

Abstract
Alkoxy radicals are versatile reactive intermediates in organic synthesis. Here, we leverage the principle of frustrated radical pair to provide convenient access to these highly reactive species directly from tertiary alcohols via oxoammonium-mediated oxidation of the corresponding alkoxides. This approach enabled various synthetically useful transformations including β-scission, radical cyclization, and remote C–H functionalization, giving rise to versatile alkoxyamines that can be further elaborated to various functionalities.
Alkoxy radicals are high-energy species because they are not stabilized by mesomeric effects or delocalization,1 and their unique reactivities have been utilized for the synthesis of complex molecules.2 Alkoxy radicals react with substrates via mainly one of three pathways: hydrogen-atom abstraction from C–H bonds, addition to unsaturated C–C π-bonds, or fragmentation via β-C–C bond scission; in each case, a carbon-centered radical is generated that can further participate in diverse transformations. Despite their synthetic potential, strategies for the convenient generation of alkoxy radicals are limited. Traditional methods rely on the preinstallation of oxygen-activating groups followed by thermolysis or UV photolysis.3 However, this approach often requires multiple steps, relatively harsh reaction conditions, and the activated precursors typically exhibit low stabilities.
In recent years, pioneering work has led to the development of alternative strategies that can afford access to alkoxy radicals directly from alcohols (Figure 1a). These reports focused on β-C–C bond cleavage of cycloalkanols via oxidation using a high-valent transition metal (Figure 1a, approach 1),4 activation using a hypervalent iodine agent (approach 2),5 light-induced ligand-to-metal charge transfer (approach 3),6 and proton-coupled electron transfer by means of photo-catalysis (approach 4) or electrochemistry (approach 5).7 These approaches have been successful in providing alkoxy radicals directly from free alcohol substrates, although approaches 1 and 2 predominantly require that the alcohol substrates contain strained ring systems and pendant electron-rich arene groups, respectively, which limits their scope. Approaches 3–5 are more general platforms for accessing alkoxyl chemistry from alcohols and have been elegantly applied in C–H functionalization or β-scission reactions. However, the downstream transformations of the incipient alkyl radical are currently limited to trapping with Michael acceptor alkenes (approaches 3 and 4), H-atom-donating thiols (approach 4), or halides and alkoxides (approach 5). In addition, challenges associated with the efficient delivery of photons at large scales may limit the scalability of the photochemical methods using traditional batch reactors.8 Thus, there remains a need for a generally applicable, operationally convenient, and scalable approach for accessing alkoxy radicals.
Figure 1.
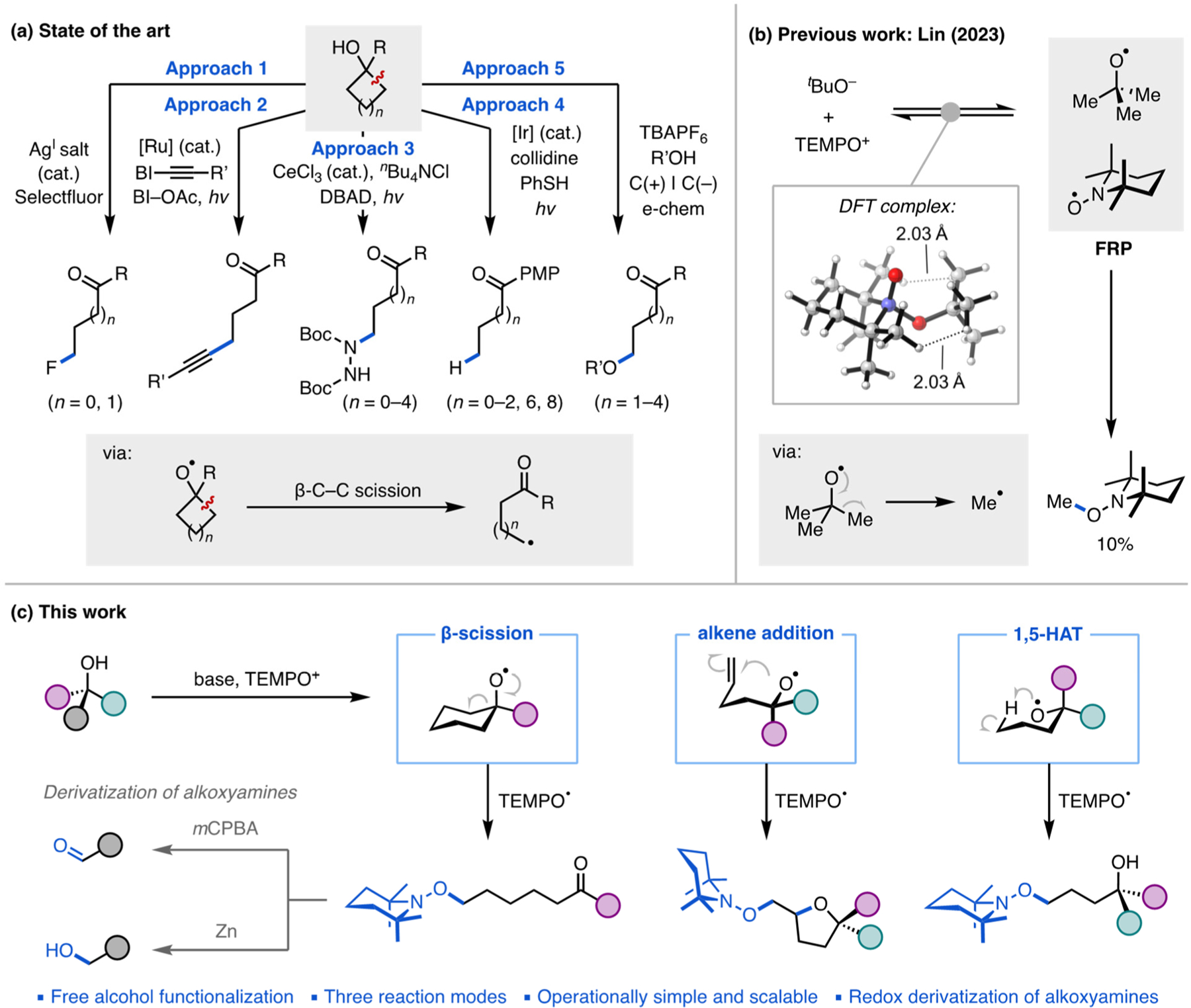
(a) Established routes for accessing alkoxy radicals directly from alcohols (Approach 1: oxidation using a high-valent transition metal; Approach 2: activation using a hypervalent iodine agent; Approach 3: light-induced ligand-to-metal charge transfer; Approach 4: proton-coupled electron transfer). [Ru] = [Ru(bpy)3](PF6)2; [Ir] = [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6; BI–OAc = 1-acetoxy-1,2-benziodoxol-3(1H)-one; DBAD = di-tert-butyl azodicarboxylate. (b) Prior work from our laboratory suggesting the prospect of using FRPs to access alkoxy radicals. (c) Synthetic approach developed in the present work.
In recent work, our group demonstrated that frustrated radical pairs (FRPs)—in this case, generated through a single-electron transfer event between a disilazide donor and an N-oxoammonium acceptor—can be used to achieve aliphatic C–H functionalization in a regioselective manner.9 In the course of this work, we found that potassium tert-butoxide (KOtBu) can be oxidized by TEMPO+BF4− to form a tBuO•/TEMPO• frustrated radical pair (Figure 1b). Density functional theory calculations carried out on an tBuO–TEMPO adduct predicted that it would feature an exceedingly weak N–O bond (BDE: 11.6 kcal/mol) and spontaneously dissociate in a homolytic fashion to form the radical pair. Significant steric repulsion between the two components (closest H···H contacts are ∼2.1 Å) is also anticipated to contribute to the low stability of the adduct and its spontaneous dissociation (ΔGdis = −7.5 kcal/mol). In the absence of an alkane substrate, a small amount of Me–TEMPO product was detected via β-scission of tBuO• (Figure 1b).10 This finding suggested to us the possibility of employing such FRP chemistry to access alkoxyl radicals and the transformations that they enable. We envisioned that alkoxy radicals could be conveniently generated from free tertiary alcohols through a deprotonation–single-electron transfer sequence (Figure 1c). Upon formation of the alkoxy radical and subsequent β-scission, the incipient carbon-centered radical would be captured by simultaneously generated TEMPO•, installing a versatile aminoxyl group that could be readily converted to various functionalities, including carbonyl and hydroxyl motifs using well-established methods.9,11 Of note, although TEMPO+ and related oxoammonium ions have been extensively studied in the oxidation of primary and secondary alcohols,12 the reactivity of oxoammonium ions with tertiary alcohols has not been established.
We set out to explore this proposed reaction strategy in the context of radical-induced C–C bond cleavage using 1-phenylcyclohexanol as a model substrate (1a; Table 1), both as a proof-of-principle and to facilitate a comparison with the established routes described above (Figure 1a). We initially evaluated conditions similar to those we previously reported for C–H functionalization using radical pairs, namely potassium hexamethyldisilazide (KHMDS) as the base, TEMPO+PF6− as the electron-acceptor species, in benzotri-fluoride (PhCF3) solvent. In this system, KHMDS deprotonates the alcohol, and single-electron transfer from the alkoxide intermediate to the oxoammonium generates the desired alkoxy radical–TEMPO• FRP (Figure 1c). This type of O-centered radical α to a saturated carbocycle is known to undergo facile ring-opening via β-scission,13 and the resulting carbon-centered radical can be trapped by TEMPO• to furnish a new C–O bond.
Table 1.
Conditions Explored for Reaction Optimizationa

| |||
|---|---|---|---|
| Entry | Additive | Yield of 1b | Yield of 1c |
| 1 | none | 14% | 18% |
| 2 | THF | 13% | 12% |
| 3 | 18-crown-6 | 3% | 2% |
| 4 | Ph3N | 9% | 13% |
| 5 | pyridine | 7% | 9% |
| 6 | 1,10-phen | 9% | 1% |
| 7 | HMPA | 9% | 8% |
| 8 | benzophenone | 32% | ND |
| 9 | benzophenoneb | 57% | 2% |
| 10 c | benzophenone | 72% | 2% |
Optimization was conducted on a 0.1 mmol scale. Yield was determined by 1H NMR using mesitylene internal standard.
0.25 equiv of benzophenone.
2.5 equiv of TEMPO+PF6−, 2.25 equiv of KHMDS, 0.45 equiv of benzophenone.
Under these initial conditions, 14% of the desired β-scission product 1b was formed, featuring an appended aminoxyl group (Table 1, Entry 1). However, we also isolated side product 1c in 18% yield, featuring additional functionalization of the α-position of the carbonyl group, presumably through a polar pathway involving α-deprotonation and nucleophilic addition to TEMPO+. We hypothesized that the single-electron oxidation of the alkoxide could be facilitated—and side product formation reduced—with the addition of a Lewis base that would coordinate to the potassium counterion. However, addition of tetrahydrofuran (THF), triphenylamine (Ph3N), pyridine, and hexamethylphosphoramide (HMPA) did not improve the reaction outcome (Entries 2, 4, 5, 7), while readily oxidized 18-crown-6 was detrimental (Entry 3). Addition of 1,10-phenanthroline was effective in suppressing side product formation, although the overall yield of 1b was low in this case (9%; Entry 6). Interestingly, the use of benzophenone as an additive successfully suppressed side product formation and afforded the desired product in 32% yield (Entry 8). Preliminary NMR data suggest that benzophenone may also help attenuate the basicity of KHMDS and thus suppress the formation of 1c (see the SI for details). Further optimization of the reaction by changing the KHMDS/benzophenone ratio from 1:1 to 5:1 improved conversion to 57% (Entry 9), and finally, increasing the equivalence of TEMPO+PF6− and KHMDS further improved yield to 72% (Entry 10).14
With these optimized conditions, we explored the scope of the FRP-mediated functionalization of tertiary alcohols via β-scission with a diverse array of cycloalkanol substrates. On a synthetically relevant scale (0.2 mmol), the reaction proceeded smoothly with 1-phenylcyclohexanol (1a in 81% yield), 1-phenylcyclopentanol (2a in 74% yield), and 1-phenyl-cyclobutanol (3a in 72% yield) (Figure 2). Other electronically differentiated arenes α to the hydroxyl group, such as 4-Cl-C6H4 (4a), 4-F-C6H4 (5a), 4-t-Bu-C6H4 (6a) were well-tolerated (66–77% yield), although the product yield from a substrate containing electron-rich 4-OMe-C6H4 (7a) was slightly lower (52%). A substrate featuring pyridine as a substituent (8a) gave the product in a relatively low yield (33%), presumably due to limited solubility of the substrate in PhCF3. Notably, good reactivity was also found with 1-methyl substituted cycloalkanols (9a and 10a), indicating that 1-aryl substitution leading to conjugated ketone product is not a prerequisite for this reaction, as is the case for some earlier methods.7a,c,15 Functionalities such as difluoro (11a), acetal (12a), trifluoromethyl (13a), and tetrahydropyran (14a) were also compatible. Derivatives of adamantanone (15a), camphor (16a), and norcamphor (17a) containing bridged tri- and bicyclic structures all furnished the desired β-scission product, with substrate 16a showing a preference for the generation of a secondary over primary carbon-radical (regioisomeric ratio, r.r., of 2.5:1).
Figure 2.

Scope of β-scission reactions explored in this work. When a diastereomeric ratio (d.r.) is given, a * symbol indicates the carbon at which the diastereoisomerism is concerned. aElimination side-product observed (see the SI). bYield was determined by 1H NMR. For substrates 7a and 18a, the alkoxide was preformed by mixing the alcohol with KHMDS and benzophenone followed by addition of TEMPO+PF6− (see SI); these substrates were found to undergo side reactions with TEMPO+ directly, and pre-mixing with the base to form the alkoxide reduces the influence of this process.
Natural terpene compounds sabinene hydrate (18a) and globuol (19a), which contain fused ring systems, were also viable substrates; these yielded products in which one of the two possible β-C–C bonds was preferentially cleaved, presumably the one which leads to the more stable carbon-centered radical. A similar result was found when estrone derivative 21a was used (i.e., β-C–C bond cleavage preferentially generated a tertiary versus a primary carbon-centered radical), while for cholesterol derivative 20a, there was no distinction between the two β-C–C bonds, which would both form primary radicals. Finally, our method can also accommodate acyl radical generation, as evidenced from the success of reactions carried out using the essential oil 2-hydroxy-3-pinanone (22a) and an isatin derivative 23a, which yielded acyl aminoxyl derivatives 22b and 23b, respectively. Finally, acyclic tertiary alcohols 24a and 25a also underwent β-scission to yield the desired alkoxyamine products.
We next went on to explore the utility of our approach for carrying out other transformations known for alkoxy radicals. For example, we investigated several tertiary alcohol substrates featuring pendent alkenes that could undergo 3-exo-trig and 5-exo-trig cyclizations via homolytic alkoxyl addition to the double bond (Figure 3a). For substrates with 1,1-dimethyl substitutions (26a and 27a), three-membered epoxide and five-membered tetrahydrofuran rings were formed in 81% and 85% yield, respectively, without further optimization of the conditions developed for the β-scission reaction. Interestingly, cycloalkanols 28a and 29a exclusively underwent radical cyclization without any sign of β-scission, furnishing spirocyclic products. When protected sclareol 30a was used as a substrate, the base-sensitive acetate group was tolerated under the reaction conditions. When the acyclic monoterpene linalool 32a was employed, 5-exo-trig radical cyclization was favored over the 3-exo-trig pathway (products formed in an 8.0:1 ratio). Notably, a sesquiterpene alcohol α-bisabolol 31a exclusively reacted at the prenyl alkene and did not yield any cyclization product with the endocyclic alkene, presumably due to unfavorable geometric constraints.
Figure 3.

(a) Scope of radical cyclization. (b) Scope of remote C–H functionalization. When a diastereomeric ratio (d.r.) is given, a * symbol indicates the carbon at which the diastereoisomerism is concerned. aStereochemistry undetermined due to the small amount of product obtained (for 32c) or the flexible nature of 7-membered rings (for 37a). bBis-aminoxylated side-product observed (see SI).
Another transformation of interest is intramolecular H-atom transfer (HAT), wherein a radical center is effectively transposed from oxygen to a carbon atom. Alkoxy radicals are known to undergo highly regioselective δ-C–H bond abstraction (1,5-HAT) via a six-membered ring transition state.16 We explored the utility of our approach for achieving such reactivity, starting with simple, linear substrates containing activated benzylic (34a) and allylic C–H bonds (33a) (Figure 3b). In both cases, δ-functionalized products were obtained albeit in moderate yields, which we attribute to the fact that the alcohol functionalities in the substrate and product are not effectively distinguished in the linear scaffold. In further scope study, we found that the use of rigidified substrates (35a–38a) resulted in improved yields (52–61%) even though the reactive C–H bonds exhibit higher bond dissociation energies. The nuclear magnetic resonance spectra of these products revealed deshielding of the O–H proton presumably through hydrogen bonding with the adjacent alkoxyamine group, which thus protected it from competing with the substrate’s hydroxy group. Other strained substrates containing benzylic (39a and 40a) and allylic δ-C–H bonds (41a) were also tolerated. Notably, starting from sesquiterpene cedrol (42a), 1,5-HAT was found to take place at a predisposed methyl group to furnish δ C–H aminoxylation product, while a side-reaction also generated a ring-opening product through β-scission. Finally, we note that primary and secondary alcohols are currently not suitable substrates for this reaction strategy due to the competing alcohol oxidation reactivity of TEMPO+.12
To demonstrate the synthetic utility of our FRP-mediated approach for accessing alkoxy radicals, we performed a gram-scale reaction with 2-hydroxy-3-pinanone (22a) (Figure 4a). Notably, the product was obtained in good yield (72%) without any optimization. Furthermore, this and other alkoxyamine products could be transformed to diverse useful functionalities under simple oxidizing or reducing conditions (Figure 4b). Treatment of the O-acyl TEMPO product 22b, which features a remote acetyl group, with diisobutylaluminum hydride (DIBAL-H)17 resulted in the selective formation of the corresponding hydroxy aldehyde without further reduction to a diol. The C–H aminoxylation product generated from cedrol (42b) was readily converted to natural product cedranediol (42d)18 via zinc mediated reductive N–O bond cleavage.19 This facile synthetic route stands in contrast to a previously reported synthesis of this cedranoid sesquiterpene compound, which required 10 steps and yielded a racemic mixture.20 We also demonstrated the oxidation of alkoxyamine products to carbonyl compounds using 3-chloroperxybenzoic acid (mCPBA) as an oxidant. When sclareol derivative 30b was used as a substrate, the epoxide ring was preserved and aldehyde 30d was obtained in good yield. Under the same conditions, the major ring-opening product from (−)-globulol (19b) was efficiently converted into 1,10-dioxotayloriane 19d,21 affording another concise late-stage approach to natural product synthesis from abundant precursors.17
Figure 4.

(a) Gram-scale reaction. (b) Product derivatization reactions.
In summary, we have described a convenient and scalable method for accessing alkoxy radical chemistry from free alcohols. The simple deprotonation–single-electron transfer sequence can be used to furnish three different alkoxy radical-mediated transformations (β-scission, radical cyclization, and 1,5-HAT) using a broad scope of feedstock and complex alcohol substrates. The aminoxyl functionality of the products can be further converted to C–H, C–OH, and C=O groups. This approach thus provides convenient access to rare terpenes from abundant natural products.
Supplementary Material
ACKNOWLEDGMENTS
Research was supported by the NIH (R01GM134088), Genentech, and Dreyfus Foundation (Teacher-Scholar Award). M.J. was supported by the NIH (F32GM142264). S. Lee was supported by the Kwanjeong Educational Foundation. We thank Zhipeng Lu (Cornell University) for contributions during the initial stage of the project; Dr. Jack A. Terrett and Elisia Villemure (Genentech) for helpful discussion; Dr. Ivan Keresztes (Cornell University) for his help in NMR experiments; Dr. Dave B. Collum, and Jesse Spivey (Cornell University) for providing a sample of recrystallized KHMDS; Dr. Zhiqiang Wei (Cornell University) for reproducing experiments; Dr. Yeosan Lee (Cornell University) for synthesizing substrates.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c07125.
Experimental procedures and supporting characterization data and spectra (PDF)
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.4c07125
Contributor Information
Minsoo Ju, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States.
Sukwoo Lee, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States.
Halle M. Marvich, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States
Song Lin, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States.
REFERENCES
- (1).For representative reviews, see: (a) Gray P; Williams A The Thermochemistry and Reactivity of Alkoxyl Radicals. Chem. Rev 1959, 59, 239–328. [Google Scholar]; (b) Tsui E; Wang H; Knowles RR Catalytic Generation of Alkoxy Radicals from Unfunctionalized Alcohols. Chem. Sci 2020, 11, 11124–11141. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chang L; An Q; Duan L; Feng K; Zuo Z Alkoxy Radicals See the Light: New Paradigms of Photochemical Synthesis. Chem. Rev 2022, 122, 2429–2486. [DOI] [PubMed] [Google Scholar]
- (2).For selected examples in total synthesis, see: (a) Renata H; Zhou Q; Dünstl G; Felding J; Merchant RR; Yeh C-H; Baran PS Development of a Concise Synthesis of Ouabagenin and Hydroxylated Corticosteroid Analogues. J. Am. Chem. Soc 2015, 137, 1330–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hussain N; Morgan DO; White CR; Murphy JA Alkoxy Radicals in Organic Synthesis. A Novel Approach to a Key Intermediate in Milbemycin Chemistry. Tetrahedron Lett 1994, 35, 5069–5072. [Google Scholar]; (c) Yamashita S; Naruko A; Nakazawa Y; Zhao L; Hayashi Y; Hirama M Total Synthesis of Limonin. Angew. Chem., Int. Ed 2015, 127, 8658–8661. [DOI] [PubMed] [Google Scholar]; (d) Suginome H; Yamada S A Simple New General Synthesis of Macrocyclic Ketones: A New Entry to the Synthesis of Exaltone and (±)-Muscone. Tetrahedron Lett 1987, 28, 3963–3966. [Google Scholar]; (e) El Gehani AAMA; Maashi HA; Harnedy J; Morrill LC Chem. Commun 2023, 59, 3655–3664. [DOI] [PubMed] [Google Scholar]
- (3).For examples of preformed O–X alkoxy radical precursor, see: (a) Walling C; Clark RT Reactions of Primary and Secondary Alkoxy Radicals Derived from Hypochlorites. J. Am. Chem. Soc 1974, 96, 4530–4534. [Google Scholar]; (b) Walling C; Bristol D δ-Chloro Alcohols and Tetrahydrofurans from Primary and Secondary Alkyl Hypochlorites. J. Org. Chem 1972, 37, 3514–3516. [Google Scholar]; (c) Beckwith J; Hay BP; Williams GM Generation of Alkoxyl Radicals from O-Alkyl Benzenesulphenates. Chem. Commun 1989, 17, 1202–1202. [Google Scholar]; (d) Hartung J; Gallou F Ring Closure Reactions of Substituted 4-Pentenyl-1-Oxy Radicals. The Stereoselective Synthesis of Functionalized Disubstituted Tetrahydrofurans. J. Org. Chem 1995, 60, 6706–6716. [Google Scholar]; (e) Barton DHR; Beaton JM; Geller LE; Pechet MM A New Photochemical Reaction. J. Am. Chem. Soc 1960, 82, 2640–2641. [Google Scholar]
- (4).For selected examples using Ag, see: (a) Zhao H; Fan X; Yu J; Zhu C Silver-Catalyzed Ring-Opening Strategy for the Synthesis of β- and γ-Fluorinated Ketones. J. Am. Chem. Soc 2015, 137, 3490–3493. [DOI] [PubMed] [Google Scholar]; (b) Zhou X; Ding H; Chen P; Liu L; Sun Q; Wang X; Wang P; Lv Z; Li M Radical Dehydroxymethylative Fluorination of Carbohydrates and Divergent Transformations of the Resulting Reverse Glycosyl Fluorides. Angew. Chem., Int. Ed 2020, 59, 4138–4144. [DOI] [PubMed] [Google Scholar]; (c) Ren S; Feng C; Loh T-P Iron- or Silver-Catalyzed Oxidative Fluorination of Cyclopropanols for the Synthesis of β-Fluoroketones. Org. Biomol. Chem. 2015, 13, 5105–5109. Using Cu: [DOI] [PubMed] [Google Scholar]; (d) Ren H; Song J-R; Li Z-Y; Pan W-D Oxazoline-/Copper-Catalyzed Alkoxyl Radical Generation: Solvent-Switched to Access 3a,3a′-Bisfuroindoline and 3-Alkoxyl Furoindoline. Org. Lett 2019, 21, 6774–6778. [DOI] [PubMed] [Google Scholar]; (e) Jiang C; Wang L; Zhang H; Chen P; Guo Y-L; Liu G Enantioselective Copper-Catalyzed Trifluoromethylation of Benzylic Radicals via Ring Opening of Cyclopropanols. Chem 2020, 6, 2407–2419. [Google Scholar]; (f) Wu L; Wang L; Chen P; Guo Y-L; Liu G Enantioselective Copper-Catalyzed Radical Ring-Opening Cyanation of Cyclopropanols and Cyclopropanone Acetals. Adv. Synth. Catal 2020, 362, 2189–2194. Using Mn: [Google Scholar]; (g) Ren R; Zhao H; Huan L; Zhu C Manganese-Catalyzed Oxidative Azidation of Cyclobutanols: Regiospecific Synthesis of Alkyl Azides by C–C Bond Cleavage. Angew. Chem., Int. Ed 2015, 54, 12692–12696. [DOI] [PubMed] [Google Scholar]; (h) Ren R; Wu Z; Xu Y; Zhu C C–C Bond-Forming Strategy by Manganese-Catalyzed Oxidative Ring-Opening Cyanation and Ethynylation of Cyclobutanol Derivatives. Angew. Chem., Int. Ed 2016, 128, 2916–2919. [DOI] [PubMed] [Google Scholar]; (i) Wang D; Ren R; Zhu C Manganese-Promoted Ring-Opening Hydrazination of Cyclobutanols: Synthesis of Alkyl Hydrazines. J. Org. Chem 2016, 81, 8043–8049. [DOI] [PubMed] [Google Scholar]; (j) Allen BDW; Hareram MD; Seastram AC; McBride T; Wirth T; Browne DL; Morrill LC Org. Lett 2019, 21, 9241–9246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Jia K; Zhang F; Huang H; Chen Y Visible-Light-Induced Alkoxyl Radical Generation Enables Selective C(sp3)–C(sp3) Bond Cleavage and Functionalizations. J. Am. Chem. Soc 2016, 138, 1514–1517. [DOI] [PubMed] [Google Scholar]; (b) Wu X; Zhang H; Tang N; Wu Z; Wang D; Ji M; Xu Y; Wang M; Zhu C Metal-Free Alcohol-Directed Regioselective Heteroarylation of Remote Unactivated C(sp3)–H Bonds. Nat. Commun 2018, 9, 3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(c) Li G-X; Hu X; He G; Chen G Photoredox-Mediated Remote C(sp3)–H Heteroarylation of Free Alcohols. Chem. Sci 2019, 10, 688–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(d) Jia K; Pan Y; Chen Y Selective Carbonyl–C(sp3) Bond Cleavage to Construct Ynamides, Ynoates, and Ynones by Photoredox Catalysis. Angew. Chem., Int. Ed 2017, 56, 2478. [DOI] [PubMed] [Google Scholar]
- (8).(e) Jia K; Li J; Chen Y Selective P–C(sp3) Bond Cleavage and Radical Alkynylation of α-Phosphorus Alcohols by Photoredox Catalysis. Chem.—Eur. J 2018, 24, 3174. [DOI] [PubMed] [Google Scholar]
- (9).(f) Wang D; Mao J; Zhu C Visible Light-Promoted Ring-Opening Functionalization of Unstrained Cycloalkanols via Inert C–C Bond Scission. Chem. Sci 2018, 9, 5805–5809. Reference 5f shows two examples of substrates without a pendant aryl group α to the alcohol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Zhang K; Chang L; An Q; Wang X; Zuo Z Dehydroxymethylation of Alcohols Enabled by Cerium Photocatalysis. J. Am. Chem. Soc 2019, 141, 10556–10564. [DOI] [PubMed] [Google Scholar]; (b) An Q; Wang Z; Chen Y; Wang X; Zhang K; Pan H; Liu W; Zuo Z Cerium-Catalyzed C–H Functionalizations of Alkanes Utilizing Alcohols as Hydrogen Atom Transfer Agents. J. Am. Chem. Soc 2020, 142, 6216–6226. [DOI] [PubMed] [Google Scholar]; (c) Guo J-J; Hu A; Chen Y; Sun J; Tang H; Zuo Z Photocatalytic C–C Bond Cleavage and Amination of Cycloalkanols by Cerium (III) Chloride Complex. Angew. Chem., Int. Ed 2016, 55, 15319–15322. [DOI] [PubMed] [Google Scholar]
- (11).(a) Yayla HG; Wang H; Tarantino KT; Orbe HS; Knowles RR Catalytic Ring-Opening of Cyclic Alcohols Enabled by PCET Activation of Strong O–H Bonds. J. Am. Chem. Soc 2016, 138, 10794–10797. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ota E; Wang H; Frye NL; Knowles RR A Redox Strategy for Light-Driven, Out-of-Equilibrium Isomerizations and Application to Catalytic C–C Bond Cleavage Reactions. J. Am. Chem. Soc 2019, 141, 1457–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhao K; Yamashita K; Carpenter JE; Sherwood TC; Ewing WR; Cheng PTW; Knowles RR Catalytic Ring Expansions of Cyclic Alcohols Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc 2019, 141, 8752–8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(d) Tsui E; Metrano AJ; Tsuchiya Y; Knowles RR Catalytic Hydroetherification of Unactivated Alkenes Enabled by Proton-Coupled Electron Transfer. Angew. Chem., Int. Ed 2020, 59, 11845–11849. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hareram MD; El Gehani AAMA; Harnedy J; Seastram AC; Jones AC; Burns M; Wirth T; Browne DL; Morrill LC Electrochemical Deconstructive Functionalization of Cycloalkanols via Alkoxy Radicals Enabled by Proton-Coupled Electron Transfer. Org. Lett 2022, 24, 3890–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Harnedy J; Maashi HA; El Gehani AAMA; Burns M; Morrill LC Deconstructive Functionalization of Unstrained Cycloalkanols via Electrochemically Generated Aromatic Radical Cations. Org. Lett 2023, 25, 1486–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Bonfield HE; Knauber T; Lévesque F; Moschetta EG; Susanne F; Edwards LJ Photons as a 21st Century Reagent. Nat. Commun 2020, 11, 804. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Buglioni L; Raymenants F; Slattery A; Zondag SDA; Noël T Technological Innovations in Photo-chemistry for Organic Synthesis: Flow Chemistry, High-Throughput Experimentation, Scale-Up, and Photoelectrochemistry. Chem. Rev 2022, 122, 2752–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lu Z; Ju M; Wang Y; Meinhardt JM; Martinez Alvarado JI; Villemure E; Terrett JA; Lin S Regioselective Aliphatic C–H Functionalization Using Frustrated Radical Pairs. Nature 2023, 619, 514–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Nakamura T; Watanabe Y; Suyama S; Tezuka H Study of Alkyl Radicals Fragmentation from 2-Alkyl-2-Propoxyl Radicals. J. Chem. Soc. Perk. T 2002, 2, 1364–1369. [Google Scholar]; (b) Vasilopoulos A; Krska SW; Stahl SS C(sp3)–H Methylation Enabled by Peroxide Photosensitization and Ni-Mediated Radical Coupling. Science 2021, 372, 398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).(a) Hartmann M; Li Y; Studer A Transition-Metal-Free Oxyarylation of Alkenes with Aryl Diazonium Salts and TEMPONa. J. Am. Chem. Soc 2012, 134, 16516–16519. [DOI] [PubMed] [Google Scholar]
- (17).(b) Zhu Q; Gentry EC; Knowles RR Catalytic Carbocation Generation Enabled by the Mesolytic Cleavage of Alkoxyamine Radical Cations. Angew. Chem., Int. Ed 2016, 55, 9969–9973. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kielty P; Farràs P; Smith DA; Aldabbagh F 2-(Fluoromethyl)-4,7-Dimethoxy-1-Methyl-1H-Benzimidazole. Molbank 2020, 2020, M1129. [Google Scholar]; (d) Norcott PL; Hammill CL; Noble BB; Robertson JC; Olding A; Bissember AC; Coote ML TEMPO–Me: An Electrochemically Activated Methylating Agent. J. Am. Chem. Soc 2019, 141, 15450–15455. [DOI] [PubMed] [Google Scholar]
- (18).Nutting JE; Rafiee M; Stahl SS Tetramethylpiperidine N-Oxyl (TEMPO), Phthalimide N-Oxyl (PINO), and Related N-Oxyl Species: Electrochemical Properties and Their Use in Electrocatalytic Reactions. Chem. Rev 2018, 118, 4834–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).(a) Suárez E; Rodriguez MS β-Fragmentation of Alkoxyl Radicals: Synthetic Applications. In Radicals in Organic Synthesis; Renaud P, Sibi MP, Eds.; Wiley, 2001; pp 440–454. [Google Scholar]; (b) Murakami M; Ishida N β-Scission of Alkoxy Radicals in Synthetic Transformations. Chem. Lett 2017, 46, 1692–1700. [Google Scholar]
- (20).Additional details of the reaction optimization are described in the Supporting Information (SI). [Google Scholar]
- (21).(a) Wang D; Mao J; Zhu C Visible Light-Promoted Ring-Opening Functionalization of Unstrained Cycloalkanols via Inert C–C Bond Scission. Chem. Sci 2018, 9, 5805–5809. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huang L; Ji T; Rueping M Remote Nickel-Catalyzed Cross-Coupling Arylation via Proton-Coupled Electron Transfer-Enabled C–C Bond Cleavage. J. Am. Chem. Soc 2020, 142, 3532–3539. [DOI] [PubMed] [Google Scholar]; (c) Wu X; Wang M; Huan L; Wang D; Wang J; Zhu C Tertiary-Alcohol-Directed Functionalization of Remote C(sp3)–H Bonds by Sequential Hydrogen Atom and Heteroaryl Migrations. Angew. Chem., Int. Ed 2018, 57, 1640–1644. [DOI] [PubMed] [Google Scholar]
- (22).(a) Čeković Ž Reactions of δ-Carbon Radicals Generated by 1,5-Hydrogen Transfer to Alkoxyl Radicals. Tetrahedron 2003, 59, 8073–8090. [Google Scholar]; (b) Stateman LM; Nakafuku KM; Nagib DA Remote C–H Functionalization via Selective Hydrogen Atom Transfer. Synthesis 2018, 50, 1569–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Inokuchi T; Kawafuchi H Reactivity of TEMPO Anion as a Nucleophile and Its Applications for Selective Transformations of Haloalkanes or Acyl Halides to Aldehydes. Tetrahedron 2004, 60, 11969–11975. [Google Scholar]
- (24).Sadgrove NJ; Klepp J; Legendre SVA-M; Lyddiard D; Sumby CJ; Greatrex BW Revision of the Phytochemistry of Eremophila Sturtii and E. Mitchellii. J. Nat. Prod 2018, 81, 405–409. [DOI] [PubMed] [Google Scholar]
- (25).Dinca E; Hartmann P; Smrcek J; Dix I; Jones PG; Jahn U General and Efficient α-Oxygenation of Carbonyl Compounds by TEMPO Induced by Single-Electron-Transfer Oxidation of Their Enolates. Eur. J. Org. Chem 2012, 2012, 4461–4482. [Google Scholar]
- (26).Ihara M; Makita K; Takasu K Facile Construction of the Tricyclo[5.2.1.01,5]Decane Ring System by Intramolecular Double Michael Reaction: Highly Stereocontrolled Total Synthesis of (±)-8,14-Cedranediol and (±)-8,14-Cedranoxide. J. Org. Chem 1999, 64, 1259–1264. [Google Scholar]
- (27).(a) Liu H; Wu C; Becker H; Zapp J Sesquiterpenoids and Diterpenoids from the Chilean Liverwort Lepicolea Ochroleuca. Phytochemistry 2000, 53, 845–849. [DOI] [PubMed] [Google Scholar]; (b) Chen J-J; Bai W; Gobu F-R; Wu C-H; Zeng J; Sun M; Gao K Sesquiterpenoids from the Roots of Vladimiria Muliensis. J. Asian Nat. Prod. Res 2015, 17, 1188–1195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.