

INTRODUCTION
Childhood interstitial and diffuse lung disease (chILD) is an umbrella term that encompasses numerous heterogeneous, diffuse lung disorders. Most chILD disorders are rare or ultra-rare, but collectively there are many affected infants and children, often with severe lung disease. The true incidence and prevalence of chILD are unknown.
During the 1990s to early 2000s, it was recognized that the etiologies of chILD disorders differed from those of adult interstitial lung diseases. Some chILD disorders are primary lung disorders, while some chILD disorders are secondary to a systemic etiology. While chILD disorders most frequently affect the interstitial compartment of the lungs, often multiple compartments are involved including the alveoli, airways, and pulmonary vasculature. In 2006, the term chILD was coined, and a working clinical definition of chILD was established. ChILD syndrome is defined as 3 of the following 4 criteria in the absence of another etiology: (1) respiratory signs including, but not limited tachypnea, retractions, clubbing, crackles, wheezing, and failure to thrive; (2) respiratory symptoms including, but not limited to, cough, increased work of breathing, and exercise intolerance; (3) hypoxemia either at rest, with activity, or during sleep; and (4) diffuse abnormalities on chest imaging. After the identification of chILD disorders, the chILD research network was formed and developed a pediatric-specific classification scheme that still frames our thinking of chILD disorders.1–3 (Fig. 1) represents the current classification scheme that has evolved from the intitial descriptions of chILD classification.
Fig. 1.
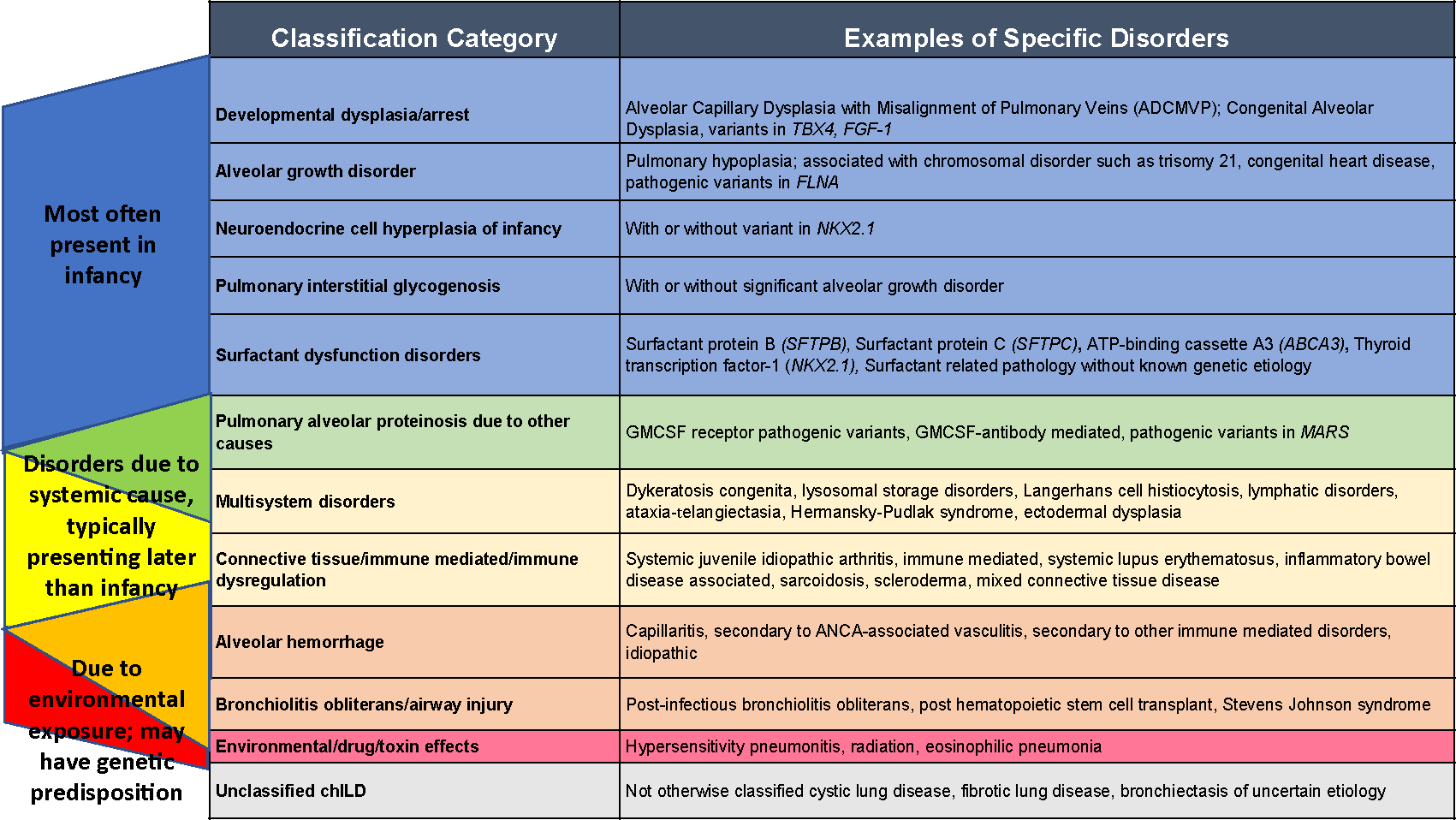
Classification categories of chILD disorders. There is overlap between conditions presenting most often in infancy, those secondary to a systemic disease process, and conditions due to environmental exposures. Diagnosis and management involve synthesis of the patient’s clinical context, histopathologic classification, and underlying disease processes.
ChILD disorders continue to have high morbidity and mortality, and further collaborative research is essential. Through national and international collaborations and innovations, the care of affected infants and children has advanced. This article will focus on recent innovations in chILD multidisciplinary clinical care team expansion, research network collaborations, registry investigations, genetic advancements, understanding of disease mechanisms, and therapeutic developments.
PRESENTATION AND DIAGNOSTIC EVALUATION OF CHILDHOOD INTERSTITIAL AND DIFFUSE LUNG DISEASE
Recognizing chILD disorders can be challenging, and the diagnosis is often delayed or missed. There is significant overlap in symptoms between chILD disorders and other more common respiratory disorders in infants and children. The key to recognizing chILD disorders is to look for respiratory symptoms that are disproportionately severe or persist as compared to more common diseases such as premature lung disease, cystic fibrosis, primary ciliary dyskinesia, chronic aspiration, congenital heart disease, lower respiratory tract infections, or asthma. Infants and young children are most likely to present with hypoxemia and increased work of breathing; many infants present with failure to thrive and only some have crackles or wheezing on physical examination.1 Approximately half of older children with chILD disorders present with hypoxemia and increased work of breathing but are more likely to present with dyspnea, exercise intolerance, or cough.4
The chILD diagnostic evaluation is complex and can involve extensive testing to arrive at the diagnosis (see Fig. 1). Making the correct diagnosis involves taking a detailed medical history including presentation, episodes of worsening, progression, current symptoms, and limitations. History should include a detailed family history of interstitial lung disorders, unexplained neonatal respiratory failure, adults with chronic respiratory failure or pulmonary fibrosis, and disorders including immune and autoimmune diseases. Patients will often require several clinical studies including laboratory evaluations, genetic testing, high-resolution chest computed tomography (CT), pulmonary function testing, bronchoscopy with bronchoalveolar lavage, and for some children lung biopsy. If possible, chest CT and lung biopsy should not be performed during exacerbations, in the setting of recent lower respiratory tract infections, or close to treatment courses of steroids as illness or steroid treatment can alter histopathologic findings. Guidelines exist for the diagnosis of chILD during infancy,2 but not for older children. Establishment of a chILD diagnosis can end the diagnostic odyssey for children and their families, limit unnecessary diagnostic testing, and inform disease surveillance, genetic counseling, and in some cases therapeutic interventions. Prompt treatment of lung disease may limit or even reverse lung damage, improve quality of life, and reduce associated morbidities and mortality.
CURRENT CHILDHOOD INTERSTITIAL AND DIFFUSE LUNG DISEASE DIAGNOSTIC CATEGORIES
Childhood interstitial and diffuse lung disorders are currently classified into the following diagnostic categories (see Fig. 1): surfactant dysfunction disorders (includes pathogenic variants in SFTPB, SFTPC, ABCA3, and NKX2-1), suspected surfactant dysfunction without identified surfactant gene variants; pulmonary alveolar proteinosis due to other causes (includes granulocyte-macrophage colony stimulating factor [GM-CSF] antibody-mediated and pathogenic variants in methionyl-tRNA synthetase [MARS]); lung developmental dysplasia (includes alveolar capillary dysplasia [ACD] with misalignment of the pulmonary veins [MPV], pathogenic variants in FOXF1 and TBX4); alveolar growth disorders (includes lung disease associated with trisomy 21, FLNA, and congenital heart disease); pulmonary interstitial glycogenosis (PIG; with and without significant alveolar growth disorders); neuroendocrine cell hyperplasia of infancy (NEHI); bronchiolitis obliterans (BO; includes postinfectious, post-hematopoietic stem cell transplantation [HSCT], postlung transplantation, and unknown etiologies); alveolar hemorrhage (includes capillaritis, secondary to antineutrophil cytoplasmic antibody [ANCA]-associated vasculitis, secondary to other immune-mediated disorders, and idiopathic); chILD associated with connective tissue or immune-mediated disorders (includes secondary to autoimmune, connective tissues, immune dysregulation disorders, and associated with other inborn errors of immunity); chILD due to other specific or multisystem disorders (such as lymphatic disorders, dyskeratosis congenita, ataxia-telangiectasia, Hermansky–Pudlak, ectodermal dysplasia, or secondary to storage disorders such as Niemann–Pick); environmental/toxic/drug-related chILD (includes hypersensitivity, radiation-induced, and eosinophilic pneumonitis); or unclassified chILD (includes not otherwise classified cystic lung disease, fibrotic lung disease, and bronchiectasis of uncertain etiology).5 Some chILD disorders are primary pulmonary disorders and others are secondary to a systemic disorder. Many chILD disorders have an underlying genetic etiology and have provided important insights into human lung development and function. While a full discussion of each of these diagnostic categories is beyond the scope of this article, we will discuss some of the most common chILD disorders including NEHI, PIG, genetic disorders of surfactant dysfunction, ACD, chILD associated with immune dysregulation or connective tissue disorders, and BO.
NEUROENDOCRINE CELL HYPERPLASIA OF INFANCY
NEHI was among the first chILD disorders to be described clinically and histopathologically.6,7 Patients with NEHI typically present during infancy (first 2 years of life) with tachypnea, hypoxemia, retractions, and respiratory crackles; radiographic features include hyperinflation, interstitial markings, and ground glass opacities (Fig. 2A, B).6,7 NEHI has a characteristic appearance on chest CT, with ground glass opacity often most prominent in the right middle lobe and lingula; air trapping with mosaic attenuation can also be seen (Fig. 2C, D).8 Sensitivity and specificity of CT for NEHI diagnosis are 78% and 100%, respectively, often obviating a biopsy.9 Other imaging abnormalities include increased anteroposterior chest diameter.10
Fig. 2.
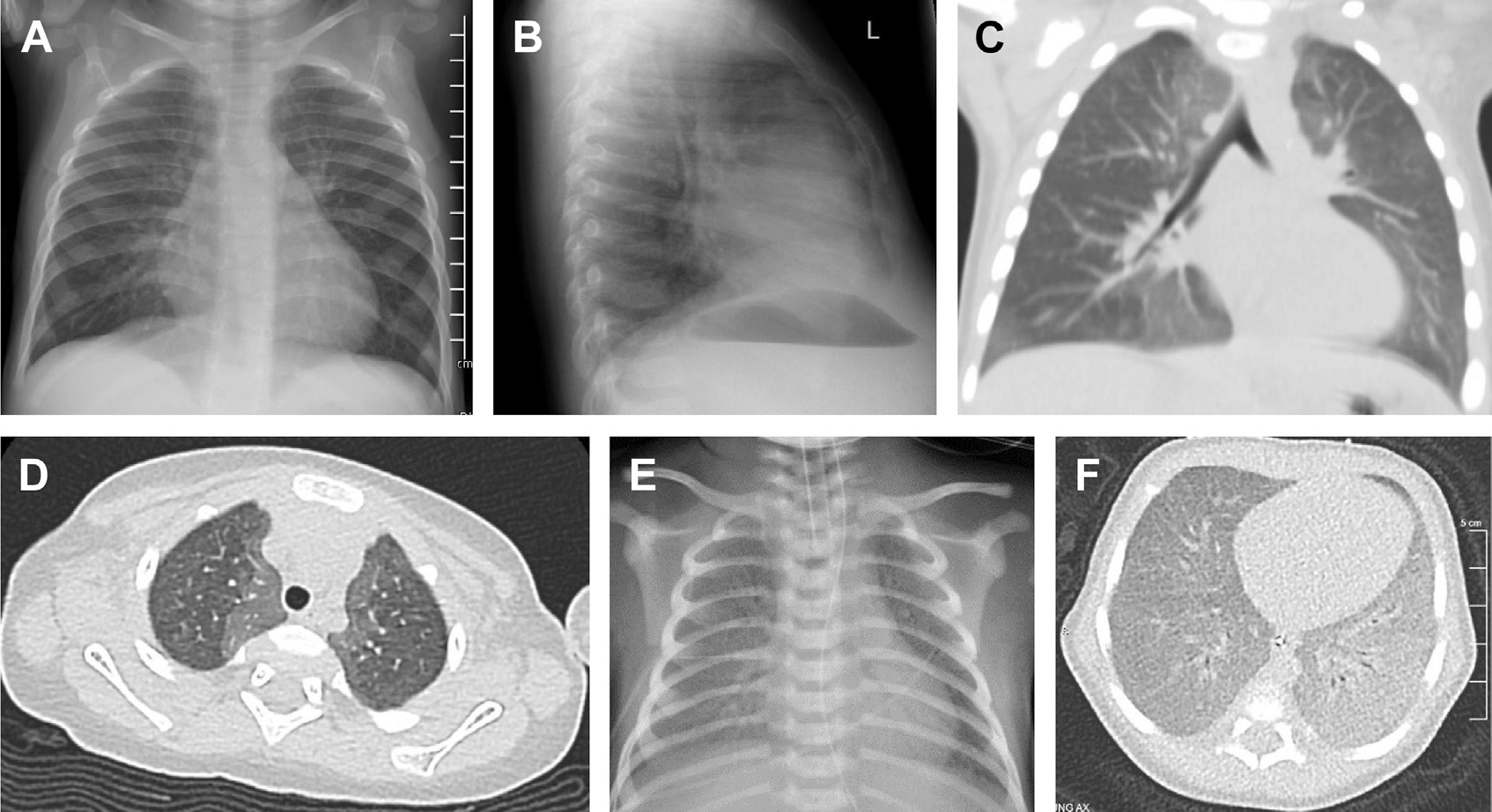
A 10 month old female infant presented with 5 months history of tachypnea, recurrent lower respiratory tract infections associated with respiratory exacerbations, and inability to wean from supplemental oxygen for chronic hypoxemia. Chest radiograph (A and B) demonstrates mild central perihilar interstitial opacities, nonspecific findings. Chest CT (C and D) demonstrates diffuse ground glass opacities in a well-demarcated, geographic pattern that is central/perihilar, and most significant in the right middle lobe and lingula. A 5 day old male infant presented with respiratory distress at birth requiring intubation, mechanical ventilation, and high supplemental oxygen (FiO2 1.0). Administration of exogenous surfactant resulted in transient improvements in oxygenation and ventilation. Chest radiograph (E) demonstrates diffuse ground glass opacities and air bronchograms. Genetic testing identified biallelic pathogenic variants in SFTPB. Chest CT (F) demonstrates diffuse ground glass opacities.
Histologic features of NEHI include increased alveolar macrophages and airway smooth muscle hyperplasia, without other structural abnormalities. Notably, there is increased staining of bombesin and serotonin, as well as increased numbers of pulmonary neuroendocrine cells (PNEC) in the airways.1,7 Although increased PNEC may not be specific for NEHI, PNEC density correlates with airway obstruction on infant pulmonary function testing.11 In a murine model, increased PNECs and neuropeptide secretion led to excess lung fluid and impaired gas exchange.12 Biomarkers in serum13 and bronchoalveolar lavage fluid14,15 can differentiate NEHI from other forms of chILD and pediatric lung disease, although this use is limited to research. While a specific genetic cause for NEHI has not been elucidated, some patients with familial NEHI have pathogenic variants in NKX2-1 which encodes thyroid transcription factor-1 (TTF-1).16 Typically, patients with NEHI, in addition to the symptoms described earlier, demonstrate poor growth and failure to thrive is associated with longer duration of supplemental oxygen use.17 A NEHI Clinical score was developed and applied to a retrospective cohort of nearly 200 children and had a sensitivity of 87%, performing equally well in patients diagnosed by lung biopsy or solely by chest CT.18 Among patients with NEHI, comorbidities are common and include gastroesophageal reflux, aspiration, and immune system abnormalities.18 Though NEHI often improves clinically over time, including eventual resolution of supplemental oxygen use, both pulmonary function and radiographic abnormalities can persist into adulthood.19
PULMONARY INTERSTITIAL GLYCOGENOSIS
PIG is a rare condition of early infancy, presenting with hypoxemia, respiratory distress, and diffuse interstitial opacities and hyperinflation on chest radiograph, often associated with respiratory symptoms out of proportion to what would be expected for the clinical scenario.20 Generally, PIG is considered a condition that does not persist beyond infancy, perhaps representing a time-limited developmental phenotype, although a patient has been reported with biopsy-proven PIG at the age of 14 months.21 PIG can be diffuse or patchy and associated with lung growth abnormalities and/or alveolar simplification, often in conjunction with chromosomal anomalies, pulmonary vascular disease, and congenital heart disease.22,23 In the initial application of infant lung biopsy classification by the chILD Research Network, 40% of patients with alveolar growth disorders had patchy PIG on biopsy.1 The initial histopathologic description of PIG is of glycogen-filled, spindle-shaped cells widening the alveolar interstitium, positive for the mesenchymal cell marker vimentin.20 In this series of 7 infants, 6 survived, and 5 received corticosteroids, including an extremely preterm infant with bronchopulmonary dysplasia who ultimately died.20 The surviving patient who did not receive steroids gradually improved with supportive care.20 Lipofibroblasts are also present in biopsy specimens of patients with PIG, suggestive of a process of aberrant or delayed development.1 Recently, it has been proposed that PIG cells may be lung-resident mesenchymal stem cells, based on consistent biomarker staining.24
In a cohort of 24 infants with biopsy-diagnosed PIG, most patients were born late preterm or term, required ventilatory support, and had structural heart disease.25 On biopsy, most had evidence of alveolar growth abnormalities and/or pulmonary hypertension.25 One-third of patients required respiratory support on discharge.25 Over 90% of patients were treated with systemic steroids, but response was difficult to quantify.25 Twenty-two patients had CT imaging, with ground glass opacities most common.25 Radiographic abnormalities are variable and nonspecific and overlap with other forms of chILD, such as surfactant dysfunction disorders. In a single-institution radiographic review, ground glass opacities and cystic lucencies were the most common CT findings in PIG, with septal thickening and architectural distortion seen less frequently.26 Steroid treatment of PIG is controversial. Anecdotally, some patients seem to have a rapid and sustained improvement following systemic steroids,27,28 while others show little or equivocal improvement.25 Prognosis is variable and depends on the clinical context. Of note, mortality risk is increased in patients who have accompanying alveolar growth abnormalities. In the cohort of 24 patients described earlier, at median 3 years follow-up, half of the patients no longer required respiratory support, 1 patient died, and 1 patient underwent lung transplantation.25
ChILD ASSOCIATED WITH INBORN ERRORS OF IMMUNITY, AUTOIMMUNE DISORDERS, AND CONNECTIVE TISSUE DISORDERS
In addition to primary pulmonary disorders, chILD has also been associated with inborn errors of immunity, autoimmune, and connective tissues disorders. Patients with these systemic disorders may have multiple pulmonary complications including infectious, postinfectious, and noninfectious. The pulmonary complications including chILD depend on the specific underlying disorder, but there can be significant clinical overlap, with more than one complication arising. ChILD associated with these systemic disorders may exhibit slow progression or fulminant onset.29 In monitoring for chILD associated with systemic disorders, pediatric pulmonologists, immunologists, and rheumatologists should maintain a high index of suspicion and recognize subtle respiratory signs and symptoms. Making a timely diagnosis and understanding the underlying genetics and pathobiologic mechanisms may assist with screening for complications, identifying targeted therapies, and determining prognosis and recurrence risk.
A broad range of interstitial lung disease (ILD) histopathologic patterns can be associated with immune, autoimmune, and rheumatologic disorders. Patterns of chILD that should raise suspicion for systemic disorders include follicular bronchiolitis, lymphocytic interstitial pneumonitis, granulomatous lymphocytic interstitial lung disease, and granulomatous “sarcoid like” disease, all of which may have overlapping histopathologic features. Other patterns associated with systemic disorders include organizing pneumonia, hypersensitivity pneumonitis, desquamative interstitial pneumonitis with extensive alveolar infiltration of macrophages, nonspecific interstitial pneumonia, and pulmonary fibrosis without another known etiology. Alveolar involvement may be present including alveolar hemorrhage with inflammation or vasculitis of the arterioles, venules, or septal capillaries or autoimmune pulmonary alveolar proteinosis due to autoimmunity or immune-mediated macrophage dysfunction.30 These pathologic processes are characterized by patchy or diffuse inflammation in the interstitial, alveolar, pulmonary vascular, pleural, or airway compartments. This chronic inflammation may lead to fibrosis and scarring in these compartments, resulting in significant morbidity and substantial risk for mortality.
As increased susceptibilities to infections are not the only manifestations of immunologic disorders, the term “inborn errors of immunity” (IEI) instead of “immunodeficiency” is currently favored. There are 10 groups of IEI disorders based on the International Union of Immunological Societies Expert Report.31 Patients with diseases of immune dysregulation and autoinflammatory disorders are at higher risk of presenting with chILD.31 In addition, autoimmune and inflammatory manifestations including chILD are also present in the categories of combined immunodeficiencies or complement deficiencies. The immune dysregulation and autoinflammatory disorders have overlap with or may confer susceptibility to rheumatologic disorders presenting with chILD as well. Rheumatologic disease may be due to underlying genetic immune dysregulation or autoinflammatory IEIs.32–34 As we learn more about these rare disorders, the clinical spectrum, including the range of pulmonary manifestations, continues to expand. There is a continuum of immune dysregulation due to abnormal immune tolerance from multiple mechanisms that includes both monogenic IEIs and polygenic disorders. Some IEIs, particularly the immune dysregulation disorders, display incomplete penetrance and variable expressivity, suggesting other genetic, epigenetic, or environmental factors (eg, infectious, microbiome, and environmental toxins) may modify disease presentation.32
ChILD and other pulmonary manifestations can be associated with many systemic autoimmune and connective tissue disorders, including systemic sclerosis, juvenile dermatomyositis, systemic lupus erythematosus, mixed connective tissue disease, Sjögren’s syndrome, sarcoidosis, ANCA-associated vasculitis, and systemic juvenile idiopathic arthritis. While the disorders in this category are considered “acquired” or “polygenic,” there are some monogenic disorders that meet the diagnostic criteria for these disorders (eg, coatomer protein complex subunit alpha [COPA] syndrome or stimulator of interferon genes [STING]-associated vasculopathy of infancy [SAVI] presenting as ANCA-associated vasculitis35 or patients with pathogenic variants in genes encoding complement or nucleases presenting with systemic lupus erythematosus).36
BRONCHIOLITIS OBLITERANS
BO is a rare complication of lower respiratory tract insults that results in narrowing or complete obstruction of the bronchioles through excessive inflammation and fibrosis of the airway mucosa and lumen.37,38 BO may be patchy or extensive and is thought to result from severe airway damage, abnormal repair mechanisms, or likely both.39 The associated lower respiratory tract insults that are associated with BO include infections, inhalational injuries, aspiration, and immunologic dysfunction including post-HSCT, graft versus host disease, lung transplantation, and Stevens Johnson syndrome. The most common cause in children is postinfectious BO (PIBO) with adenovirus and mycoplasma most frequently associated. PIBO can develop at any age but is most common in children aged less than 3 years. BO can also be associated with Stevens Johnson syndrome, and these cases are typically more severe. Signs and symptoms signaling the presence of BO include a more severe initial illness, incomplete recovery from the initial insult, need for prolonged respiratory support, shortness of breath, exercise intolerance, tachypnea, increased work of breathing, persistent coughing and/or wheezing, hypoxemia, and respiratory crackles. Prognosis of BO is variable and depends on progression and extent of pathology. Referral for lung transplant is rarely indicated, and death, while uncommon, has been reported. Morbidity is significant, with many children requiring extensive and prolonged respiratory support and treatments.40,41
Diagnosing BO includes having a high index of suspicion in the setting of a respiratory insult known to trigger this condition. Lung biopsy is the gold standard but is not required if the clinical history, imaging, and pulmonary function testing support this diagnosis. Pulmonary function testing includes fixed airway obstruction with normal or decreased forced vital capacity (FVC) and decreased forced expiratory volume in one second (FEV1) with incomplete or little response to bronchodilators. Patients with BO typically have normal total lung capacity, but high residual volume and air trapping. Diffusion abnormalities may be present in more advanced cases.42,43 Over time, both FVC and FEV1 increase after the development of BO, but the FEV1/FVC ratio declines more than expected, suggesting a mismatch in the growth of the airway and lung parenchyma (dysanaptic growth).42 Patients with BO may have hypoxemia, reduced capacity, and flow limitation with air trapping, and they may develop dynamic hyperinflation with exercise.44 To diagnose and characterize BO, it is important to obtain both inspiratory and expiratory high-resolution chest CT (HRCT) images. Classic findings include mosaic perfusion, vascular attenuation, areas of air-trapping that worsen on exhalation, ground glass opacities, bronchial wall thickening, and in some cases bronchiectasis.40,45,46 Findings may be more unilateral in appearance, and this pattern is referred to as Swyer–James syndrome.47
Treatment data are limited and mostly based on expert opinion, case series, and observational studies, and they depend to an extent on the underlying etiology. Supportive care, regardless of the etiology, is essential and includes managing gas exchange abnormalities, treating comorbidities, and limiting further lower airway damage, including prevention of both lower respiratory tract infections and toxic inhalant exposures. Anti-inflammatory therapies to limit airway inflammation and further progression of fibrosis are frequently pursued for all forms of BO. If bronchiectasis is present, airway clearance should be initiated to prevent exacerbations and progression. Depending on disease severity, PIBO is most often treated with corticosteroids, often given as a monthly pulse,48 and immunomodulatory dosing of intravenous immunoglobulin,43,49 but the duration of therapy is unclear. In PIBO, tiotropium, a long-acting muscarinic antagonist, resulted in significant improvement in multiple lung function parameters compared with placebo.50 For all forms of BO, combination therapy has been extrapolated from treatment of post-HSCT BO and is composed of inhaled steroids to provide local anti-inflammatory effects, leukotriene inhibitors to reduce cellular homing, activation, and possibly to block fibroblast proliferation and collagen deposition, and azithromycin to provide antimicrobial effects, immunomodulation, and to reduce neutrophils and interleukin (IL)-8.51–53 In BO after HSCT or lung transplantation, systemic corticosteroid therapy is used during acute episodes of worsening but is not recommended chronically due to lack of benefit and risk of both toxicities and infectious complications. Review of treatment guidelines for BO after HSCT or lung transplantation are beyond the scope of this review, but secondline treatment approaches include extracorporeal photopheresis,54–61 the Bruton’s tyrosine kinase inhibitor ibrutinib,62,63 the Janus kinase (JAK) inhibitor ruxolitinib,64–68 the Rho-associated coiled-coil kinase 2 inhibitor belumosudil,69–71 low-dose IL-2,72–74 and the tumor necrosis factor inhibitor etanercept.75,76 The role of antifibrotic therapies in BO is under investigation and not yet established.
GENETIC CHILDHOOD INTERSTITIAL AND DIFFUSE LUNG DISEASE DISORDERS
While neonatal respiratory distress syndrome (RDS) is common among preterm infants and results from a developmental insufficiency of pulmonary surfactant production, the identification of term infants with severe RDS prompted investigation for genetic causes of surfactant deficiency. Pathogenic variants in several genes that encode proteins required for surfactant assembly, function, and homeostasis were the first genetic causes of chILD to be identified.77–79 With the increased availability of exome and whole genome sequencing, additional genetic causes of chILD have been elucidated. As many chILD disorders have overlapping clinical and histologic features, identification of the underlying molecular etiology is critical to understanding the disease severity and progression, other affected organ systems, potential treatments, inheritance, and recurrence risk. While genetic causes of chILD are individually rare, their identification has informed mechanisms of human lung development and function. Here, we review the most frequently identified genetic causes of chILD, disease mechanisms, and associated clinical features. With continued application of exome and whole genome sequencing to infants and children with suspected chILD,80–82 this list will undoubtedly continue to expand.
Surfactant Protein B Deficiency
Surfactant protein B (SP-B) deficiency was the first identified genetic cause of severe neonatal RDS among term infants, but it remains a rare cause of genetic surfactant dysfunction.77 SP-B is a 79 amino acid, extremely hydrophobic protein that is synthesized by alveolar type II cells and required for the surface tension lowering properties of pulmonary surfactant.83 Infants with biallelic pathogenic variants in the gene (SFTPB) that encodes SP-B typically present with severe respiratory failure shortly after birth and die within the first few months of life without lung transplantation.77,84,85 The chest radiograph and chest CT from affected infants demonstrates diffuse granular opacities (Fig. 2E, F), similar to premature infants with a developmental insufficiency of surfactant production.77 The most common pathogenic SFTPB variant is a frameshift variant c.361delinsGAA; p.Pro121Glnfs*95 and is identified in more than two-thirds of affected infants.84,85 This SFTPB variant results in a premature stop codon and undetectable levels of SP-B in lung tissue and bronchoalveolar lavage fluid, as well as misprocessing of the surfactant protein-C proprotein (proSP-C) and accumulation of aberrant surfactant protein-C (SP-C) propeptides that may contribute to alveolar epithelial cell dysfunction.86 This variant is present in approximately 1 out of 2900 individuals in gnomAD(87) (gnomAD v4.0.0, accessed April 2024) and with increased frequency in some ethnicities.87 The incidence of SP-B deficiency is estimated at 1 per million births, and the diagnosis is most frequently made with genetic testing including gene panel, exome, or whole genome sequencing. Survival beyond the first year of life has been reported for some infants with SFTPB variants that permit some amount of SP-B function, including missensevariants.88,89 SFTPB variants are typically inherited from heterozygous (carrier), healthy parents without obvious lung dysfunction.77,85,90 Infants with SP-B deficiency may demonstrate a transient response to exogenous surfactant administration with improvement in oxygenation and ventilation; however, this response is typically short-lived and diminishes with time.77,90 Lung transplant, performed at a few centers in the United States, remains the definitive treatment option for progressive respiratory failure due to SP-B deficiency.84,91 Gene therapy using a modified adenoviral vector was used to rescue SP-B deficiency in a murine model.92 While promising as a potential therapy for human infants with SP-B deficiency, clinical trials will be challenging, given the high mortality in the first few months of life and extremely rare disease incidence.92–95
Surfactant Protein C-related Lung Disease
SP-C is a small and highly hydrophobic protein that is synthesized exclusively by alveolar epithelial type II (AECII) cells.96,97 SP-C is synthesized as a 191 or 197 amino acid proprotein (proSP-C) that undergoes cleavage at both the amino and carboxy termini to yield the 34 amino acid mature protein.98–100 SP-C contributes to the surface-tension lowering qualities of surfactant and stabilization of surfactant at low long volumes, but it is not required for surfactant function, lamellar body formation, or survival as demonstrated by normal pulmonary function among SP-C–deficient mice.101 Pathogenic variants in SFTPC act in an autosomal dominant manner with variable penetrance and can cause diverse pulmonary phenotypes including neonatal respiratory failure, chILD, and adult pulmonary fibrosis.102 Approximately half of disease-associated SFTPC variants are inherited, and half arise de novo. Even among family members that share the same SFTPC variant, there are differences in age of presentation and disease severity, with some individuals remaining healthy for most of their lives.102 The reasons for variable penetrance are not clearly defined, may be related to other genetic (eg, ABCA3 variant) or environmental (eg, prematurity, smoking) modifiers,103–105 and thereby complicate estimates of disease incidence. By reviewing the gnomAD database87 for disease-associated SFTPC variants, we estimate a disease incidence of 1 per 60,000 individuals.
The majority of disease-associated SFTPC variants are located in the BRICHOS domain, which contains the terminal 100 amino acids of the proprotein.106 The BRICHOS domain shares homology with a set of proteins associated with familial dementias and is required for proper trafficking of the extremely hydrophobic proprotein through the AECII.106,107 Cell-based and mouse models demonstrate that SP-C BRICHOS domain variants result in a toxic gain of function with abnormal proSP-C trafficking, retention in the endoplasmic reticulum, and activation of the unfolded protein response.107,108 Variants in the non-BRICHOS or linker domain including the most common pathogenic variant p.Ile73Thr (~30% of affected individuals) are misrouted to the endosomes and plasma membrane and result in increased mitophagy.109,110 Interestingly, very few disease-associated variants have been identified in the mature SP-C peptide, emphasizing that the mechanism of action is a toxic gain of function. As the mutant proSP-C protein interferes with processing of the wild-type proSP-C, the therapeutic approach for SP-C–associated lung disease is likely to be directed at inactivating the mutant allele rather than gene addition approaches.94,95
ATP-binding Cassette A3 Deficiency
ATP-binding cassette A3 (ABCA3) is transmembrane protein that localizes to the membrane of lamellar bodies, specialized intracellular organelles that assemble and store surfactant in AEC2 cells. ABCA3 transports phospholipids from the cytoplasm into the lamellar bodies that combine with surfactant proteins B and C to make surfactant. ABCA3 belongs to a large superfamily of transporter proteins with similar structures, the most well characterized of which is cystic Fibrosis transmembrane receptor (CFTR) also known as ABCC7.111,112 ABCA3 deficiency is the most common genetic cause of surfactant dysfunction and most commonly presents as neonatal respiratory failure, and less commonly as chILD or adult pulmonary fibrosis.78,113–116 In contrast to SP-B and SP-C, ABCA3 is a large 1704 amino acid protein that is expressed in multiple tissues including the kidney and brain117; however, only pulmonary phenotypes have been definitively associated with biallelic pathogenic ABCA3 variants. Over 300 pathogenic ABCA3 variants have been reported, including loss-of-function variants (frameshift and nonsense), missense, splicing, small insertion/deletions, large intergenic deletions, and uniparental disomy.113–115,118 Monoallelic or heterozygous ABCA3 variants have been associated with neonatal RDS among term or late preterm infants, which is typically reversible with neonatal intensive care. Monoallelic ABCA3 variants may modify disease severity among preterm infants with RDS or bronchopulmonary dysplasia or may be associated with mild chILD.119–121 A genotype–phenotype association has been demonstrated for infants with biallelic loss-of-function variants in ABCA3 presenting with neonatal respiratory failure and dying in the first few months of life without lung transplantation.113,115 The age of presentation, disease severity, and progression for infants with missense, splicing, and small insertion/deletions are more difficult to predict and complicate discussions regarding lung transplantation. Functional characterization of ABCA3 missense variants has demonstrated 2 classes: those that disrupt intracellular trafficking and are retained in the endoplasmic reticulum and those that traffic to the lamellar bodies but have reduced phospholipid transport.122–126 The specific variant class (mistrafficking vs impaired phospholipid transport), residual ABCA3 function, and activation of intracellular stress pathways likely contribute to disease presentation. The most common pathogenic ABCA3 variant is p.Glu292Val that is present in 0.45% of individuals in gnomAD(87) and enriched in some ethnic groups, is most often associated with interstitial lung disease and adult pulmonary fibrosis, rather than neonatal respiratory failure,113,114,116 and likely permits some residual ABCA3 function.122 Based on loss-of-function and known pathogenic ABCA3 variants in gnomAD, the incidence of ABCA3 deficiency is estimated at approximately 1 per 45,000 births, approximately 90 affected infants per year in the United States.
Given the similarities in structure, transport function, and variant classes between ABCA3 and CFTR, the CFTR potentiator ivacaftor has been studied in vitro using a human pulmonary epithelial cell line (A549) that transiently expresses individual ABCA3 variants. Ivacaftor improves uptake of fluorescently labeled phosphatidylcholine (TopF-PC), suggesting an improved phospholipid transport and a potential role for CFTR potentiators to correct phospholipid transport ABCA3 variants.112 However, not all phospholipid transport ABCA3 variants demonstrated similar results,112 suggesting that future therapies are likely to be variant-specific or may require combination therapies. While small molecules may be helpful for individuals with chILD due to ABCA3 missense variants, infants with biallelic loss-of-function ABCA3 variants and severe neonatal respiratory failure may be candidates for gene replacement strategies.94,95
Thyroid Transcription Factor 1-related Lung Disease
The NKX2–1 gene encodes TTF-1, which is expressed in the lung, thyroid, hypothalamus, and basal ganglia and is critical for lung branching and epithelial cell morphogenesis as well as embryonic development of the thyroid and portions of the brain.127,128 TTF-1 regulates the transcription of several surfactant-associated genes including SFTPB, SFTPC, and ABCA3, among others. Individuals with pathogenic variants or gene deletions of NKX2–1 may display phenotypic features of “brain-lung-thyroid” syndrome, which includes neurologic abnormalities (most frequently movement disorders), neonatal respiratory failure or chILD, and central hypothyroidism.129,130 Affected individuals may have involvement in one or more organ systems, and approximately 50% have abnormalities in all 3. NKX2–1 variants can be inherited or arise de novo and include single nucleotide variants as well as whole gene deletions.129 Similar to individuals with SFTPC variants, there is variable penetrance, even among family members with the same NKX2–1 variant in terms of phenotype/organ involvement, disease severity, and progression.129–132 The lung histology from affected individuals most frequently identifies growth abnormalities with alveolar enlargement and simplification.129 The pathogenic mechanism of lung disease related to NKX2–1 variants is likely related to haploinsufficiency, although other mechanisms may contribute to the observed phenotypic spectrum. Pathogenic variants in NKX2–1 should be suspected among term infants with respiratory failure and neurologic or thyroid disease.
Alveolar Capillary Dysplasia with Misalignment of the Pulmonary Veins
ACD was first identified by Janney and colleagues in a term infant who presented with progressive respiratory failure and severe pulmonary hypertension shortly after birth and died at 2 days of age.133 Lung tissue at autopsy demonstrated failure of formation and ingrowth of the alveolar capillaries, abnormal air–blood barriers, and anomalous veins in the bronchovascular bundles and termed congenital ACD.133 Since that early report, more than 200 infants with ACD have been described in the medical literature. Most affected infants are born at term and develop respiratory symptoms shortly after birth. Infants develop progressive respiratory failure and severe pulmonary hypertension refractory to medical therapies. Chest radiograph findings are typically nonspecific, and family history is usually negative. Extrapulmonary anomalies involving the heart, gastrointestinal tract (eg, malrotation), and genitourinary system are identified in the majority of affected infants.134 Delayed or less fulminant presentations of ACD-MPV have been reported, with some of these infants surviving with lung transplantation.135,136 Histopathologic findings include decreased alveolar capillaries with displacement from the alveolar epithelium and malposition of the pulmonary veins within the same bronchovascular bundles as the small pulmonary arteries, medial arterial hypertrophy, lobular maldevelopment, and lymphangiectasis.1,135,137–139 Of note, the misaligned pulmonary veins have now been identified as intrapulmonary shunt vessels.140
Pathogenic variants in or deletions including or upstream of FOXF1 have been identified in more than 90% of infants with ACDMPV. The pathogenic mechanism is that of haploinsufficiency. While most cases arise from de novo FOXF1 variants, familial cases have been reported.134 FOXF1 encodes a transcription factor that is expressed in visceral mesoderm and is involved in lung, intestinal, and cardiac development.141 Recently, a compound transcellular activator of nuclear FOXF1 expression (TanFe) that activates and stabilizes FOXF1 has demonstrated increased angiogenesis in human organoid and murine models of ACDMPV, suggesting a potential therapy for this disorder that is lethal without lung transplantation.142
Immune Dysregulation Disorders Associated with Childhood Interstitial and Diffuse Lung Disease
Pathogenic variants in genes encoding proteins involved in the regulation of interferon signaling have been associated with immune dysregulation disorders, and chILD is often a prominent disease manifestation. COPA syndrome and SAVI are 2 classic examples of immune dysregulation disorders involving interferon signaling. COPA syndrome is caused by pathogenic variants in the COPA gene. COPA encodes the alpha subunit of coatomer complex I (COP-I), a major component of vesicular trafficking machinery that facilitates retrograde transport between the endoplasmic reticulum and Golgi.143 Mutant COPA results in endoplasmic reticulum stress and cytokine upregulation.143 The majority of COPA pathogenic variants are inherited with incomplete penetrance and located within exons 8 to 9, which encodes the WD40 repeat region that is important for cargo protein recognition.143–145 In addition to arthritis, affected children often present with cough, tachypnea, hypoxemia, and acute and chronic pulmonary hemorrhage.144
Pathogenic variants in STING1 (TMEM173) that encode STING, an endoplasmic reticulum membrane protein, result in STING-associated vasculopathy with onset in infancy. Affected infants typically present with failure to thrive, systemic inflammation including fever, violaceous, scaling rash of the face and extremities, and chILD. STING1 variants typically arise de novo, although familial cases have been reported.146–149 STING activates interferon regulatory factor 3 through binding cyclic guanosine monophosphate-adenosine monophosphate.150 Mutant STING is constitutively active and resulting in increased expression of interferon-1 and subsequent inflammation.150
Aminoacyl-transfer RNA Synthetase Genes Associated with Childhood Interstitial and Diffuse Lung Disease
Interstitial lung disease can be the presenting or prominent feature for infants and children with biallelic pathogenic variants in genes that encode aminoacyl-tRNA synthetases, including FARSA, FARSB, LARS1, MARS1, and MARS2.151–154 Affected children often have disease beyond the lung including failure to thrive, neurodevelopmental delays, liver dysfunction, hypoalbuminemia, and anemia. The mechanism by which pathogenic variants in these genes result in chILD is related to impaired protein synthesis in specific tissues including the lung. Dietary supplementation with added methionine may augment enzyme function and may alter disease severity or progression.153
ADVANCES IN MULTIDISCIPLINARY CARE OF PATIENTS WITH CHILDHOOD INTERSTITIAL AND DIFFUSE LUNG DISEASE
Supporting families through the diagnosis of a chILD disorder and the associated complex clinical care requires the collaboration of a multidisciplinary care team with expertise in caring for patients with chILD disorders.155 As reported for other pediatric chronic respiratory disorders, dedicated multidisciplinary care promotes quality of care, education, and family support and improves patient experience and clinical outcomes.156–158 Team members may include pediatric pulmonologists, geneticists and genetic counselors, nursing, respiratory therapists, nutritionists, physical therapists, social workers, neonatologists, rheumatologists, immunologists, gastroenterologists, surgeons, pathologists, radiologists, palliative care specialists, and transplant pulmonologists, among others.155 Ideally, centers providing this care couple clinical care with education and research so that patients and families can learn about family support networks, participate in education and advocacy efforts, be included in national registries for clinical research, offered participation in available clinical trials, and access opportunities to advance the field in collaboration with providers. These care teams may not be accessible to children living far away from pediatric hospitals and centers with chILD expertise. To provide more equitable care of patients with chILD disorders, experienced centers may provide education for pediatric pulmonologists and pediatricians, and many offer second opinion services or patient clinical review.
TREATMENT INNOVATIONS
Treatments for chILD disorders are limited due to lack of clinical trials and evidence-based data to guide clinical decision-making. Much of the care is based on expert recommendations and clinical experience. Historically, chILD disorders with underlying inflammation have been treated with a wide spectrum of immunosuppressive medications. While most medications (eg, corticosteroids, azithromycin, and hydroxychloroquine) are used empirically, some targeted therapies are available including selective JAK kinase inhibitors for immune dysregulation disorders COPA and SAVI.145,159–161 The development of additional targeted therapies will be based on knowledge of the underlying disease mechanisms, genetics, and shared histopathologic features.2,113,162–170 Supportive therapy is the mainstay of chILD care and include correcting gas exchange abnormalities, optimizing nutrition, diagnosing and treating comorbid conditions, promoting healthy lung growth, and preventing additional lung insults.18,171,172 The importance of avoiding respiratory tract infections for patients with chILD is underscored by the findings that most acute exacerbations in chILD are triggered by infection, and mortality in chILD is highly associated with an acute exacerbation.169 Families should be encouraged to keep vaccinations up to date unless contraindicated and avoid exposure to inhalant toxins such as cigarettes, vaping products, and environmental pollutants. Exercise was shown in an adult retrospective multicentered international cohort of adult patients with fibrotic interstitial lung disease to lower risk of death or lung transplant,173 and thus, it is likely that exercise has benefit among affected children as well and should be encouraged with appropriate support of oxygenation and ventilation. In patients with severe or worsening chILD, poor quality of life despite optimal medical or surgical management, or in those with an underlying diagnosis with known lethality (eg, ACD-MPV, biallelic loss-of-function variants in ABCA3 or SFTPB), expeditious discussions with the family regarding lung transplantation should be pursued.84,113
Although there are no currently approved treatments for chILD, international research networks demonstrated the feasibility of conducting an international prospective double-blind, randomized clinical trial to investigate the pharmacokinetics and safety of nintedanib, a tyrosine kinase inhibitor used to treat adult pulmonary fibrosis, in children with progressive fibrosing interstitial lung disease (InPedILD).174,175 Nintedanib has demonstrated clinical benefit in several trials for adult fibrotic ILD, including idiopathic pulmonary fibrosis,176 systemic sclerosis-associated ILD,177 and other chronic progressive ILDs.178 In these trials, nintedanib slowed the progression of decline in lung function, and nintedanib is approved for use in adults in several countries including the United States. In the InPedILD randomized, controlled clinical trial, patients aged 6 to 17 years with fibrosing chILD received nintedanib or placebo for 24 weeks; weight-based dosing resulted in similar pharmacokinetics as adults and an acceptable safety profile.174,175 Similar to the results from the adult trials, nintedanib may slow progression of lung disease in pediatric patients, as the adjusted mean changes in percentage predicted FVC at baseline in comparison to week 24 were smaller in the treated group in comparison to placebo.175 In addition to showing safety and possible efficacy, the InPedILD study also defined and characterized the underlying chILD disorders and contributing clinical factors that lead to pediatric pulmonary fibrosis. The open label extension study continues, and there will be additional studies of pediatric pulmonary fibrosis because of this impressive international collaboration.
Given the rarity of chILD disorders, multicentered, randomized clinical trials will continue to be challenging and costly. Alternative study designs such as “N of 1” when a single patient is given a treatment or placebo with a crossover design may have more ultility in chILD disorders. With improved understanding of the underlying disease mechanisms, more targeted and specific therapies will become available. This personalized approach has been particularly useful in patients with monogenic immune regulatory disorders leading to chILD. In these disorders, mechanistically targeted therapies can halt or even reverse associated ILD.179 This approach underscores the importance of continued genetic testing and functional studies of known and novel genes to advance treatments of chILD. Obtaining insurance approval, counseling of risks and benefits, practicing shared decision-making, and monitoring for clinical efficacy and side effects are paramount.
IMAGING INNOVATIONS
Radiologists and pathologists are essential members of the chILD multidisciplinary care team. Considerable advances have been made in understanding clinical radiographic and pathologic correlations in chILD.180 Imaging and histopathologic review with experienced radiologists and pathologists can identify the presence of a chILD disorder, establish a differential diagnosis, assist with planning the specific location for lung biopsy, and offer pathologic mechanisms to guide treatment. The current chILD classification system was developed through retrospective review of lung biopsy proven-chILD with correlation of clinical, radiographic, and histopathologic findings.1 While chILD disorders are most commonly characterized by chest radiograph or chest CT, novel CT techniques, MRI, and chest ultrasound have recently been applied to chILD disorders.180–182
Chest radiographs are most frequently performed during the initial evaluation of a suspected chILD disorder. Radiographs may demonstrate findings of diffuse lung disease including hazy or ground glass opacities or hyperinflation, but are frequently nonspecific.183 HRCT is more sensitive for detecting chILD and can be performed without sedation, even among young children.183 For some children, controlled ventilation with imaging obtained both during inhalation and exhalation is required. Older children who can lay still and hold their breath can perform controlled ventilation HRCT without sedation, but for infants, younger children, or children with atypical neurodevelopment, sedation and anesthesia may be needed. These studies are recommended to be performed in centers with experience in lung recruitment maneuvers and coordination of imaging timing with radiology.184 Both chest radiograph and HRCT may be used to establish a diagnosis of chILD, to monitor disease progression, or to follow treatment response.185
For many chILD disorders, chest imaging results can be characteristic or suggestive of specific disorders but further clinical, genetic, or histopathologic confirmation of diagnosis is needed. In NEHI, the HRCT findings are very sensitive and specific and may eliminate the need for a lung biopsy if the clinical history is also compatible.186 Of note, when the diagnosis is made by clinical history and imaging findings it is termed “NEHI syndrome” as compared to “NEHI” when biopsy proven.
Newer imaging modalities include dual-energy CT and MRI show future promise in chILD. Dual-energy or spectral CT allows for the generation of dual-energy data, can provide information on both anatomic and functional abnormalities associated with chILD, and is promising as a potential noninvasive measure of disease progression or therapeutic response.180 The use of chest MRI for chILD disorders has been more limited due to respiratory motion artifact and decreased spatial resolution as compared to chest HRCT.180 However, recently, 3T MRI adequately demonstrated consolidations, parenchymal bands, and thickening of pulmonary fissures in patients with chILD.187 HRCT remains superior for most of the characteristic imaging findings in chILD (eg, ground glass opacities, septal thickening, nodules, and cysts).187 Chest MRI alternating with HRCT can be useful for following disease progression or therapeutic response in patients with established chILD. For patients who are more sensitive to radiation (eg, patients with dyskeratosis congenita, ataxia telangiectasia, and others) exposure to HRCT may increase the risk for fibrosis or oncologic manifestations, and therefore, using MRI to investigate chILD may be pursued, acknowledging the decreased image resolution. Modern 1H ultra-short echo time MR images have been reported to provide structural information similar to HRCT, but without the ionizing radiation exposure.188 Additionally, hyperpolarized xenon 129 use with MRI is sensitive in detecting airflow obstruction in patients with obstructive lung disease and can be performed even in young children who are unable to perform spirometry.189,190 These modalities are active areas of investigation and have demonstrated utility in detecting lung abnormalities in infants and children with chILD and other chronic lung diseases.191–193
RESEARCH NETWORKS AND FAMILY FOUNDATIONS
In 2004, the US chILD Research Network (chILDRN) and chILD Foundation were established with the shared goals of advancing care for children with chILD disorders and providing support for affected families. As part of the National Institutes of Health funded Rare Lung Diseases Consortium held in 2004, Robin Deterding, MD, and Lisa Young, MD, established the chILDRN and brought together 6 families to create the chILD Foundation. Over the next decade, the chILDRN and chILD Foundation held annual conferences, assembling families, physicians, and investigators with clinical and research interests in chILD to advance collaboration and identify goals.112 Early landmark accomplishments of the US chILDRN include developing a classification scheme for diffuse lung disease in pediatric lung biopsies and application to biopsies obtained in infants aged 0 to 2 years1 and children aged 2 to 18 years.3 These publications provided a framework for grouping chILD disorders by histopathologic findings to identify similarities and differences in clinical presentation, outcomes, and mortality. More recently, in collaboration with international networks, chILD researchers from European Union (EU) and the US designed and completed an international trial for children with fibrosing chILD discussed in the treatment section. This study highlights the need not only to classify a disease process at diagnosis, but follow over time, as, for example, multiple disparate conditions may result in the final common pathway of fibrosis.
The US chILDRN meets through monthly virtual meetings to discuss difficult clinical cases, research, and recent publications. This format allows for broad participation, collaboration, and sharing expertise among centers. Currently, over 40 sites in the United States and Canada participate in the chILDRN. Recently, working groups focused on Education, Clinical Challenges, Radiology, Pathology, Genetics, and the chILDRN national patient registry have been formed to facilitate focused collaboration, research, and scholarship. The original American Thoracic Society (ATS) practice guideline regarding diagnosis and management of chILD in infancy was published in 2013 by members of the chILDRN.2 Including input from multiple specialties and countries, this study emphasized excluding more common conditions and a systematic approach, including detailed family history, imaging, and genetic studies. Recent accomplishments of the chILDRN include publication of the initial enrollment cohort of the chILD national registry,113 an interest group supported by the ATS, multiple clinical and research abstracts at the annual ATS international meeting, several publications,88,89 webinars, and educational materials for pediatric pulmonologists, pediatricians, patients, and families. Many of these activities have been performed in collaboration with the chILD Foundation (child-foundation.org). Of note, the chILD Foundation is a key partner in clinical and translation research and has funded several research projects within and outside of the chILDRN, fostering discovery and ongoing collaboration.12,19,194,195
PATIENT REGISTRIES
The US chILDRN, EU chILD, and Australia and New Zealand (AustralANZ), and others have published reports describing their individual patient registries. A recent US registry report, with both retrospective and prospective multicentered data collection from participating specialized chILD centers included demographic, clinical, pulmonary function testing, genetic, radiographic, and histopathologic data.113 In this cohort of 683 registry participants, the median current age was 11 years, with median age at diagnosis of 22 months. The most frequent diagnosis (22.7% of registry participants) was NEHI, followed by connective tissue or immune-mediated chILD disorders (16.5%) and “unclassified ILD” (11%). Respiratory morbidity at time of enrollment was assessed using the Fan severity of illness score for ILD,114 as reported by the enrolling center for each participant. While a quarter of participants were asymptomatic, the lifetime morbidity experienced by children in this cohort is substantial with a total mortality of 6% in those with available outcome data. Overall, 63% of participants had a history of home supplemental oxygen use and 5.1% were symptomatic with pulmonary hypertension. Longitudinal data collection is ongoing, providing opportunities for future clinical research, according to diagnosis (eg, NEHI) and outcome (eg, fibrosis).
The initial report of the chILD EU international registry includes 575 participants and multiple chILD diagnoses. Of note, multidisciplinary review was conducted for 376 participants by the chILD EU collaborative, with the diagnosis compared to that of the referring center. For 13% of participants, the diagnosis was changed following this collaborative review.115 The most frequent diagnoses were surfactant related (21.7%), infant conditions of undefined etiology (18.5%), and diffuse lung disease associated with a systemic disease process (13.6%).115 To date, the EU registry has enrolled more than 1000 children.116 The EU collaborative defined and evaluated the impact of acute exacerbations in 719 chILD registry participants.169 Exacerbations were associated with tachypnea, dyspnea, and increased supplemental oxygen requirement, as well as a decline in pulmonary function that did not return to baseline.169 Viral illness triggered most exacerbations. During the study period, 81 registry participants died, and exacerbation was associated with mortality in 60% of these participants.169 The EU collaborative also reported longitudinal outcome data for 79 participants with chILD resulting from ABCA3 deficiency, including participants who have reached adulthood.196 Additional publications include development and validation of a health-related quality of life instrument specific to chILD,197 a randomized controlled trial of hydroxychloroquine for participants with surfactant dysfunction disorders,198 and a report detailing high health care resource utilization for chILD disorders.199
An initial registry report from chILD AustralANZ included 108 participants with chILD from 8 centers.200 Disease prevalence during the period was estimated as 1.5 affected per million children aged 0 to 18 years.200 Of those with defined diagnoses, NEHI was the most common (13%), and overall mortality was 6.9%.200 Recently, this group evaluated the use of trio exome sequencing for 36 participants with chILD and identified 5 pathogenic variants in genes associated with chILD disorders, including 4 genes not typically included on clinically available panels, yielding a genetic diagnostic rate of 14%.81 The chILD group from Turkey recently published their registry experience, which began in 2021 and includes 416 participants from 19 centers.201 Children were divided by age group (0–2 years, 2–18 years) which permitted clinical comparisons between those diagnosed in infancy and those diagnosed later in childhood. Participants diagnosed during infancy were more likely to have a lower weight Z score and ground glass opacities on chest imaging, whereas older children were more likely to have bronchiectasis, mosaic attenuation, and nodular opacities and receive steroids.201 Increased collaboration of the international chILD networks remains an ongoing priority to advance our collective understanding of these rare disorders and develop effective therapies.
FUTURE DIRECTIONS
Given the morbidity and mortality of chILD disorders, improved therapies are desperately needed. Innovations in understanding the natural history of specific chILD disorders as well as genetic and disease mechanisms will lead to more specific and targeted therapies, improve quality of life, and reduce mortality, as has recently been demonstrated for individuals with cystic fibrosis. The care of children with chILD can continue to thrive through raised awareness, reducing delays in diagnosis, and referral to chILDRN centers, inclusion of all participants in registry studies, and support of clinical, basic, and translational research.
KEY POINTS.
Childhood interstitial and diffuse lung disease (chILD) is an umbrella term that encompasses numerous heterogeneous, diffuse lung disorders. Most chILD disorders are rare or ultra-rare, but collectively there are many affected infants and children, often with severe lung disease. The true incidence and prevalence of chILD are unknown.
During the 1990s to early 2000s, it was recognized that the etiologies of chILD disorders differed from those of adult interstitial lung diseases. Some chILD disorders are primary lung disorders, while some chILD disorders are secondary to a systemic etiology.
While chILD disorders most frequently affect the interstitial compartment of the lungs, often multiple compartments are involved including the alveoli, airways, and pulmonary vasculature.
In 2006, the term chILD was coined, and a working clinical definition of chILD was established. ChILD syndrome is defined as 3 of the following 4 criteria in the absence of another etiology: (1) respiratory signs including, but not limited to, tachypnea, retractions, clubbing, crackles, wheezing, and failure to thrive; (2) respiratory symptoms including, but not limited to, cough, increased work of breathing, and exercise intolerance; (3) hypoxemia either at rest, with activity, or during sleep; and (4) diffuse abnormalities on chest imaging. After the identification of chILD disorders, the chILD research network was formed and developed a pediatric-specific classification scheme that still frames our thinking of chILD disorders.
CLINICS CARE POINTS.
ChILD are a diverse group of rare disorders that primarily affect the alveolar and interstitial space but can involve multiple compartments of the lung leading to significant morbidity and mortality.
In recent years, significant advancements have been made in chILD including classification, genetic discovery, understanding the disease mechanisms of specific disorders, and therapeutic developments.
ChILD research networks and patient registries have provided a framework for innovation in standardizing classification, developing diagnostic guidelines, understanding the pathobiology and disease mechanisms, discovering genetic etiologies, establishing infrastructure for clinical trials, and facilitating natural history studies, multicenter collaboration, patient and family education, and advocacy.
ACKNOWLEDGMENTS
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, United States, NHGRI, United States, NHLBI, United States, NIDA, United States, NIMH, United States, and NINDS, United States. The data used for the analyses described in this article were obtained from the GTEx Portal in April 2024.
Footnotes
DISCLOSURE
Dr A. Casey has no disclosures. Drs E.K. Fiorino and J. Wambach developed continuing medical education in childhood interstitial lung disease for Simumetrix. Support: National Institutes of Health, United States (R01–149853- JAW), Washington University School of Medicine, United States/St. Louis Children’s Hospital Children’s Discovery Institute (JAW).
REFERENCES
- 1.Deutsch GH, Young LR, Deterding RR, et al. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med 2007;176(11):1120–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kurland G, Deterding RR, Hagood JS, et al. An official american thoracic society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med 2013;188(3):376–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langston C, Dishop MK. Diffuse lung disease in infancy: a proposed classification applied to 259 diagnostic biopsies. Pediatr Dev Pathol 2009;12(6):421–37. [DOI] [PubMed] [Google Scholar]
- 4.Fan LL, Dishop MK, Galambos C, et al. Diffuse lung disease in biopsied children 2 to 18 years of age. Application of the child classification scheme. Ann Am Thorac Soc 2015;12(10):1498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nevel RJ, Deutsch GH, Craven D, et al. The US national registry for childhood interstitial and diffuse lung disease: report of study design and initial enrollment cohort. Pediatr Pulmonol; 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deterding RR, Pye C, Fan LL, et al. Persistent tachypnea of infancy is associated with neuroendocrine cell hyperplasia. Pediatr Pulmonol 2005;40(2):157–65. [DOI] [PubMed] [Google Scholar]
- 7.Deterding RR, Fan LL, Morton R, et al. Persistent tachypnea of infancy (pti)–a new entity. Pediatr Pulmonol 2001;Suppl 23:72–3. [PubMed] [Google Scholar]
- 8.Brody AS, Guillerman RP, Hay TC, et al. Neuroendocrine cell hyperplasia of infancy: diagnosis with high-resolution ct. AJR Am J Roentgenol 2010;194(1):238–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Connor MG, Wurth M, Young LR. Rare becomes more common: recognizing neuroendocrine cell hyperplasia of infancy in everyday pulmonary consultations. Ann Am Thorac Soc 2015;12(11):1730–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mastej EJ, DeBoer EM, Humphries SM, et al. Lung and airway shape in neuroendocrine cell hyperplasia of infancy. Pediatr Radiol 2018;48(12):1745–54. [DOI] [PubMed] [Google Scholar]
- 11.Young LR, Brody AS, Inge TH, et al. Neuroendocrine cell distribution and frequency distinguish neuroendocrine cell hyperplasia of infancy from other pulmonary disorders. Chest 2011;139(5):1060–71. [DOI] [PubMed] [Google Scholar]
- 12.Xu J, Xu L, Sui P, et al. Excess neuropeptides in lung signal through endothelial cells to impair gas exchange. Dev Cell 2022;57(7):839–853 e836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doan ML, Elidemir O, Dishop MK, et al. Serum kl-6 differentiates neuroendocrine cell hyperplasia of infancy from the inborn errors of surfactant metabolism. Thorax 2009;64(8):677–81. [DOI] [PubMed] [Google Scholar]
- 14.Deterding RR, Wagner BD, Harris JK, et al. Pulmonary aptamer signatures in children’s interstitial and diffuse lung disease. Am J Respir Crit Care Med 2019;200(12):1496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Popler J, Wagner BD, Tarro HL, et al. Bronchoalveolar lavage fluid cytokine profiles in neuroendocrine cell hyperplasia of infancy and follicular bronchiolitis. Orphanet J Rare Dis 2013;8:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young LR, Deutsch GH, Bokulic RE, et al. A mutation in ttf1/nkx2.1 is associated with familial neuroendocrine cell hyperplasia of infancy. Chest 2013;144(4):1199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nevel RJ, Garnett ET, Schaudies DA, et al. Growth trajectories and oxygen use in neuroendocrine cell hyperplasia of infancy. Pediatr Pulmonol 2018;53(5):656–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liptzin DR, Pickett K, Brinton JT, et al. Neuroendocrine cell hyperplasia of infancy. Clinical score and comorbidities. Ann Am Thorac Soc 2020;17(6):724–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nevel RJ, Garnett ET, Worrell JA, et al. Persistent lung disease in adults with nkx2.1 mutation and familial neuroendocrine cell hyperplasia of infancy. Ann Am Thorac Soc 2016;13(8):1299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Canakis AM, Cutz E, Manson D, et al. Pulmonary interstitial glycogenosis: a new variant of neonatal interstitial lung disease. Am J Respir Crit Care Med 2002;165(11):1557–65. [DOI] [PubMed] [Google Scholar]
- 21.Liptzin DR, Udoko MN, Pinder M, et al. Pulmonary interstitial glycogenosis after the first year. Pediatr Pulmonol 2021;56(9):3056–8. [DOI] [PubMed] [Google Scholar]
- 22.Deutsch GH, Young LR. Pulmonary interstitial glycogenosis: Words of caution. Pediatr Radiol 2010;40(9):1471–5. [DOI] [PubMed] [Google Scholar]
- 23.Castillo M, Vade A, Lim-Dunham JE, et al. Pulmonary interstitial glycogenosis in the setting of lung growth abnormality: radiographic and pathologic correlation. Pediatr Radiol 2010;40(9):1562–5. [DOI] [PubMed] [Google Scholar]
- 24.Galambos C, Wartchow E, Weinman JP, et al. Pulmonary interstitial glycogenosis cells express mesenchymal stem cell markers. Eur Respir J 2020;56(4):2000853. [DOI] [PubMed] [Google Scholar]
- 25.Liptzin DR, Baker CD, Darst JR, et al. Pulmonary interstitial glycogenosis: diagnostic evaluation and clinical course. Pediatr Pulmonol 2018;53(12):1651–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weinman JP, White CJ, Liptzin DR, et al. High-resolution ct findings of pulmonary interstitial glycogenosis. Pediatr Radiol 2018;48(8):1066–72. [DOI] [PubMed] [Google Scholar]
- 27.Deutsch GH, Young LR. Histologic resolution of pulmonary interstitial glycogenosis. Pediatr Dev Pathol 2009;12(6):475–80. [DOI] [PubMed] [Google Scholar]
- 28.Ross MK, Ellis LS, Bird LM, et al. Pulmonary interstitial glycogenosis in a patient ultimately diagnosed with noonan syndrome. Pediatr Pulmonol 2014;49(5):508–11. [DOI] [PubMed] [Google Scholar]
- 29.Saper VE, Chen G, Deutsch GH, et al. Emergent high fatality lung disease in systemic juvenile arthritis. Ann Rheum Dis 2019;78(12):1722–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCarthy C, Carey BC, Trapnell BC. Autoimmune pulmonary alveolar proteinosis. Am J Respir Crit Care Med 2022;205(9):1016–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tangye SG, Al-Herz W, Bousfiha A, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J Clin Immunol 2022;42(7):1473–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walter JE, Ziegler JB, Ballow M, et al. Advances and challenges of the decade: the ever-changing clinical and genetic landscape of immunodeficiency. J Allergy Clin Immunol Pract 2023;11(1):107–15. [DOI] [PubMed] [Google Scholar]
- 33.Long A, Kleiner A, Looney RJ. Immune dysregulation. J Allergy Clin Immunol 2023;151(1):70–80. [DOI] [PubMed] [Google Scholar]
- 34.Sogkas G, Witte T. The link between rheumatic disorders and inborn errors of immunity. EBioMedicine 2023;90:104501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wobma H, Shin DS, Chou J, et al. Dysregulation of the cgas-sting pathway in monogenic autoinflammation and lupus. Front Immunol 2022;13:905109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hiraki LT, Silverman ED. Genomics of systemic lupus erythematosus: insights gained by studying monogenic young-onset systemic lupus erythematosus. Rheum Dis Clin North Am 2017;43(3):415–34. [DOI] [PubMed] [Google Scholar]
- 37.Barker AF, Bergeron A, Rom WN, et al. Obliterative bronchiolitis. N Engl J Med 2014;370(19):1820–8. [DOI] [PubMed] [Google Scholar]
- 38.Kurland G, Michelson P. Bronchiolitis obliterans in children. Pediatr Pulmonol 2005;39(3):193–208. [DOI] [PubMed] [Google Scholar]
- 39.King T. Bronchiolitis. In: Fishman’s pulmonary diseases and disorders. New York: McGraw-Hill; 1998. [Google Scholar]
- 40.Jerkic SP, Brinkmann F, Calder A, et al. Postinfectious bronchiolitis obliterans in children: diagnostic workup and therapeutic options: a workshop report. Can Respir J 2020;2020:5852827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flanagan F, Casey A, Reyes-Mugica M, et al. Postinfectious bronchiolitis obliterans in children. Paediatr Respir Rev 2022;42:69–78. [DOI] [PubMed] [Google Scholar]
- 42.Colom AJ, Maffey A, Garcia Bournissen F, et al. Pulmonary function of a paediatric cohort of patients with postinfectious bronchiolitis obliterans. A long term follow-up. Thorax 2015;70(2):169–74. [DOI] [PubMed] [Google Scholar]
- 43.Moonnumakal SP, Fan LL. Bronchiolitis obliterans in children. Curr Opin Pediatr 2008;20(3):272–8. [DOI] [PubMed] [Google Scholar]
- 44.Frohlich LF, Vieira PJ, Teixeira PJ, et al. Exercise capacity in adolescent and adult patients with post infectious bronchiolitis obliterans. Pediatr Pulmonol 2014;49(9):911–8. [DOI] [PubMed] [Google Scholar]
- 45.Colom AJ, Teper AM. Clinical prediction rule to diagnose post-infectious bronchiolitis obliterans in children. Pediatr Pulmonol 2009;44(11):1065–9. [DOI] [PubMed] [Google Scholar]
- 46.Fischer GB, Sarria EE, Mattiello R, et al. Post infectious bronchiolitis obliterans in children. Paediatr Respir Rev 2010;11(4):233–9. [DOI] [PubMed] [Google Scholar]
- 47.Hamada K, Oishi K, Hirano T, et al. Swyer-james syndrome. Am J Respir Crit Care Med 2018;197(1):130–1. [DOI] [PubMed] [Google Scholar]
- 48.Tomikawa SO, Adde FV, da Silva Filho LV, et al. Follow-up on pediatric patients with bronchiolitis obliterans treated with corticosteroid pulse therapy. Orphanet J Rare Dis 2014;9:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yilmaz AI, Gul Y, Kapakli H, et al. Successful treatment of postinfectious bronchiolitis obliterans with gamma globulin in a tertiary center: 10 years of experience. Pediatr Pulmonol 2023;58(10):2769–76. [DOI] [PubMed] [Google Scholar]
- 50.Teixeira MFC, Rodrigues JC, Leone C, et al. Acute bronchodilator responsiveness to tiotropium in postinfectious bronchiolitis obliterans in children. Chest 2013;144(3):974–80. [DOI] [PubMed] [Google Scholar]
- 51.Penack O, Marchetti M, Ruutu T, et al. Prophylaxis and management of graft versus host disease after stem-cell transplantation for haematological malignancies: updated consensus recommendations of the european society for blood and marrow transplantation. Lancet Haematol 2020;7(2):e157–67. [DOI] [PubMed] [Google Scholar]
- 52.Norman BC, Jacobsohn DA, Williams KM, et al. Fluticasone, azithromycin and montelukast therapy in reducing corticosteroid exposure in bronchiolitis obliterans syndrome after allogeneic hematopoietic sct: a case series of eight patients. Bone Marrow Transplant 2011;46(10):1369–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Williams KM, Cheng GS, Pusic I, et al. Fluticasone, azithromycin, and montelukast treatment for new-onset bronchiolitis obliterans syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016;22(4):710–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brownback KR, Simpson SQ, Pitts LR, et al. Effect of extracorporeal photopheresis on lung function decline for severe bronchiolitis obliterans syndrome following allogeneic stem cell transplantation. J Clin Apher 2016;31(4):347–52. [DOI] [PubMed] [Google Scholar]
- 55.Del Fante C, Galasso T, Bernasconi P, et al. Extracorporeal photopheresis as a new supportive therapy for bronchiolitis obliterans syndrome after allogeneic stem cell transplantation. Bone Marrow Transplant 2016;51(5):728–31. [DOI] [PubMed] [Google Scholar]
- 56.Del Fante C, Scudeller L, Oggionni T, et al. Long-term off-line extracorporeal photochemotherapy in patients with chronic lung allograft rejection not responsive to conventional treatment: a 10-year single-centre analysis. Respiration 2015;90(2):118–28. [DOI] [PubMed] [Google Scholar]
- 57.Greer M, Dierich M, De Wall C, et al. Phenotyping established chronic lung allograft dysfunction predicts extracorporeal photopheresis response in lung transplant patients. Am J Transplant 2013;13(4):911–8. [DOI] [PubMed] [Google Scholar]
- 58.Hefazi M, Langer KJ, Khera N, et al. Extracorporeal photopheresis improves survival in hematopoietic cell transplant patients with bronchiolitis obliterans syndrome without significantly impacting measured pulmonary functions. Biol Blood Marrow Transplant 2018;24(9):1906–13. [DOI] [PubMed] [Google Scholar]
- 59.Jaksch P, Scheed A, Keplinger M, et al. A prospective interventional study on the use of extracorporeal photopheresis in patients with bronchiolitis obliterans syndrome after lung transplantation. J Heart Lung Transplant 2012;31(9):950–7. [DOI] [PubMed] [Google Scholar]
- 60.Lucid CE, Savani BN, Engelhardt BG, et al. Extracorporeal photopheresis in patients with refractory bronchiolitis obliterans developing after allo-sct. Bone Marrow Transplant 2011;46(3):426–9. [DOI] [PubMed] [Google Scholar]
- 61.Morrell MR, Despotis GJ, Lublin DM, et al. The efficacy of photopheresis for bronchiolitis obliterans syndrome after lung transplantation. J Heart Lung Transplant 2010;29(4):424–31. [DOI] [PubMed] [Google Scholar]
- 62.Stadler M, Ahlborn R, Kamal H, et al. Limited efficacy of imatinib in severe pulmonary chronic graft-versus-host disease. Blood 2009;114(17):3718–9. author reply 3719–3720. [DOI] [PubMed] [Google Scholar]
- 63.Watanabe S, Waseda Y, Kimura H, et al. Imatinib for bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant 2015;50(9):1250–2. [DOI] [PubMed] [Google Scholar]
- 64.Schoettler M, Duncan C, Lehmann L, et al. Ruxolitinib is an effective steroid sparing agent in children with steroid refractory/dependent bronchiolitis obliterans syndrome after allogenic hematopoietic cell transplantation. Bone Marrow Transplant 2019;54(7):1158–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Streiler C, Shaikh F, Davis C, et al. Ruxolitinib is an effective steroid sparing agent in bronchiolitis obliterans due to chronic graft-versus-host-disease. Bone Marrow Transplant 2020;55(6):1194–6. [DOI] [PubMed] [Google Scholar]
- 66.Zeiser R, Polverelli N, Ram R, et al. Ruxolitinib for glucocorticoid-refractory chronic graft-versus-host disease. N Engl J Med 2021;385(3):228–38. [DOI] [PubMed] [Google Scholar]
- 67.Zhang MY, Zhao P, Zhang Y, et al. Efficacy and safety of ruxolitinib for steroid-refractory graft-versus-host disease: systematic review and meta-analysis of randomised and non-randomised studies. PLoS One 2022;17(7):e0271979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao Y, OuYang G, Shi J, et al. Salvage therapy with low-dose ruxolitinib leads to a significant improvement in bronchiolitis obliterans syndrome in patients with cgvhd after allogeneic hematopoietic stem cell transplantation. Front Pharmacol 2021;12:668825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cutler c, lee sj, arai s, et al. Belumosudil for chronic graft-versus-host disease after 2 or more prior lines of therapy: the rockstar study. Blood. 2021;138(22):2278–2289. Blood 2022;139(11):1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Defilipp z, kim ht, yang z, et al. Clinical response to belumosudil in bronchiolitis obliterans syndrome: a combined analysis from 2 prospective trials. Blood adv. 2022;6(24):6263–6270. Blood Adv 2023;7(22):7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jagasia M, Lazaryan A, Bachier CR, et al. Rock2 inhibition with belumosudil (kd025) for the treatment of chronic graft-versus-host disease. J Clin Oncol 2021;39(17):1888–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim HT, Koreth J, Whangbo J, et al. Organ-specific response after low-dose interleukin-2 therapy for steroid-refractory chronic graft-versus-host disease. Blood Adv 2022;6(15):4392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koreth J, Kim HT, Jones KT, et al. Efficacy, durability, and response predictors of low-dose interleukin-2 therapy for chronic graft-versus-host disease. Blood 2016;128(1):130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Whangbo JS, Kim HT, Mirkovic N, et al. Dose-escalated interleukin-2 therapy for refractory chronic graft-versus-host disease in adults and children. Blood Adv 2019;3(17):2550–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Borthwick LA, Corris PA, Mahida R, et al. Tnfalpha from classically activated macrophages accentuates epithelial to mesenchymal transition in obliterative bronchiolitis. Am J Transplant 2013;13(3):621–33. [DOI] [PubMed] [Google Scholar]
- 76.Yanik GA, Mineishi S, Levine JE, et al. Soluble tumor necrosis factor receptor: enbrel (etanercept) for subacute pulmonary dysfunction following allogeneic stem cell transplantation. Biol Blood Marrow Transplant 2012;18(7):1044–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nogee LM, de Mello DE, Dehner LP, et al. Brief report: deficiency of pulmonary surfactant protein b in congenital alveolar proteinosis. N Engl J Med 1993;328(6):406–10. [DOI] [PubMed] [Google Scholar]
- 78.Shulenin S, Nogee LM, Annilo T, et al. Abca3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med 2004;350(13):1296–303. [DOI] [PubMed] [Google Scholar]
- 79.Nogee LM, Dunbar AE 3rd, Wert SE, et al. A mutation in the surfactant protein c gene associated with familial interstitial lung disease. N Engl J Med 2001;344(8):573–9. [DOI] [PubMed] [Google Scholar]
- 80.Group NIS, Krantz ID, Medne L, et al. Effect of whole-genome sequencing on the clinical management of acutely ill infants with suspected genetic disease: a randomized clinical trial. JAMA Pediatr 2021;175(12):1218–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Temple SEL, Ho G, Bennetts B, et al. The role of exome sequencing in childhood interstitial or diffuse lung disease. Orphanet J Rare Dis 2022;17(1):350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang H, Pan J, Spielberg DR, et al. A dominant negative variant of rab5b disrupts maturation of surfactant protein b and surfactant protein c. Proc Natl Acad Sci U S A 2022;119(6). e2105228119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.O’Reilly MA, Weaver TE, Pilot-Matias TJ, et al. In vitro translation, post-translational processing and secretion of pulmonary surfactant protein b precursors. Biochim Biophys Acta 1989;1011(2–3):140–8. [DOI] [PubMed] [Google Scholar]
- 84.Eldridge WB, Zhang Q, Faro A, et al. Outcomes of lung transplantation for infants and children with genetic disorders of surfactant metabolism. J Pediatr 2017;184:157–164 e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nogee LM, Wert SE, Proffit SA, et al. Allelic heterogeneity in hereditary surfactant protein b (sp-b) deficiency. Am J Respir Crit Care Med 2000;161(3 Pt 1):973–81. [DOI] [PubMed] [Google Scholar]
- 86.Wert SE, Whitsett JA, Nogee LM. Genetic disorders of surfactant dysfunction. Pediatr Dev Pathol 2009;12(4):253–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581(7809):434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lopez-Andreu JA, Hidalgo-Santos AD, Fuentes-Castello MA, et al. Delayed presentation and prolonged survival of a child with surfactant protein b deficiency. J Pediatr 2017;190:268–270 e261. [DOI] [PubMed] [Google Scholar]
- 89.Dunbar AE 3rd, Wert SE, Ikegami M, et al. Prolonged survival in hereditary surfactant protein b (sp-b) deficiency associated with a novel splicing mutation. Pediatr Res 2000;48(3):275–82. [DOI] [PubMed] [Google Scholar]
- 90.Wambach JA, Wegner DJ, Kitzmiller J, et al. Homozygous, intragenic tandem duplication of sftpb causes neonatal respiratory failure. Am J Respir Cell Mol Biol 2024;70(1):78–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Palomar LM, Nogee LM, Sweet SC, et al. Long-term outcomes after infant lung transplantation for surfactant protein b deficiency related to other causes of respiratory failure. J Pediatr 2006;149(4):548–53. [DOI] [PubMed] [Google Scholar]
- 92.Kang MH, van Lieshout LP, Xu L, et al. A lung tropic aav vector improves survival in a mouse model of surfactant b deficiency. Nat Commun 2020;11(1):3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Peers de Nieuwburgh M, Wambach JA, Griese M, et al. Towards personalized therapies for genetic disorders of surfactant dysfunction. Semin Fetal Neonatal Med 2023;28(6):101500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cooney AL, Wambach JA, Sinn PL, et al. Gene therapy potential for genetic disorders of surfactant dysfunction. Front Genome 2021;3:785829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sitaraman S, Alysandratos KD, Wambach JA, et al. Gene therapeutics for surfactant dysfunction disorders: targeting the alveolar type 2 epithelial cell. Hum Gene Ther 2022;33(19–20):1011–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Glasser SW, Korfhagen TR, Bruno MD, et al. Structure and expression of the pulmonary surfactant protein sp-c gene in the mouse. J Biol Chem 1990;265(35):21986–91. [PubMed] [Google Scholar]
- 97.Korfhagen TR, Glasser SW, Wert SE, et al. Cis-acting sequences from a human surfactant protein gene confer pulmonary-specific gene expression in transgenic mice. Proc Natl Acad Sci U S A 1990;87(16):6122–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mulugeta S, Nguyen V, Russo SJ, et al. A surfactant protein c precursor protein brichos domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am J Respir Cell Mol Biol 2005;32(6):521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Beers MF, Lomax C. Synthesis and processing of hydrophobic surfactant protein c by isolated rat type ii cells. Am J Physiol 1995;269(6 Pt 1): L744–53. [DOI] [PubMed] [Google Scholar]
- 100.Beers MF, Kim CY, Dodia C, et al. Localization, synthesis, and processing of surfactant protein sp-c in rat lung analyzed by epitope-specific antipeptide antibodies. J Biol Chem 1994;269(32):20318–28. [PubMed] [Google Scholar]
- 101.Glasser SW, Burhans MS, Korfhagen TR, et al. Altered stability of pulmonary surfactant in sp-c-deficient mice. Proc Natl Acad Sci U S A 2001;98(11):6366–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Thomas AQ, Lane K, Phillips J, et al. Heterozygosity for a surfactant protein c gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med 2002;165(9):1322–8. [DOI] [PubMed] [Google Scholar]
- 103.Bullard JE, Nogee LM. Heterozygosity for abca3 mutations modifies the severity of lung disease associated with a surfactant protein c gene (sftpc) mutation. Pediatr Res 2007;62(2):176–9. [DOI] [PubMed] [Google Scholar]
- 104.Coghlan MA, Shifren A, Huang HJ, et al. Sequencing of idiopathic pulmonary fibrosis-related genes reveals independent single gene associations. BMJ Open Respir Res 2014;1(1):e000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wambach JA, Yang P, Wegner DJ, et al. Surfactant protein-c promoter variants associated with neonatal respiratory distress syndrome reduce transcription. Pediatr Res 2010;68(3):216–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nerelius C, Martin E, Peng S, et al. Mutations linked to interstitial lung disease can abrogate anti-amyloid function of prosurfactant protein c. Biochem J 2008;416(2):201–9. [DOI] [PubMed] [Google Scholar]
- 107.Mulugeta S, Maguire JA, Newitt JL, et al. Misfolded brichos sp-c mutant proteins induce apoptosis via caspase-4- and cytochrome c-related mechanisms. Am J Physiol Lung Cell Mol Physiol 2007;293(3):L720–9. [DOI] [PubMed] [Google Scholar]
- 108.Katzen J, Wagner BD, Venosa A, et al. An sftpc brichos mutant links epithelial er stress and spontaneous lung fibrosis. JCI Insight 2019;4(6):e126125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nureki SI, Tomer Y, Venosa A, et al. Expression of mutant sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J Clin Invest 2018;128(9):4008–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Alysandratos KD, Russo SJ, Petcherski A, et al. Patient-specific ipscs carrying an sftpc mutation reveal the intrinsic alveolar epithelial dysfunction at the inception of interstitial lung disease. Cell Rep 2021;36(9):109636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kinting S, Hoppner S, Schindlbeck U, et al. Functional rescue of misfolding abca3 mutations by small molecular correctors. Hum Mol Genet 2018;27(6):943–53. [DOI] [PubMed] [Google Scholar]
- 112.Kinting S, Li Y, Forstner M, et al. Potentiation of abca3 lipid transport function by ivacaftor and genistein. J Cell Mol Med 2019;23(8):5225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wambach JA, Casey AM, Fishman MP, et al. Genotype-phenotype correlations for infants and children with abca3 deficiency. Am J Respir Crit Care Med 2014;189(12):1538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Klay D, Platenburg M, van Rijswijk R, et al. Abca3 mutations in adult pulmonary fibrosis patients: a case series and review of literature. Curr Opin Pulm Med 2020;26(3):293–301. [DOI] [PubMed] [Google Scholar]
- 115.Kroner C, Wittmann T, Reu S, et al. Lung disease caused by abca3 mutations. Thorax 2017;72(3):213–20. [DOI] [PubMed] [Google Scholar]
- 116.Tomer Y, Wambach J, Knudsen L, et al. The common abca3(e292v) variant disrupts at2 cell quality control and increases susceptibility to lung injury and aberrant remodeling. Am J Physiol Lung Cell Mol Physiol 2021;321(2):L291–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mulugeta S, Gray JM, Notarfrancesco KL, et al. Identification of lbm180, a lamellar body limiting membrane protein of alveolar type ii cells, as the abc transporter protein abca3. J Biol Chem 2002;277(25):22147–55. [DOI] [PubMed] [Google Scholar]
- 118.Hamvas A, Nogee LM, Wegner DJ, et al. Inherited surfactant deficiency caused by uniparental disomy of rare mutations in the surfactant protein-b and atp binding cassette, subfamily a, member 3 genes. J Pediatr 2009;155(6):854–859 e851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wambach JA, Wegner DJ, Depass K, et al. Single abca3 mutations increase risk for neonatal respiratory distress syndrome. Pediatrics 2012;130(6):e1575–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Naderi HM, Murray JC, Dagle JM. Single mutations in abca3 increase the risk for neonatal respiratory distress syndrome in late preterm infants (gestational age 34–36 weeks). Am J Med Genet A 2014;164A(10):2676–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wittmann T, Frixel S, Hoppner S, et al. Increased risk of interstitial lung disease in children with a single r288k variant of abca3. Mol Med 2016;22:183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wambach JA, Yang P, Wegner DJ, et al. Functional characterization of atp-binding cassette transporter a3 mutations from infants with respiratory distress syndrome. Am J Respir Cell Mol Biol 2016;55(5):716–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wambach JA, Yang P, Wegner DJ, et al. Functional genomics of abca3 variants. Am J Respir Cell Mol Biol 2020;63(4):436–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Matsumura Y, Ban N, Inagaki N. Aberrant catalytic cycle and impaired lipid transport into intracellular vesicles in abca3 mutants associated with nonfatal pediatric interstitial lung disease. Am J Physiol Lung Cell Mol Physiol 2008;295(4):L698–707. [DOI] [PubMed] [Google Scholar]
- 125.Matsumura Y, Ban N, Ueda K, et al. Characterization and classification of atp-binding cassette transporter abca3 mutants in fatal surfactant deficiency. J Biol Chem 2006;281(45):34503–14. [DOI] [PubMed] [Google Scholar]
- 126.Matsumura Y, Sakai H, Sasaki M, et al. Abca3-mediated choline-phospholipids uptake into intracellular vesicles in a549 cells. FEBS Lett 2007;581(17):3139–44. [DOI] [PubMed] [Google Scholar]
- 127.Minoo P, Hamdan H, Bu D, et al. Ttf-1 regulates lung epithelial morphogenesis. Dev Biol 1995;172(2):694–8. [DOI] [PubMed] [Google Scholar]
- 128.Kimura S, Hara Y, Pineau T, et al. The t/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev 1996;10(1):60–9. [DOI] [PubMed] [Google Scholar]
- 129.Hamvas A, Deterding RR, Wert SE, et al. Heterogeneous pulmonary phenotypes associated with mutations in the thyroid transcription factor gene nkx2–1. Chest 2013;144(3):794–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Krude H, Schutz B, Biebermann H, et al. Choreoathetosis, hypothyroidism, and pulmonary alterations due to human nkx2–1 haploinsufficiency. J Clin Invest 2002;109(4):475–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Thorwarth A, Schnittert-Hubener S, Schrumpf P, et al. Comprehensive genotyping and clinical characterisation reveal 27 novel nkx2–1 mutations and expand the phenotypic spectrum. J Med Genet 2014;51(6):375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Patel NJ, Jankovic J. In: Adam MP, Feldman J, Mirzaa GM, et al. , editors. Nkx2–1-related disorders. Seattle (WA): Genereviews; 1993–2024. [PubMed] [Google Scholar]
- 133.Janney CG, Askin FB, Kuhn C 3rd. Congenital alveolar capillary dysplasia–an unusual cause of respiratory distress in the newborn. Am J Clin Pathol 1981;76(5):722–7. [DOI] [PubMed] [Google Scholar]
- 134.Sen P, Yang Y, Navarro C, et al. Novel foxf1 mutations in sporadic and familial cases of alveolar capillary dysplasia with misaligned pulmonary veins imply a role for its DNA binding domain. Hum Mutat 2013;34(6):801–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Towe CT, White FV, Grady RM, et al. Infants with atypical presentations of alveolar capillary dysplasia with misalignment of the pulmonary veins who underwent bilateral lung transplantation. J Pediatr 2018;194:158–164 e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Edwards JJ, Murali C, Pogoriler J, et al. Histopathologic and genetic features of alveolar capillary dysplasia with atypical late presentation and prolonged survival. J Pediatr 2019;210:214–219 e212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bishop NB, Stankiewicz P, Steinhorn RH. Alveolar capillary dysplasia. Am J Respir Crit Care Med 2011;184(2):172–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sen P, Thakur N, Stockton DW, et al. Expanding the phenotype of alveolar capillary dysplasia (acd). J Pediatr 2004;145(5):646–51. [DOI] [PubMed] [Google Scholar]
- 139.Langston C Misalignment of pulmonary veins and alveolar capillary dysplasia. Pediatr Pathol 1991;11(1):163–70. [DOI] [PubMed] [Google Scholar]
- 140.Galambos C, Sims-Lucas S, Abman SH. Three-dimensional reconstruction identifies misaligned pulmonary veins as intrapulmonary shunt vessels in alveolar capillary dysplasia. J Pediatr 2014;164(1):192–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stankiewicz P, Sen P, Bhatt SS, et al. Genomic and genic deletions of the fox gene cluster on 16q24.1 and inactivating mutations of foxf1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet 2009;84(6):780–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pradhan A, Che L, Ustiyan V, et al. Novel foxf1-stabilizing compound tanfe stimulates lung angiogenesis in alveolar capillary dysplasia. Am J Respir Crit Care Med 2023;207(8):1042–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Watkin LB, Jessen B, Wiszniewski W, et al. Copa mutations impair er-golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet 2015;47(6):654–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ban N, Matsumura Y, Sakai H, et al. Abca3 as a lipid transporter in pulmonary surfactant biogenesis. J Biol Chem 2007;282(13):9628–34. [DOI] [PubMed] [Google Scholar]
- 145.Fremond ML, Nathan N. Copa syndrome, 5 years after: where are we? Joint Bone Spine 2021;88(2):105070. [DOI] [PubMed] [Google Scholar]
- 146.Jeremiah N, Neven B, Gentili M, et al. Inherited sting-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest 2014;124(12):5516–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Fremond ML, Crow YJ. Sting-mediated lung inflammation and beyond. J Clin Immunol 2021;41(3):501–14. [DOI] [PubMed] [Google Scholar]
- 148.Fremond ML, Hadchouel A, Berteloot L, et al. Overview of sting-associated vasculopathy with onset in infancy (savi) among 21 patients. J Allergy Clin Immunol Pract 2021;9(2):803–818 e811. [DOI] [PubMed] [Google Scholar]
- 149.Li J, An S, Du Z. Familial interstitial lung disease caused by mutation of the sting1 gene. Front Pediatr 2020;8:543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Liu Y, Jesus AA, Marrero B, et al. Activated sting in a vascular and pulmonary syndrome. N Engl J Med 2014;371(6):507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schuch LA, Forstner M, Rapp CK, et al. Fars1-related disorders caused by bi-allelic mutations in cytosolic phenylalanyl-trna synthetase genes: look beyond the lungs. Clin Genet 2021;99(6):789–801. [DOI] [PubMed] [Google Scholar]
- 152.Zadjali F, Al-Yahyaee A, Al-Nabhani M, et al. Homozygosity for farsb mutation leads to phe-trna synthetase-related disease of growth restriction, brain calcification, and interstitial lung disease. Hum Mutat 2018;39(10):1355–9. [DOI] [PubMed] [Google Scholar]
- 153.Lenz D, Stahl M, Seidl E, et al. Rescue of respiratory failure in pulmonary alveolar proteinosis due to pathogenic mars1 variants. Pediatr Pulmonol 2020;55(11):3057–66. [DOI] [PubMed] [Google Scholar]
- 154.Rips J, Meyer-Schuman R, Breuer O, et al. Mars variant associated with both recessive interstitial lung and liver disease and dominant charcotmarie-tooth disease. Eur J Med Genet 2018;61(10):616–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Vece TJ, Gower WA, Davis SD, et al. Building a pediatric rare lung disease program: it takes a community of villages. Pediatr Pulmonol 2022;57(11):2583–8. [DOI] [PubMed] [Google Scholar]
- 156.Ruiz AG, Bhatt JM, DeBoer EM, et al. Demonstrating the benefits of a multidisciplinary aerodigestive program. Laryngoscope 2020;130(2):521–5. [DOI] [PubMed] [Google Scholar]
- 157.Gardner AJ, Gray AL, Self S, et al. Strengthening care teams to improve adherence in cystic fibrosis: a qualitative practice assessment and quality improvement initiative. Patient Prefer Adherence 2017;11:761–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shapiro AJ, Zariwala MA, Ferkol T, et al. Diagnosis, monitoring, and treatment of primary ciliary dyskinesia: pcd foundation consensus recommendations based on state of the art review. Pediatr Pulmonol 2016;51(2):115–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sanchez GAM, Reinhardt A, Ramsey S, et al. Jak1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest 2018;128(7):3041–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Krutzke S, Rietschel C, Horneff G. Baricitinib in therapy of copa syndrome in a 15-year-old girl. Eur J Rheumatol 2020;7(Suppl1):S78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Fremond ML, Rodero MP, Jeremiah N, et al. Efficacy of the janus kinase 1/2 inhibitor ruxolitinib in the treatment of vasculopathy associated with tmem173-activating mutations in 3 children. J Allergy Clin Immunol 2016;138(6):1752–5. [DOI] [PubMed] [Google Scholar]
- 162.Young LR, Vece TJ, Guillerman RP. Response. Chest 2016;149(6):1579–80. [DOI] [PubMed] [Google Scholar]
- 163.Spagnolo P, Bush A. Interstitial lung disease in children younger than 2 years. Pediatrics 2016;137(6):e20152725. [DOI] [PubMed] [Google Scholar]
- 164.Bush A, Pavord I. Look back with (some) anger, and a lot of pleasure. Thorax 2015;70(9):819–21. [DOI] [PubMed] [Google Scholar]
- 165.McDougal J, Gettys A, Hagood JS. Child family education. Pediatr Allergy Immunol Pulmonol 2010;23(1):87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Das S, Langston C, Fan LL. Interstitial lung disease in children. Curr Opin Pediatr 2011;23(3):325–31. [DOI] [PubMed] [Google Scholar]
- 167.Vece TJ, Fan LL. Diagnosis and management of diffuse lung disease in children. Paediatr Respir Rev 2011;12(4):238–42. [DOI] [PubMed] [Google Scholar]
- 168.Krivchenia K, Hawkins SM, Iyer NP, et al. 2019 Clinical practice guideline summary for clinicians: home oxygen therapy for children. Ann Am Thorac Soc 2019;16(7):781–5. [DOI] [PubMed] [Google Scholar]
- 169.Seidl E, Schwerk N, Carlens J, et al. Acute exacerbations in children’s interstitial lung disease. Thorax 2022;77(8):799–804. [DOI] [PubMed] [Google Scholar]
- 170.Forstner M, Lin S, Yang X, et al. High-content screening identifies cyclosporin a as a novel abca3-specific molecular corrector. Am J Respir Cell Mol Biol 2022;66(4):382–90. [DOI] [PubMed] [Google Scholar]
- 171.Liptzin DR, McGraw MD, Stidham T, et al. Noninvasive management of infants with sftpc pathogenic variants. Pediatr Pulmonol 2024;59(2):488–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Liptzin DR, Patel T, Deterding RR. Chronic ventilation in infants with surfactant protein c mutations: an alternative to lung transplantation. Am J Respir Crit Care Med 2015;191(11):1338–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Guler SA, Hur SA, Stickland MK, et al. Survival after inpatient or outpatient pulmonary rehabilitation in patients with fibrotic interstitial lung disease: a multicentre retrospective cohort study. Thorax 2022;77(6):589–95. [DOI] [PubMed] [Google Scholar]
- 174.Deterding R, Young LR, DeBoer EM, et al. Nintedanib in children and adolescents with fibrosing interstitial lung diseases. Eur Respir J 2022;61(2):2201512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Deterding R, Young LR, DeBoer EM, et al. Nintedanib in children and adolescents with fibrosing interstitial lung diseases. Eur Respir J 2023;61(2):2251512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014;370(22):2071–82. [DOI] [PubMed] [Google Scholar]
- 177.Distler O, Gahlemann M, Maher TM. Nintedanib for systemic sclerosis-associated interstitial lung disease. Reply. N Engl J Med 2019;381(16):1596–7. [DOI] [PubMed] [Google Scholar]
- 178.Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med 2019;381(18):1718–27. [DOI] [PubMed] [Google Scholar]
- 179.Wobma H, Perkins R, Bartnikas L, et al. Genetic diagnosis of immune dysregulation can lead to targeted therapy for interstitial lung disease: a case series and single center approach. Pediatr Pulmonol 2022;57(7):1577–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.DiPrete O, Winant AJ, Vargas SO, et al. Advances in imaging of the child - childhood interstitial lung disease. Radiol Clin North Am 2022;60(6):1003–20. [DOI] [PubMed] [Google Scholar]
- 181.DeBoer EM, Weinman JP, Ley-Zaporozhan J, et al. Imaging of pulmonary fibrosis in children: a review, with proposed diagnostic criteria. Pediatr Pulmonol 2024;59(4):845–54. [DOI] [PubMed] [Google Scholar]
- 182.Spielberg DR, Weinman J, DeBoer EM. Advancements in imaging in child. Pediatr Pulmonol 2023. [DOI] [PubMed] [Google Scholar]
- 183.Guillerman RP. Imaging of childhood interstitial lung disease. Pediatr Allergy Immunol Pulmonol 2010;23(1):43–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Semple T, Winant AJ, Lee EY. Childhood interstitial lung disease: imaging guidelines and recommendations. Radiol Clin North Am 2022;60(1):83–111. [DOI] [PubMed] [Google Scholar]
- 185.Liang T, Vargas SO, Lee EY. Childhood interstitial (diffuse) lung disease: pattern recognition approach to diagnosis in infants. AJR Am J Roentgenol 2019;212(5):958–67. [DOI] [PubMed] [Google Scholar]
- 186.Balinotti JE, Maffey A, Colom A, et al. Clinical, functional, and computed tomography findings in a cohort of patients with neuroendocrine cell hyperplasia of infancy. Pediatr Pulmonol 2021;56(6):1681–6. [DOI] [PubMed] [Google Scholar]
- 187.Sodhi KS, Sharma M, Lee EY, et al. Diagnostic utility of 3t lung mri in children with interstitial lung disease: a prospective pilot study. Acad Radiol 2018;25(3):380–6. [DOI] [PubMed] [Google Scholar]
- 188.Torres L, Kammerman J, Hahn AD, et al. Structure-function imaging of lung disease using ultrashort echo time mri. Acad Radiol 2019;26(3):431–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Thomen RP, Walkup LL, Roach DJ, et al. Hyperpolarized (129)xe for investigation of mild cystic fibrosis lung disease in pediatric patients. J Cyst Fibros 2017;16(2):275–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Walkup LL, Myers K, El-Bietar J, et al. Xenon-129 mri detects ventilation deficits in paediatric stem cell transplant patients unable to perform spirometry. Eur Respir J 2019;53(5):1801779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Walkup LL, Myers KC, Willmering MM, et al. Modern lung magnetic resonance imaging to screen for pulmonary complications in patients with dyskeratosis congenita. Am J Respir Crit Care Med 2021;204(11):1340–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Roach DJ, Willmering MM, Plummer JW, et al. Hyperpolarized (129)xenon mri ventilation defect quantification via thresholding and linear binning in multiple pulmonary diseases. Acad Radiol 2022;29(Suppl 2):S145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hahn AD, Malkus A, Kammerman J, et al. Effects of neonatal lung abnormalities on parenchymal r(2) * estimates. J Magn Reson Imaging 2021;53(6):1853–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ikonomou L, Herriges MJ, Lewandowski SL, et al. The in vivo genetic program of murine primordial lung epithelial progenitors. Nat Commun 2020;11(1):635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Neehus AL, Carey B, Landekic M, et al. Human inherited ccr2 deficiency underlies progressive polycystic lung disease. Cell 2024;187(2):390–408 e323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Li Y, Seidl E, Knoflach K, et al. Abca3-related interstitial lung disease beyond infancy. Thorax 2023;78(6):587–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Niemitz M, Schwerk N, Goldbeck L, et al. Development and validation of a health-related quality of life questionnaire for pediatric patients with interstitial lung disease. Pediatr Pulmonol 2018;53(7):954–63. [DOI] [PubMed] [Google Scholar]
- 198.Griese M, Kappler M, Stehling F, et al. Randomized controlled phase 2 trial of hydroxychloroquine in childhood interstitial lung disease. Orphanet J Rare Dis 2022;17(1):289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Seidl E, Schwerk N, Carlens J, et al. Healthcare resource utilisation and medical costs for children with interstitial lung diseases (child) in europe. Thorax 2022;77(8):781–9. [DOI] [PubMed] [Google Scholar]
- 200.Saddi V, Beggs S, Bennetts B, et al. Childhood interstitial lung diseases in immunocompetent children in Australia and New Zealand: a decade’s experience. Orphanet J Rare Dis 2017;12(1):133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Nayir-Buyuksahin H, Emiralioglu N, Kilinc AA, et al. Childhood interstitial lung disease in Turkey: first data from the national registry. Eur J Pediatr 2024;183(1):295–304. [DOI] [PubMed] [Google Scholar]