

Abstract
The protozoan parasite Leishmania (Viannia) braziliensis is among Latin America’s most widespread Leishmania species and is responsible for tegumentary leishmaniasis (TL). This disease has multiple clinical presentations, with cutaneous leishmaniasis (CL) being the most frequent. It manifests as one or a few localized skin ulcers, which can spread to other body areas. Hence, early diagnosis and treatment, typically with pentavalent antimonials, is critical. Traditional diagnostic methods, like parasite culture, microscopy, or the polymerase chain reaction (PCR) for detection of the parasite DNA, have limitations due to the uneven distribution of parasites in biopsy samples. Nonetheless, studies have revealed high levels of parasite-specific anti-α-Gal antibodies in L. (V.) braziliensis-infected patients. Previously, we demonstrated that the neoglycoprotein NGP28b, consisting of the L. (Leishmania) major type-2 glycoinositolphospholipid (GIPL)-3-derived trisaccharide Galpα1,6Galpα1,3Galfβ conjugated to bovine serum albumin (BSA) via a linker, acts as a reliable serological biomarker (BMK) for L. (V.) braziliensis infection in Brazil. This indicates the presence of GIPL-3 or a similar structure in this parasite, and its terminal trisaccharide either functions as or is part of an immunodominant glycotope. Here, we explored whether extending the trisaccharide with a mannose unit would enhance its efficacy as a biomarker for the serological detection of L. (V.) braziliensis. We synthesized the tetrasaccharide Galpα1,6Galpα1,3Galfβ1,3Manpα(CH2)3SH (G31SH) and conjugated it to maleimide-functionalized BSA to afford NGP31b. When we assessed the efficacy of NGP28b and NGP31b by chemiluminescent enzyme-linked immunosorbent assay on a cohort of CL patients with L. (V.) braziliensis infection from Bolivia and Argentina against a healthy control group, both NGPs exhibited similar or identical sensitivity, specificity and accuracy. This finding implies that the mannose moiety at the reducing end is not part of the glycotope recognized by the parasite-specific anti-α-Gal antibodies in patients’ sera, nor does it exert a relevant influence on the terminal trisaccharide’s conformation. Moreover, the mannose does not seem to inhibit glycan-antibody interactions. Therefore, NGP31b is a viable and dependable BMK for the serodiagnosis of CL caused by L. (V.) braziliensis.
Keywords: Oligosaccharide synthesis, neoglycoprotein, Leishmania (Viannia) braziliensis, biomarker, anti-alpha-Gal antibody, chemiluminescent enzyme-linked immunosorbent assay
Graphical Abstract
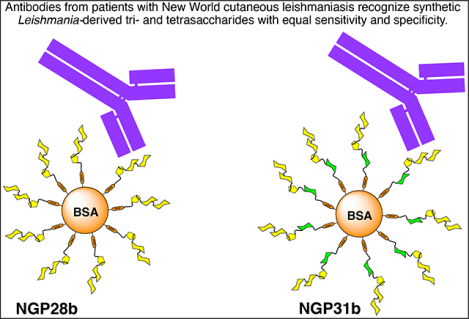
1. Introduction
1.1. Background information on Leishmania (Viannia) braziliensis, L. (Leishmania) major, and L. (V.) panamensis type-2 GIPLs
The protozoan parasite Leishmania (V.) braziliensis is among the dominant Leishmania species in Central and South America, regions where it is endemic. It causes tegumentary leishmaniasis (TL) in humans, dogs, and other mammals and is predominantly transmitted by bloodsucking Lutzomyia sandflies [1]. TL manifests in various clinical forms: cutaneous (CL), mucosal (ML), disseminated (DL), and subclinical asymptomatic leishmaniasis (SL). Among these, CL is the most common and is characterized by one or a few localized skin ulcers. However, the parasites can metastasize to the mucosa, leading to tissue-destroying mucosal leishmaniasis (ML) [2]. Disseminated leishmaniasis (DL) is another metastatic TL clinical form, characterized by a large number (>100) of acneiform or ulcerated lesions [3, 4], and up to a quarter of DL patients are simultaneously inflicted with ML [5]. Pentavalent antimonials are the standard treatment for all clinical presentations of leishmaniasis; however, it is critical to diagnose and treat patients during the disease’s early stages. Prompt intervention ensures complete parasite eradication and prevents progression to more severe forms like ML, which results in mucosal tissue damage, or DL, which poses treatment challenges [6]. Accurately diagnosing CL can be intricate since its skin lesions often resemble those of other diseases, including Hansen’s disease, syphilis, or skin cancer. A common method for diagnosing CL involves culturing parasites from biopsy specimens. Additionally, the polymerase chain reaction (PCR) targeting the parasite’s DNA is used, albeit less frequently. While both approaches offer high specificity, their sensitivity can be compromised due to the heterogeneous distribution of parasites within tissues [7]. An alternative diagnostic method is the enzyme-linked immunosorbent assay (ELISA), which detects antibodies (Abs) against the parasite by serology. The efficacy of ELISA hinges on the applied technique and the chosen antigens. Over time, several antigens have been pinpointed as effective for diagnostic purposes [8–12].
A promising avenue for CL diagnosis via serology centers on utilizing certain carbohydrate antigens derived from the parasite’s cell surface. Leishmania parasites express species-specific long-chain lipophosphoglycans (LPGs) and low molecular weight glycoinositolphospholipids (GIPLs). Certain Leishmania species, such as L. (L.) major, L. (L.) mexicana, and L. (V.) panamensis express Gal-rich type-2 GIPLs [13–16]. These GIPLs feature a terminal β-galactofuranose (β-Galf) (GIPL-1) or an α-galactopyranose (α-Galp) unit (GIPL-2 and −3) situated at the glycan’s non-reducing end (Figure 1) [15–18]. The glycan structures found in type-2 GIPLs are entirely absent in humans and exhibit strong antigenic and immunogenic properties [17–21]. Accordingly, patients infected with L. (L.) major, L. (L.) mexicana, or L. (V.) braziliensis exhibit increased levels of anti-β-Galf and anti-α-Gal Abs, which strongly recognize type-2 GIPLs, or structures resembling them [22–24].
Figure 1.
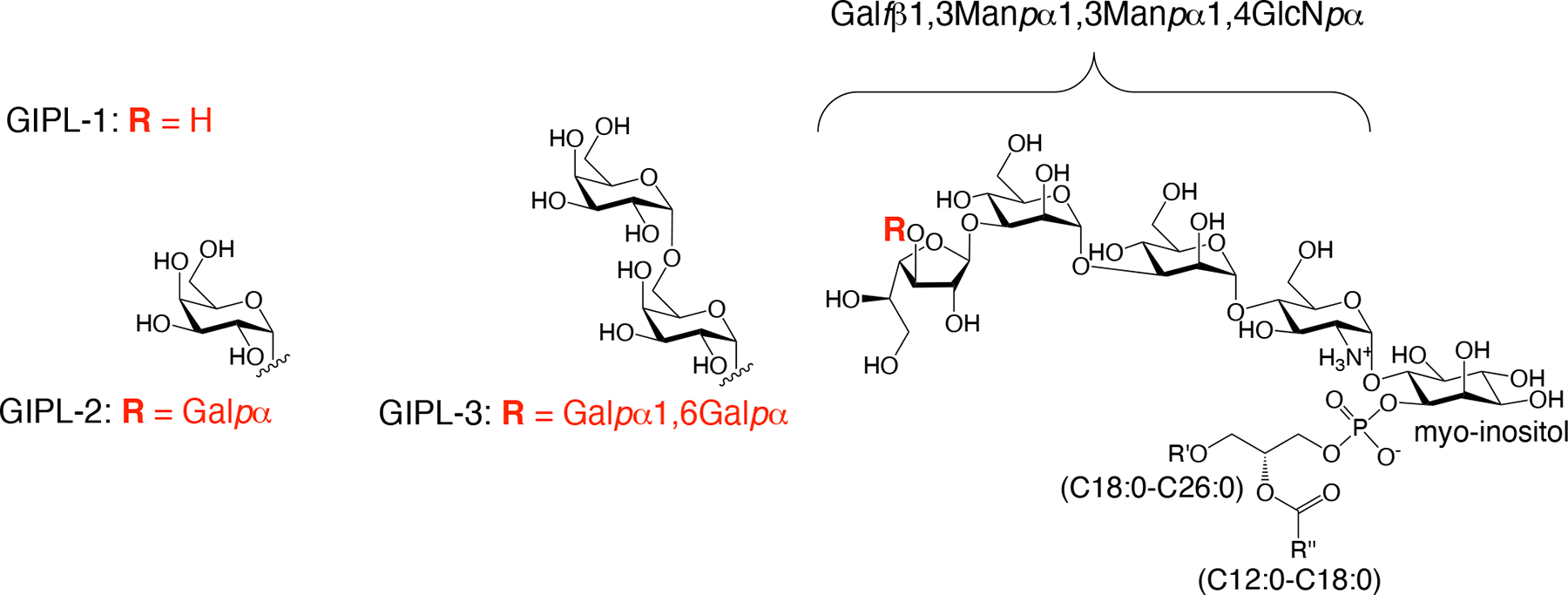
Structures of GIPL-1, −2, and −3, belonging to the galactose-rich type-2 GIPL family, as described for L. (L.) major and L. (L.) mexicana.
1.2. α-Gal-based BMK discovery by reversed immunoglycomics
We recently utilized reversed immunoglycomics, a bottom-up approach, to identify small α-Gal-containing oligosaccharides as diagnostic biomarkers (BMKs) for L. (L.) major infection. This process entails the synthesis of neoglycoproteins (NGPs), consisting of α-Gal-containing oligosaccharides, including terminal structures of different type-2 GIPLs, covalently conjugated to bovine serum albumin (BSA). These NGPs were then evaluated using chemiluminescent ELISA (cELISA) with sera from CL patients, which allowed for the identification of NGPs with high antibody reactivity [25]. Meanwhile, the natural anti-α-Gal Abs targeting enterobacteria lipopolysaccharides, which are consistently present in normal human serum (NHS) of all healthy individuals [26–29], displayed minimal or no cross-reactivity with a particular NGP [25]. For any α-Gal-containing antigen to be used as a dependable diagnostic BMK for infections by Old- or New-World Leishmania species that express type-2 GIPLs or analogous molecules, this is a crucial requirement.
While not every Leishmania species expresses type-2 GIPLs, several reports suggest that L. (V.) braziliensis parasites do. Evidence indicates that sera of patients infected with this species contain Abs that strongly recognize α-Gal-containing structures [23, 24, 30]. The challenge in discovering α-Gal-based BMK candidates for the serodiagnosis of New-World TL lies in the potential cross-reactivities with Abs present in patients with Chagas disease (CD), caused by the protozoan parasite Trypanosoma cruzi. Given that TL and CD often coexist in several Latin American regions, an accurate differential diagnosis becomes crucial [12, 31, 32]. Since the mammal-dwelling infective trypomastigote form of T. cruzi expresses glycosylphosphatidylinositol (GPI)-anchored mucin-like glycoproteins (tGPI-MUC) with terminal, non-reducing α-Gal residues, CD patients have high titers of parasite-specific anti-α-Gal Abs (Ch or CD anti-α-Gal Abs), both in the acute and chronic phases of the disease [17, 20, 22, 33–37]. The sole completely characterized α-Gal-containing glycan in tGPI-MUC is the linear trisaccharide Galpα1,3Galpβ1,4GlcNAcpα. However, most tGPI-MUC glycans are branched, and their precise structures remain elusive [36]. We have shown that Ch anti-α-Gal Abs present in the sera of CD patients react with several synthetic α-Gal-containing antigens, especially with glycans that contain terminal Galpα1,3Galpβ at the non-reducing end [38–40], and with the branched trisaccharide Galα(1,2)[Galα(1,6)]Galβ [41].
We recently identified NGP28b, consisting of the GIPL-3-derived trisaccharide Galpα1,6Galpα1,3Galfβ conjugated to BSA, as a promising BMK for diagnosing TL caused by L. (V.) braziliensis with minimal cross-reactivity with CD [30]. NGP28b effectively diagnosed TL across all clinical presentations, exhibiting an overall specificity of 85% and a sensitivity of 92%. When specifically diagnosing CL caused by L. (V.) braziliensis, NGP28b maintained the same specificity and presented a marginally reduced sensitivity of 88% [30]. Notably, NGP28b emerged as a potential prognostic BMK, given the observed decline in Ab levels post-chemotherapy, which suggests a successful parasite elimination [30]. While NGP28b stands out as a promising BMK for the L. (V.) braziliensis infection, the exact glycotope eliciting the immune response in infected individuals remains unidentified. To gain deeper insights into the structural requirements of a glycotope and potentially improve the BMK, it is valuable to study Ab responses against unique, structurally similar glycans.
Here, we present the synthesis of the Leishmania GIPL-3-derived tetrasaccharide Galα1,6Galα1,3Galfβ1,3Manpα(CH2)3SH (G31SH), its conjugation to BSA to generate NGP31b, and its direct comparison with the trisaccharide-integrated NGP28b as a diagnostic BMK for CL caused by L. (V.) braziliensis. The structure of NGP31b differs from NGP28b in having an α-Man unit added at the non-reducing end of the glycan. There is a precedence that a larger oligosaccharide can be a better serological biomarker than a smaller analog [42]. In the context of CD, we recently examined Ab responses to NGPs containing a di- or tetrasaccharide derivative of tGPI-MUC. Specifically, we studied NGP29b, containing Galfβ1,3Manpα, and NGP32b, bearing Galfβ1,3Manpα1,2-[Galfβ1,3]Manpα, using sera from chronic CD (CCD) patients. NGP29b diagnosed CCD with a sensitivity of 79% and a specificity of 93%. On the other hand, NGP32b exhibited enhanced diagnostic parameters with a sensitivity of 85% and a specificity of 100% [42]. It remained uncertain if the diagnostic efficacy of NGP28b for L. braziliensis infection could be enhanced by using an NGP with a longer GIPL-3-derived glycan moiety. This inquiry formed the core of the current study.
2. Results
2.1. Synthesis of the Galpα1,6Galpα1,3Galfβ1,3Manpα-bearing neoglycoprotein NGP31b
The pronounced Ab response to NGP28b in the sera of TL patients indicates that L. (V.) braziliensis could express GIPL-3 or a similar structure [30]. In this study, we utilized the reversed immunoglycomics strategy [25] for identifying a potential BMK that might be more accurate than NGP28b [25] for diagnosing New-World CL caused by L. (V.) braziliensis. We leveraged the established structural data of the Leishmania type-2 GIPL-3 (Figure 1) [16], and incorporated the synthesis of an NGP featuring Galpα1,6Galpα1,3Galfβ1,3Manpα, known as NGP31b, with serological evaluations using sera from patients. Our initial synthetic target was the Leishmania type-2 GIPL-3-derived tetrasaccharide Galα1,6Galα1,3Galfβ1,3Manpα(CH2)3SH (G31SH) (Scheme 1), an extended structure of the previously described trisaccharide Galα1,6Galα1,3Galfβ(CH2)3SH (G28SH) [25]. Both G28SH and G31SH were designed as 3-propylthiol glycosides, facilitating their conjugation to maleimide-functionalized BSA. The synthesis of G31SH was performed in a manner analogous to G28SH, utilizing the orthogonal protecting group and glycosylation strategy shown in Scheme 1. The 4,6-di-tert-butylsilylene-galactosyl trichloroacetimidate donor 1 (“Kiso donor”)[43] was used to glycosylate the galactofuranosyl thioglycoside acceptor 2 [44–46] under TMSOTf catalysis, which occurred with the expected high α-selectivity despite the presence of a benzoyl protecting group at position 2 [47], to afford fully protected disaccharide 3. Removal of the di-tert-butylsilylene group with hydrofluoric acid-pyridine complex gave acceptor 4, which was glycosylated with Kiso donor 1 to furnish trisaccharide 5 as the major glycosylation product with a 45% yield. Trisaccharide donor 5 was used to glycosylate mannosyl acceptor 6 [48] to yield tetrasaccharide 7, which occurred under high regio- and stereoselectivity. The di-tert-butylsilylene protecting group was cleaved off with hydrofluoric acid-pyridine complex to give the partially deprotected tetrasaccharide 8, and acid-catalyzed hydrolysis of the acetal and ketal protecting groups furnished tetrasaccharide 9. Upon radical addition of thioacetic acid to the allyl glycoside 9 in the presence of radical starter 2,2-dimethoxy-2-phenylacetophenone (DPAP), tetrasaccharide thioester 10 was obtained. Then, global deprotection under Zemplén conditions afforded the 3-thiopropyl glycoside G31SH, which oxidized to the disulfide (G31S)2 on air.
Scheme 1.
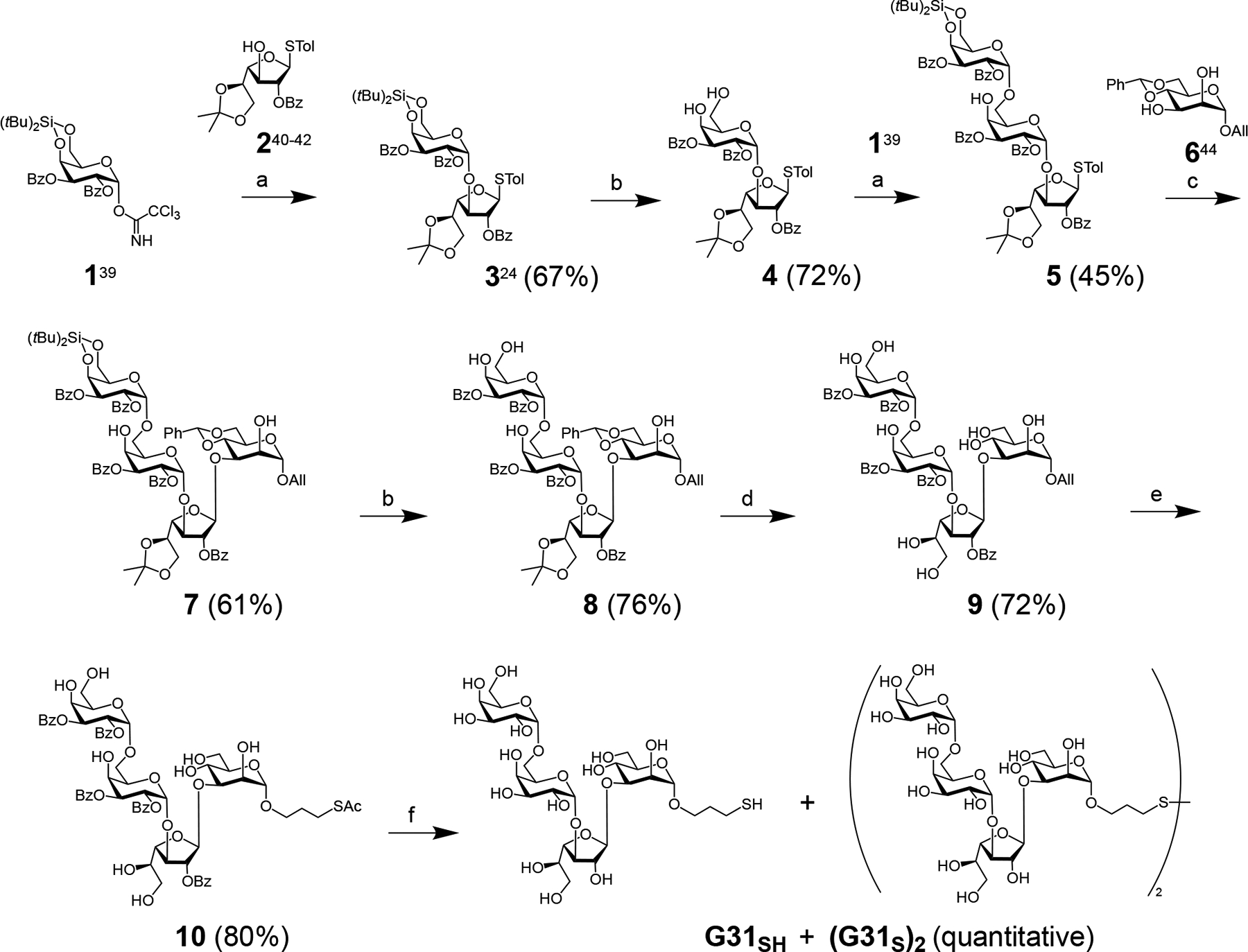
Synthesis of 3-thiopropyl tetrasaccharide G31SH and its disulfide oxidation product (G31S)2: a) TMSOTf, DCM, 0°C to rt, then 1h at rt; b) HF-pyridine, THF, 30 min at 0°C, then 30 min - 1h at rt; c) NIS, TfOH, DCM, −20°C, 2h and 15 min; d) TFA, H2O, DCM (1:1:10), rt, 1h; e) AcSH, DPAP, DCM, 30 min, UV light (350 nm); f) NaOMe, MeOH, rt, 1h.
To generate an antigen for serological testing that can adhere to the polystyrene wells of a microplate for cELISA [37], we generated an NGP, which entailed reducing (G31S)2 with TCEP-HCl to G31SH, and immediately conjugating it to commercially available BSA-maleimide at near neutral pH to give NGP31b (Figure 2A). The average number of glycan units including linkers (GU) per BSA molecule was 21, as determined by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS) (Figure 2B).
Figure 2.
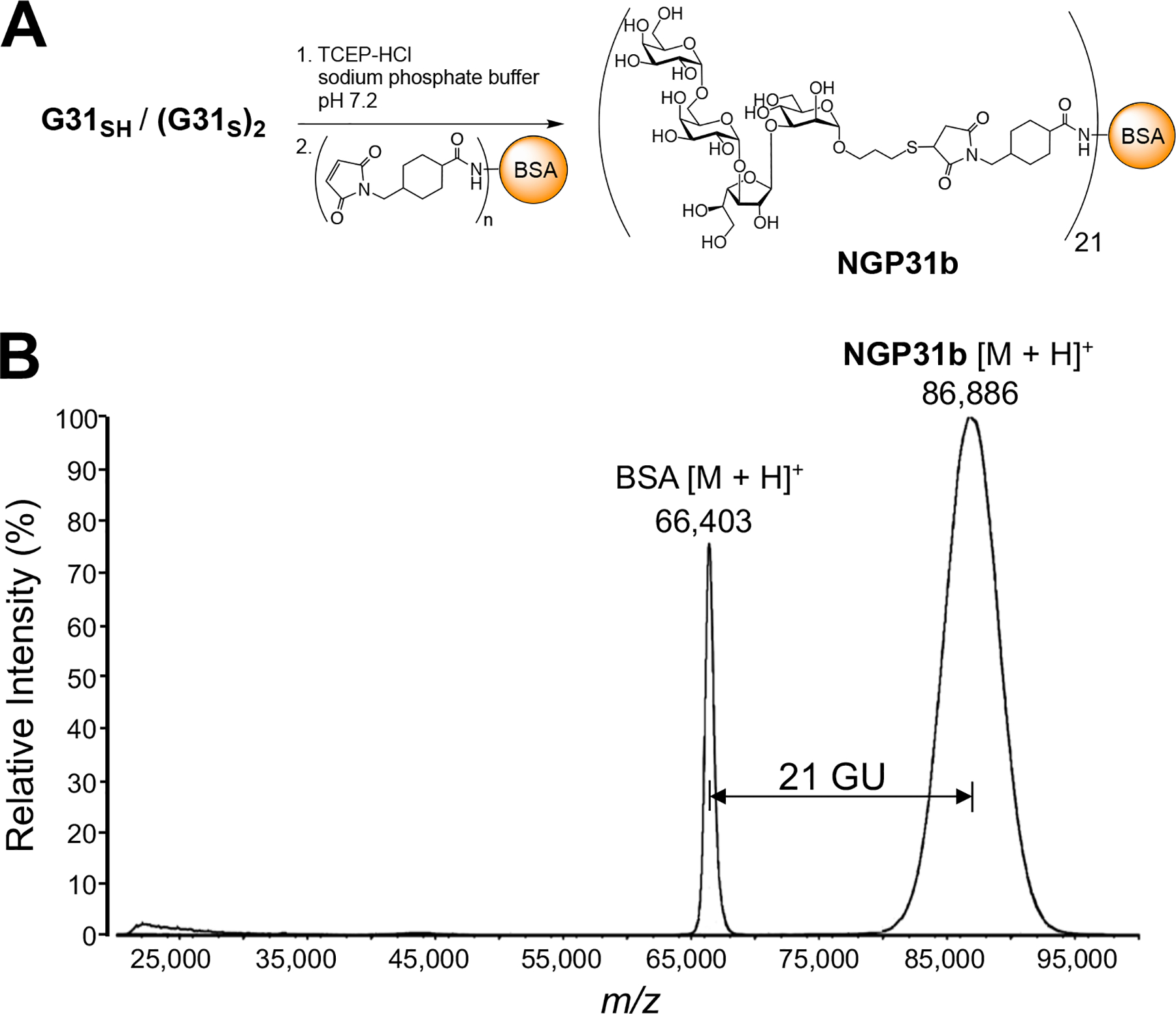
(A) Conjugation of G31SH to maleimide-derivatized BSA to furnish NGP31b. (B) Overlaid MALDI-TOF-MS of singly-charged molecular ions of BSA ([M + H]+) and NGP31b ([M + H]+). GU, average number of glycan units (including linkers) per BSA; m/z, mass to charge ratio.
To shine some light on the glycotope scaffold involved in Ab elicitation and antigen recognition, we compared NGP31b to the previously described BMK NGP28b [25]. Importantly, the average number of GUs per BSA molecule could potentially influence the Ab response in the cELISA. Therefore, for a direct comparison of the two glycans, we used an NGP28b, in which the average number of GU (21) per BSA molecule matched that of NGP31b (Suppl. Fig. S1).
2.2. Immunological Evaluation of NGP31b and comparison with NGP28b
Equipped with both NGP31b and the earlier reported NGP28b [25, 30], we could compare the antigenicity of the two NGPs by cELISA with sera from patients with L. (V.) braziliensis CL or CD, and negative controls (non-CL, non-CD). Moreover, to test whether Abs could bind to the linker or BSA portion of the NGP, we conjugated 2-mercaptoethanol (2ME) to commercially available maleimide-BSA which produced 2ME-BSA [25]. Initially, we performed cELISA using NGP28b, NGP31b, and 2ME-BSA as antigens immobilized at different quantities (ng/well), and pooled sera from patients with CL caused by L. (V.) braziliensis (CLP), and healthy individuals (non-CL, non-CD) as negative controls (NCP), at serum dilutions of 1:400 and 1:800. These cross-titrations showed a strong reactivity of the CLP to both NGP31b and NGP28b, in an antigen concentration-dependent manner (Suppl. Fig. 2). The NCP exhibited minimal cross-reactivity with both NGPs. None of the sera showed any significant Ab reactivity to 2ME-BSA, indicating that there was no significant Ab binding to the 4-(N-succinimidomethyl)-cyclohexanecarboxamide linker, or to BSA itself (Suppl. Fig. S2).
Next, we examined the differential serum reactivity of pooled sera (n=10 each) from patients with L. (V.) braziliensis CL (CLP), chronic CD patients (CDP), and negative controls (NCP), at two different serum dilutions. The differences in serum reactivities were determined by initially conducting an NGP-serum cross-titration (Figure 3A,B). The CDP demonstrated essentially the same cross-reactivity when compared to NCP in both serum dilutions. Subsequently, the CLP/NCP and CDP/NCP ratios were calculated across different NGP concentrations per well and serum dilutions (Figure 3C,D). The most marked differences in serum reactivity between CLP and NCP (33-fold for NGP31b and 39-fold for NGP28b) were noted at approximately 12.5 ng/well for both NGPs, based on the non-linear fitted curve of the assay’s response, and a serum dilution of 1:800 (Figure 3C,D, arrows).
Figure 3.
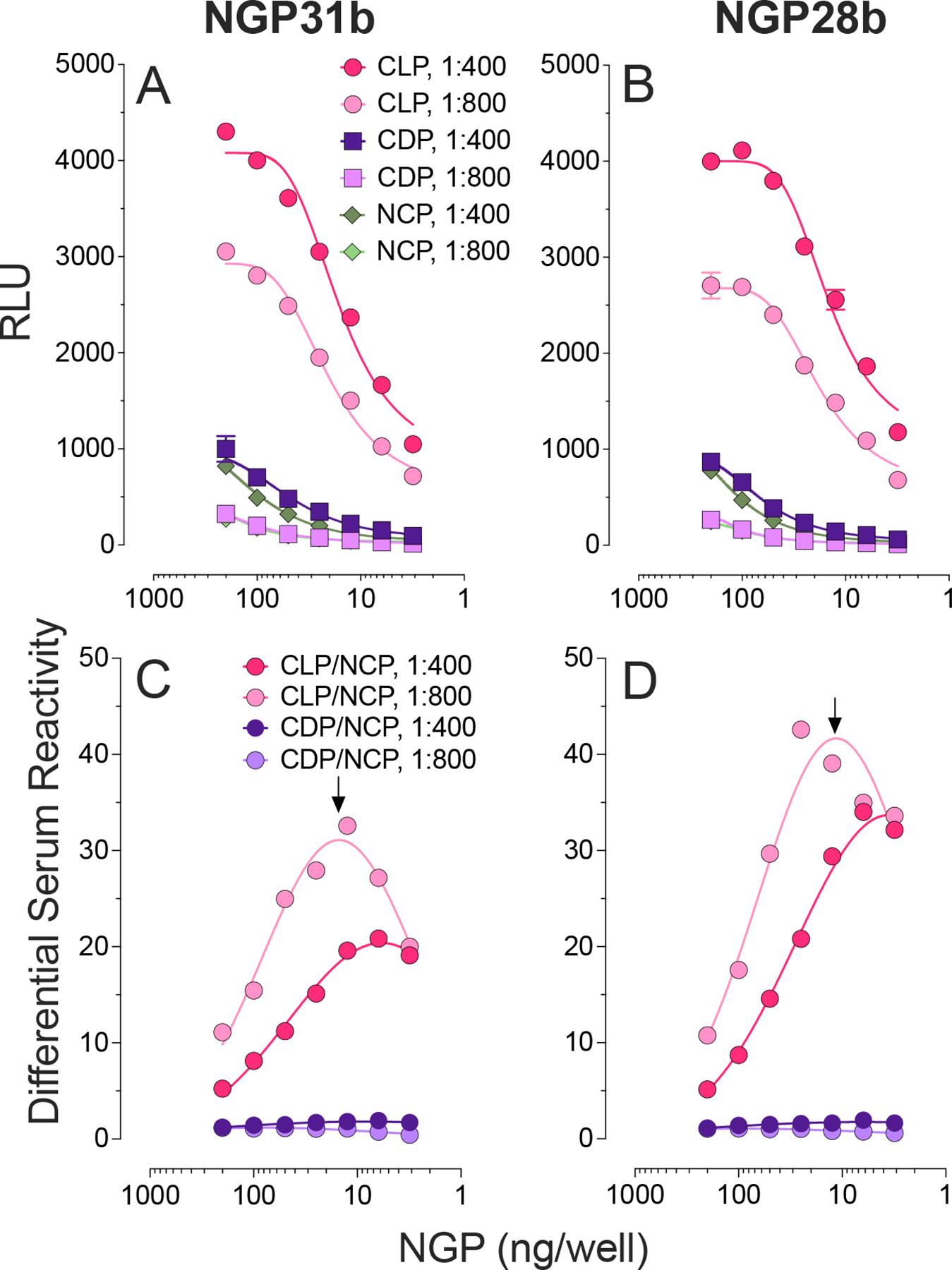
cELISA of serum-antigen cross-titration and differential serum reactivity to NGP31b and NGP28b. Each NGP was immobilized at 200, 100, 50, 25, 12.5, 6.25, and 3.125 ng/well, and assayed with pools of sera (n=10 each) from individuals with cutaneous leishmaniasis (CLP) or Chagas disease (CDP), or healthy negative controls (NCP) from an endemic area for CD and CL, at 1:400 and 1:800 dilutions. (A and B) Ab reactivity to NGP31b and NGP31b. RLU, relative luminescence units. (C and D) Differential reactivity to NGP31b and NGP28b. The differential reactivity was determined by calculating the ratio of the mean relative luminescent units (RLU), obtained in triplicate, of CLP or CD sample to that of NCP sample. Arrow, maximum differential reactivity.
For NGP31b to be effective as a diagnostic BMK, it must accurately distinguish sera from individual L. (V.) braziliensis CL patients from those of healthy individuals. To better evaluate the sensitivity and specificity of NGP31b as a diagnostic BMK for L. (V.) braziliensis CL, we measured antibody-binding responses using cELISA for 28 CL patient sera (from Argentina and Bolivia), 28 sera from CD patients from Bolivia, and 28 sera from NC individuals (without CL or CD) from an endemic area for CL and CD in Bolivia, as depicted in Figure 4A. Using NGP31b as an antigen in cELISA, we found that 23/28 (82.1%) of the CL sera displayed cELISA titers greater than the initial cutoff (Ci = 1.000). In contrast, 27/28 (96.4%) of the NC and CD sera had cELISA titers below the Ci (Table 1, Figure 4A). No significant difference was observed between the CD and NC sera reactivities with NGP31b. Our results suggest that NGP31b offers good sensitivity and outstanding specificity as a potential BMK for L. (V.) braziliensis CL. Comparing the antigenicity of NGP28b with NGP31b, we noted a marginally higher sensitivity (89.3%, 25/28) for NGP28b, while the specificity for both was identical (27/28, 96.4%) (Tables 1 and 2).
Figure 4.
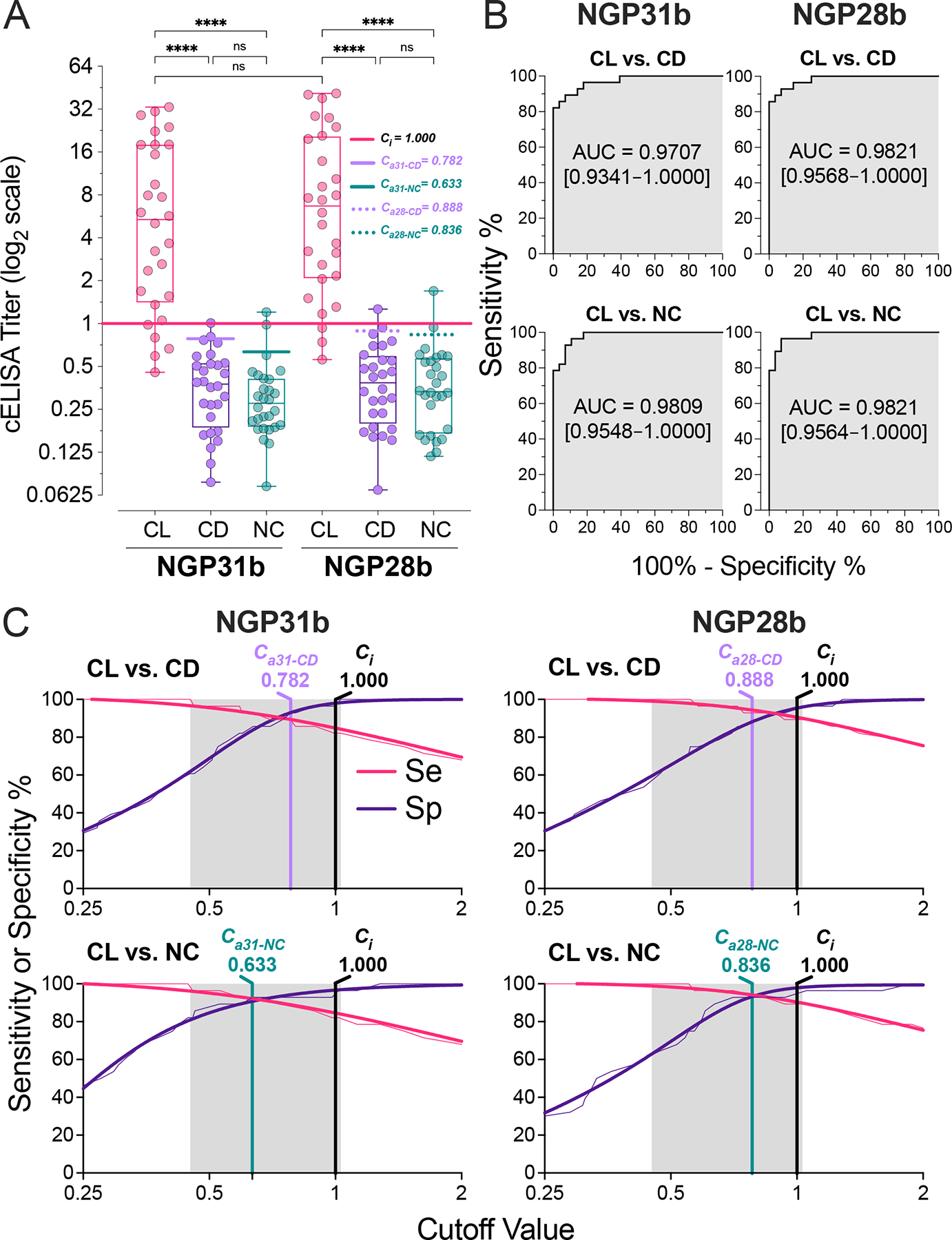
Serological assessment by cELISA of NGP31b and NGP28b. (A) IgG cELISA Titers. Box-and-whiskers plots of individual sera from patients with cutaneous leishmaniasis (CL; n=28) or Chagas disease (CD; n=28), or negative control individuals from endemic areas for CD and CL (NC; n=28). Immunoassay conditions: NGPs at 25 ng/well and 1:800 serum dilution. Each plotted value represents the mean of triplicate RLU values normalized to an NC serum pool (n=10), assayed in nine replicates per microplate. The solid horizontal black line represents the initial cutoff (Ci = 1.000), calculated as detailed in the Experimental section. Adjusted cutoffs (Ca) for CD and NC with NGP31b (Ca31-CD and Ca31-NC, respectively) and NGP28b (Ca28-CD and Ca28-NC, respectively) are indicated. Statistical analysis: Kruskal Wallis with Dunn’s multiple comparison tests. ****p<0.0001; ns, non-significant. (B) Receiver-operating characteristic (ROC) analysis for NGP31b and NGP28b. Curves illustrate IgG reactivity differences for CL vs. CD and CL vs. NC. The shaded gray area denotes the area under the curve (AUC), and the 95% confidence interval (CI) values are shown in brackets. (C) Two-graph (TG)-ROC analysis for NGP31b and NGP28b. The analysis plots ROC data for sensitivity (Se, pink) and specificity (Sp, indigo) against NGP28b when comparing CL patients with CD or NC. Thin lines represent Se and Sp raw data, while thick ones denote nonlinear fitted data. Gray regions show the cELISA titer range where Se or Sp might achieve 100%. Vertical black lines mark the initial cutoff (Ci = 1.000), vertical magenta lines the adjusted cutoffs for CL vs. CD (Ca31-CD and Ca28-CD), and vertical teal lines for CL vs. NC (Ca31-NC and Ca28-NC) for both NGPs.
Table 1.
Immunoreactivity of sera from L. (V.) braziliensis CL and CD patients, and NC individuals with NGP31b and NGP28b
| Disease/Control | n | NGP31b | NGP28b | ||||
|---|---|---|---|---|---|---|---|
| Cutoff | Positive | Negative | Cutoff | Positive | Negative | ||
| Original Values a | |||||||
| L. (V.)braziliensis CL | 28 | C i = 1.000 | 23 (82.1%) | 5 (17.9%) | C i = 1.000 | 25 (89.3%) | 3 (10.7%) |
| CD | 28 | C i = 1.000 | 1 (3.6%) | 27 (96.4%) | C i = 1.000 | 1 (3.6%) | 27 (96.4%) |
| Negative (non-CD, non-CL) | 28 | C i = 1.000 | 1 (3.6%) | 27 (96.4%) | C i = 1.000 | 1 (3.6%) | 27 (96.4%) |
| Post-TG-ROC analysis b | |||||||
| L. (V.) braziliensis CL | 28 | C a31-NC = 0.633 | 26 (92.3%) | 2 (7.7%) | C a28-NC = 0.836 | 26 (92.3%) | 2 (7.7%) |
| CD | 28 | C a31-CD = 0.782 | 2 (7.7%) | 26 (92.3%) | C a28-CD = 0.888 | 2 (7.7%) | 26 (92.3%) |
| Negative (non-CD, non-CL) | 28 | C a31-NC = 0.633 | 2 (7.7%) | 26 (92.3%) | C a28-NC = 0.836 | 2 (7.7%) | 26 (92.3%) |
Table 2.
Sensitivity, specificity, and other diagnostic parameters of NGP31b and NGP28b.
| Parametera, b | NGP31b | NGP28b |
|---|---|---|
| % | ||
| Original Values a | ||
| Sensitivity | 82.1 | 89.3 |
| Specificity | 96.4 | 96.4 |
| Accuracy | 89.3 | 92.9 |
| Post-TG-ROC analysis b | ||
| Sensitivity | 92.9 | 92.9 |
| Specificity | 92.9 | 92.9 |
| Accuracy | 92.9 | 92.9 |
Calculated based on immunoreactivity of CL (n=28) serum and NC (n=28) serum samples to NGP31b and NGP28b (Figure 4).
Sensitivity (Se) = true positive (TP)/(TP + false negative (FN)) × 100; Specificity (Sp) = true negative (TN)/(TN + false positive (FP)) × 100; False-positive rate (FPR) = 100 − specificity; Accuracy = (TP+TN)/(TP+TN+FP+FN) × 100
We then employed receiver-operating characteristics (ROC) curves to compare the sensitivity and specificity of NGP31b and NGP28b (Figure 4B). As per the area under the curve (AUC) values from the ROC analysis, NGP31b (AUC = 0.9707) displayed a marginally reduced specificity compared to NGP28b (AUC = 0.9891) when evaluating CL vs. CD. Since the AUC values for NGP31b and NGP28b were in the 0.9707–0.9821 range (Figure 4B), which is considered outstanding for any diagnostic test with an AUC > 0.9 [49]. Regarding sensitivity (CL vs. NC comparison), the two NGPs demonstrated nearly equivalent results without significant differences (Figure 4A,B). Overall, both antigens showed an excellent measure of separability and were able to distinguish between the positive (CL) and negative (NC) or heterologous (CD) cases with high accuracy. However, in particular NGP31b, should be further validated as a reliable biomarker for CL using a large and more diversified serum panel (including other confounding heterologous diseases), and considered alongside other diagnostic performance metrics.
To refine the initial titer cutoff value (Ci = 1.000; see Figure 4A, Table 1) for NGP31b and NGP28b, we performed a two-graph ROC (TG-ROC) analysis. This involved plotting the relationship between sensitivity (Se) and specificity (Sp), given by the ROC curves, against different cutoff values, following the method outlined by Greiner et al. [50] (Figure 4C). Selecting the optimal cutoff value requires balancing Se and Sp, which varies based on the intended clinical application of the diagnostic biomarker. In South America, CL caused by L. (V.) braziliensis is prevalent in areas that may geographically coincide with CD [31, 32]. Therefore, there is a critical need for a diagnostic test to accurately distinguish between the two conditions, requiring high sensitivity and specificity (both >90%) [51], to ensure proper treatment and management. We thus optimized the titer cutoff values for NGP31b to Ca31-NC and Ca31-CD of 0.633 and 0.782, respectively, which considerably enhanced Se to 92.3% (an improvement from 82.1%), while specificity was slightly reduced to 92.3% (a decrease from 96.4%) as shown in Table 2, following the post-TG-ROC analysis. For NGP28b, setting the cutoff value at a Ca28-CD of 0.888 resulted in a modest reduction in Sp to 92.3% (down from 96.4%), as indicated in Figure 4A,C, and Tables 1 and 2. A similar pattern was observed for Se when the Ca28-NC was set to 0.836. The Post-TG-ROC analysis confirmed that, despite their structural differences at the reducing end, both NGP31b and NGP28b have equivalent sensitivity, specificity, and accuracy diagnostic parameters.
3. Discussion
Prior research and our current study indicate that L. (V.) braziliensis expresses the L. (L.) major type-2 GIPL-3 or structures akin to it. These glycan structures seem potently immunogenic and elicit a robust specific anti-α-Gal Ab response in patients with CL caused by L. (V.) braziliensis [30]. The size, conformation, and binding mode of the α-Gal-bearing glycotope involved in the molecular recognition by anti-α-Gal Abs is unknown. Still, insights may be obtainable from other oligosaccharide/Ab binding phenomena. For example, Gildersleeve and Wright studied the molecular recognition of blood group A antigens, where the minimal trisaccharide determinant [GalNAcα1–3(Fucα1–2)Galβ-], which is part of larger glycans of glycolipids and glycoproteins in nature, was elongated with different saccharides at the reducing end [52]. Glycoarray binding studies with several commercial anti-blood group A IgM and IgG Abs revealed that the glycan size and chain extension had a significant influence on Ab binding. The authors suggest that a small di- or trisaccharide may provide all the intermolecular contacts needed to achieve tight binding and that the extended glycan chain is either tolerated or not by the Ab [52]. Another rationale is that monosaccharide residues beyond the blood group A trisaccharide provide additional interactions with the Ab, or influence the conformation of the trisaccharide which can affect binding. Another factor that may also affect the binding affinity is the density at which the glycan antigen is presented [52].
Crystallographic structures of the disaccharide Galpα1,3Galp complexed with the monoclonal anti-α-Gal Ab M86 (a mouse IgM), and human natural anti-α-Gal Abs present in NHS provided insights into the binding at the molecular level [53], which is achieved mainly by a combination of hydrogen bonds and carbon pi interactions. The binding pocket is lined mostly by the complementary determining region (CDR1) and the complementary determining region 3 (CDR3) of the heavy chain variable domain (VH) and CDR3 of the light chain variable domain (VL). The carbon pi interactions occur between VH CDR1 Trp 33 and the α-face of the reducing Gal, and VL Tyr32 and the non-reducing α-Gal residue. When compared to a Galpα1,3Galp/Ab complex of a human anti-α-Gal Ab clone, HKB7, from a patient with mammalian meat allergy, the overall CDRH1 binding mode was similar to that of M86. The structures of the Galpα1,3Galp/anti-α-Gal Ab complexes studied suggest that the disaccharide Galpα1,3Galp fills out the binding cleft and the anomeric hydroxyl of the reducing β-Gal points away from the binding groove. Thus, the authors suggested that in the case of the binding of the Galili epitope (Galpα1,3Galpβ1,4GlcNAcp) to M86 or HKB7, the GlcNAc residue at the reducing end of the Galili epitope does not contribute significantly to the binding. This conclusion is in agreement with a report by Galili and Matta who showed that the binding of polyclonal NHS anti-α-Gal Abs to Galpα1,3Galpβ1,4GlcNAcp-expressing porcine endothelial cells can be more effectively inhibited with the trisaccharide Galpα1,3Galpβ1,4GlcNAc than with the disaccharide Galpα1,3Galp in vitro [54]. However, the affinity of the trisaccharide to anti-α-Gal Abs, as measured by equilibrium dialysis, was only seven-fold higher than that of the disaccharide. Such a small difference in affinity suggests that the reducing GlcNAc moiety is either involved in a minor secondary intermolecular contact with the Ab or stabilizes the binding conformation of the disaccharide it is attached to. These data also agree with our finding that anti-α-Gal Abs present in the sera of CD patients recognize the disaccharide Galpα1,3Galpβ equally well as the trisaccharide Galpα1,3Galpβ1,4Glcp [38, 40].
The molecular recognition motifs in the binding of the trisaccharide Galα1,6Galα1,3Galfβ (G28) to L. (V.) braziliensis CL-specific anti-α-Gal Abs remain uncertain, especially in comparison to the previously described Galpα1,3Galp/anti-α-Gal Ab complexes. Yet, our findings suggest that extending Galα1,6Galα1,3Galfβ with a Manαp unit at the reducing end to produce Galα1,6Galα1,3Galfβ1,3Manα (G31) neither enhances nor compromises molecular recognition. Essentially, all diagnostic parameters (sensitivity, specificity, etc.) for L. (V.) braziliensis infection remain unchanged. This implies that the binding sites of the polyclonal CL anti-α-Gal Abs might not fully accommodate the entire tetrasaccharide. This critical question awaits resolution in subsequent studies, ideally employing highly-purified monospecific polyclonal or recombinant L. (V.) braziliensis CL-specific anti-α-Gal Abs.
4. Conclusion:
We synthesized a tetrasaccharide from the type-2 GIPL found in L. (L.) major and L. mexicana and conjugated it to BSA as a carrier protein, producing NGP31b. This and the previously synthesized NGP28b, containing a trisaccharide as glycan moiety, were tested as BMKs for New-World CL using cELISA with sera from patients infected with L. (V.) braziliensis. Both NGPs were specifically recognized by the L. (V.) braziliensis CL-specific anti-α-Gal Abs, with minimal cross-reactivity with NHS or CD anti-α-Gal Abs. The diagnostic properties such as sensitivity and specificity for both NGPs as BMKs were virtually identical, indicating that the trisaccharide in NGP28b is sufficient for effective Ab recognition and distinction from NHS and CD anti-α-Gal Abs. Adding an αManp unit to the reducing end, as seen in NGP31b, did not enhance its performance as BMK nor hinder its recognition by the L. (V.) braziliensis CL-specific anti-α-Gal Abs. Both NGP28b and NGP31b have proven to be accurate BMKs for New-World CL attributed to L. (V.) braziliensis.
5. Experimental
5.1. Synthesis of G31SH and its precursors
5.1.1. p-Tolyl 2,3-di-O-benzoyl-α-d-galactopyranosyl-(1→3)-2-O-benzoyl-5,6-O-isopropylidene-1-thio-β-d-galactofuranoside (4)
Fully protected disaccharide 3 (200 mg, 0.21 mmol) was dissolved in a mixture of HF-pyr/dry THF (400 μL/40 mL) in a plastic conical tube and stirred for 30 min at 0°C and then 30 min at rt under Ar. The reaction mixture was cooled down again to 0°C and quenched with saturated NaHCO3. Then, diluted and extracted with EtOAc, washed with water and brine, dried over MgSO4, concentrated and purified by column chromatography in silica gel (hexanes/EtOAc = 1:1) to give 4 (122 mg, 72%) as a white powder. Rf 0.37 (hexanes/EtOAc = 1:1). 1H NMR (400 MHz, CDCl3) δ 1.15 (s, 3H, CH3); 1.31 (s, 3H, CH3); 2.33 (s, 3H, arom.CH3); 3.75–3.86 (m, 2H, Hf-6a,b); 3.88–4.04 (m, 2H, Hp-6a,b); 4.16 (dd, J = 6.7, J = 4.3 Hz, 1H, Hf-5); 4.29 (dd, J = 5.3, J = 1.5 Hz, 1H, Hp-5); 4.33–4.41 (m, 2H, Hf-3, Hf-4); 4.52 (s, 1H, Hp-4); 5.29 (solvent CH2Cl2); 5.52 (br. s., 1H, Hp-1); 5.58 (d, 3J1,2 = 1.3 Hz, 1H, Hf-1); 5.67 (t, J = 2.0 Hz, 1H, Hf-2); 5.73 (br. s., 2H, Hp-2, Hp-3); 7.11 (d, J = 7.9 Hz, 2H, arom.); 7.32–7.62 (m, 11H, arom.); 7.97–8.09 (m, 6H, arom.) ppm. 13C NMR (101 MHz, CDCl3, 300K) δ 21.1 (arom.CH3); 24.8 (CH3); 26.0 (CH3); 53.4 (solvent CH2Cl2); 63.2 (Cp-6); 65.2 (Cf-6); 68.5 (Cp-2); 69.5 (Cp-4); 70.6; 71.0 (Cp-3); 74.1 (Cf-5); 82.0 (Cf-2); 82.2; 83.2 (Cp-5); 91.1 (Cf-1); 97.4 (Cp-1); 109.7 (Cq-isop.); 128.4 (C-arom.); 128.5 (C-arom.); 128.9 (Cq, arom.); 129.2 (Cq, arom.); 129.3 (Cq, arom.); 129.7 (Cq, arom.); 129.7 (C-arom.); 129.8 × 3 (C-arom.); 132.8 (C-arom.); 133.2 (C-arom.); 133.3 (C-arom.); 133.7 (C-arom.); 137.9 (Cq); 165.6 (C=O); 165.8 (C=O); 165.9 (C=O) ppm. ESI-TOF HRMS: m/z [M+Na]+ calcd for C43H44NaO13S 823.2400, found 823.2411.
5.1.2. p-Tolyl 2,3-di-O-benzoyl-4,6-O-di-tert-butylsilylene-α-d-galactopyranosyl-(1→6)-2,3-di-O-benzoyl-α-d-galactopyranosyl-(1→3)-2-O-benzoyl-5,6-O-isopropylidene-1-thio-β-d-galactofuranoside (5)
To a solution of disaccharide acceptor 4 (107 mg, 0.13 mmol) and Galp donor 1 (120 mg, 0.18 mmol) in anhydrous DCM (18 mL), freshly activated MS 4Ǻ was added and stirred under Ar for 1 h at rt. Then, the solution was cooled down to 0°C and TMS-OTf (6.5 μL, 0.036 mmol) was added dropwise. The solution was gradually brought to rt and after 1 h, the reaction mixture was quenched by addition of Et3N, filtered, and washed with water and brine. The organic layers were dried over MgSO4, concentrated, and purified by flash column chromatography on silica gel (hexanes/EtOAc = 3:1) to give the trisaccharide 5 as a beige powder (70 mg, 45%). Rf = 0.30 (hexanes/EtOAc = 3:1). 1H NMR (400 MHz, CDCl3) δ 7.93–8.03 (m, 9H, arom.); 7.93–8.03 (m, 9H, arom.); 7.31–7.57 (m, 15H, arom.); 7.20 (d, J = 7.3 Hz, 1H, arom.); 7.08–7.15 (m, 2H, arom.); 7.04 (d, J = 8.1 Hz, 2H, arom.); 5.67–5.77 (m, 4H); 5.54–5.65 (m, 2H); 5.48 (d, J = 3.8 Hz, 1H); 5.33 (d, J = 3.5 Hz, 1H); 4.79 (d, J = 2.8 Hz, 1H); 4.50–4.56 (m, 1H); 4.37–4.43 (m, 2H); 4.30 (br. s., 2H); 4.20 (d, J = 5.0 Hz, 1H); 4.03–4.16 (m, 3H); 3.72–3.80 (m, 3H); 2.30 (s, 3H, arom.CH3); 1.26 (s, 3H, CH3); 1.12 (s, 12H, tBu, CH3); 0.91 (s, 9H, tBu) ppm. 13C NMR (101 MHz, CDCl3, 300K) δ 166.0 (C=O); 165.9 (C=O); 165.8 (C=O); 165.5 (C=O); 165.4 (C=O); 163.5 (Cq); 137.9 (Cq); 133.5 (C-arom.); 133.3 (C-arom.); 133.1 (C-arom.); 133.0 (C-arom.); 132.9 (C-arom.); 132.8 (C-arom.); 129.8 × 2 (C-arom.); 129.7 × 2 (C-arom.); 129.6 × 2 (C-arom.); 129.3 (Cq, arom.); 129.2 (Cq, arom.); 129.1 (Cq, arom.); 129.0 (Cq, arom.); 128.6 (C-arom.); 128.5 (C-arom.); 128.4 (C-arom.); 128.3 (C-arom.); 128.2 (C-arom.); 109.7 (Cq-isop.); 97.6 (CH); 97.3 (CH); 91.8 (Cq); 91.4 (CH); 84.1 (CH); 82.4 (CH); 81.9 (CH); 74.1 (CH); 71.1 (CH); 71.0 (CH); 70.1 (CH); 69.2 (CH); 68.6 (CH); 68.5 (CH); 68.4 (CH); 67.3 (CH2); 67.1 (CH); 67.0 (CH2); 65.2 (CH2); 27.4 (tBu); 27.2 (tBu); 26.0 (CH3); 24.8 (CH3); 23.2 (Cq-tBu); 21.1 (arom.CH3); 20.7 (Cq-tBu) ppm. ESI-TOF HRMS: m/z [M+Na]+ calcd for C71H78NaO20SSi 1333.4474, found 1333.4535.
5.1.3. Allyl 2,3-di-O-benzoyl-4,6-O-di-tert-butylsilylene-α-d-galactopyranosyl-(1→6)-2,3-di-O-benzoyl-α-d-galactopyranosyl-(1→3)-2-O-benzoyl-5,6-O-isopropylidene-β-d-galactofuranosyl-(1→3)-4,6-O-benzylidene-α-d-mannopyranoside (7)
To a solution of the Man acceptor 6 (73 mg, 0.24 mmol) and trisaccharide donor 5 (156 mg, 0.12 mmol) in anhydrous DCM (16 mL), NIS (40 mg, 0.18 mmol) was added, and the mixture was stirred in the presence of molecular sieves (4Å) for 1h at rt under Ar, and then cooled to −20°C. A TfOH solution was prepared by diluting TfOH 100-fold with DCM. Five 20 μL aliquots of this solution (100 μL in total containing 0.012 mmol TfOH) were added to the reaction mixture (one aliquot every 15 min). After 1 h, the reaction was quenched with Et3N, filtered, and concentrated. Flash column chromatography on silica gel (hexanes/EtOAc = 2:1) gave the desired fully protected tetrasaccharide 7 (108 mg, 61%), as a white powder. Rf 0.27 (hexanes/EtOAc = 3:1). [α]D24= +25.03 (c = 0.1 in CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.93–7.98 (m, 4H, arom.); 7.87–7.91 (m, 2H, arom.); 7.77–7.85 (m, 4H, arom.); 7.49–7.58 (m, 5H, arom.); 7.27–7.45 (m, 11H, arom.); 7.19–7.25 (m, 4H, arom.); 5.87–5.99 (m, 2H); 5.84 (d, J = 5.1, 2H); 5.56–5.69 (m, 4H); 5.53 (d, J = 3.3, 1H); 5.40–5.46 (m, 1H); 5.35 (dd, J = 17.2, J = 1.6 Hz, 1H); 5.25 (dd, J = 10.4, J = 1.3 Hz, 1H); 4.99 (d, J = 8.2 Hz, 1H); 4.18–4.46 (m, 9H); 3.96–4.09 (m, 4H); 3.61–3.92 (m, 7H); 3.44–3.51 (m, 1H); 3.36–3.43 (m, 1H); 2.05 (d, J = 3.4, 1H); 1.15 (s, 3H, CH3); 1.10 (s, 9H, tBu); 1.06 (s, 3H, CH3); 0.89 (s, 9H, tBu) ppm. 13C NMR (101 MHz, CDCl3, 300K) δ 166.9 (C=O); 166.0 (C=O); 165.8 (C=O); 165.7 (C=O); 165.4 (C=O); 138.2 (Cq, arom.); 133.9 (C-arom.); 133.5 (C-arom.); 133.4 (C-arom.); 133.1 (C-arom.); 132.8 (C-arom.); 130.0 (Cq, arom.); 129.9 (Cq, arom.); 129.8 (C-arom.); 129.7 × 2 (C-arom.); 129.6 (C-arom.); 129.2 (Cq, arom.); 129.1 (Cq, arom.); 128.9 (Cq, arom.); 128.7 (C-arom.); 128.5 (C-arom.); 128.4 × 2 (C-arom.); 128.3 (C-arom.); 128.2 (C-arom.); 128.1 (C-arom.); 126.5 (C-arom.); 117.1 (C-c); 109.4 (Cq-isop.); 102.5 (CH, C-1); 102.1 (CH, C-1); 99.7 (CH, C-1); 97.8 (CH, C-1); 96.1 (CH, C-1); 84.8 (CH); 83.2 (CH); 79.8 (CH); 77.5 (CH); 77.2 (CH); 74.9 (CH); 72.1 (CH); 72.0 (CH); 71.2 (CH); 70.7 (CH); 69.3 (CH); 69.0 (CH); 68.8 (CH); 68.4 (CH); 68.2 (CH2); 67.4 (CH2); 67.3 (CH); 67.0 (CH2); 65.3 (CH2); 63.8 (CH); 29.7 (CH2); 27.5 (tBu); 27.2 (tBu); 26.0 (CH3); 23.2 (Cq-tBu); 25.1 (CH3); 20.7 (Cq-tBu) ppm. ESI-TOF HRMS: m/z [M+Na]+ calcd for C80H90NaO26Si 1517.5387, found 1517.5383.
5.1.4. Allyl 2,3-di-O-benzoyl-α-d-galactopyranosyl-(1→6)-2,3-di-O-benzoyl-α-d-galactopyranosyl-(1→3)-2-O-benzoyl-5,6-O-isopropylidene-β-d-galactofuranosyl-(1→3)-4,6-O-benzylidene-α-d-mannopyranoside (8)
Fully protected tetrasaccharide 7 (108 mg, 0.07 mmol) was dissolved in a mixture ratio (1:100 HF-pyr/dry THF) (222 μL/ 22 mL) in a plastic conical tube and stirred for 30 min at 0°C and then 1h at rt under Ar. The reaction mixture was cooled down again to 0°C and quenched with saturated NaHCO3. Then, diluted and extracted with EtOAc, washed with water and brine, dried over MgSO4, concentrated, and purified by column chromatography in silica gel (hexanes/EtOAc = 1:2) to give 8 (74 mg, 76%) as a white powder. Rf 0.38 (hexanes/EtOAc = 1:2). [α]D24 = +23.07 (c = 0.1 in CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.97 (m, 6H, arom.), 7.81 (m, , 4H, arom.), 7.57 – 7.47 (m, 6H, arom.), 7.43 – 7.30 (m, 10H, arom.), 7.30 – 7.26 (m, 2H, arom.), 7.25 – 7.21 (m, 2H, arom.), 5.95 – 5.85 (m, 1H, H-b), 5.81 (s, 1H, OCHPh), 5.74 (d, J = 1.3 Hz, 1H), 5.67 – 5.64 (m, 3H, including an H-1 ), 5.57 (d, J = 2.6 Hz, 1H, H-1), 5.52 – 5.45 (m, 2H, including an H-1), 5.31 (dq, J = 17.3, 1.7 Hz, 1H, H-c), 5.23 (dq, J = 10.4, 1.4 Hz, 1H, H-c), 4.89 – 4.83 (m, 1H), 4.44 (t, J = 5.0 Hz, 1H), 4.36 (dd, J = 9.3, 4.9 Hz, 2H), 4.27 – 3.95 (m, 13H, including an H-1), 3.92 – 3.69 (m, 6H), 3.47 (dd, J = 8.4, 6.7 Hz, 1H, H-a), 3.26 (dd, J = 8.3, 6.9 Hz, 2H, 1H+ H-a), 3.07 (s, 1H, OH), 2.41 (s, 1H, OH), 1.18 (s, 3H, CH3), 1.02 (s, 3H, CH3) ppm. 13C NMR (101 MHz, CDCl3) δ 166.7 (C=O), 166.5 (C=O), 165.8 (C=O), 165.7 (C=O), 165.5 (C=O), 138.1 (Cq), 133.8 (C-arom.), 133.7 (C-arom.), 133.4 (C-arom.), 133.38 (C-arom.), 133.3 (C-arom.), 132.9 (C-arom.), 129.9 (C-arom.), 129.73 (C-arom.), 129.71(C-arom.), 129.6 (C-arom.), 129.4 (Cq), 129.3 (Cq), 129.2(Cq), 128.9 (C-arom.), 128.7 (C-arom.), 128.5 (C-arom.), 128.4 (C-arom.), 128.3 (C-arom.), 128.1(C-arom.), 126.5 (C-arom.), 117.3 (CH2, C-c), 109.3 (CH), 102.7 (CH, C-1), 102.1(CH, OCHPh), 99.8 (CH, C-1), 98.1(CH, C-1), 96.3 (CH, C-1), 84.84 (CH), 81.9 (CH), 80.8 (CH), 77.3 (CH), 74.4 (CH), 72.4 (CH), 70.9 (CH), 70.2 (CH), 69.9 (CH), 69.8 (CH), 68.9 (CH), 68.8 (CH2), 68.6 (CH), 68.4 (CH), 68.2 (CH2), 66.9 (CH2), 65.1 (CH2), 63.9 (CH), 62.9 (CH2), 25.9 (CH3), 25.0 (CH3) ppm. ESI-TOF HRMS: m/z [M+Na]+ calcd for C72H74NaO26 1377.4366, found 1377.4356.
5.1.5. Allyl 2,3-di-O-benzoyl-α-d-galactopyranosyl-(1→6)-2,3-di-O-benzoyl-α-d-galactopyranosyl-(1→3)-2-O-benzoyl-β-d-galactofuranosyl-(1→3)-α-d-mannopyranoside (9)
Tetrasaccharide 8 (60 mg, 0.02 mmol) was dissolved in DCM (6 mL). While stirring at rt, H2O (0.5 mL) and TFA (0.5 mL) were sequentially added, and the reaction proceeded for 1 h. After the starting material was consumed based on TLC, the reaction mixture was quenched with Et3N. The solution was concentrated and dried under vacuum. Purification by column chromatography on silica gel (DCM/MeOH = /DCM = 13:1) yielded 9 (39 mg, 72%) as a white powder. Rf 0.30 (DCM/MeOH = 13:1); [α]D24 = +17.11 (c = 0.1 in CHCl3) ; 1H NMR (400 MHz, CDCl3) δ 8.00 – 7.85 (m, 10H, arom.), 7.55 – 7.40 (m, 4H, arom.), 7.39 – 7.27 (m, 8H, arom.), 7.25 (d, J = 1.5 Hz, 1H, arom.), 7.23 – 7.14 (m, 2H, arom.), 5.81 (m, 1H, H-b), 5.72 (dd, J = 10.6, 2.9 Hz, 1H), 5.65 (d, J = 2.5 Hz, 2H), 5.58 (dd, J = 10.5, 3.8 Hz, 1H), 5.54 (d, J = 3.8 Hz, 1H, H-1), 5.51 (d, J = 2.9 Hz, 1H), 5.41 (d, J = 2.6 Hz, 1H, H-1), 5.28 (s, 1H, H-1), 5.22 (dq, J = 17.2, 1.6 Hz, 1H, H-c), 5.13 (dq, J = 10.4, 1.3 Hz, 1H, H-2), 4.75 (d, J = 1.6 Hz, 1H, H-1), 4.60 – 4.51 (m, 2H), 4.44 (d, J = 12.2 Hz, 2H), 4.39 – 4.33 (m, 2H), 4.26 (d, J = 4.9 Hz, 2H), 4.20 (s, 1H), 4.08 (ddt, J = 12.4, 5.2, 1.6 Hz, 3H), 4.04 – 3.81 (m, 8H), 3.74 – 3.56 (m, 5H), 3.45 (s, 1H, OH), 2.98 (d, J = 25.8 Hz, 2H, OH), 2.82 (d, J = 4.1 Hz, 1H, OH) ppm. 13C NMR (101 MHz, CDCl3) δ 166.4 (C=O), 166.2 (C=O), 166.1 (C=O), 166.0 (C=O), 165.9 (C=O), 133.8 (C-arom.), 133.7 (C-arom.), 133.5 (C-arom.), 133.4 (C-arom.), 133.2 (C-arom.), 133.1 (C-arom.), 129.9 (C-arom.), 129.82 (C-arom.), 129.75 (C-arom.), 129.7 (C-arom.), 129.4 (C-arom.), 129.3 (C-arom.), 129.2 (C-arom.), 128.9 (C-arom.), 128.8 (C-arom.), 128.6 (C-arom.), 128.5 (C-arom.), 128.37 (C-arom.), 128.35 (C-arom.), 128.4 (C-arom.), 117.5 (CH2, C-c), 103.9 (CH), 98.9 (CH), 97.7 (CH), 97.5 (CH), 83.6 (CH), 82.4 (CH), 81.5 (CH), 77.9 (CH), 77.2 (CH), 72.4 (CH), 71.1 (CH), 70.8 (CH), 70.7 (CH), 69.2 (CH), 68.98 (CH), 68.92 (CH), 68.7 (CH), 68.3 (CH), 68.0 (CH2), 67.8 (CH), 66.6 (CH2), 65.4 (CH), 63.8 (CH2), 62.5 (CH2), 61.91 (CH2) ppm. ESI-TOF HRMS: m/z [M+Na]+ calcd for C62H66NaO26 1249.3740, found 1249.3739.
5.1.6. (Acetylthio)propyl-2,3-di-O-benzoyl-α-d-galactopyranosyl-(1→6)-2,3-di-O-benzoyl-α-d-galactopyranosyl-(1→3)-2-O-benzoyl-β-d-galactofuranosyl-(1→3)-α-d-mannopyranoside (10)
To a solution of allyl tetrasaccharide 9 (26.2 mg, 0.02 mmol) and DPAP (109 μL of a solution of 5 mg DPAP in 1mL DCM, 0.0021 mmol) in anhydrous DCM (700 μL) under Ar, thioacetic acid (8 μL, 0.10 mmol) was added, and the mixture was stirred under water cooling (~ 25 °C) for 30 min in a Rayonet UV reactor equipped with 350 nm lamps. The solution was then concentrated to near dryness. The crude product was purified by PTLC on silica gel (DCM/MeOH = 20:1) to afford the acyl-protected tetrasaccharide 10 (21.4 mg, 80%) as a white solid. Rf 0.22 (DCM/MeOH = 15:1). 1H NMR (400 MHz, CDCl3-d) δ 7.98 – 7.86 (m, 11H, arom.), 7.53 – 7.43 (m, 4H, arom.), 7.38 – 7.28 (m, 8H, arom.), 7.22 – 7.17 (m, 2H, arom.), 5.72 (dd, J = 10.5, 2.9 Hz, 1H), 5.65 (d, J = 2.6 Hz, 2H), 5.58 (dd, J = 10.5, 3.7 Hz, 1H), 5.53 (d, J = 3.7 Hz, 1H, H-1), 5.51 (d, J = 2.5 Hz, 1H), 5.42 (d, J = 2.7 Hz, 1H, H-1), 5.30 (s, 1H, H-1), 4.69 (d, J = 1.6 Hz, 1H, H-1), 4.61 – 4.53 (m, 2H), 4.51 – 3.49 (m, 25H), 3.35 – 3.25 (m, 1H), 2.88 (t, J = 7.2 Hz, 2H, OCH2CH2CH2SAc), 2.78 (s, 1H), 2.28 (s, 3H, −SCOCH3), 1.85 – 1.72 (m, 2H, OCH2CH2CH2SAc) ppm. 13C NMR (101 MHz, CDCl3) δ 196.1 (−SC=OCH3), 166.4 (C=O), 166.14 (C=O), 166.06 (C=O), 165.99 (C=O), 165.95 (C=O), 133.8 (C-arom.), 133.5 (C-arom.), 133.4 (C-arom.), 133.3 (C-arom.), 133.2 (C-arom.), 129.89 (C-arom.), 129.82 (C-arom.), 129.8 (C-arom.), 129.7 (C-arom.), 129.4 (C-arom.), 129.3 (C-arom.), 129.2 (C-arom.), 128.9 (C-arom.), 128.7 (C-arom.), 128.6 (C-arom.), 128.5 (C-arom.), 128.39 (C-arom.), 128.36 (C-arom.), 128.3 (C-arom.), 103.8 (C-1), 99.7 (C-1), 97.8 (C-1), 97.5 (C-1), 83.8 (CH), 82.4 (CH), 81.63 (CH), 77.7 (CH), 77.2 (CH), 72.5 (CH), 71.1 (CH), 70.9 (CH), 70.7 (CH), 69.2 (CH), 69.0 (CH), 68.9 (CH), 68.7 (CH), 68.2 (CH), 67.7 (CH), 66.6 (CH2), 65.7 (CH2), 65.3 (CH), 63.8 (CH2), 62.5 (CH2), 61.9 (CH2), 30.6 (SCOCH3), 29.2 (−OCH2CH2CH2SAc), 25.9 (−OCH2CH2CH2SAc) ppm. ESI-TOF HRMS: m/z [M+Na]+ calcd for C64H70NaO27S 1325.3723, found 1325.3733.
5.1.7. Thiopropyl-α-d-galactopyranosyl-(1→6)-α-d-galactopyranosyl-(1→3)-β-d-galactofuranosyl-(1→3)-α-d-mannopyranoside (G31SH)
To a flask containing 10 (21.4 mg, 0.016 mmol) in 2 mL of MeOH, 226 μL of 0.1M NaOMe was added under Ar, and stirred at rt for 1 h. HRMS indicated complete removal of the protecting groups, and all material was present as a mixture of thiol and disulfide. Drops of water were added under stirring until a pH of 7 was achieved, followed by concentration and lyophilization. Initially, the unprotected mercaptopropyl tetrasaccharide G31SH is produced, which oxidizes by handling on air within hours to the disulfide (G31S)2 (12.3 mg, quant.), which was obtained as a white powder. [α]D24 = −10.01 (c = 0.1 in H2O). 1H NMR (400 MHz, D2O) δ 5.13 (s, 1H, H-1), 5.03 (d, J = 3.4 Hz, 1H, H-1), 4.95 (d, J = 3.7 Hz, 1H, H-1), 4.87 (s, 1H, H-1), 4.39 (s, 1H), 4.27 (dt, J = 16.3, 5.9 Hz, 2H), 4.15 – 3.56 (m, 23H), 2.84 (t, 2H, OCH2CH2CH2S), 2.18 – 1.79 (m, 2H, OCH2CH2CH2S) ppm. 13C NMR (101 MHz, D2O) δ 104.7 (C-1), 99.64 (C-1), 99.59 (C-1), 98.5 (C-1), 84.6 (CH), 82.0 (CH), 79.5 (CH), 75.4 (CH), 72.9 (CH), 71.08 (CH), 71.01(CH), 69.6 (CH), 69.46 (CH), 69.45 (CH), 69.3 (CH), 69.2 (CH), 68.3 (CH), 68.2 (CH), 67.0 (CH), 66.9 (CH), 66.1 (CH), 65.1 (CH), 62.9 (CH), 61.3 (CH), 61.0 (CH), 34.9 (CH2), 28.1 (CH2) ppm. ESI-TOF HRMS: m/z [M+Na]+ calcd for C27H48NaO21S 763.2306, found 763.2340; for and for C54H94NaO42S2 1501.4559, found 1501.5159.
5.2. Conjugation of G31SH to BSA to produce NGP31b
The thiol-containing glycan (G31S)2 was conjugated to BSA using the Imject™ Maleimide-Activated BSA kit (catalog #77116, Thermo Fisher Scientific). The conjugation followed the manufacturer’s instructions, as previously described [55]. To initiate the procedure, a 0.05 M TCEP solution was obtained by diluting the proprietary Bond-Breaker 0.5 M TCEP solution (catalog #77720, Thermo Fisher Scientific,), using the kits’ conjugation buffer supplied containing 83 mM sodium phosphate buffer, 0.1 M EDTA, 0.9 M sodium chloride, and 0.02% sodium azide at pH 7.2. Upon mixing the diluted 0.9 equiv TCEP solution with disulfide (G31S)2 in a 1.5-mL microcentrifuge tube, the mixture was agitated for 30 min on a shaker, resulting in the thiol G31SH. Meanwhile, the maleimide-activated BSA (2 mg, 15–25 moles of maleimide per mole of BSA) was rehydrated using 200 μL of conjugation buffer to achieve a 10 mg/mL solution. Following this, the disaccharide solution was added to the BSA solution, and the combined solution was agitated at room temperature for 2–3 h. The solution was desalted by diluting the conjugation mixture with 1 mL HPLC-grade water, followed by centrifugation (20 min at 4,000 × g, rt) using the Amicon Ultra 3K (Millipore) centrifugal filter. This step was repeated twice. This desalting step was crucial to remove any residual salts and impurities. To ensure purity and minimize aggregation of the NGP, the solution was further processed through a 2-mL Zeba™ spin (7K MWCO, catalog # 89882, Thermo Fisher Scientific) desalting column that had undergone prior washing steps. The treated solution was then lyophilized, ensuring its long-term storage (minimum of 6 months) at −50 °C until use. The concentration of the resulting NGP31b was determined by preparing a solution in ultrapure water and assessing its concentration with the Pierce BCA Protein Assay (Thermo Fisher Scientific). Lastly, comparative MALDI-TOF MS spectra were taken to determine the conjugation efficiency, which provided insights into the average number of G31SH units attached per BSA molecule (Figure 2B).
5.3. MALDI-TOF-MS of NGP31b
To determine the average molecular masses of BSA and NGP31b, 1 μL of BSA solution (0.4 mg/mL in H2O) was mixed with 1 μL of NGP31b solution (1 mg/mL in H2O) and 2 μL of a matrix solution (10 mg/mL sinapinic acid in 50% acetonitrile with 0.1% TFA) in a 1.5-mL microcentrifuge tube. Subsequently, 2 μL of this sample-matrix mixture was applied onto a 48-well steel MALDI plate and allowed to crystallize at room temperature for approximately 20 min. Mass spectra were obtained by matrix-assisted laser/desorption ionization (MALDI-TOF-MS) using a Shimadzu MALDI-8020 MS configured in linear mode. The instrument employed dithering across a scan range of 10,000 to 100,000 m/z. Parameters for data acquisition were set as follows: laser power at 115 units, repetition rate at 50 Hz, with 5 accumulated shots and 2 blast shots to clean the target plate before acquisition, and 200 profiles were averaged per spectrum. Pulse extraction was optimized to a value of 66,431, and a blanking mass was implemented at m/z 15,000 to exclude background ions below this threshold. For data processing, the Threshold Apex software provided by Shimadzu was utilized. The processing parameters were standardized and included a set threshold for peak detection, application of Gaussian smoothing with a filter width of 400, and a set peak width of 2 to refine peak detection. The MALDI-TOF-MS was calibrated using the Pierce Bovine Serum Albumin (BSA) Standard Ampules at a concentration of 2 mg/mL (Thermo Fisher Scientific, catalog number 23209). Internal calibration was performed by setting the [BSA + H]+ ion at m/z 66,402, with a stringent mass tolerance of 5 ppm to ensure accuracy in mass measurement.
5.4. Ethics Statement
This study was conducted according to the regulations of the International Ethical Guidelines for Biomedical Research Involving Human Subjects, the Good Clinical Practice guidelines, and the Declaration of Helsinki. Panels of positive serum samples from patients with cutaneous leishmaniasis (CL) or chronic Chagas disease (CD), or from healthy individuals from endemic areas for both CL and CL (negative controls, NC) used in the study were reviewed and approved by the Institutional Review Board (IRB) of the University of Texas at El Paso, under protocol # 1590350, and by the IRB committees of the original institutions: Universidad Mayor de San Simón, Faculty of Medicine, and Fundación CEADES, Cochabamba, Bolivia; and Universidad Nacional de Salta, Consejo Nacional de Investigaciones Científicas y Técnicas, Salta, Argentina. Each participant at the original institutions voluntarily signed an informed consent form. All the serum samples used in this study were de-identified and coded using a number assigned by the principal investigator at the original institution. At UTEP, the samples received a separate code assigned by the personnel involved in the study. At no time were the PI or the personnel at UTEP able to identify any patient(s), nor were they able to identify any sample(s). Additionally, the personnel involved in the study performed at UTEP had to sign a mandatory confidentiality agreement.
5.5. Cohort Description
Serum samples from adult patients with cutaneous leishmaniasis (CL) (n=28, total) caused by L. (V.) braziliensis were from Universidad Nacional de Salta, Consejo Nacional de Investigaciones Científicas y Técnicas, Salta, Argentina (n=19), and Universidad Mayor de San Simón, Faculty of Medicine, and Fundación CEADES, Cochabamba, Bolivia (n=9). CL was diagnosed by evaluating the patient’s medical history and histological analysis of the smear of dermal lesions [12, 56]. Molecular diagnosis by PCR was also carried out in CL patients from Salta, confirming the patients were infected with L. (V.) braziliensis [57]. All patients from Salta and Cochabamba exhibited typical CL lesions. Serum samples from patients with chronic Chagas disease (CD; n=28) and from individuals seronegative for CD (negative controls, NC) (n=28) from Universidad Mayor de San Simón, Faculty of Medicine, and Fundación CEADES, Cochabamba, Bolivia. T. cruzi infections were diagnosed by the conventional serology using a conventional ELISA kit (CHAGATEK ELISA, Laboratorio Lemos SRL, Buenos Aires Argentina) and a recombinant ELISA kit (Chagatest ELISA recombinante, V3.0, Wiener Lab, Rosario, Argentina). The province of Salta, Argentina, and the department of Cochabamba, Bolivia, are endemic areas for CD and CL, and present zones with co-infection cases [58–60]. Table 3 shows the gender distribution of the study cohort.
Table 3.
Gender distribution of the study cohort.
| Gender | CD (n=28) | CL (n=28) | NC (n=28) |
|---|---|---|---|
| n (%) | |||
| Female | 11 (39%) | 4 (14%) | 14 (50%) |
| Male | 17 (61%) | 24 (86%) | 14 (50%) |
| Total | 28 (100%) | 28 (100%) | 28 (100%) |
5.6. Chemiluminescent ELISA (cELISA)
The cELISA was performed exactly as previously described [30].
Supplementary Material
Highlights.
The tetrasaccharide Galpα1,6Galpα1,3Galfβ1,3Manpα derived from a Leishmania type-2 glycoinositolphospholipid was synthesized and conjugated to the carrier protein bovine serum albumin, resulting in a novel α-Gal-containing neoglycoprotein, NGP31b.
NGP31b is an excellent biomarker for New-World or American cutaneous leishmaniasis, effectively diagnosing Leishmania (V.) braziliensis infection and differentiating between healthy individuals and those with Trypanosoma cruzi infection (Chagas disease).
The tetrasaccharide in NGP31b is unnecessarily long. A direct serological comparison with a similar neoglycoprotein, NGP28b, which comprises the trisaccharide Galpα1,6Galpα1,3Galfβ, reveals that their diagnostic attributes, such as sensitivity, specificity, and accuracy are equivalent. This indicates that the Manpα unit in NGP31b is redundant.
Acknowledgments
This work was supported by NIH/NIAID grants # 1R21AI137890-01 (to KM), U01AI129783 (to ICA), and ANPCyT, Argentina grant # PICT-2014-1579 (to JDM). ALM and IV are grateful for a Dr. Keelung Hong Graduate Research Fellowship, UTEP. ILE was supported by the UTEP Graduate Excellence Fellowship. The authors thank the Biomolecule Analysis and Omics Unit (BAOU), at BBRC/UTEP, supported by grant # 5U54MD007592 (to Robert A. Kirken), from the National Institute on Minority Health and Health Disparities (NIMHD), a component of the National Institutes of Health (NIH), for the full access to the MALDI-TOF-MS and other BAOU instruments used in this study.
References
- [1].Hong A, Zampieri RA, Shaw JJ, Floeter-Winter LM, Laranjeira-Silva MF, Pathogens, 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ramírez JL, Guevara P, Mem Inst Oswaldo Cruz, 92 (1997) 333–338. [DOI] [PubMed] [Google Scholar]
- [3].Scorza BM, Carvalho EM, Wilson ME, Int J Mol Sci, 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hashiguchi Y, Gomez EL, Kato H, Martini LR, Velez LN, Uezato H, Trop Med Health, 44 (2016) 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Turetz ML, Machado PR, Ko AI, Alves F, Bittencourt A, Almeida RP, Mobashery N, Johnson WD Jr., Carvalho EM, J Infect Dis, 186 (2002) 1829–1834. [DOI] [PubMed] [Google Scholar]
- [6].Machado PR, Rosa ME, Guimarães LH, Prates FV, Queiroz A, Schriefer A, Carvalho EM, Clin Infect Dis, 61 (2015) 945–949. [DOI] [PubMed] [Google Scholar]
- [7].Gomes CM, Paula NA, Morais OO, Soares KA, Roselino AM, Sampaio RN, An Bras Dermatol, 89 (2014) 701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Goto H, Lindoso JA, Expert Rev Anti Infect Ther, 8 (2010) 419–433. [DOI] [PubMed] [Google Scholar]
- [9].Maia Z, Lírio M, Mistro S, Mendes CM, Mehta SR, Badaro R, PLoS Negl Trop Dis, 6 (2012) e1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sánchez-Ovejero C, Benito-Lopez F, Díez P, Casulli A, Siles-Lucas M, Fuentes M, Manzano-Román R, J Proteomics, 136 (2016) 145–156. [DOI] [PubMed] [Google Scholar]
- [11].Zanetti ADS, Sato CM, Longhi FG, Ferreira SMB, Espinosa OA, Rev Inst Med Trop Sao Paulo, 61 (2019) e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bracamonte ME, Alvarez AM, Sosa AM, Hoyos CL, Lauthier JJ, Cajal SP, Juarez M, Uncos RE, Sanchez-Valdez FJ, Acuna L, Diosque P, Basombrio MA, Nasser JR, Hashiguchi Y, Korenaga M, Barroso PA, Marco JD, PLoS One, 15 (2020) e0232829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zawadzki J, Scholz C, Currie G, Coombs GH, McConville MJ, J Mol Biol, 282 (1998) 287–299. [DOI] [PubMed] [Google Scholar]
- [14].McConville MJ, Collidge TA, Ferguson MA, Schneider P, J Biol Chem, 268 (1993) 15595–15604. [PubMed] [Google Scholar]
- [15].McConville MJ, Ferguson MAJ, Biochem. J, 294 (1993) 305–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].McConville MJ, Homans SW, Thomas-Oates JE, Dell A, Bacic A, J Biol Chem, 265 (1990) 7385–7394. [PubMed] [Google Scholar]
- [17].de Lederkremer RM, Giorgi ME, Marino C, ACS Infect Dis, 8 (2022) 2207–2222. [DOI] [PubMed] [Google Scholar]
- [18].de Lederkremer RM, Colli W, Glycobiology, 5 (1995) 547–552. [DOI] [PubMed] [Google Scholar]
- [19].McConville MJ, Bacic A, J Biol Chem, 264 (1989) 757–766. [PubMed] [Google Scholar]
- [20].Travassos LR, Almeida IC, Springer Semin Immunopathol, 15 (1993) 183–204. [DOI] [PubMed] [Google Scholar]
- [21].Avila JL, Subcell Biochem, 32 (1999) 173–213. [DOI] [PubMed] [Google Scholar]
- [22].Avila JL, Rojas M, Galili U, J Immunol, 142 (1989) 2828–2834. [PubMed] [Google Scholar]
- [23].Avila JL, Rojas M, Acosta A, J Clin Microbiol, 29 (1991) 2305–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Avila JL, Rojas M, J Clin Microbiol, 28 (1990) 1530–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Montoya AL, Austin VM, Portillo S, Vinales I, Ashmus RA, Estevao I, Jankuru SR, Alraey Y, Al-Salem WS, Acosta-Serrano Á, Almeida IC, Michael K, JACS Au, 1 (2021) 1275–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Galili U, Shohet SB, Kobrin E, Stults CL, Macher BA, J. Biol. Chem, 263 (1988) 17755–17762. [PubMed] [Google Scholar]
- [27].Galili U, Mandrell RE, Hamadeh RM, Shohet SB, Griffis JM, Infect. Immun, 56 (1988) 1730–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Galili U, Rachmilewitz EA, Peleg A, Flechner I, J. Exp. Med, 160 (1984) 1519–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Galili U, The Natural Anti-Gal Antibody As Foe Turned Friend In Medicine, Elsevier Academic Press, London, San Diego, Cambridge, Oxford, 2018. [Google Scholar]
- [30].Viana SM, Montoya AL, Carvalho AM, de Mendonça BS, Portillo S, Olivas JJ, Karimi NH, Estevao IL, Ortega-Rodriguez U, Carvalho EM, Dutra WO, Maldonaldo RA, Michael K, de Oliveira CI, Almeida IC, Emerg Microbes Infect, 11 (2022) 2147–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dedet JP, Pratlong F, J Eukaryot Microbiol, 47 (2000) 37–39. [DOI] [PubMed] [Google Scholar]
- [32].Martinez DY, Verdonck K, Kaye PM, Adaui V, Polman K, Llanos-Cuentas A, Dujardin JC, Boelaert M, PLoS Negl Trop Dis, 12 (2018) e0006125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Milani SR, Travassos LR, Braz J Med Biol Res, 21 (1988) 1275–1286. [PubMed] [Google Scholar]
- [34].Almeida IC, Milani SR, Gorin PA, Travassos LR, J Immunol, 146 (1991) 2394–2400. [PubMed] [Google Scholar]
- [35].Gazzinelli RT, Pereira ME, Romanha A, Gazzinelli G, Brener Z, Parasite Immunol, 13 (1991) 345–356. [DOI] [PubMed] [Google Scholar]
- [36].Almeida IC, Ferguson MAJ, Schenkman S, Travassos LR, Biochem. J, 304 (1994) 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Almeida IC, Covas DT, Soussumi LM, Travassos LR, Transfusion, 37 (1997) 850–857. [DOI] [PubMed] [Google Scholar]
- [38].Ashmus RA, Schocker NS, Cordero-Mendoza Y, Marques AF, Monroy EY, Pardo A, Izquierdo L, Gállego M, Gascon J, Almeida IC, Michael K, Org. Biomol. Chem, 11 (2013) 5579–5583. [DOI] [PubMed] [Google Scholar]
- [39].Schocker NS, Portillo S, Brito CR, Marques AF, Almeida IC, Michael K, Glycobiology, 26 (2016) 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schocker NS, Portillo S, Ashmus RA, Brito CRN, Silva IE, Cordero-Mendoza Y, Marques AF, Monroy EY, Pardo A, Izquierdo L, Gállego M, Gascon J, Almeida IC, Michael K, in: Witzczak ZJ, Bielski R (Eds.) Coupling and Decoupling of Diverse Molecular Units in Glycosciences, Springer International Publishing AG, Cham, Switzerland, 2018, pp. 195–211. [Google Scholar]
- [41].Montoya AL, Carvajal EG, Ortega-Rodriguez U, Estevao IL, Ashmus RA, Jankuru SR, Portillo S, Ellis CC, Knight CD, Alonso-Padilla J, Izquierdo L, Pinazo MJ, Gascon J, Suarez V, Watts DM, Malo IR, Ramsey JM, Alarcón De Noya B, Noya O, Almeida IC, Michael K, Molecules, 27 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Montoya AL, Gil ER, Heydemann EL, Estevao IL, Luna BE, Ellis CC, Jankuru SR, Alarcón de Noya B, Noya O, Zago MP, Almeida IC, Michael K, Molecules, 27 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Imamura A, Kimura A, Ando H, Ishida H, Kiso M, Chem. Eur. J, 12 (2006) 8862–8870. [DOI] [PubMed] [Google Scholar]
- [44].Completo GC, Lowary TL, 73 (2008). [DOI] [PubMed] [Google Scholar]
- [45].Bai Y, Lowary TL, J. Org. Chem, 71 (2006) 9658–9671. [DOI] [PubMed] [Google Scholar]
- [46].Deng LM, Liu X, Liang XY, Yang JS, J. Org. Chem, 77 (2012) 3025–3037. [DOI] [PubMed] [Google Scholar]
- [47].Imamura A, Matsuzawa N, Sakai S, Udagawa T, Nakashima S, Ando H, Ishida H, Kiso M, J. Org. Chem, 81 (2016) 9086–9104. [DOI] [PubMed] [Google Scholar]
- [48].Winnik FM, Carver JP, Krepinsky JJ, J. Org. Chem, 47 (1982) 2701–2707. [Google Scholar]
- [49].Mandrekar JN, J Thorac Oncol, 5 (2010) 1315–1316. [DOI] [PubMed] [Google Scholar]
- [50].Greiner M, Pfeiffer D, Smith RD, Prev Vet Med, 45 (2000) 23–41. [DOI] [PubMed] [Google Scholar]
- [51].Power M, Fell G, Wright M, Evid Based Med, 18 (2013) 5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gildersleeve JC, Wright WS, Glycobiology, 26 (2016) 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Langley DB, Schofield P, Nevoltris D, Jackson J, Jackson KJL, Peters TJ, Burk M, Matthews JM, Basten A, Goodnow CC, van Nunen S, Reed JH, Christ D, Proc Natl Acad Sci U S A, 119 (2022) e2123212119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Galili U, Matta KL, Transplantation, 62 (1996) 256–262. [DOI] [PubMed] [Google Scholar]
- [55].Ortega-Rodriguez U, Portillo S, Ashmus RA, Duran JA, Schocker NS, Iniguez E, Montoya AL, Zepeda BG, Olivas JJ, Karimi NH, Alonso-Padilla J, Izquierdo L, Pinazo M-J, Noya B.A.d., Noya O, Maldonado RA, Torrico F, Gascón J, Michael K, Almeida IC, Methods Mol. Biol, 1955 (2019) 287–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Torrico MC, Fernandez-Arevalo A, Ballart C, Solano M, Rojas E, Ariza E, Tebar S, Lozano D, Abras A, Gascon J, Picado A, Munoz C, Torrico F, Gallego M, Transbound Emerg Dis, 69 (2022) 2242–2255. [DOI] [PubMed] [Google Scholar]
- [57].Marco JD, Barroso PA, Mimori T, Locatelli FM, Tomatani A, Mora MC, Cajal SP, Nasser JR, Parada LA, Taniguchi T, Korenaga M, Basombrio MA, Hashiguchi Y, BMC Infect Dis, 12 (2012) 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hoyos CL, Cajal SP, Juarez M, Marco JD, Alberti D’Amato AM, Cayo M, Torrejon I, Cimino RO, Diosque P, Krolewiecki AJ, Nasser JR, Gil JF, Biomed Res Int, 2016 (2016) 6456031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Frank FM, Fernandez MM, Taranto NJ, Cajal SP, Margni RA, Castro E, Thomaz-Soccol V, Malchiodi EL, Parasitology, 126 (2003) 31–39. [DOI] [PubMed] [Google Scholar]
- [60].Bastrenta B, Mita N, Buitrago R, Vargas F, Flores M, Machane M, Yacsik N, Torrez M, Le Pont F, Breniere F, Mem Inst Oswaldo Cruz, 98 (2003) 255–264. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.