using FoldAffinity available from the eSPC online data analysis platform (48,49,50). All binding affinity experiments were performed in triplicate, KD values closest to the Tm of the apo-protein and with the smallest confidence interval are reported.
ITC
ITC was performed on a MicroCal iTC 200 titration calorimeter (Malvern Panalytical, Malvern, United Kingdom) in Ca2+ free 20 mM HEPES (pH 7.3) and 150 mM NaCl buffer. apo-Net-1 (29.9 μM) was loaded into the measurement cell and the reference cell was filled with water. The experiment was performed at 25°C with a mixing speed of 600 rpm, and a reference power of 6 μcal/s in the high gain mode. The sample was titrated with 250 μM CaCl2, with the first injection of 0.4 μL over 0.8 s, followed by 19 injections of 2 μL over 4 s, and 200 s between injections. A reference experiment was performed under the same conditions, except that the measurement cell was filled with buffer rather than apo-Net-1. Data analysis, reference subtraction, and fitting to a one set of sites binding model was performed in the MicroCal supplied Origin 7.0 software package (OriginLab, Northampton, Massachusetts). Fitting values are reported as the mean ± 1 standard deviation of three replicate experiments.
Results
Structural examination of Net-1 and its Ca2+ binding site
In our investigation to understand the structural features of Net-1 and the binding of Ca2+ at the LN domain Ca2+ binding loop, we collected structural data through x-ray diffraction experiments. For this, a construct of Net-1 from Mus musculus consisting of the LN and the three LE domains was designed and crystallized. This construct is able to bind the Net-1 receptors, DCC, NEO, and UNC5B, while being more amenable to crystallization (27,31,51). The crystals were then soaked with samarium(III) acetate to produce the Net-1-Sm3+ complex. Sm3+ is an electron-dense heavy atom used to bind at and displace Ca2+ from Ca2+ binding sites without altering the general protein structure (52). Moreover, the high electron density of Sm3+ allows for the detection of potentially low-occupied Ca2+ binding sites, making it an ideal choice for our structural examinations. The resulting structure was determined to have a resolution of 3.4 Å (Fig. 1). As observed previously, the resulting structure contains N-glycans on residues N95, N131, and N417, but not on N116 of the Ca2+ biding loop (31,53). In addition, a Net-1 dimer can be observed as a symmetry mate where various residue side chains stabilize the interaction. The general structure of this construct is characteristic of other Net-1 structures across various species with slight variations in multiple loop regions on the LN domain (27,31,54).
Figure 1.

Structural examination of Net-1 in complex with metal ions. (A) X-ray diffraction structure model of Net-1 in complex with Sm3+ ions. Three Sm3+ ions are coordinated through crystallographic contacts (indicated with asterisk ∗), while one Sm3+ ion is bound to the Ca2+ binding loop of Net-1 (PDB: 8SNP). (B) Amino acid metal coordination of Sm3+ in the Ca2+ binding loop of Net-1. The ion is coordinated by side chain and backbone functional groups of the residues F107, D110, N112, T118, and S277. The electron density level of the 2Fo-Fc map is set to 0.522 e/Å3 (8 sigma contour level). (C) Ca2+ coordination in the Ca2+ binding loop of Net-1 (PDB: 4OVE) (31) with the indirect coordination of Ca2+ by D278 through a hydrogen bonded water molecule. To see this figure in color, go online.
The crystal structure reveals four Sm3+ binding sites on Net-1 (Fig. 1 A). Three of the four Sm3+ ions have only a few coordinating ligands from Net-1 and rely on coordinating residues of Net-1 symmetry mates. For these reasons we conclude that these three Sm3+ ions are crystallographic artifacts rather than true Ca2+ binding sites.
The fourth Sm3+ ion is bound to the Net-1 LN domain at the equivalent Ca2+ binding location of other netrin and laminin structures (Fig. 1 C) (28,29,30,31,55). This LN domain Ca2+ binding site consists of a short helix-loop segment packed against the edge of β strand 8 of the LN domain β sandwich. Most of the Sm3+ coordinating ligands come from the helix-loop segment, while a backbone carbonyl ligand from the β strand links the helix-loop segment to the β sandwich core through the Sm3+ ion. The Sm3+ is directly coordinated by the side chains of D110, N112, and T118 and backbone carbonyl oxygens from T118, F207, and S277. The side chain of D278 is oriented in a position to coordinate the Sm3+ through a hydrogen bonded water molecule; however, this water is not observable in the 2Fo-Fc map due to the large overlapping signal from the Sm3+ ion and therefore was not added to the model (Fig. 1 B). The coordination geometry of Sm3+ is identical to the Ca2+ coordination geometry of the previously published mouse Net-1 crystal structure (PDB: 4OVE) (31), which includes a water molecule coordinating Ca2+ through D278 (Fig. 1 C).
Biophysical examination of the Net-1-Ca2+ interaction
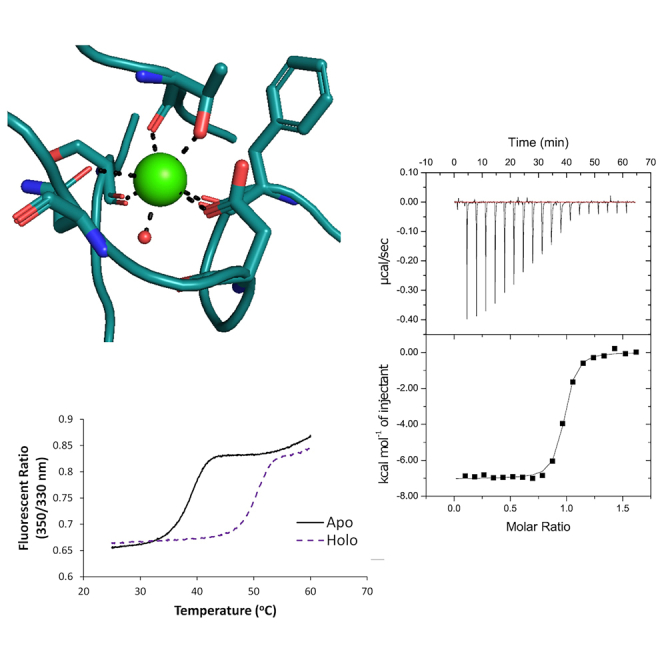
To evaluate the biophysical properties of the Net-1-Ca2+ binding interaction we began by characterizing the thermal stability of apo- and holo-Net-1 using a DSF-based TSA. The principle behind this assay is that holo-Net-1 will exhibit a higher melting point (Tm) compared with apo-Net-1, due to the stabilizing effects of Ca2+ binding. Measuring the Net-1 Tm in the presence and absence of Ca2+ can therefore be used to detect Ca2+ binding (48,49). Ca2+ free buffer and apo-Net-1 were prepared using Chelex-100 cation exchange resin. The Tm of Net-1 after treatment with the cation exchange resin is 5.4°C lower than the untreated Net-1, and the addition of EDTA up to 50 mM does not cause a measurable decrease in Tm of the treated Net-1 (Fig. 2 A). This demonstrates that the cation exchange treatment effectively removes Ca2+ from the buffer and protein to produce apo-Net-1. The same protocol was used to prepare apo-Lam-γ (Fig. 2 B). Next, we used this TSA to measure the thermal stability of holo-Net-1 at a physiological extracellular Ca2+ concentration of 1.4 mM (56). Under these conditions holo-Net-1 is considerably more thermally stable, with a Tm of 50.6°C, compared with the 38.8°C Tm of apo-Net-1 (Fig. 2 A). The same analysis for the Lam-γ reveals a Tm of 52.8°C in the presence of 1.4 mM Ca2+, compared with a Tm of 42.6°C for apo-Lam-γ (Fig. 2 B). These results clearly demonstrate the important role of Ca2+ in stabilizing Net-1 and Lam-γ structures at physiological extracellular Ca2+ concentrations.
Figure 2.

Characterization of the biophysical properties of the LN domain Ca2+ binding site. Thermal shift assay characterization of Net-1 (A) and Lam-γ (B) in 20 mM HEPES (pH 7.3) and 150 mM NaCl. Melt curve of holo-protein at a physiological extracellular Ca2+ concentration of 1.4 mM shown in a purple short dashed line (Net-1 Tm of 50.6°C, Lam-γ Tm of 52.8°C). Melt curve of Chelex-100-treated apo-protein shown in solid black lines and in the presence of 1.6–50 mM EDTA shown in orange dotted lines (Net-1 Tm of 38.8°C, Lam-γ Tm of 42.6°C), demonstrating Chelex-100 treatment results in the complete removal of Ca2+ to produce apo-protein. Melt curve before treatment with Chelex-100 resin (untreated) shown in a gray long-dashed line (Net-1 Tm of 44.2°C, Lam-γ Tm of 48.2°C). Isothermal calorimetric titration of apo-Net-1 with Ca2+ (C). apo-Net-1 (29.9 μM) in 20 mM HEPES (pH 7.3) and 150 mM NaCl was titrated with 250 μM CaCl2 at 25°C. The binding constant (Ka) is measured to be 1.46 ± 0.20 × 107 M−1 with a stoichiometry of 0.942 ± 0.022. The enthalpy change (ΔH) is −7040 ± 200 cal mol−1 and the entropy change (ΔS) is 9.16 ± 0.96 cal mol−1 K−1. ITC values are reported as the mean ± 1 standard deviation of three experimental replicates. To see this figure in color, go online.
We then used the TSA to estimate the affinity of Net-1 for Ca2+ following a previously described method (48,49). The resulting KD of the Net-1-Ca2+ interaction was determined to be 1.36 μM at 42°C, assuming a 1 to 1 binding model (Table 1; Fig. S1). Using this assay we also tested the affinity of the previously characterized Lam-γ-Ca2+ interaction and found the KD of the interaction to be 22.4 μM at 47°C, which is in appropriate agreement with the previously reported value (57) (Table 1; Fig. S1). Finally, we used the TSA to screen a variety of divalent and trivalent cations for their ability to bind Net-1 (Table S3). Of the cations screened besides Ca2+, only Sr2+, Ba2+, and Mg2+ exhibited measurable binding affinities (Table 1). Toxic metals such as Pb2+ and Cd2+ exert their toxic effects, in part by binding certain Ca2+ binding proteins (58,59); however, their binding to Net-1 was not detectable. Finally, Net-1 in the presence of Sm3+ did not produce a sigmoidal transition in the fluorescent signal that could be attributed to protein melting, indicating that Sm3+ denatures Net-1 at the concentrations used in the assay (Table S3). These results show that Net-1 exhibits a high degree of specificity for Ca2+ over other divalent cations, including biologically relevant cations such as Mg2+.
Table 1.
Divalent cation binding affinities for Net-1 and Lam-γ measured by a DSF-based TSA
| Interaction | KD of metal binding at given temperature | Asymmetric 95% CI [lower, upper] | Tm of apo-protein (°C) |
|---|---|---|---|
| Net-1-Ca2+ | 1.36 μM at 42°C | [0.925 μM, 1.94 μM] | 38.8 |
| Net-1-Sr2+ | 25.9 μM at 42°C | [12.7 μM, 53.3 μM] | 38.8 |
| Net-1-Mg2+ | 981 μM at 42°C | [151 μM, 491 mM] | 38.8 |
| Net-1-Ba2+ | 1.03 mM at 42°C | [119 μM, 513 mM] | 38.8 |
| Lam-γ-Ca2+ | 22.4 μM at 47°C | [20.4 μM, 24.6 μM] | 42.6 |
To characterize the thermodynamics of the Net-1-Ca2+ binding interaction we used ITC to measure the enthalpy change (ΔH), entropy change (ΔS), binding constant (Ka), and stoichiometry of the interaction (Fig. 2 C). ITC reveals that Net-1 strongly binds Ca2+ with a binding constant of 1.46 ± 0.20 × 107 M−1 and a stoichiometry of 1. The high affinity of the interaction, coupled with the stoichiometry of 1, indicates that the measured binding interaction is attributed to the canonical Ca2+ binding loop of Net-1. These ITC results also show that the high affinity Net-1-Ca2+ interaction is driven by favorable enthalpic and entropic contributions, assuming buffer effects and proton coupling interactions are negligible or absent (Fig. 2 C). The discrepancy in affinity of the Net-1-Ca2+ interaction measured by ITC and TSA is expected because the concentration of protein used in the TSA is much higher than the KD of the interaction. TSA experiments are known to produce inaccurate results when the KD of the interaction is much lower than the concentration of protein used in the TSA (48,49). We therefore take the affinity measured by ITC to be closest to the true value for the Net-1-Ca2+ interaction. Net-1 titrated with excess Ca2+ did not produce a measurable heat signal, further indicating that the three other Sm3+ binding sites in the Net-1-Sm3+ crystal structure are crystallographic artifacts rather than low-affinity Ca2+ binding sites.
Molecular features of the Net-1-Ca2+ interaction
To elaborate on the biophysical results and gain insights into the molecular basis and dynamic aspects of the Net-1-Ca2+ interaction, MD simulations of holo-Net-1 were performed on the energy minimized Net-1 crystal structure. For this, all Sm3+ ions were removed and a Ca2+ was placed at the location of Sm3+ in the LN domain Ca2+ binding loop. Over the course of a 100 ns MD simulation, the root mean-square deviation and Rg of the Net-1-Ca2+ complex are stable and retain an overall similar structure to the crystal structure (Fig. S2). Throughout the MD simulation, Net-1 coordinates Ca2+ in a pentagonal bipyramidal geometry with seven coordinating ligands including a water molecule that forms hydrogen bonds with D278 and the backbone carbonyl of D110 (Fig. 3 A). The decomposed interaction energies for residues near the LN domain Ca2+ binding loop were determined using the MMPBSA approach (Fig. 3 B). D110, which directly coordinates Ca2+ via its side chain and indirectly through the water hydrogen bonding with its backbone carbonyl, is found to make the greatest energetic contribution to Ca2+ binding. In addition, T118 forms a bidentate coordination with Ca2+ via its side chain and backbone carbonyl to make the second largest energy contribution. The backbone carbonyl of F107, S277, and the side chain of N112 also make considerable energetic contributions to Ca2+ binding. Finally, the side chain of D278 and the carbonyl backbone of L108 make a measurable energetic contribution by hydrogen bonding with the coordinating water ligand. To further evaluate the importance of the T118 and D110 residues, the MMPBSA approach was applied to mutants of Net-1 (Fig. S3). Mutation of threonine 118 to alanine significantly decreased the interaction between Net-1 and Ca2+ throughout the course of the 100 ns simulation, while mutation of aspartate 110 to alanine resulted in a near-zero interaction energy due to the dissociation of Ca2+ from the Ca2+ binding loop (Fig. S3).
Figure 3.

Molecular dynamics simulation analysis of the Ca2+ binding loop of Net-1. (A) Structural model of the Net-1 Ca2+ binding loop bound to Ca2+. Interacting residues and water over the 100 ns molecular dynamics trajectory are shown. (B) MMPBSA residue decomposition analysis of the ligand interaction. D110 is the major contributor to the interaction while L108 is only minimally involved via its carbonyl backbone group. Error bars are based on the standard deviation of the mean energy for each residue. (C) Sequence logo showing conservation of the Ca2+ binding interface for 831 Net-1 sequences from various species. The residue positions that contribute to the Ca2+ binding through their side chain functional groups are highly conserved. To see this figure in color, go online.
We then used a bioinformatics approach to explore the importance of individual residues involved in Ca2+ coordination to explore this binding region in more detail. Generation and analysis of a multiple sequence alignment of 831 Net-1-related sequences from various species with a minimum sequence coverage of 75% of the target sequence indicates a clear pattern of residue conservation (Fig. 3 C). The multiple sequence alignment reveals that the D110 and T118 residues are also the most highly conserved as Ca2+ coordinating residues. N112, despite coordinating Ca2+ via the carbonyl of its amide side chain, is poorly conserved (Fig. 3 C), but is substituted for amino acids containing carboxyl side chains such as glutamate and aspartate, which collectively constitute the majority of residues found at position 112. Amino acids such as F107 and S277, which coordinate through their backbone carbonyl groups, are also found to be poorly conserved. This observation is not unexpected, considering that any amino acid can coordinate Ca2+ through its backbone carbonyl, as long as it does not interfere with protein folding. Finally, D278 is reasonably well conserved and is commonly substituted for other amino acids with carboxyl or amide containing side chains such as glutamate or asparagine, which could also serve to hydrogen bond with the coordinating water. The degree of sequence conservation shown in the MSA correlates strongly with the MMPBSA calculated per residue energy contributions, with highly conserved residues such as D110 and T118 also making the largest energetic contribution to Ca2+ binding.
To expand this analysis of the Net-1 LN domain to other netrin and laminin LN domains, 150 sequences of the LN domain Ca2+ binding loops for each of the NTN1, NTN3, NTN4, NTNG1, NTNG2, LAMA5, LAMB1, and LAMC1 genes were aligned (Fig. S4). These genes were chosen since, apart from NTN3 (netrin-3), crystal structures for the expressed proteins have previously been solved. Most of these proteins are known to bind Ca2+ in their LN domain Ca2+ binding loop except for laminin subunit α-5 and laminin subunit β-1 which do not bind Ca2+ (28,29,30,31,55,60), and netrin-3, which has not been crystallized or tested for Ca2+ binding ability. From these sequence alignments it is apparent that each of the known Ca2+ binding LN domains contain conserved aspartate and threonine residues (equivalent to Net-1 D110 and T118) separated by seven amino acids, while laminin subunit β-1 and α-5 lack these conserved residues (Fig. S4). Examination of the crystal structures of each of these Ca2+ binding LN domains confirms the importance of these conserved residues in coordinating Ca2+ (28,29,30,31,55). All Ca2+ binding LN domains also have a conserved asparagine or glutamate in the helix-loop segment (equivalent to Net-1 N112) except for netrin-4, which has a conserved serine that does not coordinate Ca2+ (28). MMPBSA analysis of the Net-1 Ca2+ binding residues shows that N112 makes the smallest energy contribution of all directly coordinating amino acids. Together this demonstrates that the asparagine or glutamate of the helix-loop segment are less important for Ca2+ binding compared with the conserved aspartate and threonine. All LN domains have a conserved aspartate or asparagine on the β strand involved in Ca2+ coordination (equivalent to Net-1 D278).
Discussion
In this work, we combine with computational, biophysical, and bioinformatic techniques to determine the structural and functional basis of LN domain Ca2+ binding using Net-1 as a model. The Net-1 LN domain Ca2+ binding site is composed of a short helix-loop segment that provides five Ca2+ coordinating ligands. The last β strand of the LN domain provides the remaining two coordinating ligands that link the helix-loop segment to the of the LN domain β sandwich through the coordinated Ca2+. In this manner, the Ca2+ binding site of the Net-1 LN domain is a noncontinuous Ca2+ binding site, dissimilar to previously characterized Ca2+ binding motifs and domains (61,62,63). Sequence alignment of Net-1-related proteins, as well as other LN domain-containing proteins, reveals that amino acids involved in Ca2+ coordination are highly conserved in Net-1 and other Ca2+ binding LN domains. In particular, the conserved aspartate and threonine of the helix-loop segment are present in all known Ca2+ binding LN domains and absent in LN domains that do not bind Ca2+ (28,29,30,31,55,60). Based on the conserved LN domain Ca2+ binding sequence identified here, we propose that netrin-3, which also has this conserved sequence, including the aspartate and threonine, would be capable of binding Ca2+.
Bioinformatic sequence alignments and MMPBSA analysis demonstrate that aspartate and threonine residues of the helix-loop segment are critical for LN domain Ca2+ binding. This bidentate coordination of Ca2+ by threonine is relatively uncommon but is known to occur in other Ca2+ binding proteins such as thermolysin (62,64). Interestingly, several members of the F5/F8 type C domain family, which share the β sandwich jelly roll fold topology with the LN domain, also contain a Ca2+ binding site formed by a helix-loop segment adjacent to the β sandwich core (65,66,67,68,69). Comparison of several crystal structures of these Ca2+ binding F5/F8 type C domains to the LN domain reveals a remarkably similar method of Ca2+ ion coordination. Like the LN domain, Ca2+ is coordinated by an aspartate and bidentate threonine in the helix-loop segment. The pentagonal bipyramidal coordination sphere of the Ca2+ is completed by backbone carbonyl groups of the helix-loop segment, and a backbone carbonyl and glutamate side chain of a β strand of the β sandwich.
To explore the biophysical properties and function of the Net-1 LN domain Ca2+ binding site we utilized several biophysical techniques. ITC and TSA results reveal that the Ca2+ binding loop of Net-1 and Lam-γ LN domains bind a single Ca2+ ion with a low micromolar to nanomolar affinity. Because of this high affinity and the high concentration of free Ca2+ in the extracellular environment, these LN domain Ca2+ binding sites would be fully saturated under physiological conditions (56). Further biophysical analysis of the Net-1 and Lam-γ-Ca2+ interactions by TSA reveals a considerable increase in the Tm of these proteins in the presence of physiological Ca2+ concentrations compared with the apo-proteins. This result demonstrates the role of Ca2+ in the structural stabilization of the LN domain.
Previous studies exploring the effects of Ca2+ on Lam-γ and Net-1 ligand binding have shown that Ca2+ is required for structural stabilization of the Ca2+ binding loop, rather than facilitating direct contacts between Ca2+ and the ligand (18,27). Ca2+ is used for structural stabilization purposes by many other extracellular proteins including E-cadherins and fibrillin-1, which, like the LN domains studied here, also bind Ca2+ with high affinity (10,11). The role of E-cadherin Ca2+ binding is particularly well studied and has been shown to stabilize the structural conformation of E-cadherins and reduce their susceptibility to proteases (10), while mutations in the Ca2+ binding sites of E-cadherins cause disease (4). Given the substantial increase in the Tm of both Net-1 and Lam-γ, it would seem likely that Ca2+ binding to the LN domain would similarly stabilize the protein structure and reduce susceptibility to proteolytic degradation. Interestingly, although not all LN domains bind Ca2+ they therefore are not strictly required to stabilize all LN folds.
The coordination of Ca2+ among the laminin family thus seems to be important for structural stability and, in specific cases, for functional purposes. The data generated by this study present the unique Ca2+ coordination in a highly conserved loop region of known Ca2+ binding laminin proteins. The structural and biophysical approach combined with computational techniques allowed for the comprehensive characterization of the molecular basis for protein Ca2+ interactions among the laminin protein family.
Author contributions
S.L. and F.H. contributed equally to this work. All authors were involved in study design. J.S. and M.M. prepared Net-1-Sm3+ crystals and collected diffraction data. F.H. processed the diffraction data, built the crystallographic model, and prepared the figures. M.K. produced cell lines and established protein expression protocols. S.L., H.G., F.R., and G.P. produced proteins. S.L. removed Ca2+ from proteins and buffers. S.L. performed TSA, ITC, and BLI experiments, analyzed data, and prepared the figures and tables. F.H. performed MD and MMPBSA simulations, analyzed data, and prepared the figures. F.H. performed the bioinformatic analysis of Net-1 sequences, analyzed data, and prepared the figures. S.L. and F.H. wrote the manuscript. All authors contributed to the editorial process of the final manuscript.
Acknowledgments
J.S. is a Tier-1 Canada Research Chair and received additional support from the Canadian Institutes of Health Research (CIHR-PJT 152935). The authors would like to thank the Swiss Light Source (Villigen, Switzerland) for assistance in data collection.
Declaration of interests
The authors declared no competing interests.
Editor: Erik Lindahl.
Footnotes
Scott Legare and Fabian Heide contributed equally to this work.
Supporting material can be found online at https://doi.org/10.1016/j.bpj.2024.06.005.
Contributor Information
Scott Legare, Email: legares@myumanitoba.ca.
Jörg Stetefeld, Email: jorg.stetefeld@umanitoba.ca.
Supporting material
References
- 1.Carafoli E., Santella L., et al. Brini M. Generation, control, and processing of cellular calcium signals. Crit. Rev. Biochem. Mol. Biol. 2001;36:107–260. doi: 10.1080/20014091074183. [DOI] [PubMed] [Google Scholar]
- 2.Clapham D.E. Calcium Signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 3.Brown E.M., MacLeod R.J. Extracellular calcium sensing and extracellular calcium signaling. Physiol. Rev. 2001;81:239–297. doi: 10.1152/physrev.2001.81.1.239. [DOI] [PubMed] [Google Scholar]
- 4.Handschuh G., Candidus S., et al. Becker K.-F. Tumour-associated E-cadherin mutations alter cellular morphology, decrease cellular adhesion and increase cellular motility. Oncogene. 1999;18:4301–4312. doi: 10.1038/sj.onc.1202790. [DOI] [PubMed] [Google Scholar]
- 5.Zhang K., Chen J. The regulation of integrin function by divalent cations. Cell Adhes. Migrat. 2012;6:20–29. doi: 10.4161/cam.18702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shikamoto Y., Morita T., et al. Mizuno H. Crystal structure of Mg2+- and Ca2+-bound Gla domain of factor IX complexed with binding protein. J. Biol. Chem. 2003;278:24090–24094. doi: 10.1074/jbc.M300650200. [DOI] [PubMed] [Google Scholar]
- 7.Freedman S.J., Blostein M.D., et al. Furie B. Identification of the phospholipid binding site in the vitamin K-dependent blood coagulation protein factor IX. J. Biol. Chem. 1996;271:16227–16236. doi: 10.1074/jbc.271.27.16227. [DOI] [PubMed] [Google Scholar]
- 8.McGettrick A.J., Knott V., et al. Handford P.A. Molecular effects of calcium binding mutations in Marfan syndrome depend on domain context. Hum. Mol. Genet. 2000;9:1987–1994. doi: 10.1093/hmg/9.13.1987. [DOI] [PubMed] [Google Scholar]
- 9.Maurer P., Hohenester E. Structural and functional aspects of calcium binding in extracellular matrix proteins. Matrix Biol. 1997;15:569–581. doi: 10.1016/s0945-053x(97)90033-0. [DOI] [PubMed] [Google Scholar]
- 10.Pokutta S., Herrenknecht K., et al. Engel J. Conformational changes of the recombinant extracellular domain of E-cadherin upon calcium binding. Eur. J. Biochem. 1994;223:1019–1026. doi: 10.1111/j.1432-1033.1994.tb19080.x. [DOI] [PubMed] [Google Scholar]
- 11.Smallridge R.S., Whiteman P., et al. Downing A.K. EGF-like domain calcium affinity modulated by N-terminal domain linkage in human fibrillin-1. J. Mol. Biol. 1999;286:661–668. doi: 10.1006/jmbi.1998.2536. [DOI] [PubMed] [Google Scholar]
- 12.Scott D.L., White S.P., et al. Sigler P.B. Interfacial catalysis: the mechanism of phospholipase A2. Science. 1990;250:1541–1546. doi: 10.1126/science.2274785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ng K.K., Drickamer K., Weis W.I. Structural Analysis of Monosaccharide Recognition by Rat Liver Mannose-binding Protein. J. Biol. Chem. 1996;271:663–674. doi: 10.1074/jbc.271.2.663. [DOI] [PubMed] [Google Scholar]
- 14.Hohenester E., Yurchenco P.D. Laminins in basement membrane assembly. Cell Adhes. Migrat. 2013;7:56–63. doi: 10.4161/cam.21831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cirulli V., Yebra M. Netrins: beyond the brain. Nat. Rev. Mol. Cell Biol. 2007;8:296–306. doi: 10.1038/nrm2142. [DOI] [PubMed] [Google Scholar]
- 16.Rajasekharan S., Kennedy T.E. The netrin protein family. Genome Biol. 2009;10:239. doi: 10.1186/gb-2009-10-9-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKee K.K., Hohenester E., et al. Yurchenco P.D. Organization of the laminin polymer node. Matrix Biol. 2021;98:49–63. doi: 10.1016/j.matbio.2021.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kulczyk A.W., McKee K.K., et al. Yurchenco P.D. Cryo-EM reveals the molecular basis of laminin polymerization and LN-lamininopathies. Nat. Commun. 2023;14:317. doi: 10.1038/s41467-023-36077-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fahey B., Degnan B.M. Origin and Evolution of Laminin Gene Family Diversity. Mol. Biol. Evol. 2012;29:1823–1836. doi: 10.1093/molbev/mss060. [DOI] [PubMed] [Google Scholar]
- 20.Leclère L., Rentzsch F. Repeated Evolution of Identical Domain Architecture in Metazoan Netrin Domain-Containing Proteins. Genome Biol. Evol. 2012;4:883–899. doi: 10.1093/gbe/evs061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serafini T., Kennedy T.E., et al. Tessier-Lavigne M. The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell. 1994;78:409–424. doi: 10.1016/0092-8674(94)90420-0. [DOI] [PubMed] [Google Scholar]
- 22.Kennedy T.E., Serafini T., et al. Tessier-Lavigne M. Netrins are diffusible chemotropic factors for commissural axons in the embryonic spinal cord. Cell. 1994;78:425–435. doi: 10.1016/0092-8674(94)90421-9. [DOI] [PubMed] [Google Scholar]
- 23.Wang H., Copeland N.G., et al. Tessier-Lavigne M. Netrin-3, a Mouse Homolog of Human NTN2L, Is Highly Expressed in Sensory Ganglia and Shows Differential Binding to Netrin Receptors. J. Neurosci. 1999;19:4938–4947. doi: 10.1523/JNEUROSCI.19-12-04938.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakashiba T., Nishimura S., et al. Itohara S. Complementary expression and neurite outgrowth activity of netrin-G subfamily members. Mech. Dev. 2002;111:47–60. doi: 10.1016/s0925-4773(01)00600-1. [DOI] [PubMed] [Google Scholar]
- 25.Nakashiba T., Ikeda T., et al. Itohara S. Netrin-G1: a Novel Glycosyl Phosphatidylinositol-Linked Mammalian Netrin That Is Functionally Divergent from Classical Netrins. J. Neurosci. 2000;20:6540–6550. doi: 10.1523/JNEUROSCI.20-17-06540.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koch M., Murrell J.R., et al. Burgeson R.E. A novel member of the netrin family, beta-netrin, shares homology with the beta chain of laminin: identification, expression, and functional characterization. J. Cell Biol. 2000;151:221–234. doi: 10.1083/jcb.151.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu K., Wu Z., et al. Nikolov D.B. Neural migration. Structures of netrin-1 bound to two receptors provide insight into its axon guidance mechanism. Science. 2014;344:1275–1279. doi: 10.1126/science.1255149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reuten R., Patel T.R., et al. Koch M. Structural decoding of netrin-4 reveals a regulatory function towards mature basement membranes. Nat. Commun. 2016;7 doi: 10.1038/ncomms13515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brasch J., Harrison O.J., et al. Shapiro L. Crystal Structure of the Ligand Binding Domain of Netrin G2. J. Mol. Biol. 2011;414:723–734. doi: 10.1016/j.jmb.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seiradake E., Coles C.H., et al. Jones E.Y. Structural basis for cell surface patterning through NetrinG–NGL interactions. EMBO J. 2011;30:4479–4488. doi: 10.1038/emboj.2011.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grandin M., Meier M., et al. Stetefeld J. Structural decoding of the Netrin-1/UNC5 interaction and its therapeutical implications in cancers. Cancer Cell. 2016;29:173–185. doi: 10.1016/j.ccell.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 32.Moya-Torres A., Gupta M., et al. Stetefeld J. Homogenous overexpression of the extracellular matrix protein Netrin-1 in a hollow fiber bioreactor. Appl. Microbiol. Biotechnol. 2021;105:6047–6057. doi: 10.1007/s00253-021-11438-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heide F., Legare S., et al. Stetefeld J. Heparins mediate the multimer assembly of secreted Noggin. Protein Sci. 2022;31 [Google Scholar]
- 34.Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agirre J., Atanasova M., et al. Yamashita K. The CCP4 suite: integrative software for macromolecular crystallography. Acta Crystallogr. D Struct. Biol. 2023;79:449–461. doi: 10.1107/S2059798323003595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCoy A.J., Grosse-Kunstleve R.W., et al. Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P., Lohkamp B., et al. Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liebschner D., Afonine P.V., et al. Adams P.D. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019;75:861–877. doi: 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abraham M.J., Murtola T., et al. Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. [Google Scholar]
- 40.Huang J., MacKerell A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013;34:2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mark P., Nilsson L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A. 2001;105:9954–9960. [Google Scholar]
- 42.Valdés-Tresanco M.S., Valdés-Tresanco M.E., et al. Moreno E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theor. Comput. 2021;17:6281–6291. doi: 10.1021/acs.jctc.1c00645. [DOI] [PubMed] [Google Scholar]
- 43.Boratyn G.M., Camacho C., et al. Zaretskaya I. BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 2013;41:W29–W33. doi: 10.1093/nar/gkt282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boratyn G.M., Schäffer A.A., et al. Madden T.L. Domain enhanced lookup time accelerated BLAST. Biol. Direct. 2012;7:1–14. doi: 10.1186/1745-6150-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ovchinnikov S., Kamisetty H., Baker D. Robust and accurate prediction of residue–residue interactions across protein interfaces using evolutionary information. Elife. 2014;3 doi: 10.7554/eLife.02030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sievers F., Wilm A., et al. Higgins D.G. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crooks G.E., Hon G., et al. Brenner S.E. WebLogo: A sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bai N., Roder H., et al. Karanicolas J. Isothermal analysis of ThermoFluor data can readily provide quantitative binding affinities. Sci. Rep. 2019;9:2650. doi: 10.1038/s41598-018-37072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Niebling S., Burastero O., et al. García-Alai M. FoldAffinity: binding affinities from nDSF experiments. Sci. Rep. 2021;11:9572. doi: 10.1038/s41598-021-88985-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burastero O., Niebling S., et al. Garcia Alai M.M. eSPC: an online data-analysis platform for molecular biophysics. Acta Crystallogr. D Struct. Biol. 2021;77:1241–1250. doi: 10.1107/S2059798321008998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson R.A., Griffiths S.C., et al. Siebold C. Simultaneous binding of guidance cues NET1 and RGM blocks extracellular NEO1 signaling. Cell. 2021;184:2103–2120.e31. doi: 10.1016/j.cell.2021.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carvin D., Islam S.A., et al. Blundell T.L. In: International Tables for Crystallography Volume F: Crystallography of Biological Macromolecules. Arnold E., Himmel D.M., Rossmann M.G., editors. Wiley; Chichester: 2012. The preparation of heavy-atom derivatives of protein crystals for use in multiple isomorphous replacement and anomalous scattering. [Google Scholar]
- 53.Meier M., Gupta M., et al. Stetefeld J. The dynamic nature of netrin-1 and the structural basis for glycosaminoglycan fragment-induced filament formation. Nat. Commun. 2023;14:1226. doi: 10.1038/s41467-023-36692-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Priest J.M., Ozkan E. Structure of C. elegans UNC-6 LamN and EGF domains. RCSB Protein Data Bank. 2023 https://www.rcsb.org/ [Google Scholar]
- 55.Carafoli F., Hussain S.-A., Hohenester E. Crystal Structures of the Network-Forming Short-Arm Tips of the Laminin β1 and γ1 Chains. PLoS One. 2012;7 doi: 10.1371/journal.pone.0042473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schaafsma G. In: Calcium in Human Biology. ILSI Hman Nutrition Reviews. Nordin B.E.C., editor. Springer; London: 1988. Calcium in extracellular fluid: Homeostasis; pp. 241–259. [Google Scholar]
- 57.Paulsson M. The role of Ca2+ binding in the self-aggregation of laminin-nidogen complexes. J. Biol. Chem. 1988;263:5425–5430. [PubMed] [Google Scholar]
- 58.Choong G., Liu Y., Templeton D.M. Interplay of calcium and cadmium in mediating cadmium toxicity. Chem. Biol. Interact. 2014;211:54–65. doi: 10.1016/j.cbi.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 59.Goyer R.A. Nutrition and metal toxicity. Am. J. Clin. Nutr. 1995;61:646S–650S. doi: 10.1093/ajcn/61.3.646S. [DOI] [PubMed] [Google Scholar]
- 60.Hussain S.-A., Carafoli F., Hohenester E. Determinants of laminin polymerization revealed by the structure of the α5 chain amino-terminal region. EMBO Rep. 2011;12:276–282. doi: 10.1038/embor.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McPhalen C.A., Strynadka N.C.J., James M.N.G. In: Advances in Protein Chemistry. Anfinsen C.B., Edsall J.T., et al.Eisenberg D.S., editors. Springer; London: 1991. Calcium-binding sites in proteins: A structural perspective; pp. 77–144. [DOI] [PubMed] [Google Scholar]
- 62.Kirberger M., Yang J.J. In: Encyclopedia of Metalloproteins. Kretsinger R.H., Uversky V.N., Permyakov E.A., editors. Springer; New York: 2013. Calcium-Binding Protein Site Types; pp. 511–521. [Google Scholar]
- 63.Elíes J., Yáñez M., et al. Campos-Toimil M. In: Calcium Signaling. Islam M., editor. Springer; London: 2020. An update to calcium binding proteins; pp. 183–213. [Google Scholar]
- 64.Holden H.M., Matthews B.W. The binding of L-valyl-L-tryptophan to crystalline thermolysin illustrates the mode of interaction of a product of peptide hydrolysis. J. Biol. Chem. 1988;263:3256–3260. doi: 10.2210/pdb3tmn/pdb. [DOI] [PubMed] [Google Scholar]
- 65.Ficko-Blean E., Stuart C.P., et al. Boraston A.B. Carbohydrate Recognition by an Architecturally Complex α-N-Acetylglucosaminidase from Clostridium perfringens. PLoS One. 2012;7 doi: 10.1371/journal.pone.0033524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ficko-Blean E., Boraston A.B. The Interaction of a Carbohydrate-binding Module from a Clostridium perfringens N-Acetyl-β-hexosaminidase with Its Carbohydrate Receptor. J. Biol. Chem. 2006;281:37748–37757. doi: 10.1074/jbc.M606126200. [DOI] [PubMed] [Google Scholar]
- 67.Feil S.C., Lawrence S., et al. Parker M.W. Structure of the Lectin Regulatory Domain of the Cholesterol-Dependent Cytolysin Lectinolysin Reveals the Basis for Its Lewis Antigen Specificity. Structure. 2012;20:248–258. doi: 10.1016/j.str.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramelot T.A., Raman S., et al. Kennedy M.A. Improving NMR protein structure quality by Rosetta refinement: A molecular replacement study. Proteins: Struct. Funct., Bioinf. 2009;75:147–167. doi: 10.1002/prot.22229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rogers M.S., Hurtado-Guerrero R., et al. McPherson M.J. Cross-Link Formation of the Cysteine 228−Tyrosine 272 Catalytic Cofactor of Galactose Oxidase Does Not Require Dioxygen. Biochemistry. 2008;47:10428–10439. doi: 10.1021/bi8010835. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.