

Abstract
The actin cytoskeleton governs the dynamic functions of cells, ranging from motility to phagocytosis and cell division. To elucidate the molecular mechanism, in vitro reconstructions of the actin cytoskeleton and its force generation process have played essential roles, highlighting the importance of efficient purification methods for actin-binding proteins. In this study, we introduce a unified purification method for actin-binding proteins, including capping protein (CP), cofilin, ADF, profilin, fascin, and VASP, key regulators in force generation of the actin cytoskeleton. Exploiting a His-Strep-tag combined with a TEV protease cleavage site, we purified these diverse actin-binding proteins through a simple two-column purification process: initial purification through a Strep-Tactin column and subsequent tag removal through the reverse purification by a Ni-NTA column. Biochemical and microscopic assays validated the functionality of the purified proteins, demonstrating the versatility of the approach. Our methods not only delineate critical steps for the efficient preparation of actin-binding proteins but also hold the potential to advance investigations of mutants, isoforms, various source species, and engineered proteins involved in actin cytoskeletal dynamics.
-
•
Unified purification method for various actin-binding proteins.
-
•
His-Strep-tag and TEV protease cleavage for efficient purification.
-
•
Functional validation through biochemical and microscopic assays.
Method name: A Unified Purification Method for Actin-Binding Proteins using a TEV-cleavable His-Strep-tag
Keywords: His-tag, Strep-tag, Actin cytoskeleton, Capping protein, Cofilin, ADF, Profilin, Fascin, VASP
Graphical abstract
Specifications table
| Subject area: | Biochemistry, Genetics and Molecular Biology |
| More specific subject area: | Protein purification methods |
| Name of your method: | A Unified Purification Method for Actin-Binding Proteins using a TEV-cleavable His-Strep-tag |
| Name and reference of original method: | N/A |
| Resource availability: | N/A |
Background
The force of actin cytoskeleton is pivotal for various dynamic functions of cells, such as cell motility, deformation, vesicle transport, and phagocytosis [[1], [2], [3], [4], [5]]. To decipher the underlying mechanism, in vitro reconstitution studies have played fundamental roles in identifying minimal components and provide biophysical models [[6], [7], [8], [9], [10]]. In particular, pioneering work by Cortese et al. and Miyata et al. demonstrated that actin filaments serve as polymerization motors by showing that de novo actin polymerization in cell-sized liposomes can deform the lipid bilayer membrane from within [[11], [12], [13]]. Bottom-up studies further reconstituted actin treadmilling-based motility of Listeria and beads coated with actin nucleation promoting factor (NPF) in a solution of purified actin and actin-binding proteins, including Arp2/3, profilin, cofilin, ADF, capping protein (CP), and VASP [[14], [15], [16], [17], [18], [19], [20]]. These reconstitution studies which identified minimal factors have been extended to artificial cell studies which utilize the force of actin cytoskeleton for mechanical functions to dynamic morphological changes of the compartments [[21], [22], [23]].
To characterize actin cytoskeleton in a reconstituted system, the purification of actin-binding proteins is prerequisite. Initially, these proteins were isolated from native sources [[24], [25], [26], [27], [28]]. Purification methods for recombinant, tag-free proteins have broadened the accessibility of actin studies through common expression hosts [[29], [30], [31], [32]]. However, the methods often require multiple rounds of chromatography. Additionally, ammonium sulfate precipitation used in those methods varies in the reagent amount and the required precautions depending on the type of protein, making it cumbersome when dealing with multiple types of proteins. While affinity tags have significantly improved the yield and efficiency of these purification procedures, these methods still have typically been tailored for individual proteins [[33], [34], [35], [36], [37]].
Several reconstitution studies have demonstrated the purification of multiple components using a consistent purification tag such as His-tag, His-MBP-tag, and FLAG-tag [[38], [39], [40]]. A notable example is the PURE system, an in vitro transcription-translation system involving 36 proteins [41]. A recent study established the OnePot method, where all 36 E. coli clones producing His-tagged proteins are cocultured and induced in a single flask, followed by a unified purification step using a Ni-NTA column [42]. This led to 14-fold reduction in the cost of system preparation. Thus, establishing a similar purification method using a unified tag and protocol for the actin cytoskeleton system could greatly streamline the preparation processes.
In this study, we introduce a new purification method for recombinant actin-binding proteins. Leveraging the selective interaction of the Twin-Strep-tag to the Strep-Tactin column [43,44], our method yielded highly purified proteins from a single-column purification. Further, the 6 × His-tag and TEV cleavage site allowed for the purification of tag-free proteins through the second column purification with Ni-NTA. Importantly, we applied our method to various actin-binding proteins, namely CP, profilin, cofilin, ADF, fascin, and VASP, and validated the generalizability of our protocol.
Method details
Materials
-
•
Strep-Tactin XT 4Flow resin (Iba, 2-5010-010).
-
•
Strep-Tactin XT 4Flow High Capacity resin (Iba, 2-5030-025).
-
•
Ni-NTA agarose (Qiagen, 30210; Wako, 141-09764).
-
•
Rabbit skeletal actin (Cytoskeleton, AKL99).
-
•
Pyrene-labeled actin (Cytoskeleton, AP05).
-
•
Arp2/3 (Cytoskeleton, RP01P).
-
•
GST-VCA (Cytoskeleton, VCG03).
-
•
Cofilin (Cytoskeleton, CF01).
-
•
Biotin (Wako, 021-08712).
-
•
Luria-Bertani (LB) medium powder (Nacalai Tesque, 20068-75).
-
•
Ampicillin sodium salt (Nacalai Tesque, 19769-22).
-
•
Kanamycin Monosulfate (Nacalai Tesque, 19757-14).
-
•
Chloramphenicol (Nacalai Tesque, 08027-72).
-
•
Phenylmethylsulfonyl fluoride (PMSF) (Nacalai Tesque, 27327-94).
-
•
Ultrapure IPTG (M&S TechnoSystems, GEN-S-02122-25G).
-
•
Benzonase (Sigma-Aldrich, E1014).
-
•
cOmplete (EDTA-free) (Sigma-Aldrich, 11873580001).
-
•
His-tagged TEV protease (Nippon Gene, 314-09311).
-
•
Precast gels for SDS-PAGE (FUJIFILM, SuperSepTM Ace; Bio-Rad, Criterion™ TGX).
-
•
Dialysis tube (Visking, dialysis cellulose tubes, 5 nm pore size, approximately 12–14 kDa).
-
•
Coverslips (Matsunami C022221 and C022401, 22 × 22 mm² and 22 × 40 mm²).
Equipment
-
•
Shaking incubator (Thermo Scientific, MaxQ 6000).
-
•
Centrifuge (Kubota, Hybrid High Speed Cooling Centrifuge 6200).
-
•
OD measurement (WPA, CO8000 Cell Density Meter).
-
•
Ultracentrifuge (Hitachi, himac CS150GX II).
-
•
Sonicator (Qsonica, Misonix S4000 Ultrasonic Liquid Processor).
-
•
Rotary shaker (EYELA, MRM-1000).
-
•
UV-Vis spectrophotometer (Shimadzu Corporation, Biospec-Nano).
-
•
Chromatography system (Bio-Rad, NGC Quest 10).
-
•
Ultrafiltration (Sartorius, VivaspinTurbo15 10K MWCO).
-
•
Fluorometer (Jasco, FP-8550).
-
•
Temperature controller (Jasco, CTU-100).
-
•
Plasma cleaner (Harrick Plasma, PDC-32G).
-
•
Microscope (Olympus, IX71).
-
•
Objective lens (Evident, UAPON150XO, N2709600).
-
•
Red laser (Vortran, STRADUS 639-160).
-
•
EM-CCD camera (Andor, iXon3).
Buffers
*Note that β-mercaptoethanol, PMSF, cOmplete, and Benzonase should be added immediately before use.
[Protein Expression and Purification]
-
•
CP lysis buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM NaCl, 1 mM EDTA, 14 mM β-mercaptoethanol, 1 mM PMSF, 1 × complete, 1/1000 Benzonase)
-
•
CP wash buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM NaCl, 1 mM EDTA, 14 mM β-mercaptoethanol)
-
•
CP elution buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM NaCl, 1 mM EDTA, 14 mM β-mercaptoethanol, 50 mM biotin)
-
•
CP dialysis buffer (25 mM Tris-HCl (pH 7.5 at RT), 150 mM NaCl, 7 mM β-mercaptoethanol)
-
•
TEV protease lysis buffer (50 mM Tris-HCl (pH 7.5 at RT), 250 mM NaCl, 5 mM MgCl2, 7 mM β-mercaptoethanol)
-
•
TEV protease dialysis buffer (25 mM Tris-HCl (pH 7.5 at RT), 200 mM NaCl, 7 mM β-mercaptoethanol)
-
•
Cofilin lysis buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM DTT, 10 % glycerol, 1 mM EDTA, 1 mM PMSF, 100 µg/mL DNase I)
-
•
Cofilin wash buffer (25 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM DTT, 10 % glycerol)
-
•
Cofilin elution buffer (25 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM DTT, 10 % glycerol, 50 mM biotin)
-
•
Cofilin dialysis buffer (25 mM Tris-HCl (pH 7.5 at RT), 50 mM KCl, 1 mM DTT, 10 % glycerol)
-
•
ADF lysis buffer (50 mM Tris-HCl (pH 7.5, RT), 1 mM MgCl2, 150 mM KCl, 1 mM DTT, 10 % glycerol, 1 mM PMSF, 1/1000 Benzonase, ×1 Protease inhibitor cocktail V)
-
•
ADF wash buffer (50 mM Tris-HCl (pH 7.5, RT), 1 mM MgCl2, 150 mM KCl, 1 mM DTT, 10 % glycerol)
-
•
ADF elution buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM MgCl2, 1 mM DTT, 1 mM PMSF, 10 % glycerol, 50 mM biotin)
-
•
ADF dialysis buffer (25 mM Tris-HCl (pH 7.5 at RT), 50 mM KCl, 1 mM DTT, 10 % glycerol)
-
•
Profilin lysis buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM DTT, 10 % glycerol, 1 mM EDTA, 1 mM PMSF, 100 µg/mL DNase I)
-
•
Profilin wash buffer (25 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM DTT, 10 % glycerol)
-
•
Profilin elution buffer (25 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM DTT, 10 % glycerol, 50 mM biotin)
-
•
Profilin dialysis buffer (25 mM Tris-HCl (pH 7.5 at RT), 50 mM KCl, 1 mM DTT, 10 % glycerol)
-
•
Fascin lysis buffer (50 mM Tris-HCl (pH 7.5 RT), 1 mM MgCl2, 150 mM KCl, 1 mM DTT, 10 % glycerol, 1 mM PMSF, 1/1000 Benzonase, × 1 Protease inhibitor cocktail V)
-
•
Fascin wash buffer (50 mM Tris-HCl (pH 7.5 RT), 1 mM MgCl2, 150 mM KCl, 1 mM DTT, 10 % glycerol)
-
•
Fascin elution buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM MgCl2, 1 mM DTT, 1 mM PMSF, 10 % glycerol, 50 mM biotin)
-
•
Fascin dialysis buffer (25 mM Tris-HCl (pH 7.5 at RT), 50 mM KCl, 1 mM DTT, 10 % glycerol)
-
•
VASP lysis buffer (50 mM Tris-HCl (pH 7.5 RT), 1 mM MgCl2, 150 mM KCl, 1 mM DTT, 10 % glycerol, 1 mM PMSF, 1/1000 Benzonase, × 1 Protease inhibitor cocktail V)
-
•
VASP wash buffer (50 mM Tris-HCl (pH 7.5 RT), 1 mM MgCl2, 150 mM KCl, 1 mM DTT, 10 % glycerol)
-
•
VASP elution buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM MgCl2, 1 mM DTT, 1 mM PMSF, 10 % glycerol, 50 mM biotin)
-
•
VASP dialysis buffer (25 mM Tris-HCl (pH 7.5 at RT), 50 mM KCl, 1 mM DTT, 10 % glycerol)
[Pyrene-Actin Polymerization Assay]
-
•
G-buffer (5 mM Tris-HCl (pH 7.5 at RT), 0.2 mM CaCl2)
-
•
10E/1M (10 mM EGTA, 1 mM MgCl2)
-
•
G-Mg (2 mM Tris-HCl (pH 7.5 at RT), 0.2 mM ATP, 0.1 mM MgCl2, 0.5 mM DTT, 1 mM NaN3)
-
•
10 × KMEI (100 mM imidazole (pH 7.0 at RT), 500 mM KCl, 10 mM MgCl2, 10 mM EGTA)
-
•
1 × KMEI (10 mM imidazole (pH 7.0 at RT), 50 mM KCl, 1 mM MgCl2, 1 mM EGTA)
[Sedimentation Assay]
*Note that the pH of Tris-HCl in general actin buffer and cofilin/ADF buffer should be adjusted at 8.0 because the cleavage activity of cofilin/ADF are different by pH.
-
•
General actin buffer (GAB: 5 mM Tris-HCl (pH 8.0 at RT), 0.2 mM CaCl2)
-
•
Cofilin/ADF buffer (150 mM Tris-HCl (pH 8.0 at RT), 50 mM KCl, 2 mM MgCl2, 1 mM ATP)
-
•
Profilin buffer (25 mM Tris-HCl (pH 7.5 at RT), 50 mM KCl, 1 mM DTT, 10 % glycerol)
-
•
10 × APB (100 mM Tris-HCl (pH 7.5 at RT), 500 mM KCl, 20 mM MgCl2, 10 mM ATP)
[TIRF Imaging]
-
•
Polymerization solution (50 mM KCl, 1 mM MgCl2, 1 mM EGTA, 10 mM imidazole (pH 7.0 at RT), 1 mM ATP, 1 mg/mL glucose oxidase, 0.2 mg/mL catalase, 2 mg/mL BSA, 3 mg/mL glucose)
Plasmids
-
•
6 × His-TEV protease plasmid, pET28-2 × Strep-tag plasmid, 7×His-VASP(Human) plasmid [45], Cofilin-EYFP plasmid [46], GFP-Fascin plasmid were kind gifts from Dr. Yubin Zhou, Dr. Masato Kanemaki, Dr. Baoyu Chen, Dr. Kensaku Mizuno and Dr. Shigeko Yamashiro, respectively. pET3d-Capβ2 (mouse)-Capα1 (mouse) and pMW profilin (human) were kind gifts from Dr. Cécile Sykes and Dr. Julie Plastino. The human ADF sequence was purchased from GeneScript as synthetic DNA based on the reference sequence (GenBank S65738) with a silent mutation A463T to remove the BamHI site. The ORF sequences of each actin-binding proteins are summarized in supplementary table 1.
-
•
pET28-6 × His-2 × Strep: To construct the backbone vector pET28-6 × His-2 × Strep, oligo DNAs encoding 6 × His sequence (fwd: 5’- AGGAGATATACCATGGGCCATCACCATCACCATCACA-TGGGCTGGTCTCA-3’ and rev: 5’-TGAGACCAGCCCATGTGATGGTGATGGTGATGGCCC-ATGGTATATCTCCT-3’) were annealed and inserted into NcoI site of pET28-2 × Strep-tag by Gibson assembly.
-
•
pET28-6 × His-2 × Strep-TEVsite-Capβ2 (mouse)-Capα1 (mouse), pET28-6 × His-2 × Strep-TEVsite-Cofilin (human), pET28-6 × His-2 × Strep-TEVsite-Profilin (human), pET28-6 × His-2 × Strep-TEVsite-fascin (human), pET28-6 × His-2 × Strep-TEVsite-VASP (human): Each ORF region was PCR amplified with the primers and templates listed in supplementary table 2. PCR fragments were column-purified and inserted between NheI and EcoRI of pET28-6 × His-2 × Strep by Gibson assembly. Accordingly, the amino acid sequence, MGHHHHHHMGWSHPQFEKGGGSGGGSGGGSWSHPQFEKGAGAGAGAGAAS-ENLYFQ/SD (6 × His-tag, Strep-tag II, and TEV cleavage site are underlined), was appended to the N-terminus of Capβ2 and other proteins of interest.
-
•
pET28-6 × His-2 × Strep-TEVsite-ADF (human): Synthetic DNA fragment of human ADF was inserted between NheI and EcoRI sites of pET28- × His-2 × Strep by Gibbson assembly.
-
•
pQE80L-6 × His-MBP-TEVprotease (S219V): Ser219Val mutation was introduced by PCR with the template plasmid gifted from Dr. Yubin Zhou and the primer set of fwd: 5’-TGGTGAAACCTG-AAGAACCTTTT-3’ and rev: 5’-TTCACCATGAAAACTTTATGGCC-3’. TEV protease (S219V) was then PCR amplified with the primer set of fwd: 5’-AATTGAGCTCGATGAGCGGCCTGGTG-C-3’ and rev: 5’-AATTGGATCCTTATTGCGAGTACACCAATTCATTCATG-3’ and inserted between SalI and BamHI sites of pQE-80L MBP-SspB Nano plasmid (Addgene #60409) by using restriction digestion and T4 ligase ligation.
Liter scale expression and purification of CP
-
1.
BL21(DE3) RIL cells transformed with pET28-6 × His-2 × Strep-TEVsite-Capβ2 (mouse)-Capα1 (mouse) were grown overnight at 37 °C in LB medium containing 50 µg/mL of Kanamycin and 25 µg/mL of Chloramphenicol (LB/Kan/Cam).
-
2.
Pre-culture was inoculated to 6 × 1 L LB/Kan/Cam.
-
3.
When OD600 reached around 0.5, IPTG was added to a final concentration of 300 µM, and protein expression was allowed to proceed for 3 h at 37 °C.
-
4.
The cells were then harvested by centrifugation, rinsed once CP wash buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM NaCl, 1 mM EDTA, 14 mM β-mercaptoethanol), and stored at -80 °C.
-
5.
The cells were gently resuspended in CP lysis buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM NaCl, 1 mM EDTA, 14 mM β-mercaptoethanol, 1 mM PMSF, 1 × complete, 1/1000 Benzonase) at a ratio of 10 mL per 1 g pellet.
*Note: Vortexing should be avoided at this step because it may induce CP aggregation.
-
6.
The cells were sonicated on ice (90 cycles of 2-sec sonication with 10-sec intervals) using a Misonix Ultrasonic Liquid Processor Sonicator S4000 at an amplitude of 30.
*Note: Sonication interval should be as long as possible to avoid the shear stress and temperature increase during sonication.
-
7.
The cell lysate was centrifuged at 4 °C, 13,420 × g for 3 min. and the supernatant was kept on ice.
-
8.
The pellets were again resuspended in CP lysis buffer and sonicated.
-
9.
After repeating these centrifugation, resuspension, and sonication steps, all the lysate was centrifuged at 4 °C, 13,420 × g for 1 h (the supernatant and the precipitate from this process were examined by SDS-PAGE).
-
10.
The supernatant was then applied to 2 mL of Strep-Tactin XT 4Flow high-capacity column, equilibrated with CP lysis buffer.
-
11.
The column was washed by 2 × 10 mL of CP wash buffer and CP was eluted with 12 × 1 mL of CP elution buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM NaCl, 1 mM EDTA, 14 mM β-mercaptoethanol, 50 mM biotin).
-
12.
The fractions containing CP were collected, combined with His-tagged TEV protease (Nippon Gene) at a 30:1 (w/w) ratio, and dialyzed against CP dialysis buffer (25 mM Tris-HCl (pH 7.5 at RT), 150 mM NaCl, 7 mM β-mercaptoethanol).
-
13.
To purify tag-free CP, the sample was applied to 4 mL of Ni-NTA agarose column equilibrated with CP dialysis buffer and washed and eluted by 3 × 4 mL of CP dialysis buffer, 3 × 4 mL of CP dialysis buffer supplemented with 10 mM imidazole, and 3 × 4 mL of CP dialysis buffer supplemented with 500 mM imidazole.
-
14.
CP in flow through and 0 mM imidazole fractions were concentrated using VivaspinTurbo15 (10K MWCO, SARTORIUS) and applied to Superdex 200 increase 10/300 column on NGC chromatography system (Biorad), equilibrated with CP dialysis buffer.
-
15.
CP was then concentrated using VivaspinTurbo15, aliquoted, snap-frozen with liquid nitrogen, and stored at -80 °C.
The expression and purification protocols for other proteins followed a procedure similar to that described for CP. Specific buffer conditions, modifications, and additional details are described below.
Expression and purification of 6 × His-MBP-TEV Protease (S219V)
BL21(DE3) RIL cells transformed with pQE80L-6 × His-MBP-TEVprotease (S219V) were cultured overnight in LB broth supplemented with 100 µg/mL Ampicillin and 25 µg/mL Chloramphenicol (LB/Amp/Cam). Pre-culture was inoculated to LB/Amp/Cam and cultured at 37 °C until OD600 reached around 0.4. Protein expression was induced by adding 300 µM IPTG and allowed to proceed for 17 hours at 18 °C. The cells were re-suspended with TEV protease lysis buffer (50 mM Tris-HCl (pH 7.5 at RT), 250 mM NaCl, 5 mM MgCl2, 7 mM β-mercaptoethanol) and lysed by microfluidizer. The protein was purified by Ni-NTA, eluted with 60–100 mM imidazole, and then dialyzed against TEV protease dialysis buffer (25 mM Tris-HCl (pH 7.5 at RT), 200 mM NaCl, 7 mM β-mercaptoethanol). The elution was concentrated by Amicon Ultra (10 kDa cutoff) and further dialyzed against TEV protease dialysis buffer supplemented with 50 % glycerol (25 mM Tris-HCl (pH 7.5 at RT), 200 mM NaCl, 7 mM β-mercaptoethanol, 50 % glycerol). The protein was stored at -20 °C.
Expression and purification of cofilin
BL21-CodonPlus (DE3)-RIL was transformed with pET28-6 × His-2 × Strep-TEVsite-Cofilin (human). The bacteria were cultured overnight in LB/Kan/Cam. Pre-culture was inoculated to 4 × 1 L LB/Kan/Cam and cultured at 37 °C until OD600 reached around 0.4–0.6. Protein expression was induced by adding 300 µM IPTG and allowed to proceed for 20 h at 18 °C. The cells were re-suspended with 80 mL of cofilin lysis buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM DTT, 10 % glycerol, 1 mM EDTA, 1 mM PMSF, 100 µg/mL DNase I) and lysed by microfluidizer (Microfluidics, Model M-110Y, 80 psi, 3 times). The protein was bound to 4 mL of Strep-Tactin XT 4 Flow column equilibrated with cofilin wash buffer (25 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM DTT, 10 % glycerol), washed with 20 mL of cofilin wash buffer, and eluted with 2 mL of cofilin elution buffer (25 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM DTT, 10 % glycerol, 50 mM biotin) once and 8 mL of cofilin elution buffer 4 times. To 12 mg tagged-cofilin, 22 µL of 6 × His-MBP-TEV protease (S219V) (13.55 mg/mL) was added (40:1 w/w), and the sample was dialyzed overnight against cofilin dialysis buffer (25 mM Tris-HCl (pH 7.5 at RT), 50 mM KCl, 1 mM DTT, 10 % glycerol). The proteins were then applied to 4 mL of Ni-NTA agarose column equilibrated with cofilin dialysis buffer and washed and eluted by 2 × 4 mL cofilin dialysis buffer, 2 × 4 mL cofilin dialysis buffer supplemented with 10 mM imidazole, and 2 × 4 mL cofilin dialysis buffer supplemented with 500 mM imidazole. Tag-free cofilin eluted in flow through and 0 mM imidazole fractions was then concentrated by Amicon (10 kDa cut off), aliquoted, snap frozen by liquid nitrogen, and stored at -80 °C.
Expression and purification of ADF
BL21(DE3)-RIL was transformed with pET100-6 × His-2 × Strep-TEVsite-ADF (human). The bacteria were cultured overnight in LB/Kan/Cam. Pre-culture was inoculated to 2 × 1 L LB/Kan/Cam and cultured at 37 °C until OD600 reached around 0.4–0.6. Protein expression was induced by adding 300 µM IPTG and allowed to proceed for 22 hours at 18 °C. For each 1 g of cell pellet, the cells were resuspended in 10 mL ADF lysis buffer (50 mM Tris-HCl (pH 7.5, RT), 1 mM MgCl2, 150 mM KCl, 1 mM DTT, 10 % glycerol, 1 mM PMSF, 1/1000 Benzonase, × 1 Protease inhibitor cocktail V) and sonicated on ice (150 cycles of 2 sec sonication with 10 sec intervals) using a Misonix Ultrasonic Liquid Processor Sonicator S4000. The protein was bound to 2 mL of Strep-Tactin XT 4 Flow column equilibrated with ADF wash buffer (50 mM Tris-HCl (pH 7.5, RT), 1 mM MgCl2, 150 mM KCl, 1 mM DTT, 10 % glycerol), washed with 10 mL × 2 of ADF wash buffer, and eluted with 1 mL of ADF elution buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM MgCl2, 1 mM DTT, 1 mM PMSF, 10 % glycerol, 50 mM biotin) once and 3.75 mL of ADF elution buffer 4 times. The sample combined with His-tagged TEV protease (Nippon Gene) at 50:1 (w/w) ratio, dialyzed for 3 h × 2 against ADF dialysis buffer (25 mM Tris-HCl (pH 7.5 at RT), 50 mM KCl, 1 mM DTT, 10 % glycerol). The proteins were then applied to 4 mL of Ni-NTA agarose column equilibrated with ADF dialysis buffer and washed and eluted by 2 × 4 mL ADF dialysis buffer, 3 × 4 mL ADF dialysis buffer supplemented with 10 mM imidazole, 3 × 4 mL ADF dialysis buffer supplemented with 20 mM imidazole, and 1 × 6 mL ADF dialysis buffer supplemented with 500 mM imidazole. Tag-free ADF eluted in flow through and 0 mM imidazole fractions was then concentrated using VivaspinTurbo15 and applied to Superdex 200 increase 10/300 column on NGC chromatography system, equilibrated with ADF dialysis buffer. ADF was then concentrated using VivaspinTurbo15 again, aliquoted, snap-frozen with liquid nitrogen, and stored at -80 °C.
Expression and purification of profilin
BL21-CodonPlus (DE3)-RIL was transformed with pET28-6 × His-2 × Strep-TEVsite-Profilin (human). The bacteria were cultured overnight in LB broth supplemented with 50 µg/mL Kanamycin and 25 µg/mL Chloramphenicol (LB/Kan/Cam). Pre-culture was inoculated to 4 × 1 L LB media supplemented with LB/Kan/Cam and cultured at 37 °C until OD600 reached around 0.6–0.7. Protein expression was induced by adding 300 μM IPTG and allowed to proceed for 20 h at 18 °C. The cells were re-suspended with 80 mL of profilin lysis buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM DTT, 10 % glycerol, 1 mM EDTA, 1 mM PMSF, 100 µg/mL Dnase I) and lysed by microfluidizer. The protein was bound to 4 mL of Strep-Tactin XT 4 Flow column equilibrated with profilin wash buffer (25 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM DTT, 10 % glycerol), washed with 20 mL of profilin wash buffer, and eluted with 2 mL of profilin elution buffer (25 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM DTT, 10 % glycerol, 50 mM biotin) once and 8 mL of profilin elution buffer 4 times. To 12 mg tagged-cofilin, 22 µL of 6 × His-MBP-TEV protease (S219V) (13.55 mg/mL) was added (40:1 w/w), and the sample was dialyzed overnight against profilin dialysis buffer (25 mM Tris-HCl (pH 7.5 at RT), 50 mM KCl, 1 mM DTT, 10 % glycerol). The proteins were then applied to 4 mL of Ni-NTA agarose column equilibrated with profilin dialysis buffer and washed and eluted by 2 × 4 mL profilin dialysis buffer, 2 × 4 mL profilin dialysis buffer supplemented with 10 mM imidazole, and 2 × 4 mL profilin dialysis buffer supplemented with 500 mM imidazole. Tag-free profilin eluted in 0 mM and 10 mM imidazole fraction was then by dialyzed against profilin dialysis buffer, concentrated by Amicon (10 kDa cut off), aliquoted, snap frozen by liquid nitrogen, and stored at -80 °C.
Expression and purification of fascin
BL21-CodonPlus (DE3)-RP was transformed with pET100-6 × His-2 × Strep-TEVsite-fascin (human). The bacteria were cultured overnight in LB broth supplemented with 50 µg/mL Kanamycin and 25 µg/mL Chloramphenicol (LB/Kan/Cam). Pre-culture was inoculated to 2 × 1 L LB media supplemented with LB/Kan/Cam and cultured at 37 °C until OD600 reached around 0.4–0.6. Protein expression was induced by adding 300 μM IPTG and allowed to proceed for 22 h at 18 °C. For each 1 g of cell pellet, the cells were resuspended in 10 mL fascin lysis buffer (50 mM Tris-HCl (pH 7.5, RT), 1 mM MgCl2, 150 mM KCl, 1 mM DTT, 10 % glycerol,1 mM PMSF, 1/1000 Benzonase, × 1 Protease inhibitor cocktail V) and sonicated on ice (150 cycles of 2 sec sonication with 10 sec intervals) using a Misonix Ultrasonic Liquid Processor Sonicator S4000. The protein was bound to 2 mL of Strep-Tactin XT 4 Flow column equilibrated with fascin wash buffer (50 mM Tris-HCl (pH 7.5, RT), 1 mM MgCl2, 150 mM KCl, 1 mM DTT, 10 % glycerol), washed with 10 mL × 2 of fascin wash buffer, and eluted with 1 mL of fascin elution buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM MgCl2, 1 mM DTT, 1 mM PMSF, 10 % glycerol, 50 mM biotin) once and 3.75 mL of fascin elution buffer 4 times. The sample combined with His-tagged TEV protease (Nippon Gene) at 60:1 (w/w) ratio, dialyzed for 3 hours × 2 against fascin dialysis buffer (25 mM Tris-HCl (pH 7.5 at RT), 50 mM KCl, 1 mM DTT, 10 % glycerol). The proteins were then applied to 4 mL of Ni-NTA agarose column equilibrated with fascin dialysis buffer and washed and eluted by 2 × 4 mL fascin dialysis buffer, 3 × 4 mL fascin dialysis buffer supplemented with 10 mM imidazole, 3 × 4 mL fascin dialysis buffer supplemented with 20 mM imidazole, and 1 × 6 mL fascin dialysis buffer supplemented with 500 mM imidazole. Tag-free fascin eluted in flow through, 0 mM, 10 mM and 20 mM (except #3) imidazole fractions was then concentrated using VivaspinTurbo15, dialyzed for 3 h × 2 against fascin dialysis buffer again. After concentration using VivaspinTurbo15, fascin was applied to Superdex 200 increase 10/300 column on NGC chromatography system, equilibrated with fascin dialysis buffer. Finally, fascin was then concentrated using VivaspinTurbo15 again, aliquoted, snap-frozen with liquid nitrogen, and stored at -80 °C.
Expression and purification of VASP
BL21-CodonPlus (DE3)-RP was transformed with pET28-6 × His-2 × Strep-TEVsite-VASP (human). The bacteria were cultured overnight in LB/Kan/Cam. Pre-culture was inoculated to 2 × 1 L LB media supplemented with LB/Kan/Cam and cultured at 37 °C until OD600 reached around 0.4-0.6. Protein expression was induced by adding 300 μM IPTG and allowed to proceed for 22 h at 18 °C. For each 1 g of cell pellet, the cells were resuspended in 10 mL VASP lysis buffer (50 mM Tris-HCl (pH 7.5, RT), 1 mM MgCl2, 150 mM KCl, 1 mM DTT, 10 % glycerol, 1 mM PMSF, 1/1000 Benzonase, × 1 Protease inhibitor cocktail V) and sonicated on ice (150 cycles of 2 sec sonication with 10 sec intervals) using a Misonix Ultrasonic Liquid Processor Sonicator S4000. The protein was bound to 2 mL of Strep-Tactin XT 4 Flow column equilibrated with VASP wash buffer (50 mM Tris-HCl (pH 7.5, RT), 1 mM MgCl2, 150 mM KCl, 1 mM DTT, 10 % glycerol), washed with 10 mL × 2 of VASP wash buffer, and eluted with 1 mL of VASP elution buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM KCl, 1 mM MgCl2, 1 mM DTT, 1 mM PMSF, 10 % glycerol, 50 mM biotin) once and 3.75 mL of VASP elution buffer 4 times. The sample combined with His-tagged TEV protease (Nippon Gene) at 50:1 (w/w) ratio, dialyzed for 3 h × 2 against VASP dialysis buffer (25 mM Tris-HCl (pH 7.5 at RT), 50 mM KCl, 1 mM DTT, 10 % glycerol). The proteins were then applied to 4 mL of Ni-NTA agarose column equilibrated with VASP dialysis buffer and washed and eluted by 2 × 4 mL VASP dialysis buffer, 3 × 4 mL VASP dialysis buffer supplemented with 10 mM imidazole, 3 × 4 mL VASP dialysis buffer supplemented with 20 mM imidazole, and 1 × 6 mL VASP dialysis buffer supplemented with 500 mM imidazole. Tag-free VASP eluted in flow through and 0 mM, 10 mM, 20 mM imidazole fractions was then concentrated using VivaspinTurbo15, dialyzed for 3 h × 2 against VASP dialysis buffer again. After concentration using VivaspinTurbo15, VASP was applied to Superdex 200 increase 10/300 column on NGC chromatography system, equilibrated with VASP dialysis buffer. Finally, VASP was then concentrated using VivaspinTurbo15 again, aliquoted, snap-frozen with liquid nitrogen, and stored at -80 °C.
Pyrene-actin polymerization assay
The reaction mixture for the pyrene actin polymerization assay was prepared according to a previously reported method with slight modifications [[47], [48], [49]]. Note that the changes in intensity of pyrene-labeled actin could be induced not only by its polymerization state but also by interacting proteins or binding nucleotides [50].
-
1.
Actin stock was prepared by mixing non-labeled and pyrene-labeled actin at a 19:1 ratio (5 % pyrene labeled) in G-buffer (5 mM Tris-HCl (pH 7.5 at RT), 0.2 mM CaCl2) and incubated at 4 °C overnight.
-
2.
Preparation of mix 1 (25 µL): Mix actin stock and 1/10 volume (of the actin stock) of 10E/1M (10 mM EGTA, 1 mM MgCl2) to give a 1 µM final concentration of actin in 50 µL reaction. Fill the mixture with G-Mg (2 mM Tris-HCl (pH 7.5 at RT), 0.2 mM ATP, 0.1 mM MgCl2, 0.5 mM DTT, 1 mM NaN3) up to 25 µL.
-
3.
Preparation of mix 2 (25 µL): Mix 2.78 µL of 10 × KMEI (100 mM imidazole (pH 7.0 at RT), 500 mM KCl, 10 mM MgCl2, 10 mM EGTA) (compensating salt concentration for mix1), with Arp2/3, GST-VCA, and CP in 1 × KMEI (10 mM imidazole (pH 7.0 at RT), 50 mM KCl, 1 mM MgCl2, 1 mM EGTA) to give 10 nM Arp2/3, 100 nM GST-VCA, and desired concentration of CP in 50 µL reaction. Fill the mixture with 1 × KMEI up to 25 µL.
-
4.
Quickly mix mix 1 to mix 2 by using a p200 pipette (set to 200 µL), pipetted well, and incubated for exactly 2 min at RT.
-
5.
50 µL of the reaction was transferred to the cuvette.
-
6.
Pyrene-actin polymerization was monitored using a fluorometer FP-8550 (Jasco) with temperature control maintained at 25 °C (CTU-100, Jasco).
-
7.
Pyrene fluorescence and its kinetics were measured by 365 nm excitation and 407 nm emission.
Sedimentation assay
When investigating the polymerization/depolymerization dynamics of actin, it is crucial to ensure the presence of ATP in the solutions. In this method, ATP comes from the lyophilized actin powder purchased from Cytoskeleton Inc.
[Cofilin and ADF]
-
1.
Actin was diluted to 2 µM in general actin buffer (GAB: 5 mM Tris-HCl (pH 8.0 at RT), 0.2 mM CaCl2).
-
2.
Cofilin and ADF were diluted to 160 µM in cofilin/ADF buffer (150 mM Tris-HCl (pH 8.0 at RT), 50 mM KCl, 2 mM MgCl2, 1 mM ATP).
-
3.
Both solutions were centrifuged at 4 °C, 20,000 × g for 10 min to remove any denatured proteins.
-
4.
To polymerize actin, 4.5 µL of 10 × APB (100 mM Tris-HCl (pH 7.5 at RT), 500 mM KCl, 20 mM MgCl2, 10 mM ATP) was added to 40.5 µL of actin in GAB.
-
5.
This mixture was incubated for 1 h at RT.
-
6.
Subsequently, 5 µL of cofilin and ADF in cofilin/ADF buffer was added, and the sample underwent further incubation at RT for 1 h, followed by ultracentrifugation at RT, 100,000 × g for 1 h.
-
7.
The resulting supernatants and pellets were subjected to SDS-PAGE followed by SYPRO Ruby staining.
[Profilin]
-
1.
Actin was diluted to 5 µM in general actin buffer (GAB: 5 mM Tris-HCl (pH 8.0 at RT), 0.2 mM CaCl2).
-
2.
Profilin was diluted to 155 µM in profilin buffer (25 mM Tris-HCl (pH 7.5 at RT), 50 mM KCl, 1 mM DTT, 10 % glycerol).
-
3.
Both solutions were centrifuged at 4 °C, 20,000 × g for 10 min to remove any denatured proteins.
-
4.
A mixture of 40.5 µL of actin and 4.5 µL of profilin was prepared, kept on ice for 1 h, and subsequently incubated at 30 °C for 30 min.
-
5.
Actin polymerization was initiated by adding 5 µL of 10 × APB (150 mM Tris-HCl (pH 8.0 at RT), 500 mM KCl, 20 mM MgCl2, 10 mM ATP) and incubated for 1 h at RT.
-
6.
The samples were ultracentrifuged at RT, 100,000 × g for 1 h.
-
7.
The resulting supernatants and pellets were subjected to SDS-PAGE followed by SYPRO Ruby staining.
TIRF imaging
TIRF imaging was performed following previously reported protocols with slight modifications [51,52].
-
1.
Two types of coverslips (22 × 22 mm² and 22 × 40 mm², Matsunami C022221 and C022401) were sonicated in Milli-Q water and methanol, treated with air plasma (Harrick Plasma, PDC-32G) for 2 min at 18 W, and incubated with Silane-PEG-5K (1 mg/mL in DMSO, Biopharma PEG Scientific MF001020-5K) overnight.
-
2.
Actin polymerization was initiated by mixing 7.5 µM actin (10 % Alexa Fluor-647 labeled) with or without fascin, VASP in polymerization solution (50 mM KCl, 1 mM MgCl2, 1 mM EGTA, 10 mM imidazole (pH 7.0 at RT), 1 mM ATP, 1 mg/mL glucose oxidase, 0.2 mg/mL catalase, 2 mg/mL BSA, 3 mg/mL glucose).
-
3.
The samples were incubated at RT between the two coverslips for 1 h.
-
4.
Images were taken by an IX71 inverted microscope (Evident) equipped with 20 mW Red laser (STRADUS 639-160, VORTRAN), UAPON150XOTIRF lens (Evident, N2709600), and iXon3 EM-CCD camera (ANDOR) driven by MetaVue (version 7.8.4.0).
Other methods
-
1.
SDS-PAGE was carried out using SuperSepTM Ace 10-20 % (FUJIFILM, 198-15041) or Criterion™ TGX precast gel (BIORAD, 5671095).
-
2.
The protein bands were visualized by staining with 0.1 % Coomassie Brilliant Blue R-250 (Nacalai Tesque, 09408-52) in 10 % acetic acid and 50 % methanol or SYPRO Ruby (Thermo Fisher Scientific, S12000).
-
3.
Protein concentrations were determined by 280 nm absorbance using BioSpec-nano (Shimadzu Corporation).
Method validation
Plasmid design and purification scheme
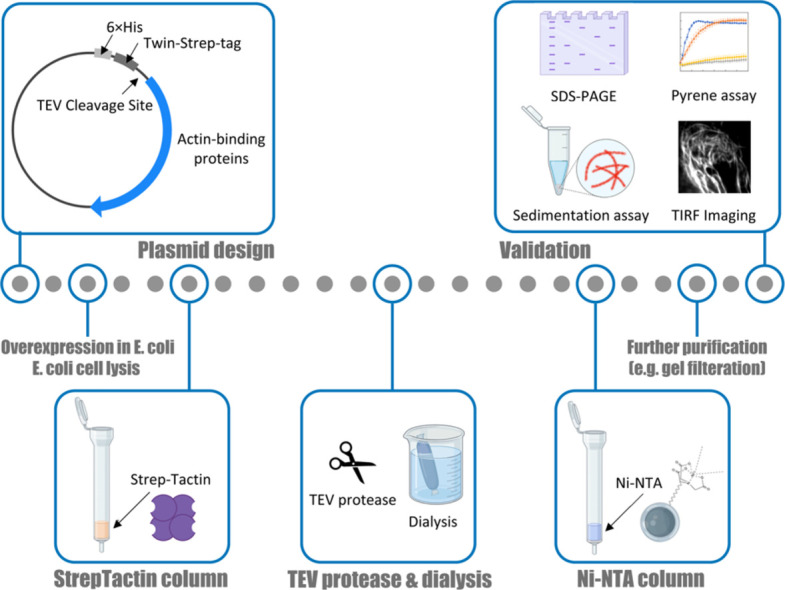
For the purification of actin-binding proteins, we utilized the Twin-Strep-tag [[53], [54], [55],43] for its highly selective binding to Strep-Tactin [55,43,44] and straightforward purification procedure. To obtain tag-free proteins, we also designed a TEV protease cleavage site and 6 × His-tag at the C- and N-terminus of the Twin-Strep-tag, respectively (Fig. 1A). This design allows for the removal of the purification tag (6 × His-Twin-Strep) and His-tagged TEV protease through Ni-NTA column. CP was purified as a hetero-dimeric complex with the purification tag fused to the N-terminus of the β2 subunit, whereas cofilin, profilin, and VASP were purified with the tag fused to the N-terminus of their open reading frames.
Fig 1.

Purification of actin-binding proteins with the cleavable His-Strep-tag. (A) Schematic representation of plasmids. 6 × His-tag, Twin-Strep-tag, and TEV cleavage sites were designed at the N-terminus of Capβ2, cofilin, profilin, ADF, fascin and VASP. Capα1 was purified as a hetero-dimer with Capβ2. (B) Schematic overview of the purification process. (C) SDS-PAGE result of the purified proteins.
As summarized in Fig. 1B, our purification protocol starts with Strep-Tactin column purification. Following the elution with biotin, the protein was typically processed with TEV protease to cleave the purification tag during dialysis, which also removes biotin from the sample solution. After Ni-NTA column purification to remove His-Strep-tag and His-tagged TEV protease, the proteins of interest can be further purified by other purification methods, such as size exclusion chromatography. Based on this method, we successfully purified CP, cofilin, profilin, ADF, fascin, and VASP (Fig. 1C). In the following sections, we describe the optimization of expression and lysis conditions for CP, liter scale expression and purification of CP, and application of our method to other actin-binding proteins.
Optimization for expression and cell lysis conditions on batch purification of CP
Among actin-binding proteins, capping protein (CP) is a key factor in understanding the structural organization and force generation mechanism of the actin cytoskeleton [[56], [57], [58], [59]]. In many studies, recombinant mouse CP has been purified based on the method developed by Soeno, et al. or its derivative protocols. However, those methods often involve repetitive chromatography and ammonium sulfate precipitation process. Thus, we first aimed to develop and test our purification method for CP.
We first tested our construct and Strep-tag purification for CP with a 10 mL culture scale. We investigated several conditions in the cell growth and cell lysis to optimize CP yield (Table. 1). Referencing earlier studies [29,34,60], we compared 37 °C 3 h and 18 °C 20 h for growth condition (Condition I). In assessing cell lysis, we examined how tolerant CP is to physical perturbations such as vortex, temperature rise, and sonication intervals (Condition II and III). We determined the solubility of CP by centrifugation of lysates and evaluated the purification yield using Strep-Tactin agarose beads.
Table. 1.
Conditions tested in batch purification. Vortex: Cell pellet was vortexed in resuspension buffer instead of gentle brushing. 37 °C incubation: Cell resuspension was incubated at 37 °C for 15 min.
| Condition | #1 | #2 | #3 | #4 | #5 | #6 | |
|---|---|---|---|---|---|---|---|
| Condition I | Growth | 18 °C, 20 hours | 18 °C, 20 hours | 18 °C, 20 hours | 18 °C, 20 hours | 37 °C, 3 hours | 37 °C, 3 hours |
| Condition II | Perturbation during cell lysis | - | vortex | 37 °C incubation | - | - | - |
| Condition III | Sonication | 2 sec ON/4 sec OFF | 2 sec ON/4 sec OFF | 2 sec ON/4 sec OFF | 2 sec ON/10 sec OFF | 2 sec ON/4 sec OFF | 2 sec ON/10 sec OFF |
Fig. 2 shows the SDS-PAGE results of the analysis. Compared with the growth at 37 °C, the growth at 18 °C yielded more soluble CP (sup/ppt in #1 and #4–6), suggesting that CP favors lower temperatures to fold into soluble protein in the bacterial expression system. However, total CP expression was higher when expressed 3 hours at 37 °C (total in #5, #6) compared to 20 h at 18 °C (total in #1–#4). Consequently, 37 °C expression resulted in a higher yield of CP after batch purification with Strep-Tactin resin (Elution in #1 and #4–6).
Fig. 2.

Batch analysis of CP expression and purification. #1–#6 corresponds to the conditions in Table 1. t: total lysate, s, p: supernatant and pellet of lysate (13,420 × g, 60 min). elution: CP bound to Strep-Tactin beads was eluted with 50 mM biotin.
We then examined CP's tolerance to physical perturbations during cell resuspension. The soluble fraction of CP decreased in sample #2 compared to #1, suggesting that vortexing may induce CP aggregation (sup/ppt in #1 and #2). Furthermore, 15-minute incubation of cell suspension at 37 °C also decreased CP solubility (sup/ppt in #1 and #3). Notably, 37 °C incubation produced an additional band of approximately 12 kDa in elution fraction (Elution #3), suggesting proteolysis in the β-globule region within the β2 subunit of CP [61].
In Condition III, we compared two sonication intervals: one with a 4-second pause and another with a 10-second pause, to balance total process duration against potential protein denaturation. Particularly for samples grown at 37 °C (#5 and #6), the longer interval improved CP solubility. This finding suggests that CP is susceptible to shear stress or temperature increases caused by short-interval sonication. Taken together, our results suggest that CP is sensitive to physical perturbations during cell resuspension and we concluded that the condition used in sample #6 yields the most soluble CP.
Batch purification experiment was conducted as follows. BL21(DE3) RIL cells transformed with pET28-6 × His-2 × Strep-TEVsite-Capβ2 (mouse)-Capα1 (mouse) were grown overnight at 37 °C in LB medium containing 50 μg/mL of Kanamycin and 25 μg/mL of Chloramphenicol (LB/Kan/Cam). Pre-culture was inoculated to 10 mL of LB/Kan/Cam and cultured at 37 °C until OD600 reached around 0.5. Protein expression was induced by adding 300 μM IPTG and allowed to proceed for 3 h at 37 °C or 20 h at 18 °C. The cells were harvested by centrifugation, and washed once with CP wash buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM NaCl, 1 mM EDTA, 14 mM β-mercaptoethanol). The pellets were resuspended with 2 mL of CP lysis buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM NaCl, 1 mM EDTA, 14 mM β-mercaptoethanol, 1 mM PMSF, 1 × complete, and 1/1000 Benzonase). Sample #1 and #3-6 were gently resuspended on ice using a brush, while sample #2 was vortexed for 30 sec. Subsequently, only sample #3 was incubated at 37 °C for 15 min. The cell lysis was performed by sonication on ice (sample #1, #2-3, #5: sixty cycles of 2-sec sonication with 4-sec intervals, sample #4, #6: sixty cycles of 2-sec sonication with 10-sec intervals) using a Misonix Ultrasonic Liquid Processor Sonicator S4000 at an amplitude of 30. The lysate was then centrifuged at 4 °C, 13420 × g for 1 h. The pellets were resuspended in 2 mL of CP lysis buffer for SDS-PAGE analysis. For purification, 1.5 mL of supernatant was added to 20 µL of Strep-Tactin XT 4Flow high-capacity resin, equilibrated with CP lysis buffer, in a 1.5 mL tube. The sample was rotated at 4 °C for 30 min, after which the resin was washed 3 times with 200 μl of CP wash buffer. Strep-tagged proteins were eluted by rotating for 30 min at 4 °C in 50 µL of CP elution buffer (50 mM Tris-HCl (pH 7.5 at RT), 150 mM NaCl, 1 mM EDTA, 14 mM β-mercaptoethanol, 50 mM biotin). The total lysate, both the supernatant and pellet, after sonication, as well as the final eluted product, were analyzed by SDS-PAGE with Coomassie Brilliant Blue staining.
CP purification from liter scale culture
Based on the small-scale purification results, we conducted liter-scale expression and purification of CP. Opting for condition #6 (Table 1) due to its overall yield, we expressed CP in 6 L culture medium and applied the cell lysate to 2 mL of Strep-Tactin XT 4Flow high-capacity resin. As shown in Fig. 3A and ‘before TEV’ lane in Fig. 3B, His-Strep-tagged CP was efficiently enriched in 50 mM biotin elution through a Strep-Tactin column purification. Double bands corresponding to His-Strep-tagged Capβ2 (36.5 kDa) and Capα1 (33.0 kDa) confirmed that CP was eluted as a heterodimeric complex.
Fig. 3.

Purification of CP from 6 L of culture. (A) SDS-PAGE analysis of cell lysate and Strep-Tactin column purification. t: total lysate. sup, ppt: supernatant and pellet of lysate (13,420 × g, 60 min). elution: CP bound to Strep-Tactin column was eluted with 50 mM biotin. (B) SDS-PAGE analysis of TEV cleavage and Ni-NTA purification of tag-cleaved CP. The purification tag was cleaved off from CP by His-tagged TEV protease and the cleaved tag and TEV protease were removed by Ni-NTA purification. Before TEV: collected elution fractions after Strep-Tactin column purification. After TEV: after overnight TEV cleavage reaction at 4 °C. f.t.: flow through of Ni-NTA purification. (C) Chromatogram of Superdex 200 increase 10/300 column gel filtration. Fraction numbers correspond to the lanes in (D). (D) SDS-PAGE analysis of gel filtration chromatography.
To obtain tag-free CP, next we treated the purified CP with TEV protease. Since CP is sensitive to temperature rise (Fig. 2), we conducted TEV cleavage reaction at 4 °C during overnight dialysis. The band shift from His-Strep-tagged Capβ2 (36.5 kDa) to tag-free Capβ2 (30.8 kDa) and the appearance of cleaved His-Strep-tag (5.9 kDa) near 10 kDa marker verified successful cleavage. A Ni-NTA column was then used to purify tag-free CP separating from the cleaved tag as well as His-tagged TEV protease. Fig. 3B shows that tag-free CP was collected in flow through and 0 mM imidazole fractions, successfully separated from the cleaved tag and His-tagged TEV protease (31.8 kDa), which were eluted in 500 mM fraction.
We then examined the purified CP on gel filtration chromatography. The 280 nm absorbance chromatogram shows a sharp peak at around 14 mL elution volume, indicating a high purity of CP (Fig. 3C). This was further supported by SDS-PAGE analysis of the peak fractions (Fig. 3D). Although the peak in the chromatogram was accompanied by a minor shoulder peak to the lower molecular weight side, this may reflect an equilibrium between multiple conformations, since SDS-PAGE of these fractions showed no significant differences. Overall, our method yielded 8.8 mg of CP from 6 L of culture.
Purification of cofilin, profilin, ADF, fascin, and VASP
Having established the purification protocol using a His-Strep-tag combined with a TEV protease cleavage site, we extended our method to other actin-binding proteins, namely cofilin, profilin, ADF, fascin and VASP. As shown in Fig. 4A, cofilin and profilin were successfully purified from a single Strep-Tactin XT column purification. The purification tag on cofilin and profilin were then removed by TEV protease and tag-free proteins were purified by Ni-NTA chromatography. While cofilin was eluted without imidazole competition, profilin showed a weak affinity to Ni-NTA resin and required 10 mM imidazole for efficient recovery from the column (Fig. 4B). Consequently, for these two proteins, we performed dialysis to remove the imidazole, which is known to cause protein precipitation upon storage in freezers [62,63].
Fig. 4.

Purification of cofilin, profilin, ADF, fascin and VASP. (A) SDS-PAGE analysis of Strep-Tactin column purification of cofilin, profilin, ADF, fascin and VASP. (B) Ni-NTA column purification of cofilin, profilin, ADF, fascin and VASP after TEV cleavage of the purification tag. (-)TEV: sample before adding TEV protease. input: collected elution fractions after Strep-Tactin column purification. f.t. flow through. Imidazole concentrations in elution buffer were indicated on the lanes. (C) Chromatogram of Superdex 200 increase 10/300 column gel filtration and SDS-PAGE analysis of gel filtration chromatography of ADF, fascin, and VASP. Orange bars indicate the fraction analyzed in SDS-PAGE (E). input: collected elution fractions after Ni-NTA purification. (D) SDS-PAGE analysis of gel filtration chromatography of ADF, fascin, and VASP.
Unlike cofilin and profilin, the Strep-Tactin elution of ADF, fascin, and VASP displayed intermediate bands in SDS-PAGE (Fig. 4A). Thus, we added a gel filtration step for these proteins. Following Strep-tag purification, proteins were processed by TEV protease during dialysis and applied to Ni-NTA chromatography, similar to cofilin and profilin. ADF was eluted without imidazole competition, meanwhile fascin and VASP exhibited affinity to Ni-NTA resin. Of note, some fraction of fascin and VASP appeared to remain on Ni-NTA even after 20 mM imidazole wash. We collected the target protein enriched fractions (ADF: 0 mM imidazole, fasctin: 0–20 mM imidazole, VASP: 0–20 mM imidazole) and applied to gel filtration. Fig. 4C and D show the chromatograms and SDS-PAGE results of gel filtration, respectively. Fascin was eluted as a single peak, while ADF and VASP showed multiple peaks. Particularly, gel filtration of VASP separated many of intermediate bands from the full-length VASP. Since the full-length VASP also appeared in the lanes of the second and later peaks (Fig. 4D), it was suggested that the intermediate proteins form complexes with the full-length VASP, likely through the C-terminal tetramerization domain [64,65], and they were co-eluted during the chromatography process.
Fig. 1C represents the final products of our purification process for CP, cofilin, profilin, ADF, fascin and VASP. The data shows our approach. Notably, our results also revealed that each protein exhibits a distinct affinity to the Ni-NTA column. These findings are crucial for reliably reproducing and fine-tuning the purification process in future studies. Collectively, these results indicate that our purification method is directly applicable to cofilin, profilin, ADF, fascin and VASP.
Activity measurements of the purified actin-binding proteins
To validate the activity of the purified proteins, we conducted a series of biochemical and microscopic assays. First, we employed the pyrene actin polymerization assay to evaluate the barbed-end capping activity of CP (Fig. 5A). The assay incorporated Arp2/3 and GST-VCA to facilitate filament nucleation [66]. We observed rapid actin polymerization in the presence of Arp2/3 and VCA. However, the addition of 2 nM CP resulted in partial inhibition of actin polymerization, while 20 nM and 200 nM CP led to complete inhibition. These results confirmed the functionality of the purified CP as an effective barbed-end capper.
Fig. 5.

Activity evaluation of the purified actin-binding proteins. (A) Pyrene actin polymerization assay and the effect of CP on barbed end capping. 1 µM actin (5 % pyrene-labeled), 10 nM Arp2/3, 100 nM GST-VCA were mixed with 0, 2, 20, 200 nM CP. Error bars represent SEM (n=4). (B, C) Sedimentation assay for cofilin, ADF, and profilin. In (B), 2 µM actin and 8 µM cofilin or ADF were applied. In (C), 5 µM actin and 15.5 µM profilin were applied. (D) TIRF imaging and actin bundling activity of fascin and VASP. 7.5 µM actin (10 % Alexa Fluor-647-labeled) was observed with the indicated concentrations of fascin and VASP. Scale bar: 5 µm.
The activity of cofilin and ADF was assessed using a co-sedimentation assay due to the high sequence similarity of these two proteins [67]. In this assay, actin filaments polymerized in a 50 mM KCl buffer were collected in the pellet after ultracentrifugation. Adding cofilin at a ratio of 2 µM actin to 8 µM cofilin, a reduction in the F-actin fraction was observed (Fig. 5B). When using ADF under the same condition, a further reduction in the F-actin fraction was observed similarly to the previous study [67]. These results indicate that purified cofilin and ADF effectively sever actin filaments.
Profilin's activity was similarly tested through a co-sedimentation assay. Initially, actin was mixed with or without profilin in a low-salt concentration buffer, favoring the monomeric G-actin state. Actin polymerization was then induced in 50 mM KCl buffer. The presence of profilin reduced the actin pellet fraction (Fig. 5C), supporting its role in suppressing actin polymerization.
Lastly, the activity of fascin and VASP was examined using Total Internal Reflection Fluorescence (TIRF) microscopy (Fig. 5D). In case TIRF is not available, individual actin filaments can be observed by fluorescence microscopy with rhodamine-phalloidin-labeling [48], and the bundling of actin filaments can be observed by phase contrast microscopy [68]. When observing actin solely, the actin filaments were distributed relatively uniformly under the evanescent field. In contrast, the addition of 1 µM fascin caused actin filament bundling, as shown in the previous study [69]. When VASP was added, actin formed mesh- and rope-like superstructures, as previously reported [64]. These observations support the actin-bundling function of the purified fascin and VASP.
Taken together, these results demonstrate that CP, cofilin, ADF, profilin, fascin and VASP were all successfully purified as functional proteins.
CRediT authorship contribution statement
Daichi Nakajima: Validation, Investigation, Visualization, Writing – original draft, Writing – review & editing. Nozomi Takahashi: Validation, Investigation, Visualization. Takanari Inoue: Writing – review & editing, Supervision, Project administration, Funding acquisition. Shin-ichiro M. Nomura: Writing – review & editing, Supervision, Project administration, Funding acquisition. Hideaki T. Matsubayashi: Conceptualization, Validation, Investigation, Visualization, Writing – original draft, Writing – review & editing, Supervision, Project administration, Funding acquisition.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We thank Yubin Zhou for TEV protease plasmid; Masato Kanemaki for pET28-2×Strep-tag plasmid; Kensaku Mizuno for Cofilin-EYFP plasmid; Baoyu Chen for 7×His-VASP plasmid; Shigeko Yamashiro for GFP-Fascin plasmid. We appreciate Shiva Razavi and Erin D. Goley for technical support of pyrene assay and insightful comments on actin preparation. We also appreciate Julie Plastino and Cecile Sykes for pET3d-Capβ2(mouse)-Capα1(mouse) plasmid, pMW-profilin plasmid, and insightful advice on protein purification. Our appreciation extends to generous support from the laboratory of Satoshi Murata and lab members. FRIS CoRE (a shared research environment in Tohoku University) is also acknowledged. We also thank Robert DeRose for proofreading the manuscript and experimental support. This study was supported by the PRESTO program of the Japan Science and Technology Agency (JPMJPR12A5 to TI, JPMJPR20KA to HTM), NIGMS (R35GM149329 to TI), NSF (000819255 to TI), and MEXT/JSPS KAKENHI Grants (23K17485, 20H05968, 20H05701, 20H05970, 20H00619 to SMN, 23K14150 and 23H04397 to HTM).
During the preparation of this work the authors used ChatGTP-4o in order to improve readability and language. After using the tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication. The graphical abstract was created with BioRender.com.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.mex.2024.102884.
Appendix. Supplementary materials
Data availability
Data will be made available on request.
References
- 1.Lappalainen P., Kotila T., Jégou A., Romet-Lemonne G. Biochemical and mechanical regulation of actin dynamics. Nat. Rev. Mol. Cell Biol. 2022 doi: 10.1038/s41580-022-00508-4. [DOI] [PubMed] [Google Scholar]
- 2.Insall R. Actin in 2021. Curr. Biol. 2021;31:R496–R498. doi: 10.1016/j.cub.2021.04.013. [DOI] [PubMed] [Google Scholar]
- 3.Chakrabarti R., Lee M., Higgs H.N. Multiple roles for actin in secretory and endocytic pathways. Curr. Biol. 2021;31:R603–R618. doi: 10.1016/j.cub.2021.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mylvaganam S., Freeman S.A., Grinstein S. The cytoskeleton in phagocytosis and macropinocytosis. Curr. Biol. 2021;31:R619–R632. doi: 10.1016/j.cub.2021.01.036. [DOI] [PubMed] [Google Scholar]
- 5.Jaumouillé V., Waterman C.M. Physical constraints and forces involved in phagocytosis. Front. Immunol. 2020;11:1–20. doi: 10.3389/fimmu.2020.01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Theriot J.A. The polymerization motor. Traffic. 2000;1:19–28. doi: 10.1034/j.1600-0854.2000.010104.x. [DOI] [PubMed] [Google Scholar]
- 7.Cáceres R., Abou-Ghali M., Plastino J. Reconstituting the actin cytoskeleton at or near surfaces in vitro. Biochim. Biophys. Acta BBA – Mol. Cell Res. 2015;1853:3006–3014. doi: 10.1016/j.bbamcr.2015.07.021. [DOI] [PubMed] [Google Scholar]
- 8.Mullins R.D., Hansen S.D. In vitro studies of actin filament and network dynamics. Curr. Opin. Cell Biol. 2013;25:6–13. doi: 10.1016/j.ceb.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vogel S.K., Schwille P. Minimal systems to study membrane–cytoskeleton interactions. Curr. Opin. Biotechnol. 2012;23:758–765. doi: 10.1016/j.copbio.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 10.Lopes Dos Santos R., Campillo C. Studying actin-induced cell shape changes using giant unilamellar vesicles and reconstituted actin networks. Biochem. Soc. Trans. 2022;50:1527–1539. doi: 10.1042/BST20220900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cortese J.D., Schwab B., Frieden C., Elson E.L. Actin polymerization induces a shape change in actin-containing vesicles. Proc. Natl. Acad. Sci. U. S. A. 1989;86:5773–5777. doi: 10.1073/pnas.86.15.5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyata H., Hotani H. Morphological changes in liposomes caused by polymerization of encapsulated actin and spontaneous formation of actin bundles. Proc. Natl. Acad. Sci. U. S. A. 1992;89:11547–11551. doi: 10.1073/pnas.89.23.11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyata H., Nishiyama S., Akashi K.-I., Kinosita K. Protrusive growth from giant liposomes driven by actin polymerization. Proc. Natl. Acad. Sci. 1999;96:2048–2053. doi: 10.1073/pnas.96.5.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cameron L.A., Footer M.J., Van Oudenaarden A., Theriot J.A. Motility of ActA protein-coated microspheres driven by actin polymerization. Proc. Natl. Acad. Sci. U. S. A. 1999;96:4908–4913. doi: 10.1073/pnas.96.9.4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.T.P. Loisel, R. Boujemaa, D. Pantaloni, M. Carlier, Reconstitution of actin-based motility of Listeria and Shigella using pure proteins, 401 (1999) 613–616. [DOI] [PubMed]
- 16.Bernheim-Groswasser A., Wiesner S., Golsteyn R.M., Carlier M.-F., Sykes C. The dynamics of actin-based motility depend on surface parameters. Nature. 2002;417:308–311. doi: 10.1038/417308a. [DOI] [PubMed] [Google Scholar]
- 17.Wiesner S., Helfer E., Didry D., Ducouret G., Lafuma F., Carlier M.F., Pantaloni D. A biomimetic motility assay provides insight into the mechanism of actin-based motility. J. Cell Biol. 2003;160:387–398. doi: 10.1083/jcb.200207148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Romero S., Le Clainche C., Didry D., Egile C., Pantaloni D., Carlier M.F. Formin is a processive motor that requires profilin to accelerate actin assembly and associated ATP hydrolysis. Cell. 2004;119:419–429. doi: 10.1016/j.cell.2004.09.039. [DOI] [PubMed] [Google Scholar]
- 19.Co C., Wong D.T., Gierke S., Chang V., Taunton J. Mechanism of actin network attachment to moving membranes: barbed end capture by N-WASP WH2 domains. Cell. 2007;128:901–913. doi: 10.1016/j.cell.2006.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akin O., Mullins R.D. Capping protein increases the rate of actin-based motility by promoting filament nucleation by the Arp2/3 complex. Cell. 2008;133:841–851. doi: 10.1016/j.cell.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dürre K., Keber F.C., Bleicher P., Brauns F., Cyron C.J., Faix J., Bausch A.R. Capping protein-controlled actin polymerization shapes lipid membranes. Nat. Commun. 2018 doi: 10.1038/s41467-018-03918-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simon C., Kusters R., Caorsi V., Allard A., Abou-Ghali M., Manzi J., Di Cicco A., Lévy D., Lenz M., Joanny J.-F., Campillo C., Plastino J., Sens P., Sykes C. Actin dynamics drive cell-like membrane deformation. Nat. Phys. 2019;15:602–609. doi: 10.1038/s41567-019-0464-1. [DOI] [Google Scholar]
- 23.Sakamoto R., Banerjee D.S., Yadav V., Chen S., Gardel M.L., Sykes C., Banerjee S., Murrell M.P. Membrane tension induces F-actin reorganization and flow in a biomimetic model cortex. Commun. Biol. 2023;6:325. doi: 10.1038/s42003-023-04684-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maruyama K., Kimura S., Ishi T., Kuroda M., Ohashi K. Beta-actinin, a regulatory protein of muscle. Purification, characterization and function. J. Biochem. (Tokyo) 1977;81:215–232. doi: 10.1093/oxfordjournals.jbchem.a131438. [DOI] [PubMed] [Google Scholar]
- 25.Bamburg J.R., Harris H.E., Weeds A.G. Partial purification and characterization of an actin depolymerizing factor from brain. FEBS Lett. 1980;121:178–182. doi: 10.1016/0014-5793(80)81292-0. [DOI] [PubMed] [Google Scholar]
- 26.Reichstein E., Korn E.D. Acanthamoeba profilin. a protein of low molecular weight from Acanpthamoeba castellanii that inhibits actin nucleation. J. Biol. Chem. 1979;254:6174–6179. doi: 10.1016/S0021-9258(18)50534-2. [DOI] [PubMed] [Google Scholar]
- 27.Yamashiro-Matsumura S., Matsumura F. Purification and characterization of an F-actin-bundling 55-kilodalton protein from HeLa cells. J. Biol. Chem. 1985;260:5087–5097. doi: 10.1016/S0021-9258(18)89183-9. [DOI] [PubMed] [Google Scholar]
- 28.Halbrügge M., Walter U. Purification of a vasodilator-regulated phosphoprotein from human platelets. Eur. J. Biochem. 1989;185:41–50. doi: 10.1111/j.1432-1033.1989.tb15079.x. [DOI] [PubMed] [Google Scholar]
- 29.Soeno Y., Abe H., Kimura S., Maruyama K., Obinata T. Generation of functional beta-actinin (CapZ) in an E. coli expression system. J. Muscle Res. Cell Motil. 1998;19:639–646. doi: 10.1023/a:1005329114263. [DOI] [PubMed] [Google Scholar]
- 30.Hawkins M., Pope B., Maciver S.K., Weeds A.G. Human actin depolymerizing factor mediates a pH-sensitive destruction of actin filaments. Biochemistry. 1993;32:9985–9993. doi: 10.1021/bi00089a014. [DOI] [PubMed] [Google Scholar]
- 31.Babcock G., Rubenstein P.A. Synthesis of mammalian profilin in Escherichia coli and its characterization. Cell Motil. Cytoskeleton. 1989;14:230–236. doi: 10.1002/cm.970140209. [DOI] [PubMed] [Google Scholar]
- 32.Edwards R.A., Herrera-Sosa H., Otto J., Bryan J. Cloning and expression of a murine fascin homolog from mouse brain. J. Biol. Chem. 1995;270:10764–10770. doi: 10.1074/jbc.270.18.10764. [DOI] [PubMed] [Google Scholar]
- 33.Shekhar S., Carlier M.-F. Enhanced depolymerization of actin filaments by ADF/cofilin and monomer funneling by capping protein cooperate to accelerate barbed-end growth. Curr. Biol. 2017;27:1990–1998. doi: 10.1016/j.cub.2017.05.036. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Eeuwen T., Boczkowska M., Rebowski G., Carman P.J., Fregoso F.E., Dominguez R. Transition state of Arp2/3 complex activation by actin-bound dimeric nucleation-promoting factor. Proc. Natl. Acad. Sci. 2023;120 doi: 10.1073/pnas.2306165120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu J.-X., Xiang Y.-F., Zhang J.-X., Ju H.-Q., Chen Z.-P., Wang Q.-L., Chen W., Peng X.-L., Han B., Wang Y.-F. Cloning, soluble expression, rapid purification and characterization of human cofilin1. Protein Expr. Purif. 2012;82:186–191. doi: 10.1016/j.pep.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 36.Sedeh R.S., Fedorov A.A., Fedorov E.V., Ono S., Matsumura F., Almo S.C., Bathe M. Structure, evolutionary conservation, and conformational dynamics of homo sapiens fascin-1, an F-actin crosslinking protein. J. Mol. Biol. 2010;400:589–604. doi: 10.1016/j.jmb.2010.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barzik M., Kotova T.I., Higgs H.N., Hazelwood L., Hanein D., Gertler F.B., Schafer D.A. Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J. Biol. Chem. 2005;280:28653–28662. doi: 10.1074/jbc.M503957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iwasaki S., Sasaki H.M., Sakaguchi Y., Suzuki T., Tadakuma H., Tomari Y. Defining fundamental steps in the assembly of the Drosophila RNAi enzyme complex. Nature. 2015;521:533–536. doi: 10.1038/nature14254. [DOI] [PubMed] [Google Scholar]
- 39.Schöneberg J., Pavlin M.R., Yan S., Righini M., Lee I.-H., Carlson L.-A., Bahrami A.H., Goldman D.H., Ren X., Hummer G., Bustamante C., Hurley J.H. ATP-dependent force generation and membrane scission by ESCRT-III and Vps4. Science. 2018;362:1423–1428. doi: 10.1126/science.aat1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murayama Y., Endo S., Kurokawa Y., Kurita A., Iwasaki S., Araki H. Coordination of cohesion and DNA replication observed with purified proteins. Nature. 2024;626:653–660. doi: 10.1038/s41586-023-07003-6. [DOI] [PubMed] [Google Scholar]
- 41.Shimizu Y., Inoue A., Tomari Y., Suzuki T., Yokogawa T., Nishikawa K., Ueda T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001;19:751–755. doi: 10.1038/90802. [DOI] [PubMed] [Google Scholar]
- 42.Lavickova B., Maerkl S.J. A simple, robust, and low-cost method to produce the PURE cell-free system. ACS Synth. Biol. 2019;8:455–462. doi: 10.1021/acssynbio.8b00427. [DOI] [PubMed] [Google Scholar]
- 43.Schmidt T.G.M., Batz L., Bonet L., Carl U., Holzapfel G., Kiem K., Matulewicz K., Niermeier D., Schuchardt I., Stanar K. Development of the Twin-Strep-tag® and its application for purification of recombinant proteins from cell culture supernatants. Protein Expr. Purif. 2013;92:54–61. doi: 10.1016/j.pep.2013.08.021. [DOI] [PubMed] [Google Scholar]
- 44.Voss S., Skerra A. Mutagenesis of a flexible loop in streptavidin leads to higher affinity for the Strep-tag II peptide and improved performance in recombinant protein purification. Protein Eng. Des. Sel. 1997;10:975–982. doi: 10.1093/protein/10.8.975. [DOI] [PubMed] [Google Scholar]
- 45.Chen X.J., Squarr A.J., Stephan R., Chen B., Higgins T.E., Barry D.J., Martin M.C., Rosen M.K., Bogdan S., Way M. Ena/VASP proteins cooperate with the WAVE complex to regulate the actin cytoskeleton. Dev. Cell. 2014;30:569–584. doi: 10.1016/j.devcel.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaji N., Muramoto A., Mizuno K. LIM Kinase-mediated cofilin phosphorylation during mitosis is required for precise spindle positioning. J. Biol. Chem. 2008;283:4983–4992. doi: 10.1074/jbc.M708644200. [DOI] [PubMed] [Google Scholar]
- 47.Doolittle L.K., Rosen M.K., Padrick S.B. Measurement and analysis of in vitro actin polymerization. Methods Mol. Biol. 2013 doi: 10.1007/978-1-62703-538-5_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hansen S.D., Zuchero J.B., Mullins R.D. In: Adhes. Protein Protoc. Coutts A.S., editor. Humana Press; Totowa, NJ: 2013. Cytoplasmic actin: purification and single molecule assembly assays; pp. 145–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun H., Luo Y., Miao Y. Purification of globular actin from rabbit muscle and pyrene fluorescent assays to investigate actin dynamics in vitro. Bio-Protoc. 2018;8 doi: 10.21769/BioProtoc.3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kouyama T., Mihashi K. Fluorimetry study of N-(1-pyrenyl) iodoacetamide-labelled F-actin. Eur. J. Biochem. 1981;114:33–38. doi: 10.1111/j.1432-1033.1981.tb06167.x. [DOI] [PubMed] [Google Scholar]
- 51.Gidi Y., Bayram S., Ablenas C.J., Blum A.S., Cosa G. Efficient one-step PEG-silane passivation of glass surfaces for single-molecule fluorescence studies. ACS Appl. Mater. Interfaces. 2018;10:39505–39511. doi: 10.1021/acsami.8b15796. [DOI] [PubMed] [Google Scholar]
- 52.Akıl C., Tran L.T., Orhant-Prioux M., Baskaran Y., Manser E., Blanchoin L., Robinson R.C. Insights into the evolution of regulated actin dynamics via characterization of primitive gelsolin/cofilin proteins from Asgard archaea. Proc. Natl. Acad. Sci. 2020 doi: 10.1073/pnas.2009167117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmidt T.G.M., Koepke J., Frank R., Skerra A. Molecular interaction between the Strep-tag affinity peptide and its cognate target, Streptavidin. J. Mol. Biol. 1996;255:753–766. doi: 10.1006/jmbi.1996.0061. [DOI] [PubMed] [Google Scholar]
- 54.Junttila M.R., Saarinen S., Schmidt T., Kast J., Westermarck J. Single-step Strep-tag purification for the isolation and identification of protein complexes from mammalian cells. Proteomics. 2005;5:1199–1203. doi: 10.1002/pmic.200400991. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt T.G., Skerra A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2007;2:1528–1535. doi: 10.1038/nprot.2007.209. [DOI] [PubMed] [Google Scholar]
- 56.Edwards M., Zwolak A., Schafer D.A., Sept D., Dominguez R., Cooper J.A. Capping protein regulators fine-tune actin assembly dynamics. Nat. Rev. Mol. Cell Biol. 2014;15:677–689. doi: 10.1038/nrm3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mullins R.D., Bieling P., Fletcher D.A. From solution to surface to filament: actin flux into branched networks. Biophys. Rev. 2018;10:1537–1551. doi: 10.1007/s12551-018-0469-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carlier M.-F., Shekhar S. Global treadmilling coordinates actin turnover and controls the size of actin networks. Nat. Rev. Mol. Cell Biol. 2017;18:389–401. doi: 10.1038/nrm.2016.172. [DOI] [PubMed] [Google Scholar]
- 59.Funk J., Merino F., Schaks M., Rottner K., Raunser S., Bieling P. A barbed end interference mechanism reveals how capping protein promotes nucleation in branched actin networks. Nat. Commun. 2021;12:5329. doi: 10.1038/s41467-021-25682-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bieling P., Hansen S.D., Akin O., Li T.-D., Hayden C.C., Fletcher D.A., Mullins R.D. WH2 and proline-rich domains of WASP-family proteins collaborate to accelerate actin filament elongation. EMBO J. 2018;37:102–121. doi: 10.15252/embj.201797039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takeda S., Minakata S., Koike R., Kawahata I., Narita A., Kitazawa M., Ota M., Yamakuni T., Maeda Y., Nitanai Y. Two distinct mechanisms for actin capping protein regulation—steric and allosteric inhibition. PloS Biol. 2010;8(7) doi: 10.1371/2Fjournal.pbio.1000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharma S., Evans D.B., Vosters A.F., Chattopadhyay D., Hoogerheide J.G., Campbell C.M. Immobilized metal affinity chromatography of bacterially expressed proteins engineered to contain an alternating-histidine domain. Methods. 1992;4:57–67. doi: 10.1016/1046-2023(92)90056-E. [DOI] [Google Scholar]
- 63.Hefti M.H., Van Vugt-Van der Toorn C.J.G., Dixon R., Vervoort J. A novel purification method for histidine-tagged proteins containing a thrombin cleavage site. Anal. Biochem. 2001;295:180–185. doi: 10.1006/abio.2001.5214. [DOI] [PubMed] [Google Scholar]
- 64.Bachmann C., Fischer L., Walter U., Reinhard M. The EVH2 domain of the vasodilator-stimulated phosphoprotein mediates tetramerization, F-actin binding, and actin bundle formation. J. Biol. Chem. 1999;274:23549–23557. doi: 10.1074/jbc.274.33.23549. [DOI] [PubMed] [Google Scholar]
- 65.Kuhnel K., Jarchau T., Wolf E., Schlichting I., Walter U., Wittinghofer A., Strelkov S.V. The VASP tetramerization domain is a right-handed coiled coil based on a 15-residue repeat. Proc. Natl. Acad. Sci. 2004;101:17027–17032. doi: 10.1073/pnas.0403069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rohatgi R., Ma L., Miki H., Lopez M., Kirchhausen T., Takenawa T., Kirschner M.W. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–231. doi: 10.1016/S0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- 67.Yeoh S., Pope B., Mannherz H.G., Weeds A. Determining the differences in actin binding by human ADF and cofilin 1 1Edited by J. Karn. J. Mol. Biol. 2002;315:911–925. doi: 10.1006/jmbi.2001.5280. [DOI] [PubMed] [Google Scholar]
- 68.Liu A.P., Richmond D.L., Maibaum L., Pronk S., Geissler P.L., Fletcher D.A. Membrane-induced bundling of actin filaments. Nat. Phys. 2008;4(10):789–793. doi: 10.1038/nphys1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Breitsprecher D., Koestler S.A., Chizhov I., Nemethova M., Mueller J., Goode B.L., Small J.V., Rottner K., Faix J. Cofilin cooperates with fascin to disassemble filopodial actin filaments. J. Cell Sci. 2011;124:3305–3318. doi: 10.1242/jcs.086934. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.