

Abstract
Forty-one derivatives of spirooxindoles, active against HCT-116 colon cancer cells, underwent pharmacophore-based 3D-QSAR analysis to understand their correlation with anti-cancer activity. The study identified a seven-point pharmacophore model (ADHHRRR1) and QSAR models, offering insights for lead optimization and novel analogue design, thus advancing anti-cancer drug discovery. This research underscores the value of molecular modeling in elucidating structure-activity relationships and enhancing drug development efforts.
Keywords: Spirooxindoles, Anti-cancer activity, Pharmacophore modelling, QSAR studies
1. Introduction
Cancer represents a significant health challenge characterized by uncontrolled cell proliferation and differentiation mechanisms, ranking as the second leading cause of death globally, following cardiovascular diseases [[1], [2], [3], [4], [5], [6], [7], [8]]. Colon cancer specifically stands as the second most fatal cancer affecting both genders worldwide, often diagnosed at an advanced stage when tumor cell dissemination has already occurred [9,10]. Recent research highlights the role of low folate and methionine levels [11], obesity [12,13], and non-steroidal anti-inflammatory drugs [14] in the development of colon cancer. Despite advancements in cancer treatment, chemotherapy resistance poses a fundamental issue in cancer therapy [[15], [16], [17], [18]]. Thus, the primary focus of organic medical chemistry is to devise and discover more effective and alternative treatment options for colorectal cancer.
Spirooxindoles have recently garnered attention as compelling synthetic targets due to their presence in various natural products and biologically active compounds [[19], [20], [21], [22]]. These compounds demonstrate a broad spectrum of pharmacological activities, including anti-cancer, local anesthetic, anti-tumor, anti-inflammatory, and anti-tubercular properties (Fig. 1) [[23], [24], [25], [26], [27], [28], [29], [30], [31]]. Given their diverse pharmacological effects, spirooxindoles hold promise as potential candidates for drug discovery. Their structure incorporates oxindoles and other heterocyclic moieties simultaneously, allowing for multifunctionality. The multiple functionalized oxindole groups can serve as both hydrogen bond donors and acceptors, facilitating interactions with biological targets. Additionally, the inclusion of a cycloalkyl or heterocyclic moiety fused at the C-3 position of oxindole offers opportunities to modulate the physicochemical properties of spirooxindoles [[32], [33], [34], [35], [36]].
Fig. 1.

Bioactive compounds incorporating spirooxindoles.
In contemporary times, the process of discovering new drugs has become increasingly costly and time-consuming. However, advancements in computing offer a promising avenue for leveraging computational chemistry tools to streamline drug design and development, thereby mitigating research expenses and enhancing the selection of compounds for in vitro testing [[37], (a), (b)]. Among the latest breakthroughs are in silico methodologies, encompassing quantitative structure-activity relationship (QSAR), molecular docking, ADME prediction techniques [[38], (a), (b)]. In continuation of the preceding discussion, quantitative structure-activity relationships (QSAR) serve as a rational framework for the development of more potent and targeted therapeutic medications by elucidating the connections between a compound's biological activity and its physicochemical properties. Quantitative Structure-Activity Relationship (QSAR) models establish mathematical equations linking chemical structures to their biological activities through a linear regression model represented as y = Xb + e. This formulation describes predictor variables (X) relative to a predicted variable (y) using a regression vector (b). In typical QSAR investigations, identifying molecular descriptors with significant impact on the desired biological activity is crucial. Various methods like Multiple Linear Regression (MLR), Genetic Algorithm (GA), Partial Least Squares (PLS), and Principal Component Analysis (PCA) are employed for variable selection to compile such descriptors. Our interest lies in exploring the pharmacophore generation and QSAR studies of spirooxindole derivatives containing the isatin moiety, as reported in the literature by a research group. These derivatives have demonstrated anti-cancer activity against the HCT-116 colon cancer cell line [39]. QSAR analysis establishes a mathematical correlation between biological activities and computable parameters such as topological, electronic, physicochemical, stereochemical, or indices. Such studies play a crucial role in the design of novel anti-cancer molecules.
2. Methods
2.1. Dataset and ligand preparation
For the preparation of common pharmacophore through 3D-QSAR studies, a set of 41 spirooxindole derivatives with well-defined anticancer activity against colon cancer cell line HCT-116 was used for the QSAR analysis. In the QSAR computations, the in vitro inhibitory concentrations (IC50) of the compounds against colon cancer cells were converted to the logarithm unit of molar grade (pIC50 = -logIC50). The 3D structures of ligands were generated using Maestro and optimized using the LigPrep module of Schrodinger (2022-1).
Maestro drew sketches of all 41 compounds. Further, the semi-empirical OPLS 2005 force field was used to optimize the geometry. Because molecules lack 3D coordinates, ionization, stereochemistry, and tautomers, ligand preparation is typically necessary. The Schrödinger Ligprep tool was used to prepare the least energy state of the ligand, which generates stereoisomers, converts 1D/2D structures to 3D, neutralizes charged structures, or establishes the most likely ionization state at a user-defined pH. The OPLS 2005 force field was then used to minimize this ligand molecule. To retain the diversity of structure and activity in both sets for the QSAR model and the creation and validation of pharmacophore, all the molecules were split into a training set and a test set.
2.2. Creation of pharmacophore sites
For the generation of pharmacophore models, the Phase software (Schrodinger 2022-1) was utilized. The dataset of ligands was classified based on their activity, with a threshold of PIC50 > 5.5 for active compounds and PIC50 < 4.7 for inactive ones, to generate a common pharmacophore hypothesis. Phase identified six inherent pharmacophoric qualities: H-bond acceptor (A), H-bond donor (D), hydrophobic group (H), negatively charged group (N), positively charged group (P), and aromatic ring (R), defining the chemical characteristics of all ligands. Following generation, the pharmacophore hypotheses were scored and rated, with six common locations identified among the compounds. Table 1 provides details on the vector, volume, site, survival, and survival active scores and rankings for the top six resulting hypotheses.
Table 1.
The score of different parameters of the hypotheses.
| ID | Survival Score | Site Score | Vector Score | Volume Score | Selectivity Score | Matches | Inactive |
|---|---|---|---|---|---|---|---|
| ADHHRRR | 6.997571 | 0.740037 | 0.982411 | 0.825500 | 3.408231 | 11 | 2.203132 |
A: acceptor; H: hydrophobic; R: aromatic ring; D: Donor.
2.3. Identifying common pharmacophores
To generate a hypothesis, the scores and alignment of active ligands were scrutinized, leading to the selection of ADHHRRR as the top pharmacophore hypothesis for further investigation (refer to Table 1). The chosen 3D pharmacophore hypothesis (depicted in Fig. 1b) comprised two hydrophobic groups (H) represented by green spheres, one hydrogen bond donor (D) depicted by a blue sphere with two arrows, one hydrogen bond acceptor (A) illustrated by a pink sphere with two arrows, and three aromatic rings (R) depicted by grey circles. The critical pharmacophoric components of this selected pharmacophore are outlined in the 2D representation in Fig. 2a: a hydroxyl group (pink sphere with two arrows) symbolizing a hydrogen bond acceptor (A), an NH group of the isatin ring (blue sphere with two arrows) signifying a hydrogen bond donor (D), the thiazolidine ring and pyrrole ring of the isatin moiety (green sphere) representing a hydrophobic group (H), and benzene side chain rings such as R9, R10, and R11. Fig. 2b represent common pharmacophoric sites of an active ligand with the labelled distance (Å unit). However in Fig. 2c and d former displays the alignment of active ligands towards the pharmacophore, while as the later displays alignment of all ligands (both active and inactive) towards the pharmacophore.
Fig. 2.

Common pharmacophore generation and validation: (a) Common pharmacophore aligned with most active ligand [three aromatic rings (dark yellow circle)], one acceptor [pink sphere with two arrows], one donor [blue sphere with two arrows], and two hydrophobic groups [green sphere]; (b) common pharmacophoric sites of an active ligand with distance. All distances are in Å unit; (c) alignment of all active ligands to the pharmacophore; and (d) alignment of all ligands (active and inactive) to the pharmacophore. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
2.4. Building 3D-QSAR models
An atom-based 3D-QSAR model was generated by using common pharmacophore hypotheses. The actual and predicted activities were correlated for the set of forty-one training molecules using PLS analysis. The training and test data set contains 70 % and 30 % data, respectively to describe the predictive capability of the model. Either atom-based or pharmacophore-based PHASE QSAR models are possible. [39] The structure-activity link in this work is explained by a QSAR model based on atoms. Atom-based QSAR models were developed for the chosen hypothesis using a rectangular grid with 1.0 Å spacing to include the area filled by the molecules in the aligned training set. van der Waals's models of the molecules in the aligned training set were put into a regular grid of cubes to create the QSAR model. In order to represent which volume elements are occupied by a van der Waals surface model of the ligand, each ligand is represented by a set of bit values (zero or one). For the purpose of building partial least-squares (PLS) QSAR models, this representation can be employed as independent variables. Predicting the behaviours of the 12 chemicals in the test set served to validate the best QSAR model. For the dataset, a four-component (PLS factor) model with good statistics was found.
2.5. Molecular docking
Molecular docking was conducted using the Schrödinger software suite (Schrödinger Release 2022-1: Maestro, Schrödinger, LLC, New York, NY). Crystal coordinates of the MDM2 protein (PDB ID 5LAW) were obtained from the Protein Data Bank (www.rcsb.org). Prior to docking, bond orders were assigned, and hydrogen atoms were added using the pre-process option, while water molecules were removed from the protein structure. Ionization of heteroatoms at physiological pH was performed using the Epic tool to reflect biological conditions. Optimization of hydrogen bonds, particularly involving histidine, aspartate, glutamate, and hydroxyl-containing amino acids, was carried out to mitigate steric clashes. The entire protein structure underwent minimization using the OPLS 2005 force field. Ligand preparation involved converting ligand structures from 1D/2D representations to 3D structures using the Ligprep tool, which included considerations of ionization, stereochemistry, and tautomeric forms. The ligand molecule underwent further minimization using the OPLS 2005 force field. Molecular grids were generated using default settings for the grid-based energy descriptor, incorporating a van der Waals radius of 1.0. The Glide ligand docking program was utilized for docking ligand molecules onto the prepared receptor grid. Favorable interactions between ligand molecules and the receptor were evaluated and scored using the Glide program in extra precision (XP) mode with the OPLS-2005 force field. This comprehensive approach facilitated the assessment of critical interactions, including hydrogen bonding, hydrophobic interactions, and π-π stacking, between the enzyme and the compounds of interest.
2.6. ADMET prediction
ADME properties of selected ligands were analyzed using the QikProp tool of the Schrodinger suite.
3. Results and discussion
The goal of this research was to generate a 3D pharmacophore for spirooxindole derivatives (Table 2) and a 3D atom-based quantitative structure-activity relationship (QSAR) model for anticancer agents to better understand the structural features of these compounds that are critical for binding and biological activity. For the QSAR studies, we have used the Phase module of the Schrodinger suite. This Phase-generated hypothesis will also show how the ligands are relative to one another when they bind to the receptor's active site. Therefore, in order to determine the general features of molecular structure that control the activity, we have created a 3D-QSAR model using the conformation indicated by the hypothesis.
Table 2.
Structures and properties of train and test ligands.
| Sr. No. | Ligand Name | QSAR Set | Activity | Predicted Activity | PLS factor |
|---|---|---|---|---|---|
| 1 |  |
Training | 5.046 | 5.309 | 4 |
| 2 | 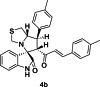 |
Training | 5.097 | 5.102 | 4 |
| 3 |  |
Training | 5.523 | 5.470 | 4 |
| 4 | 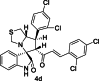 |
Test | 5.699 | 5.268 | 4 |
| 5 | 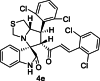 |
Test | 5.523 | 5.492 | 4 |
| 6 | 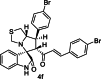 |
Training | 5.523 | 5.491 | 4 |
| 7 |  |
Training | 5.602 | 5.709 | 4 |
| 8 |  |
Training | 5.155 | 5.236 | 4 |
| 9 |  |
Training | 5.097 | 5.184 | 4 |
| 10 |  |
Training | 4.839 | 4.871 | 4 |
| 11 |  |
Training | 4.721 | 4.702 | 4 |
| 12 |  |
Training | 5.804 | 5.708 | 4 |
| 13 |  |
Training | 5.301 | 5.456 | 4 |
| 14 |  |
Training | 5.538 | 5.580 | 4 |
| 15 |  |
Test | 5.155 | 5.095 | 4 |
| 16 | 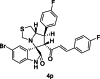 |
Training | 5.456 | 5.351 | 4 |
| 17 | 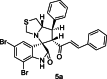 |
Training | 5.347 | 5.288 | 4 |
| 18 |  |
Training | 5.301 | 5.361 | 4 |
| 19 | 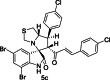 |
Training | 5.538 | 5.380 | 4 |
| 20 |  |
Training | 5.071 | 5.042 | 4 |
| 21 | 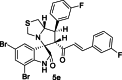 |
Training | 5.301 | 5.333 | 4 |
| 22 |  |
Training | 5.658 | 5.622 | 4 |
| 23 | 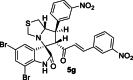 |
Training | 5.553 | 5.544 | 4 |
| 24 | 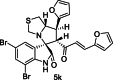 |
Training | 4.886 | 4.729 | 4 |
| 25 | 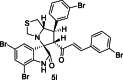 |
Test | 5.553 | 5.545 | 4 |
| 26 | 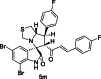 |
Test | 5.398 | 5.343 | 4 |
| 27 |  |
Training | 5.432 | 5.374 | 4 |
| 28 |  |
Test | 4.523 | 4.518 | 4 |
| 29 |  |
Training | 5.046 | 4.964 | 4 |
| 30 |  |
Test | 4.939 | 4.916 | 4 |
| 31 |  |
Test | 5.046 | 4.951 | 4 |
| 32 | 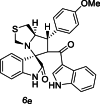 |
Training | 4.585 | 4.653 | 4 |
| 33 | 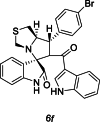 |
Test | 4.947 | 4.972 | 4 |
| 34 | 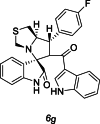 |
Training | 4.886 | 4.916 | 4 |
| 35 | 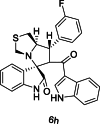 |
Test | 5.046 | 4.783 | 4 |
| 36 | 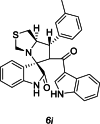 |
Test | 4.815 | 4.834 | 4 |
| 37 | 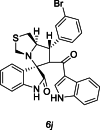 |
Training | 5.046 | 4.884 | 4 |
| 38 |  |
Training | 5.155 | 5.130 | 4 |
| 39 | 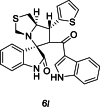 |
Training | 4.046 | 4.176 | 4 |
| 40 | 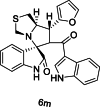 |
Training | 4.398 | 4.368 | 4 |
| 41 |  |
Test | 4.396 | 4.751 | 4 |
We employed a dataset of 29 (training set) chemicals with inhibitory activity against colon cancer cells to generate an atom-based 3D QSAR hypothesis. The model was validated using 12 (test set) compounds, which cover a wide range of anticancer activity against colon cancer cells. The alignment is generated by the best pharmacophore model (Fig. 3). For the dataset, a four-PLS factor model with solid statistics and predictive ability was generated (Table 3). Since an incremental improvement in statistical significance and predictivity was seen for each incremental increase in the included PLS components up to four, four PLS factors were included in the model development. The observed and PHASE-predicted activity can be seen in Fig. 4, Fig. 5b and is summarised in Table 2. The model expresses the 95 % variance exhibited by spirooxindole derivatives, which is close to one and indicates a close agreement of fitting points on the regression line for the observed and PHASE-predicted activity.
Fig. 3.
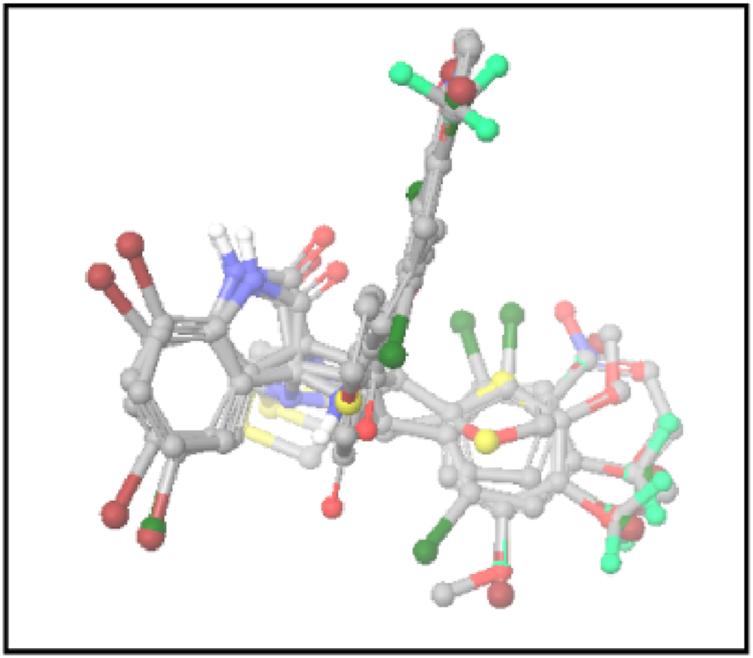
The Alignment of ligands (active and inactive) to the pharmacophore.
Table 3.
PLS statistical parameters of the selected 3D-QSAR model.
| Factors | SD | R2 | R2 Scramble | Stability | F | P | RMSE | Q2 | Pearson-r |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.2931 | 0.4807 | 0.2632 | 0.937 | 25 | 3.05E-05 | 0.28 | 0.4714 | 0.7566 |
| 2 | 0.1864 | 0.7978 | 0.5595 | 0.44 | 51.3 | 9.45E-10 | 0.26 | 0.5352 | 0.7801 |
| 3 | 0.1364 | 0.8958 | 0.669 | 0.442 | 71.7 | 2.06E-12 | 0.29 | 0.4376 | 0.6906 |
| 4 | 0.1053 | 0.9505 | 0.804 | 0.286 | 94.8 | 2.44E-14 | 0.25 | 0.6761 | 0.8844 |
SD, standard deviation of the regression; R, squared value of R2 for the regression; F, variance ratio. Large value of F indicate a more statistical regression; P, the significance level of the variance ratio. Smaller values indicate a greater degree of confidence; RMSE, root mean square error; Q, the squared value of Q2 for the predicted activities; Pearson-R, Pearson-R value for the correlation between the predicted and observed activity for the test set.
Fig. 4.

Scatter plot between observed activity versus predicted activity for training and test set compounds.
Fig. 5.

Plot between observed and predicted anti-cancer activity values of training set (a) and test set molecules (b) using the atom-based 3D-QSAR model.
The series of Spirooxindole derivatives underwent a 3D-QSAR study to determine how the spatial arrangement of structural characteristics affected their anticancer effectiveness against colon cancer cells. The regression coefficient (R2 = 0.950) of the training and cross-validation coefficient (Q2 = 0.676) demonstrated the usefulness of the model. A statistically significant regression model is shown by the large value of F (94.8), which is also supported by the low value of the variance ratio (P), a sign of a high level of confidence. The created model's capacity to predict the activity of unidentified compounds in the test set was demonstrated by the standard deviation (SD) value of 0.105 and the root mean square error (RMSE) value of 0.250. The cross-validated correlation coefficient (q2 = 0.676), which was calculated using the leave one out or leave N out technique, can be used to express the model's validity. Because it is obtained via the external validation approach by splitting the dataset into a training and test set, the q2 is a more trustworthy and robust statistical parameter than the r2.
The atoms depict the 3D properties of the ligands (atoms or pharmacophores) as they favour or inhibit action. The 3D characteristics of the QSAR model are shown in Fig. 6 as cubes that reflect the model and are coloured according to the sign of the coefficient values. By default, positive coefficients are shown as blue, and negative coefficients as red. Positive coefficients show a rise in activity whereas negative coefficients show a fall in activity. These generated cubes illustrate important features contributing to interactions between the ligand and the active site of an enzyme or receptor. The blue cubes in 3D plots of the 3D pharmacophore regions refer to ligand regions in which the Br groups at the aryl ring support this finding. It can be suggested that the substitution of electron-withdrawing groups at the aromatic ring will append the anticancer activity, whereas the addition of electron-withdrawing groups near the isatin ring will contribute to decreased receptor binding, which in turn will result in lower potency of compounds because red color cubes are for the negative specific feature is favorable for biological activity, whereas the red cubes demonstrate that particular structural feature or functional group is unfavorable for the activity. The contributions of hydrophobic (0.651), Electron Withdrawing (0.172), and hydrogen bond donor (0.017) intensities were higher which indicates their significant role in the protein-ligand interactions.
Fig. 6.

QSAR visualization of various substituents effect: (a) electron withdrawing feature; (b) hydrogen-bond donor; (c) hydrophobic features; and (d) combined effect (blue cubes showing positive potential while red cubes showing the negative potential of particular substitution). Active molecules are shown in the tube and inactive molecules are in the thin tube. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Visual analysis of Fig. 6a, reflects that the thiazolidine ring and the aryl ring both have blue cubes. Therefore, the findings indicated that the blue advantageous zones at these locations might be accountable for the elevated levels of activity. The high activity of compounds 4l, 5f, and 5l possessing the effect of the electron-withdrawing group to that particular place decreased receptor binding, which in turn will result in lower potency of compounds because red color cubes are for the negative effect of the electron-withdrawing group to that particular place. Fig. 6b illustrates that the Hydrogen bond donor characteristic is necessary at the indole ring present on spirooxindole. The N–H group of the indole ring is involved in hydrogen bonding, thus contributing to the anti-cancer activity of compounds. Fig. 6c demonstrates the effect of hydrophobic groups. It can be deduced from the figure that hydrophobic groups are well tolerated near the indole ring (blue cubes), while the substitution of hydrophobic groups at the aromatic ring is unacceptable (red cubes) or may hinder the binding of the molecules to the receptor active site and will result in decreased anticancer activity against colon cancer cells. Further, Fig. 6d showed the combined effect of all the features. The presence of NH groups (indole ring), Electron withdrawing groups on the aromatic ring, and thiazolidine ring will have a positive effect (blue cubes).
3.1. Molecular docking study
To elucidate the binding mode between the most active compound 4l and the active site amino acid residues of the enzyme, docking of compound 4l in the active site of MDM2 was conducted.
An X-ray crystal structure of tyrosine protein kinase with a resolution of 1.64 Å (MDM2, PDB ID 5LAW) [40] was retrieved from the protein data bank (www.rcsb.org). Using the Schrödinger software package (Schrödinger Release 2022-1: Maestro, Schrödinger, LLC, New York, NY), the compound 4l wes flexibly docked into the active site of MDM2. The interactions of compound with surrounding amino acids was analyzed to predict their binding modes and affinities in the active site of MDM2. The results of molecular docking investigations indicate strong binding affinity of compound 4l with the target molecule, suggesting its potential as effective agent in interacting with the chosen targets. Upon docking into the active site of MDM2, compound 4l showed two π-π interactions between bromophenyl ring of 4l and imidazole ring of HIE 96 and phenyl ring of TYR 100 (Fig. 7, Fig. 8).
Fig. 7.

Molecular docking of 4l in the active site of MDM2 (PDB ID:5LAW).
Fig. 8.

2D interaction diagram of 4l in the active site of MDM2.
3.2. ADMET prediction
The ADMET properties for the synthesized compounds can be determined in-silico by using qik prop module of Schrödinger suite 2022. The computed dipole moment of the molecule is in the range of 3.57–11.7. The solute is predicted to accept between five and eight hydrogen bonds from water molecules in an aqueous solution of the compounds. The number of likely metabolic reactions of the compounds is 2–5. The number of violations of Lipinski's rule of five is 1–2. So almost all the properties of the compounds are within the recommended values. The details of the ADMET properties for the compounds 4a-4p, 5a-5n, and 6a-6n are shown in Table 4.
Table 4.
Insilico ADMET screening for synthesized compounds.
| Entry | Dipole | Donor HB | Acceptor HB | LogPo/w | No. of Metabolite | Rule of five |
|---|---|---|---|---|---|---|
| 4a | 4.874 | 1 | 6 | 5.445 | 3 | 1 |
| 4b | 5.013 | 1 | 6 | 5.763 | 4 | 1 |
| 4c | 5.519 | 1 | 6 | 6.120 | 2 | 2 |
| 4d | 4.419 | 1 | 6 | 6.946 | 2 | 2 |
| 4e | 4.806 | 1 | 6 | 6.413 | 3 | 2 |
| 4f | 5.346 | 1 | 6 | 6.276 | 2 | 2 |
| 4g | 10.408 | 1 | 8 | 3.689 | 5 | 1 |
| 4h | 3.398 | 1 | 7.5 | 4.860 | 4 | 1 |
| 4i | 3.754 | 1 | 6 | 6.973 | 2 | 2 |
| 4j | 3.571 | 1 | 6 | 4.877 | 4 | 2 |
| 4k | 4.549 | 1 | 7 | 3.808 | 4 | 2 |
| 4l | 5.221 | 1 | 6 | 6.215 | 3 | 2 |
| 4m | 5.602 | 1 | 6 | 5.604 | 2 | 1 |
| 4n | 5.300 | 1 | 6 | 7.007 | 2 | 2 |
| 4o | 5.950 | 1 | 6 | 6.064 | 2 | 2 |
| 4p | 5.985 | 1 | 6 | 6.807 | 2 | 2 |
| 5a | 6.000 | 1 | 6 | 6.134 | 3 | 2 |
| 5b | 6.420 | 1 | 6 | 6.758 | 4 | 2 |
| 5c | 4.985 | 1 | 6 | 7.054 | 2 | 2 |
| 5d | 6.384 | 1 | 6 | 7.775 | 2 | 2 |
| 5e | 6.004 | 1 | 6 | 6.585 | 3 | 2 |
| 5f | 6.184 | 1 | 6 | 6.906 | 2 | 2 |
| 5g | 3.660 | 1 | 6 | 4.709 | 5 | 1 |
| 5k | 6.209 | 1 | 7 | 4.946 | 4 | 1 |
| 5l | 6.696 | 1 | 6 | 6.902 | 3 | 2 |
| 5m | 4.956 | 1 | 6 | 6.605 | 2 | 2 |
| 5n | 4.875 | 1 | 6 | 8.093 | 2 | 2 |
| 6a | 9.397 | 1 | 5 | 5.194 | 3 | 1 |
| 6b | 9.321 | 1 | 5 | 5.512 | 2 | 1 |
| 6c | 10.000 | 1 | 5 | 5.689 | 2 | 2 |
| 6d | 11.738 | 1 | 5 | 6.014 | 2 | 2 |
| 6e | 11.184 | 1 | 5.75 | 5.689 | 3 | 1 |
| 6f | 10.011 | 1 | 5 | 5.778 | 2 | 2 |
| 6g | 10.183 | 1 | 5 | 5.446 | 2 | 1 |
| 6h | 9.015 | 1 | 5 | 5.410 | 3 | 1 |
| 6i | 10.878 | 1 | 5 | 5.501 | 4 | 1 |
| 6j | 9.297 | 1 | 5 | 5.752 | 3 | 2 |
| 6k | 10.758 | 1 | 5 | 6.197 | 2 | 2 |
| 6l | 11.516 | 1 | 5 | 5.047 | 3 | 1 |
| 6m | 10.260 | 1 | 5.5 | 4.524 | 3 | 2 |
| 6n | 11.203 | 1 | 7.25 | 5.509 | 5 | 2 |
| Recommended values | 1–12.5 | 0–6 | 2–20 | 1–8 | Max 4 |
Dipole- Computed dipole moment of the molecule.
AccptHB- Estimated number of hydrogen bonds that would be accepted by the solute from water molecules in an aqueous solution.
logPo/w - Predicted octanol/water partition coefficient.
metab- Number of likely metabolic reactions.
Rule of Five: Number of violations of Lipinski's rule of five.
4. Conclusion
To comprehend the structural characteristics of a series of spirooxindoles derivatives with anti-cancer potential, a ligand-based pharmacophore model was developed. The pharmacophore model was generated using PHASE. A seven-point pharmacophore model (ADHHRRR1) which consists of one hydrogen bond acceptor (A), one hydrogen bond donor (D), two hydrophobic (H), and three aromatic rings (R) features were selected as the best model for further study. This work also establishes Quantitative Structure-Activity Relationships (QSAR) which helps to elucidate the 3D-structural features crucial for binding. There is strong statistical support for the QSAR model, which explains why the presence of electron-withdrawing and hydrophobic groups on the spirooxindoles moiety boosts the anti-cancer action of the compounds. In addition to above, the computational results offer a robust pharmacophore model and QSAR analysis for designing novel spirooxindole derivatives with enhanced anti-cancer activity, providing valuable insights for future drug discovery endeavors.
CRediT authorship contribution statement
Sukhmeet Kaur: Funding acquisition, Formal analysis, Data curation. Jasneet Kaur: Writing – original draft, Investigation, Formal analysis, Data curation, Conceptualization. Bilal Ahmad Zarger: Writing – review & editing, Methodology, Investigation. Nasarul Islam: Software, Resources, Project administration, Data curation, Conceptualization. Nazirah Mir: Resources.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We are thankful to P.G. Department of Chemistry, Khalsa College Amritsar for giving us the facility to carry out this work using Schrödinger software installed in the Department. N.I. would like to thank JKST&IC for financial support (JKST&IC/SRE/J/286-87).
Contributor Information
Sukhmeet Kaur, Email: sukhmeetkaur@khalsacollege.edu.in.
Jasneet Kaur, Email: jasneetkaur@khalsacollege.edu.in.
Bilal Ahmad Zarger, Email: bilal.ahmad@cihanuniversity.edu.iq, bilal.pharmacy@stephensint.com.
Nasarul Islam, Email: nasarul.chst@gmail.com.
Nazirah Mir, Email: nasarulisrat82519@jk.gov.in.
References
- 1.Youlden D.R., M. Cramb S., Yip C.H., Baade P.D. Incidence and mortality of female breast cancer in the Asia-Pacific region. Cancer Biology & Medicine. 2014;11:101–115. doi: 10.7497/j.issn.2095-3941.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang W., Hu Y., Yang Y.S., Zhang F., Zhang Y.B., Wang X.L., Tang J.F., Zhong W.Q., Zhu H.L. Design, modification and 3D QSAR studies of novel naphthalin-containing pyrazoline derivatives with/without thiourea skeleton as anticancer agents. Bioorg. Med. Chem. 2013;21:1050–1063. doi: 10.1016/j.bmc.2013.01.013. [DOI] [PubMed] [Google Scholar]
- 3.Murahari M., S.Kharkar P., Lonikar N., Mayur Y.C. Acridone-pyrimidine hybrids- design, synthesis, cytotoxicity studies in resistant and sensitive cancer cells and molecular docking studies. Eur. J. Med. Chem. 2017;130:154–170. doi: 10.1016/j.ejmech.2017.08.023. [DOI] [PubMed] [Google Scholar]
- 4.Jemal A., Siegel R., Ward E., Hao Y., Xu J., Murray T., Thun M.J., Cancer C.A. Detection of polymorphisms of DNA repair genes (XRCC1 and XPC) in Prostate Cancer J. Clin. 2008;58:71–96. [Google Scholar]
- 5.Sun X.-Q., Chen L., Li Y.-Z., Li W.-H., Liu G.-X., Tu Y.-Q., Tang Y. Structure-based ensemble-QSAR model: a novel approach to the study of the EGFR tyrosine kinase and its inhibitors. Acta Pharmacol. Sin. 2014;35:301–310. doi: 10.1038/aps.2013.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kubinyi H., Taylor J., Ramdsen C. In: Hansch C., editor. vol. 4. Pergamon; 1990. Quantitative drug design; pp. 589–643. (Comprehensive Medicinal Chemistry). [Google Scholar]
- 7.Irigaray P., Belpomme D. Basic properties and molecular mechanisms of exogenous chemical carcinogens. Carcinogenesis. 2010;31:135–148. doi: 10.1093/carcin/bgp252. [DOI] [PubMed] [Google Scholar]
- 8.Hu Z., Li M., Cao Y., Donald Akan O., Guo T., Luo F. Targeting AMPK signaling by dietary polyphenols in cancer prevention. Mol. Nutrition Food Res. 2022 doi: 10.1002/mnfr.202100732. [DOI] [PubMed] [Google Scholar]
- 9.Planche A.S., Kleandrova V.V., Luan F., Cordeiro M.N.D.S. Using multiple linear regression and artificial neural network techniques for predicting CCR5 binding affinity of substituted 1-(3, 3-diphenylpropyl)-piperidinyl amides and ureas. Bioorg. Med. Chem. 2012;20:4848–4855. [Google Scholar]
- 10.Verma R.P., Hansch C. QSAR modeling of taxane analogues against colon cancer. Eur. J. Med. Chem. 2010;45:1470–1477. doi: 10.1016/j.ejmech.2009.12.054. [DOI] [PubMed] [Google Scholar]
- 11.Schernhammer E.S., Ogino S., Fuchs C.S. Folate and vitamin B6 intake and risk of colon cancer in relation to p53 expression. Gastroenterology. 2008;135:770–780. doi: 10.1053/j.gastro.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doll R., Peto R. J. Natl. Cancer Inst. 1981;66:1191–1308. [PubMed] [Google Scholar]
- 13.Giovannucci E., Stampfer M.J., Colditz G., Rimm E.B., Willett W.C. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J. Natl. Cancer Inst. 1992;84:91–98. doi: 10.1093/jnci/84.2.91. [DOI] [PubMed] [Google Scholar]
- 14.Berkel H.J., Holcombe R.F., Middlebrooks M., Kannan K. Nonsteroidal antiinflammatory drugs and colorectal cancer. Epidemiol. Rev. 1996;18:205–217. doi: 10.1093/oxfordjournals.epirev.a017926. [DOI] [PubMed] [Google Scholar]
- 15.Gottesman M.M., Fojo, T T., Bates S.E. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 16.Cholewinski G., Dzierzbicka K., Koodziejczyk M.M. Natural and synthetic acridines/acridones as antitumor agents: their biological activities and methods of synthesis. Pharmacol. Rep. 2011;63:305–336. doi: 10.1016/s1734-1140(11)70499-6. [DOI] [PubMed] [Google Scholar]
- 17.Wu Q., Yang Z., Nie Y., Shi Y., Fan D. Multi-drug resistance in cancer chemotherapeutics: mechanisms and lab approaches. Cancer Lett. 2014;347:159–166. doi: 10.1016/j.canlet.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 18.Gottesman M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 19.Rios R. Enantioselective methodologies for the synthesis of spiro compounds. Chem. Soc. Rev. 2012;41:1060–1074. doi: 10.1039/c1cs15156h. [DOI] [PubMed] [Google Scholar]
- 20.Ball-Jones N.R., Badillo J.J., Franz A.K. Strategies for the enantioselective synthesis of spirooxindoles. Org. Biomol. Chem. 2012;10:5165–5181. doi: 10.1039/c2ob25184a. [DOI] [PubMed] [Google Scholar]
- 21.Galliford C.V., Scheidt K.A. Pyrrolidinyl-spirooxindole natural products as inspirations for the development of potential therapeutic agents. Angew Chem Int. 2007;46:8748–8758. doi: 10.1002/anie.200701342. [DOI] [PubMed] [Google Scholar]
- 22.Hong L., Wang R. Efficient indirect electrochemical synthesis of 2-substituted benzoxazoles using sodium iodide as mediator. Adv. Synth. Catal. 2013;355:1023–1052. [Google Scholar]
- 23.Zhao Y., Liu L., Sun W., Lu J., McEachern D., Li X., Yu S., Bernard D., Ochsenbein P., Ferey V., Carry J.C., Deschamps J.R., Sun D., Wang S. Diastereomeric spirooxindoles as highly potent and efficacious MDM2 inhibitors. J. Am. Chem. Soc. 2013;135:7223–7234. doi: 10.1021/ja3125417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antonchick A.P., Gerding-Reimers C., Catarinella M., Schürmann M., Preut H., Ziegler S., Rauh D., Waldmann H. Highly enantioselective synthesis and cellular evaluation of spirooxindoles inspired by natural products. Nat. Chem. 2010;2:735–740. doi: 10.1038/nchem.730. [DOI] [PubMed] [Google Scholar]
- 25.Rajesh S.M., Perumal S., Menendez J.C., Yogeeswari P., Sriram D. Antimycobacterial activity of spirooxindolo-pyrrolidine, pyrrolizine and pyrrolothiazole hybrids obtained by a three-component regio- and stereoselective 1,3-dipolar cycloaddition. Med. Chem. Commun. 2011;2:626–630. [Google Scholar]
- 26.Schönhaber J., Müller T.J.J. Luminescent bichromophoric spiroindolones – synthesis and electronic properties. Org. Biomol. Chem. 2011;9:6196–6199. doi: 10.1039/c1ob05703k. [DOI] [PubMed] [Google Scholar]
- 27.Cui C.B., Kakeya H., Osada H. Novel mammalian cell cycle inhibitors, spirotryprostatins A and B, produced by Aspergillus fumigatus, which inhibit mammalian cell cycle at G2/M phase. Tetrahedron. 1996;52:12651–12666. [Google Scholar]
- 28.Conroy H., Chakrabarti J.K. NMR Spectra of gelsemine derivatives. The Structure and biogenesis of the alkaloid gelsemine. Tetrahedron Lett. 1959;1:6–13. [Google Scholar]
- 29.Cui C.B., Kakeya H., Osada H., Spirotryprostatin B. A novel mammalian cell cycle inhibitor produced by Aspergillus fumigatus. J. Antibiot. 1996;49:832–835. doi: 10.7164/antibiotics.49.832. [DOI] [PubMed] [Google Scholar]
- 30.Vintonyak V.V., Warburg K., Kruse H., Grimme S., Hübel K., Rauh D., Waldmann H. Identification of thiazolidinones spiro-fused to indolin-2-ones as potent and selective inhibitors of the Mycobacterium tuberculosis protein tyrosine phosphatase B. Angew. Chem., Int. Ed. 2010;49:5902–5905. doi: 10.1002/anie.201002138. [DOI] [PubMed] [Google Scholar]
- 31.Tokunaga T., Hume W.E., Umezome T., Okazaki K., Ueki Y., Kumagai K., Hourai S., Nagamine J., Seki H., Taiji M., Noguchi H., Nagata R. Oxindole derivatives as orally active potent growth hormone secretagogues. J. Med. Chem. 2001;44:4641–4649. doi: 10.1021/jm0103763. [DOI] [PubMed] [Google Scholar]
- 32.Pavlovska T.L., Redkin R.G., Lipson V.V., Molecular diversity of spirooxindoles D.V. Synthesis and biological activity. Atamanuk Mol Divers. 2016;20:299–344. doi: 10.1007/s11030-015-9629-8. [DOI] [PubMed] [Google Scholar]
- 33.Ganesh M., Suraj S. Expeditious entry into carbocyclic and heterocyclic spirooxindoles. Org. Biomol. Chem. 2022;20:5651–5693. doi: 10.1039/d2ob00767c. [DOI] [PubMed] [Google Scholar]
- 34.Tantawy M.A., Nafie M.S., Elmegeed G.A. Auspicious role of the steroidal heterocyclic derivatives as a platform for anti-cancer drugs. Bioorg. Chem. 2017;73:128–146. doi: 10.1016/j.bioorg.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 35.(a) Barakat A., Islam M.S., M Ghawas H., Al-Majid A.M., El-Senduny F.F., Badria F.A., Elshaier Y.A., Ghabbour H.A. Design and synthesis of new substituted spirooxindoles as potential inhibitors of the MDM2-p53 interaction. Bioorg. Chem. 2019;86:598–608. doi: 10.1016/j.bioorg.2019.01.053. [DOI] [PubMed] [Google Scholar]; (b) Bora D., Kaushal Anjali, Shankaraiah N. Anticancer potential of spirocompounds in medicinal chemistry: a pentennial expedition. Eur. J. Med. Chem. 2021;215 doi: 10.1016/j.ejmech.2021.113263. [DOI] [PubMed] [Google Scholar]; (c) Asif M., Aqil F., Alasmary F.A., almalki A.S., Khan A.R., Nasibullah M. Lewis base-catalyzed synthesis of highly functionalized spirooxindole-pyranopyrazoles and their in vitro anticancer studies. Med. Chem. Res. 2023;32:1001–1015. [Google Scholar]
- 36.(a) Cheng D., Ishihara Y., Tan B., Barbas C.F., III Organocatalytic asymmetric assembly reactions: synthesis of spirooxindoles via organocascade strategies. ACS Catal. 2014;4:743–762. [Google Scholar]; (b) Altowyan M.S., Soliman S.M., Haukka M., Al-Shaalan N.H., Alkharboush A.A., Barakat A. Synthesis, characterization, and cytotoxicity of new spirooxindoles engrafted furan structural motif as a potential anticancer agent. ACS Omega. 2022;7:35743–35754. doi: 10.1021/acsomega.2c03790. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tabti K., Abdessadak O., Sbai A., Maghat H., Bouachrine M., Lakhlifi T. Design and development of novel spiro-oxindoles as potent antiproliferative agents using quantitative structure activity based Monte Carlo method, docking molecular, molecular dynamics, free energy calculations, and pharmacokinetics/toxicity studies. J. Mol. Str. 2023;1284 [Google Scholar]
- 37.(a) Gayathiri E., Prakash P., Kumaravel P., Jayaprakash J., Ragunathan M.G., Sankar S., Pandiaraj S., Thirumalaivasan N., Thiruvengadam M., Govindasamy R. Computational approaches for modeling and structural design of biological systems: a comprehensive review. Prog. Biophys. Mol. Biol. 2023;185:17–32. doi: 10.1016/j.pbiomolbio.2023.08.002. [DOI] [PubMed] [Google Scholar]; (b) Vijayan R.S.K., Kihlberg J., Cross J.B., Poongavanam V. Enhancing preclinical drug discovery with artificial intelligence. Drug Discov. Today. 2022;27:967–984. doi: 10.1016/j.drudis.2021.11.023. [DOI] [PubMed] [Google Scholar]
- 38.(a) Pantaleão S.Q., Fernandes P.O., Gonçalves J.E., Maltarollo V.G., Honorio K.M. Recent advances in the prediction of pharmacokinetics properties in drug design studies: a review. ChemMedChem. 2022;17 doi: 10.1002/cmdc.202100542. [DOI] [PubMed] [Google Scholar]; (b) Moulishankar A., Sundarrajan T. vol. 12. 2023. (QSAR Modeling, Molecular Docking, Dynamic Simulation and ADMET Study of Novel Tetrahydronaphthalene Derivatives as Potent Antitubercular Agents). [DOI] [Google Scholar]
- 39.(a) Barakat A., Islam M.S., Ghawas H.M., Al-Majid A.M., El-Senduny F.F., A Badria F., Elshaier Y.A.M.M., Ghabbour H.A. Design and synthesis of new substituted spirooxindoles as potential inhibitors of the MDM2-p53 interaction. Bioorg. Chem. 2019;86:598–608. doi: 10.1016/j.bioorg.2019.01.053. [DOI] [PubMed] [Google Scholar]
- 40.Gollner A., Rudolph D., Arnhof H., Bauer M., Blake S.M., Boehmelt G., Cockroft X.L., Dahmann G., Ettmayer P., Gerstberger T., Karolyi-Oezguer J., Kessler D., Kofink C., Ramharter J., Rinnenthal J., Savchenko A., Schnitzer R., Weinstabl H., Weyer-Czernilofsky U., Wunberg T., McConnell D.B. Discovery of novel spiro[3H-indole-3,2'-pyrrolidin]-2(1H)-one compounds as chemically stable and orally active inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2016;59:10147–10162. doi: 10.1021/acs.jmedchem.6b00900. [DOI] [PubMed] [Google Scholar]