

Abstract
Incorporation of C(sp3)–F bonds in biologically active compounds is a common strategy employed in medicinal and agricultural chemistry to tune pharmacokinetic and pharmacodynamic properties. Due to the limited number of robust strategies for C(sp3)–H fluorination of complex molecules, time-consuming de novo syntheses of such fluorinated analogs are typically required, representing a major bottleneck in the drug discovery process. In this work, we present a general and operationally simple strategy for site-specific β-C(sp3)–H fluorination of amine derivatives including carbamates, amides, and sulfonamides, which is compatible with a wide range of functional groups including N-heteroarenes. In this approach, an improved electrochemical Shono oxidation is used to set the site of functionalization via net α,β-desaturation to access enamine derivatives. We further developed a series of new transformations of these enamine intermediates to synthesize a variety of β-fluoro-α-functionalized structures, allowing efficient access to pertinent targets to accelerate drug discovery campaigns.
Graphical Abstract
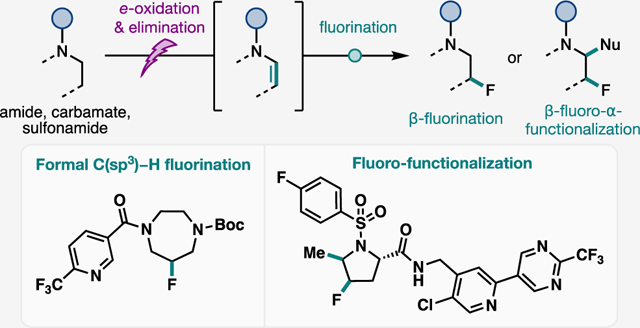
INTRODUCTION
Late-Stage C(sp3)–H Fluorination.
In medicinal chemistry, replacement of a hydrogen with a fluorine atom is routinely employed as a strategy to tune the bioactivity of drug candidates by improving potency, metabolic stability, and membrane permeability. More specifically, the addition of fluorine to the β position of cyclic amine derivatives has been shown to restrict conformations and modulate the pKa of basic moieties in several drug-like compounds (Scheme 1A).1 In 2004, researchers from Taisho Pharmaceutical conducted structure–activity relationship studies on the 2-cyanopyrrolidine core of dipeptidyl peptidase IV (DPP-IV) inhibitors, which revealed a 3-fold boost in inhibitory potency when a β-hydrogen atom was replaced with fluorine (1) due to the preferential conformation associated with the fluorinated analog.2 In 2014, researchers from AstraZeneca incorporated a β-fluorine atom in the piperidine core of a series of Mycobacterium tuberculosis (Mtb) DNA gyrase inhibitors (2) to minimize unwanted inhibition of the cardiac hERG channel. The increased selectivity arose from attenuation of the basicity of the adjacent amino group.3 In 2018, researchers from Genentech observed undesired oxidative metabolism of a proline fragment in a transient receptor potential ankyrin 1 (TRPA1) antagonist. To improve the metabolic stability of the compound, a fluorine atom was incorporated at the β position of the proline fragment (3).4 More recently, in 2022, researchers from Pfizer and Mirati Therapeutics developed a novel KRASG12D inhibitor (MRTX1133), which is currently in a phase I/II clinical trial, by introducing a β-fluorine on the pyrrolizidine core of the parent molecule (4). This modification provided an 11-fold boost in inhibitory potency, which was attributed to the decrease in basicity of the pyrrolizidine nitrogen, leading to a stronger ionic interaction of this motif with Glu62 carboxylate of KRASG12D/GDP.5
Scheme 1. (A–C) Background Information.

In the context of late-stage functionalization (LSF), strategies for the selective construction of C(sp3)–F bonds remain highly desirable, allowing for rapid diversification of bioactive molecules in drug discovery campaigns.6 While a plethora of methods emerged in recent years,7 applications in complex molecules featuring N-heteroarenes remain rare (Scheme 1B). In 2012, Groves and co-workers employed a Mn-porphyrin-oxo complex in the presence of nucleophilic fluoride, for C(sp3)–H fluorination of unactivated8 and activated9 positions. Shortly after, Britton and co-workers explored photoexcited decatungstate for the generation of carbon-centered radicals, which undergo terminal trapping with NFSI to produce C(sp3)–F bonds.10 Lectka et al. employed a polycyanoarene-based photocatalyst to effect the fluorination of C(sp3)–H bonds in the presence of Selectfluor.11 White and co-workers reported a tandem approach to achieve C(sp3)–H fluorination in complex molecules, which involves the use of iron or manganese-oxo complexes for C(sp3)–H oxidation, followed by reduction (for secondary C–H) and deoxyfluorination.12 Baran and co-workers reported the use of electrochemistry to access a radical-chain mechanism for C(sp3)–H fluorination, in which a carbon-centered radical was terminated by Selectfluor.13 More recently, Hong and co-workers employed mechano-chemistry with NFSI acting as both a HAT promoter and a fluorinating agent, similarly proceeding via a radical-chain mechanism.14 These seminal contributions, however, are not amenable to the β-fluorination of amine derivatives and would rather activate the α-position due to the lower bond dissociation energy of the α-C–H bond. Further, in each of the aforementioned examples and other C(sp3)–H fluorination methodologies, site specificity and site selectivity are often difficult to achieve when multiple C(sp3)–H bonds are available, with a mixture of fluorinated products formed (Scheme 1B).
To our knowledge, site-specific C(sp3)–H fluorination in complex settings remains underexplored.15 To this end, we sought to develop a strategy for such a transformation, focusing on β-fluorination of protected amines, given its relevance in medicinal chemistry (Scheme 1A). We envisioned that the desired site specificity could be achieved from the reaction of a protected enamine with an electrophilic fluorine source, with the resulting β-fluoro-iminium ion undergoing nucleophilic trapping with a hydride to provide the hydrofluorination product (Scheme 1C). This strategy would allow us to specifically target the β-fluorination of N-substituted amines that is difficult using current methods that rely on hydrogenatom abstraction. We also foresaw that the same β-fluoro-iminium ion could be combined with a variety of nucleophiles, allowing for the preparation of β-fluoro-α-functionalized products in a single operation. From this retrosynthetic rationale, we sought to develop a similarly general and robust strategy for the synthesis of enamines from the corresponding saturated N-substituted amines. Specifically, we aim to leverage electrochemistry to provide a chemo- and regioselective desaturation method that will enable the late-stage functionalization of complex polynitrogenated compounds that are prevalent in synthetic medicines, agrochemicals, and natural products.
Enamines as Versatile Synthetic Intermediates.
Despite the versatility of enamine derivatives, only a limited number of desaturation methods have been developed.16 For example, in 2021, Nicewicz and co-workers reported a dual copper-photoredox system to achieve desaturation of a set of piperidine and azepane derivatives via oxidation of an in situ generated α-carbon-centered radical to an iminium ion followed by deprotonation (Scheme 1C).17 Shortly after, Maulide and co-workers reported the use of triflic anhydride for the activation of electron-rich benzoyl-amides followed by deprotonation using LiHMDS.18 The combination of a potent activator and a strong base under cryogenic temperatures, however, may hinder the method’s application in LSF. In 2022, König and co-workers reported a dual iridium–nickel photocatalytic strategy, proceeding through an a-carbon-centered radical, which undergoes nickel-mediated β-hydride elimination.19 While elegant, the proposed radical mechanism dictated the necessity for an electron-withdrawing group (EWG) or a heteroatom at the γ-position relative to nitrogen, limiting the overall substrate class.
While each of these strategies provides efficient access to the enamine derivatives for substrate specific classes, a general desaturation method suitable across a broad range of amine derivatives and compatible with N-heteroarenes remains elusive. Toward addressing this challenge, we were initially inspired by Shono’s report on the elimination of N,O-acetals,20 which can be generated from electrochemical oxidation of aliphatic N-heterocyclic cores.21 Through the use of a weak acid (NH4Cl or silica) at high temperatures (100–160 °C), the corresponding enamine derivatives were generated.20 The harsh conditions required for the elimination of the N,O-acetal precluded the use of this strategy in an LSF setting. Since this initial report, structurally similar methoxy-derived N,O-acetals have been used as precursors for desaturation under the promotion of NH4Br under heating (100 °C),22 or strong Lewis acids, such as TMSOTf23 or TiCl4.24 Possibly due to the required forcing conditions, these existing methodologies have thus far only been shown to be compatible with substrates with limited structural complexity and medicinally relevant functional groups such as N-heteroarenes.
Recently, we reported a modified Shono oxidation of amine derivatives, including carbamates, amides, and sulfonamides, to access N,O-acetals from complex molecules.25 By employing trifluoroethanol (TFE) as an oxidatively resistant solvent and trifluoroacetic acid as an in situ protecting agent for basic functionalities, we were able to substantially expand the substrate scope and functional group compatibility of the Shono oxidation.26 In particular, amine derivatives bearing highly electron-deficient N-substituents, which are recalcitrant to the conventional Shono oxidation, as well as compounds containing sensitive basic nitrogen-containing groups (e.g., amines and N-heteroarenes), became compatible substrates. Coupled with subsequent activation with boron trifluoride for nucleophilic alkylation, we developed a broadly applicable α-functionalization strategy. This strategy requires the presence of an EWG attached to the amine, and tertiary amines lacking an EWG (e.g., N-benzyl-pyrrolidine and N-phenyl-pyrrolidine) do not give rise to N,O-acetal, with extensive decomposition being observed.
To further expand the utility of this methodology in the context of β-functionalization of amine derivatives, we envisioned a complementary synthetic strategy encompassing electrochemical oxidation followed by β-elimination of the ensuing N,O-acetals to achieve desaturation under mild conditions. The development of hydrofunctionalization and difunctionalization27 methods for the resultant enamine derivatives will then allow us to access a diverse range of β-fluorinated compounds in the late-stage modification of complex bioactive targets. Herein, we report an operationally simple, three-step synthetic sequence to accomplish the desired synthetic objective, which encompasses an improved electrochemical Shono oxidation followed by a mild elimination process promoted by a dynamic Lewis pair and β-fluoro-a-functionalization of the resultant enamine, both of which are specifically developed in this work.
RESULTS AND DISCUSSION
α,β-Desaturation of Saturated N-Heterocyclic Cores.
Initially, we studied the desaturation with model compound 5 (Scheme 2), featuring a Boc-protected pyrrolidine, the most common amine-protecting group and one of the most common aliphatic heterocyclic cores used in medicinal chemistry,28 as well as a pyridine, the most common N-heteroarene present in FDA-approved pharmaceuticals.29 When compound 5 was subjected to electrolysis in TFE with TFA as an additive (Scheme 2A, entry 1), a condition that we previously studied for selective oxidation of sulfonamides or amides in the presence of N-heteroarenes,25 complete N-Boc-amine deprotection was observed. Such incompatibility was a result of the strong acidity of TFA, which is a common reagent employed for Boc deprotection.30 Accordingly, we evaluated similar conditions in the absence of TFA (entry 2) and observed good efficiency (62% yield). While adjusting the current magnitude led to lower yield (entry 3), changing the electrolyte to Et4NPF6 provided improved 77% yield (entry 5).
Scheme 2. (A–D) Development of an Operationally Simple Dehydrogenation Strategy of Protected Amines.

aNMR yields. bOnly products with Boc deprotection observed. cDIPEA and BF3•Et2O were premixed prior to addition of N,O-acetal.
For oxidation of other Boc-protected amines that lack a basic N-heteroarene such as 7, we observed reduced yield for the electrochemical oxidation (Scheme 2B). Notably, the graphite anode was visibly degraded, and a significant amount of mass balance could not be accounted for (~42% for reaction with 7). This observation led us to hypothesize that the locally acidic environment generated near the anode due to proton formation during electrolysis promoted undesired Boc deprotection and caused electrode degradation. In contrast, the oxidation of compound 5 (Scheme 2A) proceeded without anode degradation. We hypothesized that the pyridine group in 5 served to buffer the solution pH, avoiding buildup of an excessively acidic environment.
With this in mind, we further optimized the electrochemical oxidation of 7; with the inclusion of 1 equiv of pyridine as a basic additive, no electrode decomposition was observed, and product 8 was formed with a better mass balance. However, additional charge was needed to achieve high conversion of 7 likely as a result of unproductive pyridine oxidation–reduction cycling between the two electrodes, providing a trade-off between Faradaic efficiency and reaction yield. General guidelines on additive use for different types of substrates are provided in the Supporting Information (see Section S9).
Next, we aimed to develop a mild elimination protocol from the TFE-derived N,O-acetals afforded in the electrooxidation (Scheme 2C). Previously, we established that iminium ions carrying a wide variety of functional groups and N-heteroarenes were accessible by exposing these N,O-acetals to BF3•Et2O.25 In order to access the corresponding enamine derivatives, we explored the use of hindered non-nucleophilic bases, which would serve to deprotonate the highly acidic β-proton of the iminium ion without quenching the Lewis acid. After initial reactivity was established with diisopropylethyl-amine (DIPEA), varying ratios of DIPEA and BF3•Et2O were explored to optimize the reaction (entries 6–9). While BF3 and DIPEA have been previously shown to form a Lewis acid–base pair,31 this adduct formation is dynamic. Indeed, treating substrate 6 with a premixed solution of DIPEA and BF3•Et2O gave similar yield under otherwise identical conditions (entry 10).
Despite this elimination method being compatible with a wide variety of functional groups (vide infra), we observed poor performance for substrates containing secondary amides such as 10 (Scheme 2C). Treatment of 10 with DIPEA and BF3•Et2O (with or without premixing, entries 11–12) led to full conversion but formation of several byproducts. We attributed this side reactivity to the deprotonation of the secondary amide promoted by coordination of amide carbonyl with BF3. Given the prevalence of secondary amides in bioactive molecules, we sought to address this issue and posited that the use of a milder and more hindered base would suppress this undesired pathway. Through screening several organic bases with 10, it was found that 2,4,6-collidine and 2,6-lutidine efficiently promoted the desired elimination (entries 13–16). Further improvements were observed upon switching MeCN to CH2Cl2 as the solvent. These alternative conditions allowed us to further expand the applicability of the desaturation approach.
To further simplify the synthetic operation, we studied different approaches to circumvent a rigorous purification of the intermediate N,O-acetal (Scheme 2D). When desaturation of 5 into 9 was conducted with a full column chromatographic isolation of N,O-acetal 6, a 2-step yield of 62% was obtained. Notably, we found that this purification process was unnecessary, as the same efficiency (65% yield) was observed by simply removing volatiles in vacuo after electrolysis and subjecting the crude to the optimal elimination conditions. Alternatively, simple filtration of the crude electrolysis mixture containing the N,O-acetal through a short silica plug resulted in 62% yield of 9 upon elimination. The procedure was adopted for the substrate scope studies given that it allows for the removal of minor side products in certain cases that may affect the subsequent formation or purification of the enamines.
With this set of optimized desaturation conditions, we first investigated the scope of this synthetic strategy for the synthesis of enamines (Scheme 3). We chose to examine substrates that cover a wide range of scaffolds, N-substituents, and functional groups. In terms of the amine core, a variety of pyrrolidine, piperidine, morpholine, piperazine, azepane, homopiperazine rings, including monocyclic and fused- and spiro-bicyclic structures, as well as acyclic amines, proved to be compatible. N-substituents that are often seen in biologically active molecules or employed as protecting groups in synthesis, such as Boc, Cbz, acyl, and sulfonyl, are well-tolerated, whereas N-aryl and N-alkyl are currently not suitable for our method. Functional groups such as esters (11, 16, 18, 19, 20, 23, 32, 35, and 38), aryl halides (16, 20, 22, and 36), alkyl fluorides (15, 30, 36, and 39), and sulfone (25) were also shown to be compatible. The stereochemistry of proline derivative 16 was not affected during the desaturation protocol, retaining >99% ee (see the SI for details). Secondary amides 11, 24, and 36 underwent the transformations smoothly upon simply replacing DIPEA with 2,6-lutidine during the elimination step. A variety of medicinally relevant N-heteroarenes were compatible, such as pyridines (9, 21, 23, 27, 34, 36, and 39), 1,2,4-oxadiazole (17), quinazoline (22), pyrazole (28), quinoline (29), and pyrimidine (30 and 36).
Scheme 3. Scope for Desaturation of Amine Derivatives.

a2,6-Lutidine instead of DIPEA. bSee the SI for details. cThe byproduct was tentatively assigned to regioisomeric N,O-acetal. d16% of unreacted SM was recovered.
Notably, this synthetic strategy displayed good to excellent site selectivity when multiple N-substituted amines with distinct electronic properties were present in the same substrate. Because the initial Shono oxidation sets the site of desaturation, the selectivity typically followed the trend of oxidation potential where the most electron-rich reacting motif underwent preferential functionalization. For example, the electrochemical oxidation of 15 happened selectively on the proline ring that is protected with a Boc group, while no reactivity of the piperidine ring was observed. The site selectivity was further investigated with the polynitrogenated compounds 26, 27, 28, 29, 30, 34, and 37. The gelatinase inhibitor fragments 39a and 39b could be obtained and easily separated, and we did not observe the formation of the third possible desaturated product 39c, which indicated that both 39a and 39b originated from oxidation of the carbamate rather than the amide. In several of these cases, the site selectivity can be rationalized by the trends of oxidation potentials of the competing N-substituted amine units (e.g., 15, 30, 34, 37, and 39; see Figure 1). In other cases, ring strain may also be at play in determining the site selectivity of the electrochemical oxidation (e.g., 26–29).
Figure 1.

Cyclic voltammetry for different protected pyrrolidines. WE: 3 mm glassy carbon disk, solvent: MeOH, electrolyte: Et4NBF4, and v = 100 mV/s. Voltammograms for Cbz- and Ns-protected pyrrolidines are not shown for clarity, which are instead presented in the SI.
When we explored the synthesis of SUAM-1221 fragments 40 and 41, a site selectivity favoring the desaturation of either the N-Cbz (40) or N-sulfonyl (41) proline ring was attained. Interestingly, by employing additional charge during the electrochemical step, the double desaturation of both proline and pyrrolidine rings was achieved (40c and 41c) in higher efficiency. We note that for the desaturation leading to 41, the product selectivity could not be predicted simply by comparing the oxidation potential of sulfonamide and tertiary amide fragments. The observation of 41a as the major product pointed to additional factors that were at play in determining the selectivity of the Shono oxidation. A possible yet currently unsubstantiated rationale for this outcome involves an intramolecular electron transfer between the two N-containing groups upon initial oxidation to a radical cation32 followed by a selectivity-determining step resulting in the loss of a proton and an additional electron.
In contrast to previously published desaturation protocols,16–19 the present strategy is agnostic of amine-protecting groups (carbamate, amide, and sulfonamide) and ring sizes (5-, 6-, and 7-membered rings or acyclic), making it a general entry into N-substituted enamines.
β-Fluorination and β,α-Fluorofunctionalization of Enamine Derivatives.
The versatility of enamines as synthons has been previously demonstrated, with the β-position having been functionalized via the reaction with electrophiles or electrophilic radicals.23,33 Related to our work, the fluorofunctionalization of N-protected enamines has been explored.33b-e Nonetheless, to the best of our knowledge, methods for the direct hydrofluorination of these nucleophiles remained unavailable. We envisioned that the reaction of these intermediates with an electrophilic fluorine source would give rise to a β-fluoro-iminium ion, which could then be reduced in the presence of a hydride source. In this approach, however, the reaction between the substrate and the iminium ion intermediate with the reagents needs to outcompete the undesired quenching of the electrophilic fluorine by the reductant. To this end, we undertook high-throughput experimentation (HTE) to survey a panel of mild hydride sources along with commercially available electrophilic fluorine reagents (NFSI and Selectfluor) across various solvents (Scheme 4A) and quickly discovered that the combination of sodium cyanoborohydride (NaBH3CN) and NFSI was efficient in all solvents tested. Further optimization of the system with respect to the ratio of NaBH3CN and NFSI led to optimal conditions A, which employed 2 equiv of each reagent in MeCN (see the SI for details). This reaction thus completed the planned synthetic sequence and furnished site-specific hydrofluorination at the β-position of an amine derivative.
Scheme 4. (A,B) Development of Hydrofluorination of Protected Enamine and the Telescoped Protocol.

aNMR yields, with dimethyl terephthalate as a reference standard.
Initial attempts also demonstrated the feasibility of a one-pot approach for the overall C(sp3)–H fluorination of N-Ts-pyrrolidine, furnishing 43 in 53% isolated yield (Scheme 4B). In this study, DIPEA employed in the elimination step proved to be incompatible with hydrofluorination conditions, but using 2,6-lutidine instead allowed the newly developed hydrofluorination to be successfully carried out on a crude reaction mixture from the desaturation.
Next, we applied this hydrofluorination method on a diverse suite of enamines derived from the above-mentioned desaturation protocol (Scheme 5), demonstrating the net β-C(sp3)–H fluorination in several complex settings. The mild conditions are compatible with several spectating functional groups such as esters (53 and 57), lactam (51), sulfone (47), and N-heteroarenes [e.g., pyridines (49, 55, and 58), 1,2,4-oxadiazole (45), and pyrimidine (51)].
Scheme 5. Scope for Hydrofluorination and β-Fluoro-α-functionalization of Enamine Derivatives.

For determination of the relative stereochemistry of products, see the SI. For compounds whose relative stereochemistry is not indicated in the scheme, we have yet to be able to assign it confidently.
In our exploration of complex substrates 49 and 50, carrying out the hydrofluorination with conditions A provided the desired β-fluorinated product, but chromatographic purification proved challenging due to the presence of polar impurities from commercial NaBH3CN solution in THF. To address this issue, we developed an alternative set of conditions using commercially available polymer-supported cyanoborohydride (PS-BH3CN) and Selectfluor (see the SI for details). Desired products were isolated in high yields without complication in the purification step.34 Due to the lower overall reactivity of PS-BH3CN vis-à-vis NaBH3CN, residual moisture in the system led to the formation of small amounts of the corresponding β-fluoro-hemiaminal via H2O trapping of the β-fluoro-iminium ion. This undesired reactivity could be suppressed with the addition of powdered molecular sieves (4 Å) as a drying agent. These alternative conditions B allowed access to more polar products conveniently and were applied to streamline the synthesis of several other products (43, 44b, 49, 50, and 58) (Scheme 5).
Finally, we expanded these findings into a strategy for the β-fluoro-α-functionalization of N-substituted amines, where the β-fluoro-iminium ion was intercepted by a compatible carbon nucleophile.35,36 A series of different nucleophiles were interrogated in combination with different electrophilic fluorine sources (see the SI for details). Three nucleophiles (TMSCN, AllylSnBu3, and Me3Al) emerged as promising candidates, providing the desired difunctionalized products. Cyano-fluorination37 was demonstrated in a set of compounds featuring acyclic (62), 5- and 6-membered rings (59, 60, and 61), and pyridine (61), where Boc-, Cbz-, and sulfonyl-protected enamines were suitable substrates. Products 59–62 are precursors to noncanonical amino acids,38 a class of molecules that has gained much attention in the discovery of new biologically active molecules. In all cases, the diastereoselectivity was controlled by the intrinsic properties of the substrates.
Allyl-fluorination could also be applied in Boc-, Cbz-, and Ts-protected enamines (63–66). We further leveraged cross olefin metathesis and subsequent functional group conversions to transform the allylated product 65 to a fluorinated analogue of the natural product bgugaine (67).39 Lastly, methyl-fluorination was performed using an excess of trimethylaluminum as the nucleophile, which allowed the synthesis of a variety of medicinally relevant products (68–74) through this formal difunctionalization strategy.40 Notably, this reaction was translated to the functionalization of an enol-ether, where an incipient fluorinated oxocarbenium ion can be intercepted by the carbon nucleophile (75).
Finally, we sought to showcase this synthetic strategy in the late-stage fluorination of synthetic targets that have shown to play beneficial effects in medicinal chemistry studies. Thus, we identified that our approach was applicable to the generation of fluorinated amide 56 (via enamide 37), which is a fragment of DPP-IV inhibitor 1 (see Scheme 1), where the additional fluorine contributed to a boost in potency.2 Notably, we also successfully demonstrated this strategy in the expedient synthesis of structurally complex TRPA1 antagonist 72 [a mixture of four diastereomers; via sulfonyl-enamine 36] by means of formal β-fluoro-α-methylation directly from the parent medicinal target. This product closely resembles compound 3 (see Scheme 1), for which the addition of β-fluorine increased metabolic stability and the addition of a-methyl provided higher potency vs the parent compound. These modifications ultimately resulted in the identification of a lead molecule that possessed a balanced combination of potency and metabolic stability. In the original work by Genentech,4 a total of 14 steps (including 4 steps to synthesize the β-fluoro-α-methylated proline core) was employed to prepare 3. This result shows how a late-stage functionalization approach could accelerate the synthesis of promising analogs directly from parent drug candidates, a pressing task in medicinal chemistry.
CONCLUSIONS
In summary, we report a new synthetic strategy to access complex β-fluorinated amine derivatives through a sequential electrochemical oxidation, elimination of the resultant N,O-acetals, and redox-mediated fluorofunctionalization of the ensuing enamines. To improve the efficiency and synthetic usefulness of this strategy, we optimized the Shono oxidation to be compatible with common structures in medicines and also developed two new protocols to accomplish desaturation and hydrofluorination. This overall approach provided convenient access to net β-C–H fluorinated products from a diverse scope of N-substituted amine precursors including complex natural products, medicinal agents, and their analogs. The synthetic versatility of enamine derivatives derived from these compounds allowed for the development of three additional difunctionalization reactions,41 yielding molecules containing a β-C–F bond with the concomitant construction of α-C–CN, C–allyl, or C–CH3 bonds.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of General Medical Sciences (R01GM130928) and Genentech. This study made use of the Cornell University NMR facility supported by the National Science Foundation (CHE-1531632). S.L. thanks the Camille and Henry Dreyfus Foundation for a Teacher-Scholar Award. J.S.K.H. acknowledges the Department of Defense for an NDSEG fellowship. We thank Prof. Kevin Moeller (Washington University in St. Louis) for helpful discussions and suggestions about the Shono oxidation, Tianwei Chen (Cornell University) for synthesis of compounds 33b and 54, and Zongbin Jia (Cornell University) for reproducing experiments.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c02548.
Experimental details, analytical data, and NMR spectra (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.4c02548
The authors declare no competing financial interest.
Contributor Information
Luiz F. T. Novaes, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14850, United States.
Justin S. K. Ho, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14850, United States.
Kaining Mao, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14850, United States.
Elisia Villemure, Department of Discovery Chemistry, Genentech, Inc., South San Francisco, California 94080, United States.
Jack A. Terrett, Department of Discovery Chemistry, Genentech, Inc., South San Francisco, California 94080, United States
Song Lin, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14850, United States.
REFERENCES
- (1).(a) Johnson BM; Shu Y-Z; Zhuo X; Meanwell NA Metabolic and Pharmaceutical Aspects of Fluorinated Compounds. J. Med. Chem 2020, 63, 6315–6386. [DOI] [PubMed] [Google Scholar]; (b) Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA Applications of Fluorine in Medicinal Chemistry. J. Med. Chem 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]; (c) Hagmann WK The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem 2008, 51, 4359–4369. [DOI] [PubMed] [Google Scholar]; (d) Böhm HJ; Banner D; Bendels S; Kansy M; Kuhn B; Müller K; Obst-Sander U; Stahl M. Fluorine in Medicinal Chemistry. ChemBioChem. 2004, 5, 637–643. [DOI] [PubMed] [Google Scholar]
- (2).Fukushima H; Hiratate A; Takahashi M; Saito M; Munetomo E; Kitano K; Saito H; Takaoka Y; Yamamoto K. Synthesis and structure-activity relationships of potent 3- or 4-substituted-2-cyanopyrrolidine dipeptidyl peptidase IV inhibitors. Bioorg. Med. Chem 2004, 12, 6053–6061. [DOI] [PubMed] [Google Scholar]
- (3).Hameed P S; Patil V; Solapure S; Sharma U; Madhavapeddi P; Raichurkar A; Chinnapattu M; Manjrekar P; Shanbhag G; Puttur J; Shinde V; Menasinakai S; Rudrapatana S; Achar V; Awasthy D; Nandishaiah R; Humnabadkar V; Ghosh A; Narayan C; Ramya VK; Kaur P; Sharma S; Werngren J; Hoffner S; Panduga V; Kumar CNN; Reddy J; Kumar KN M; Ganguly S; Bharath S; Bheemarao U; Mukherjee K; Arora U; Gaonkar S; Coulson M; Waterson D; Sambandamurthy VK; de Sousa SM Novel N-linked amino-piperidine-based gyrase inhibitors with improved hERG and in vivo efficacy against mycobacterium tuberculosis. J. Med. Chem 2014, 57, 4889–4905. [DOI] [PubMed] [Google Scholar]
- (4).Chen H; Volgraf M; Do S; Kolesnikov A; Shore DG; Verma VA; Villemure E; Wang L; Chen Y; Hu B; Lu A-J; Wu G; Xu X; Yuen P-W; Zhang Y; Erickson SD; Dahl M; Brotherton-Pleiss C; Tay S; Ly JQ; Murray LJ; Chen J; Amm D; Lange W; Hackos DH; Reese RM; Shields SD; Lyssikatos JP; Safina BS; Estrada AA Discovery of a Potent (4R,5S)-4-Fluoro-5-methylproline Sulfonamide Transient Receptor Potential Ankyrin 1 Antagonist and Its Methylene Phosphate Prodrug Guided by Molecular Modeling. J. Med. Chem 2018, 61, 3641–3659. [DOI] [PubMed] [Google Scholar]
- (5).Wang X; Allen S; Blake JF; Bowcut V; Briere DM; Calinisan A; Dahlke JR; Fell JB; Fischer JP; Gunn RJ; Hallin J; Laguer J; Lawson JD; Medwid J; Newhouse B; Nguyen P; O’Leary JM; Olson P; Pajk S; Rahbaek L; Rodriguez M; Smith CR; Tang TP; Thomas NC; Vanderpool D; Vigers GP; Christensen JG; Marx MA Identification of MRTX1133, a Noncovalent, Potent, and Selective KRASG12D Inhibitor. J. Med. Chem 2022, 65, 3123–3133. [DOI] [PubMed] [Google Scholar]
- (6).For reviews on late-stage functionalization, see:; (a) Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW The Medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev 2016, 45, 546–576. [DOI] [PubMed] [Google Scholar]; (b) Castellino NJ; Montgomery AP; Danon JJ; Kassiou M. Late-stage Functionalization for Improving Drug-like Molecular Properties. Chem. Rev 2023, 123, 8127–8153. For a more focused essay on late-stage fluorination, see: [DOI] [PubMed] [Google Scholar]; (c) Neumann CN; Ritter T. Late-Stage-Fluorination: Fancy Novelty or Useful Tool? Angew. Chem., Int. Ed 2015, 54, 3216–3221. [DOI] [PubMed] [Google Scholar]; (d) Wang Y; Dana S; Long H; Xu Y; Li Y; Kaplaneris N; Ackermann L. Electrochemical Late-Stage Functionalization. Chem. Rev 2023, 123, 11269–11335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).For additional C(sp3)–H fluorination methodologies, see:Kee CW; Chin KF; Wong MW; Tan C-H Selective fluorination of alkyl C–H bonds via photocatalysis. Chem. Commun 2014, 50, 8211–8214.Xia J-B; Zhu C; Chen C. Visible light-promoted metal-free sp3-C–H fluorination. Chem. Commun 2014, 50, 11701–11704.West JG; Bedell TA; Sorensen EJ The Uranyl Cation as a Visible-Light Photocatalyst for C(sp3)–H Fluorination. Angew. Chem., Int. Ed 2016, 55, 8923–8927.Egami H; Masuda S; Kawato Y; Hamashima Y. Photo-fluorination of Aliphatic C–H Bonds Promoted by the Phthalimide Group. Org. Lett 2018, 20, 1367–1370. Bloom S; Pitts CR; Miller DC; Haselton N; Holl MG; Urheim E; Lectka T. A Polycomponent Metal-Catalyzed Aliphatic, Allylic, and Benzylic Fluorination. Angew. Chem., Int. Ed 2012, 51, 10580–10583.Amaoka Y; Nagatomo M; Inoue M. Metal-Free Fluorination of C(sp3)–H Bonds Using a Catalytic N-Oxyl Radical. Org. Lett 2013, 15, 2160–2163. Pitts CR; Ling B; Woltornist R; Liu R; Lectka T. Triethylborane-Initiated Radical Chain Fluorination: A Synthetic Method Derived from Mechanistic Insight. J. Org. Chem 2013, 79, 8895–8899.Xia J-B; Ma Y; Chen C Vanadium-catalyzed C(sp3)–H fluorination reactions. Org. Chem. Front 2014, 1, 468–472. Zhang X; Guo S; Tang P. Transition-metal free oxidative aliphatic C–H fluorination. Org. Chem. Front 2015, 2, 806–810.Yakubov S; Stockerl WJ; Tian X; Shahin A; Mandigma MJP; Gschwind RM; Barham JP Benzoates as photo-sensitization catalysts and auxiliaries in efficient, practical, light-powered direct C(sp3)–H fluorinations. Chem. Sci 2022, 13, 14041–14051.
- (8).Liu W; Huang X; Cheng M-J; Nielsen RJ; Goddard WA; Groves JT Oxidative Aliphatic C-H Fluorination with Fluoride Ion Catalyzed by a Manganese Porphyrin. Science 2012, 337, 1322–1325. [DOI] [PubMed] [Google Scholar]
- (9).Huang X; Liu W; Ren H; Neelamegam R; Hooker JM; Groves JT Late Stage Benzylic C–H Fluorination with [18F]-Fluoride for PET Imaging. J. Am. Chem. Soc 2014, 136, 6842–6845. [DOI] [PubMed] [Google Scholar]
- (10).(a) Halperin SD; Fan H; Chang S; Martin RE; Britton R. A Convenient Photocatalytic Fluorination of Unactivated C–H Bonds. Angew. Chem., Int. Ed 2014, 53, 4690–4693. [DOI] [PubMed] [Google Scholar]; (b) Halperin SD; Kwon D; Holmes M; Regalado EL; Campeau L-C; DiRocco DA; Britton R. Development of a Direct Photocatalytic C–H Fluorination for the Preparative Synthesis of Odanacatib. Org. Lett 2015, 17, 5200–5203. [DOI] [PubMed] [Google Scholar]
- (11).Bloom S; Knippel JL; Lectka T. A photocatalyzed aliphatic fluorination. Chem. Sci 2014, 5, 1175–1178. [Google Scholar]
- (12).Elegant strategies with sequential C(sp3)–H oxidation and deoxyfluorination with DAST have been reported:Osberger TJ; Rogness DC; Kohrt JT; Stepan AF; White MC Oxidative diversification of amino acids and peptides by small-molecule iron catalysis. Nature 2016, 537, 214–219. Chambers RK; Weaver JD; Kim J; Hoar JL; Krska SW; White MC A preparative small-molecule mimic of liver CYP450 enzymes in the aliphatic C–H oxidation of carbocyclic N-heterocycles. Proc. Natl. Acad. Sci. U.SA 2023, 120, No. e2300315120.
- (13).Takahira Y; Chen M; Kawamata Y; Mykhailiuk P; Nakamura H; Peters BK; Reisberg SH; Li C; Chen L; Hoshikawa T; Shibuguchi T; Baran PS Electrochemical C(sp3)–H Fluorination. Synlett 2019, 30, 1178–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Min S; Park B; Nedsaengtip J; Hong SH Mechanochemical Direct Fluorination of Unactivated C(sp3)–H Bonds. Adv. Synth. Catal 2022, 364, 1975–1981. [Google Scholar]
- (15).A highly site-selective example has been recently disclosed: Ruskin J; Sachs RK; Wang M; Dekeyser R; Lew Z; Williams P; Hwang H; Majumdar A; Dudding T; Lectka T. Metal Ion-Induced Large Fragment Deactivation: A Different Strategy for Site-Selectivity in a Complex Molecule. Angew. Chem., Int. Ed 2024, 63, No. e202317070. [DOI] [PubMed] [Google Scholar]
- (16).In addition to the examples highlighted in scheme 1C, others reports are available for the α,β-desaturation of aliphatic N-heterocycles with a more focused substrate class.Li X; Cheng Z; Liu J; Zhang Z; Song S; Jiao N. Selective desaturation of amides: a direct approach to enamides. Chem. Sci 2022, 13, 9056–9061. Chuentragool P; Parasram M; Shi Y; Gevorgyan V. General, Mild, and Selective Method for Desaturation of Aliphatic Amines. J. Am. Chem. Soc 2018, 140, 2465–2468.
- (17).Holmberg-Douglas N; Choi Y; Aquila B; Huynh H; Nicewicz DA β-Functionalization of Saturated Aza-Heterocycles Enabled by Organic Photoredox Catalysis. ACS Catal. 2021, 11, 3153–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Spieβ P; Berger M; Kaiser D; Maulide N. Direct Synthesis of Enamides via Electrophilic Activation of Amides. J. Am. Chem. Soc 2021, 143, 10524–10529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ritu SD; Tian YM; Karl T; Jain N; König B. Photocatalyzed Dehydrogenation of Aliphatic N-Heterocycles Releasing Dihydrogen. ACS Catal. 2022, 12, 10326–10332. [Google Scholar]
- (20).Shono T; Matsumura Y; Tsubata K; Sugihara Y; Yamane S; Kanazawa T; Aoki T. Electroorganic Synthesis of Enamides and Enecarbamates and Their Utilization in Organic Synthesis. J. Am. Chem. Soc 1982, 104, 6697–6703. [Google Scholar]
- (21).Shono T; Hamaguchi H; Matsumura Y. Electroorganic chemistry. XX. Anodic oxidation of carbamates. J. Am. Chem. Soc 1975, 97, 4264–4268. [Google Scholar]
- (22).Tereshchenko OD; Perebiynis MY; Knysh IV; Vasylets OV; Sorochenko AA; Slobodyanyuk EY; Rusanov EB; Borysov OV; Kolotilov SV; Ryabukhin SV; Volochnyuk DM Electrochemical Scaled-up Synthesis of Cyclic Enecarbamates as Starting Materials for Medicinal Chemistry Relevant Building Bocks. Adv. Synth. Catal 2020, 362, 3229–3242. [Google Scholar]
- (23).(a) Trindade AF; Faulkner EL; Leach AG; Nelson A; Marsden SP Fragment-oriented synthesis: β-elaboration of cyclic amine fragments using enecarbamates as platform intermediates. Chem. Commun 2020, 56, 8802. [DOI] [PubMed] [Google Scholar]; (b) Bach T; Brummerhop H. An Expedient Synthesis of N-Acceptor-Substituted 2,3-Dihydropyrrols from the Corresponding 2-Pyrroldinones. J. Prakt Chem 1999, 341, 312–315. [Google Scholar]
- (24).Frankowski KJ; Liu R; Milligan GL; Moeller KD; Aubé J. Practical Electrochemical Anodic Oxidation of Polycyclic Lactams for Late-Stage Functionalization. Angew. Chem., Int. Ed 2015, 54, 10555–10558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Novaes LFT; Ho JSK; Mao K; Liu K; Tanwar M; Neurock M; Villemure E; Terrett JA; Lin S. Exploring Electrochemical C(sp3)-H Oxidation for the Late-Stage Methylation of Complex Molecules. J. Am. Chem. Soc 2022, 144, 1187–1197. [DOI] [PubMed] [Google Scholar]
- (26).A different approach explored in the literature to functionalize complex peptides involved the use of electroauxiliaries:Veedu DKP; Connal LA; Malins LR Tunable Electrochemical Peptide Modifications: Unlocking New Levels of Orthogonality for Side-Chain Functionalization. Angew. Chem., Int. Ed 2023, 62, No. e202215470.Shoji T; Kim S; Yamamoto K; Kawai T; Okada Y; Chiba K. Anodic Substitution Reaction of Proline Derivatives Using the 2,4,6-Trimethoxyphenyl Leaving Group. Org. Lett 2014, 16, 6404–6407.
- (27).For a review on carbofluorination of π systems, see: McKnight DA; Cadwallader D; Le CM Carbofluorination of π-Bonds and Related Reactions Involving Tandem C–C/C–F Bond Formation. Eur. J. Org. Chem 2023, 27, No. e202300017. [Google Scholar]
- (28).Roughley SD; Jordan AM The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]
- (29).Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (30).For selected applications of TFA in Boc-amine deprotection, see:Englund EA; Gopi HN; Appella DH An Efficient Synthesis of a Probe for Protein Function: 2,3-Diaminopropionic Acid with Orthogonal Protecting Groups. Org. Lett 2004, 6, 213–215. Shendage DM; Fröhlich R; Haufe G. Highly Efficient Stereoconservative Amidation and Deamidation of α-Amino Acids. Org. Lett 2004, 6, 3675–3678. For kinetic analysis of Boc deprotection, see: Ashworth IW; Cox BG; Meyrick B. Kinetics and Mechanism of N-Boc Cleavage: Evidence of a Second-Order Dependence upon Acid Concentration. J. Org. Chem 2010, 75, 8117–8125.
- (31).Iashin V; Chernichenko K; Pápai I; Repo T. Atom-Efficient Synthesis of Alkynylfluoroborates Using BF3-Based Frustrated Lewis Pairs. Angew. Chem., Int. Ed 2016, 55, 14146–14150. [DOI] [PubMed] [Google Scholar]
- (32).Related intramolecular electron-transfer has been studied for systems containing tertiary amines with electron-rich arenes.Moeller KD; Wang PW; Tarazi S; Marzabadi MR; Wong PL Anodic Amide Oxidations in the Presence of Electron-Rich Phenyl Rings: Evidence for an Intramolecular Electron-Transfer Mechanism. J. Org. Chem 1991, 56, 1058–1067.
- (33).For examples, see:; (a) Rey-Rodriguez R; Retailleau P; Bonnet P; Gillaizeau I. Iron-Catalyzed Trifluoromethylation of Enamide. Chem.–Eur. J 2015, 21, 3572–3575. [DOI] [PubMed] [Google Scholar]; (b) Gichuhi PN; Kuriyama M; Onomura O. Diastereoselective Synthesis of 3-Fluoro-2-substituted Piperidines and Pyrrolidines. Heterocycles 2014, 88, 331–346. [Google Scholar]; (c) Phipps RJ; Hiramatsu K; Toste FD Asymmetric Fluorination of Enamides: Access to α-Fluoroimines Using an Anionic Chiral Phase-Transfer Catalyst. J. Am. Chem. Soc 2012, 134, 8376–8379. [DOI] [PubMed] [Google Scholar]; (d) Dilman AD; Belyakov PA; Struchkova MI; Arkhipov DE; Korlyukov AA; Tartakovsky VA Fluorocyanation of Enamines. J. Org. Chem 2010, 75, 5367–5370. [DOI] [PubMed] [Google Scholar]; (e) Liu J; Chan J; Bryant CM; Duspara PA; Lee EE; Powell D; Yang H; Liu Z; Walpole C; Roberts E; Batey RA Effect of Acid Catalysis on the Direct Electrophilic Fluorination of Ketones, Ketals, and Enamides Using Selectfluor™. Tetrahedron Lett. 2012, 53, 2971–2975. [Google Scholar]
- (34).Hutchins RO; Natale NR; Taffer IM Cyanoborohydride Supported on an Anion Exchange Resin as a Selective Reducing Agent. J. Chem. Soc., Chem. Commun 1978, 1088–1089.
- (35).For other carbon nucleophiles explored with acyl-iminium ions, see: Mitsudo K; Yamamoto J; Akagi T; Yamashita A; Haisa M; Yoshioka K; Mandai H; Ueoka K; Hempel C; Yoshida J-I; Suga S. Stereoselective nucleophilic addition reactions to cyclic N-acyliminium ions using the indirect cation pool method: Elucidation of stereoselectivity by spectroscopic conformational analysis and DFT calculations. Beilstein J. Org. Chem 2018, 14, 1192–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).For related β-hydroxy-α-functionalizations of enamines, see: Shennan BDA; Sánchez-Alonso S; Rossini G; Dixon DJ 1,2-Redox Transpositions of Tertiary Amides. J. Am. Chem. Soc 2023, 145, 21745–21751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).For related cyano-fluorination of enamines, see:Dilman AD; Belyakov PA; Struchkova MI; Arkhipov DE; Korlyukov AA; Tartakovsky VA Fluorocyanation of Enamines. J. Org. Chem 2010, 75, 5367–5370. Feng T; Wang S; Liu Y; Liu S; Qiu Y. Electrochemical Desaturative β-acylation of cyclic N-Aryl Amines. Angew. Chem., Int. Ed 2022, 61, No. e202115178.
- (38).Hickey JL; Sindhikara D; Zultanski SL; Schultz DM Beyond 20 in the 21st Century: Prospects and Challenges of Non-canonical Amino Acids in Peptide Drug Discovery. ACS Med. Chem. Lett 2023, 14, 557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Rakba N; Melhaoui A; Loyer P; Delcros JG; Morel I; Lescoat G. Bgugaine, a pyrrolidine alkaloid from Arisarum vulgare, is a strong hepatotoxin in rat and human liver cell cultures. Toxicol. Lett 1999, 104, 239–248. [DOI] [PubMed] [Google Scholar]
- (40).Related multistep approaches for fluorofunctionalizations have been reported: Griffiths RJ; Kong WC; Richards SA; Burley GA; Willis MC; Talbot EPA Oxidative β-C–H sulfonylation of cyclic amines. Chem. Sci 2018, 9, 2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).During the final stage of manuscript writing, a relevant approach for β-chloro-α-alkylation of cyclic sulfonamides was reported: Kundu G; Lambert TH Electrochemical Vicinal C–H Difunctionalization of Saturated Azaheterocycles. J. Am. Chem. Soc 2024, 146, 1794–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.