

Abstract
Current CD33-targeted immunotherapies typically recognize the membrane-distal V-set domain of CD33. Here, we show that decreasing the distance between T cell and leukemia cell membrane increases the efficacy of CD33 chimeric antigen receptor (CAR) T cells. We therefore generated and optimized second-generation CAR constructs containing single-chain variable fragments from antibodies raised against the membrane-proximal C2-set domain, which bind CD33 regardless of whether the V-set domain is present (CD33PAN antibodies). CD33PAN CAR T cells resulted in efficient tumor clearance and improved survival of immunodeficient mice bearing human AML cell xenografts and, in an AML model with limited CD33 expression, forced escape of CD33neg leukemia. Compared to CD33V-set CAR T cells, CD33PAN CAR T cells showed greater in vitro and in vivo efficacy against several human AML cell lines with differing levels of CD33 without increased expression of exhaustion markers. CD33PAN moieties were detected at a higher frequency on human leukemic stem cells, and CD33PAN CAR T cells had greater in vitro efficacy against primary human AML cells. Together, our studies demonstrate improved efficacy with CAR T cells binding CD33 close to the cell membrane, providing the rationale to investigate CD33PAN CAR T cells further toward possible clinical application.
Keywords: MT: Novel therapeutic targets and biomarker development Special Issue, acute myeloid leukemia, AML, adoptive cell therapy, CD33, chimeric antigen receptor, CAR, immunotherapy, membrane proximity, T cells
Graphical abstract
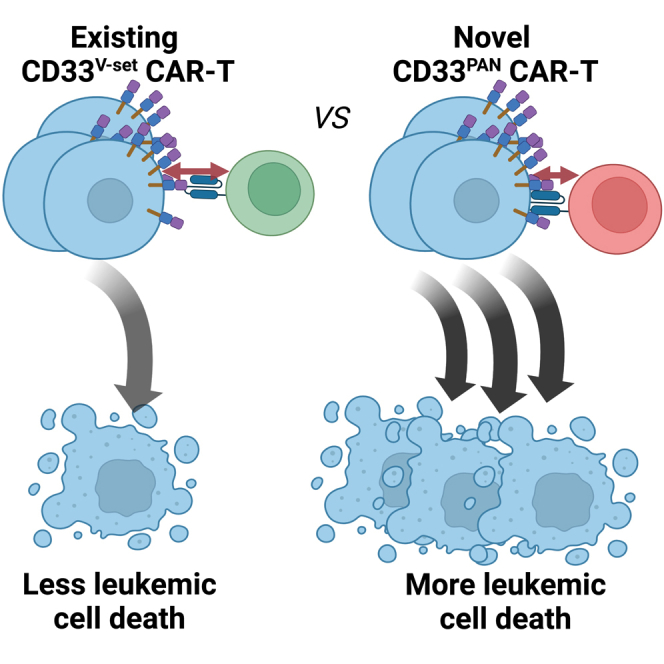
Fiorenza and colleagues found that decreasing the distance between T cell and leukemia cell membranes increases the efficacy of CD33 chimeric antigen receptor (CAR) T cells. CAR T cells targeting the membrane-proximal C2-set domain showed superior anti-leukemic activity, providing the rationale to investigate such cells further toward possible clinical application.
Introduction
CD33 (sialic acid-binding immunoglobulin-like lectin-3 [SIGLEC-3]) is physiologically expressed on maturing and mature myeloid cells, monocytes, macrophages, and dendritic cells. This glycoprotein of elusive function is also displayed on some subsets of B cells and activated T as well as natural killer cells, whereas there is controversy as to whether CD33 is expressed on normal pluripotent hematopoietic stem cells.1,2,3,4,5,6 Consistent with a myeloid differentiation antigen, almost all patients with acute myeloid leukemia (AML) have at least some CD33+ leukemia cells and on underlying leukemic stem/progenitor cells in some, although the proportion of positive cells and the CD33 antigen density per cell varies widely between and within patients.2,3,4,5,7,8,9,10,11,12,13 Because of this expression pattern, CD33 has long served as a therapeutic target in AML.2,4,8,9,10,11,12,13 Improved survival in some patients with the CD33 antibody-drug conjugate gemtuzumab ozogamicin (GO) validates CD33 as a drug target5,14 and has sparked interest in exploiting CD33-directed therapies for other diseases.15 Still, CD33 is a difficult target: several investigational drugs have failed clinically, and many patients with CD33+ AML do not benefit from GO.5 Therefore, efforts are ongoing to develop more potent therapeutics such as chimeric antigen receptor (CAR)-modified T cells.16,17,18,19,20,21,22,23 However, results in CD33-directed CAR T trials have so far been underwhelming, with no reported durable responses in patients with AML receiving such therapies.16,20
As one limitation, GO and almost all investigational CD33-directed therapeutics recognize the membrane-distal V-set domain of CD33 (Figure 1A).17 For immune effector cell-engaging therapies, this may result in the formation of a less efficient immunological synapse relative to membrane-proximal antigen binding.24,25,26,27,28,29,30 In proof-of-concept studies primarily using CD33/CD3 bispecific antibodies (BiAbs), we indeed showed that binding closer to the cell membrane enhances their cytolytic activity.31 Limited data suggested that this principle might extend to CAR T cells.31 We therefore raised monoclonal antibodies (mAbs) against the membrane-proximal C2-set domain of CD33 that binds CD33 regardless of whether the V-set domain is present (i.e., have properties of CD33PAN mAbs).31 Here, we examined how the distance between the CD33+ leukemia cell and the T cell impacts CD33 CAR T cell efficacy. In our studies, we assessed the impact of two variables, namely the distance between CD33 binding epitope and cell membrane and the length of the linker between the single-chain variable fragment (scFv) and the transmembrane domain of the CAR. Increasing evidence indicates the optimal spacer length must be determined empirically as it depends on the position of the target epitope and the level of steric hindrance.32 Demonstrating higher efficacy with binding closer to the cell membrane, we then optimized CARs built with scFvs from CD33PAN mAbs and characterized their properties in vitro and in vivo in models of human AML.
Figure 1.

Optimization of CD33V-set CAR My96
(A) Schematic of different types of CD33 antibodies binding full-length CD33 (CD33FL), a naturally occurring isoform lacking the exon 2-encoded V-set domain (CD33ΔE2), and an artificial CD33 molecule with deletion of exons 3 and 4 that results in membrane proximal relocation of the V-set domain (CD33ΔE3-4). (B) Schematic of CAR constructs with short, intermediate, and long extracellular spacers. Note the mutated CH2 domain to prevent Fc binding. Illustrations created with BioRender.com. (C) Human myeloid CD33+ ML-1, K562, or ML-1 CD33KO (by CRISPR-Cas9) cells were incubated with primary human T cells transduced with a CD33V-set My96-based CAR construct with either a short, intermediate, or long spacer at various E:T cell ratios as indicated. After 4 h, cytotoxicity was assessed by chromium-51 release. (D) Geometric mean fluorescence intensity of transduction marker tCD19 expression by flow cytometry. Experiments performed in technical triplicates. Shown are mean ± SEM in triplicate from two representative experiments from different healthy donors. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; ns (not significant) by two-way ANOVA with post-hoc Tukey correction.
Results
Membrane-proximal binding enhances CD33 CAR T cell efficacy
We previously demonstrated that binding closer to the cell membrane increases the efficacy of CD33V-set/CD3 BiAbs.31 Very limited data suggested this principle might apply to CD33V-set CAR T cells.31 To study this relationship further, we generated CD33V-set CAR T cells with two different scFvs (My96 and A33) for testing in models of CD33+ AML. In our approach, we wanted to ensure that the CD33-directed scFvs themselves did not give rise to non-specific killing. Controlling for the latter is important yet challenging in preclinical models of CAR T cell therapy. Non-specific cell killing when a CAR molecule is transduced into a T cell can arise from both intrinsic activity of the T cell and tonic signaling effects of the CAR, which may lead to the production of effector molecules and non-specific cell death. To account for both possibilities, we generally used isogenic cell lines lacking CD33 (either endogenously or via CRISPR-Cas9 gene editing) as our control of non-specific killing. In settings where such sublines were not available (most notably, immunodeficient mouse models and primary human AML cell samples), we used either CD19-directed CAR T cells or non-transduced T cells as controls. To account for the broad range of CD33 expression on the surface of human AML cells,2,4,5 we used multiple cell lines with varying expression of CD33 (Figure S1).
For My96, a CAR we focused on because of the clinical exploration of its scFv, we tested three different spacer lengths comparatively in vitro (Figure 1B). In these studies, the long spacer decreased the efficacy of My96 CAR T cells against CD33+ target cells, while CAR T cells with intermediate and short spacers were similarly effective (Figure 1C). The truncated (t)CD19 marker was expressed at similar levels in these CAR T cell products (Figure 1D), indicating that efficacy differences between long spacer and intermediate or short spacer were not due to differences in CAR expression levels. Therefore, for subsequent experiments, CAR constructs with short spacers were used. Of note, consistent with CD33-specific cytotoxicity, My96 CAR T cells were completely ineffective against CD33KO ML-1 cells (Figure 1C).
To test the relationship between CAR T cell efficacy and location of the CD33 binding epitope, we used ML-1 cells with CRISPR-Cas9-mediated deletion of endogenous CD33 loci and RS4;11 cells (CD33− human B-ALL cell line) as target cells for short-term in vitro cytotoxicity assays. In both cell lines, we expressed CD33FL or an artificial variant of CD33 in which the C2-set domain was removed and the V-set domain was placed at the membrane-proximal position (CD33ΔE3-4; Figure 1A). Engineered sublines of both cell lines showed stable expression of wild-type and shortened CD33 constructs over time, and we selected sublines with relatively similar CD33 levels for further study (Figure 2A). CD33 CAR T cells were generally more effective against CD33+ RS4;11 cells than CD33+ ML-1 cells, perhaps partly because of substantially higher CD33 expression on RS4;11 cells. While CD33V-set CAR T cells were ineffective against CD33KO ML-1 cells or parental RS4;11 cells (Figure S2A), CD33V-set CD4+ My96 CAR T cells yielded greater cytotoxicity at high, intermediate, and low effector:target (E:T) ratios against ML-1 cells displaying CD33ΔE3-4 than ML-1 cells displaying CD33FL; this was despite slightly higher CD33 expression on CD33FL cells. Statistically significantly increased cytotoxicity was also seen at an E:T ratio of 1:2 in the case of My96 CD4+ CAR T against RS4;11 cells displaying CD33ΔE3-4 relative to cells displaying CD33FL. In the case of A33 CD4+ CAR T, statistically significantly increased cytotoxicity was seen at E:T cell ratios of 1:4 and 1:8 against ML-1 CD33ΔE3-4 and RS4;11 CD33ΔE3-4 cells, respectively. A smaller increase was also seen with CD8+ CAR T cells, particularly with A33 CAR T cells (which we generated with the intermediate linker) and ML-1 cells but also, at low E:T cell ratios, CD33-expressing RS4;11 cells (Figures 2B, 2C, and S2B). Statistically significantly increased tumor necrosis factor α (TNF-α) expression was measured in CD4+ and, to a lesser extent, CD8+ CAR T cells with ML-1 and K562 cells displaying CD33ΔE3-4 compared to cells displaying CD33FL (Figure 3). In contrast, the percentage of CD4+ or CD8+ CAR T cells expressing CD107a, interferon γ (IFN-γ), or interleukin-2 (IL-2) was similar with CD33FL and CD33ΔE3-4 cells (Figure S3). Together, these observations indicated superior functionality of CAR T cells when binding CD33 proximally and provided the rationale to explore CAR T cells targeting CD33 via the C2-set domain.
Figure 2.

Effect of membrane proximity of the target epitope on the anti-tumor efficacy of CD33-directed CAR T cells
(A) ML-1 cells with CRISPR-Cas9-mediated deletion of the endogenous CD33 locus and parental CD33− RS4;11 cells were engineered to overexpress either CD33FL or CD33ΔE3-4. Relative expression of the target proteins was flow cytometrically assessed with Quantibrite-PE via a CD33V-set antibody (P67.6). (B and C) Primary human CD8+ or CD4+ T cells expressing either (B) an My96-based CAR construct or (C) an A33-based CAR construct were incubated with different target cells at an E:T cell ratio of 1:2, 1:4, or 1:8 for 16 h before flow cytometric assessment of cytotoxicity by subtracting absolute number of live tumor cells from CD33KO ML-1 (7.29 × 106 ± 8.67 × 105) or parental RS4;11 (7.77 × 104 ± 2.03 × 104) cells. Shown are mean ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; ns (not significant) by two-way ANOVA with post-hoc Tukey correction. Shown are technical triplicates of one representative out of two separate experiments.
Figure 3.

Effect of membrane proximity of the targeted CD33 epitope on TNF-α expression in CD8+ and CD4+ CAR T cells
(A) CD8+ or (B) CD4+ primary human T cells expressing either an My96-based CAR construct or an A33-based CAR construct were incubated with ML-1 or K562 cells with CRISPR-Cas9-mediated deletion of the endogenous CD33 locus (C33KO) or sublines engineered to overexpress CD33FL or CD33ΔE3-4 at various E:T cell ratios as indicated. After 12 h, the percentage of CAR T cells positive for TNF-α was assessed flow cytometrically by intracellular cytokine staining. Shown are mean ± SEM from technical triplicates. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; ns (not significant) by two-way ANOVA with post hoc Tukey correction.
Development and optimization of CD33PAN CAR T cells
Among the mAbs we raised against the C2-set domain of CD33, several bind CD33 whether or not the V-set domain is present (CD33PAN mAbs; Figure 1A).31 The latter is important considering the existence of a splice variant of CD33 that expresses only the C2-set but not the V-set.5,33,34 Using an scFv from one of these CD33PAN mAbs (CD33-1H7), we confirmed that CD33PAN CAR T cells exerted cytotoxic effects against ML-1 cells displaying CD33FL but not CD33ΔE3-4 or CD33-deleted ML-1 cells, consistent with these effects being specific to C2-set domain targeting (Figure 4A). This was unlike the effects seen with CD33V-set CAR T cells (A33 and My96), which eliminated AML cells expressing CD33ΔE3-4 more efficiently than they eliminated AML cells expressing CD33FL (Figure 4A). We then performed experiments to maximize the anti-leukemic efficacy of CD33PAN CAR T cells, focusing on different spacer lengths. In these experiments, CD8+ and CD4+ T cells expressing a 1H7 CAR with the long linker yielded significantly less cytotoxic effects and polyfunctional effector molecule expression (intracellular granzyme B, TNF-α, IFN-γ, and IL-2 combined; for gating, see Figure S4A) than cells expressing the CAR construct with the intermediate and short spacer (Figures 4B–4D). T cells expressing a CAR with the intermediate spacer induced slightly lower cytotoxic effects at some E:T cell ratios but equivalent effector molecule polyfunctionality/expression compared to cells with a CAR containing the short spacer (Figures 4B–4D and S4B). There were slight differences in cytokine secretion profiles between CD8+ and CD4+ CAR T cells and target cells. Specifically, CD8+ cells showed increased expression of TNF-α, IFN-γ, and IL-2 with the short spacer, whereas release of perforin, granzyme A, and granzyme B was higher in cells with the intermediate spacer against both ML-1 and K562 cells. In contrast, in CD4+ CAR T cells, increased TNF-α, perforin, and granzyme A and B were seen in CD4+ cells with the short spacer against ML-1 cells but not K562 cells, whereas higher IFN-γ and IL-2 expression was found in cells with the intermediate spacer (Figure S5). While expression of the short spacer transgene was higher, expression of the intermediate and long spacer transgenes was equivalent (Figure S6), implying an effect on cytotoxicity and function independent of transgene expression.
Figure 4.

Specific targeting of different CD33 proteins with linker-optimized CD33PAN and CD33V-set CAR T cells
(A) CD8+ CD33PAN (1H7) or CD33V-set (A33 and My96) CAR T cells were co-cultured with ML-1 cells with CRISPR-Cas9-mediated deletion of the endogenous CD33 locus (CD33KO) or sublines engineered to overexpress CD33FL or CD33ΔE3-4 at various E:T cell ratios as indicated and assessed for cytotoxicity by chromium-51 release 4 h later. Shown are mean ± SEM from technical triplicates. (B) CD8+ or (C) CD4+ CD33PAN (1H7) CAR T cells expressing a short, intermediate, or long extracellular spacer were co-cultured with ML-1 cells with CRISPR-Cas9-mediated deletion of the endogenous CD33 locus (CD33KO) or parental CD33+ AML cell lines as indicated and assessed for cytotoxicity by chromium-51 release 4 h later. Shown are mean ± SEM from technical triplicates of one representative out of two separate experiments for CD4+ and CD8+ CAR T cells. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; ns (not significant) by two-way ANOVA with post hoc Tukey correction. (D) Percentage of CAR T cells positive for granzyme B, IL-2, TNF-α, and/or IFN-γ as assessed by intracellular staining and multiparameter flow cytometry of 1H7 CAR T cells expressing a short, intermediate, or long extracellular spacer following 24-h co-culture with parental CD33+ AML cell lines as indicated. Shown are average intracellular expression levels in concatenated technical triplicates repeated in two separate experiments.
CD33PAN CAR T cells show effective leukemic cell clearance in mice bearing human AML cell xenografts
We next examined CD33PAN CAR T cells in vivo using three firefly luciferase-expressing AML cell lines—HL-60, KG-1a, and MOLM-14—that we demonstrated to be lysed by 1H7 CD33PAN CAR T cells in vitro (Figures 4A, 4B, and S7) and are known to reliably engraft in immunodeficient NOD.scid IL-2rg−/− (NSG) mice without radiation35; the latter is important considering radiation may enhance CAR T cell reactivity and obscure the comparison of CAR constructs.36 NSG mice were injected with 1 × 106 GFP+ HL-60 cells via tail vein. One to two weeks later, mice received 1 × 106–1 × 107 combined CD4+ and CD8+ 1H7 CAR T cells in a 1:1 ratio bearing either the short or intermediate spacer (cell doses similar to those used in other mouse studies testing CAR T cell therapies37,38,39,40,41), control CD19-directed CAR T cells (FMC63) if AML cells expressed CD19,42 or PBS control. As shown in Figure 5A, CD33PAN CAR T cells prolonged survival in a dose-dependent manner, with no deaths from AML recorded within 100 days post-tumor transfer in mice that received 1 × 107 1H7 CAR T cells. CD33PAN CAR T cells also prolonged survival in mice that received 1 × 106 HL-60 cells and a low of dose of 1 × 106 CAR T cells when compared to control CD19-directed (FMC63) CAR T cells that displayed equivalent median overall survival to PBS control (Figures 5A–5C). As demonstrated in Figure 5C, CD33PAN CAR T cells harboring the intermediate spacer showed a statistically non-significant difference in survival to animals that received CAR T cells with the short spacer.
Figure 5.

In vivo activity of 1H7 CD33PAN CAR T cells
Immunodeficient NSG mice were injected with GFP-expressing HL-60 (A–C), KG-1a (D), or MOLM-14 (E) cells. (A) Survival of mice bearing HL-60 cells that received either 2 × 106 1H7.int, 1 × 107 1H7.int, or vehicle (PBS) control 2 weeks after transplantation of 2 × 106 HL-60 cells (n = 5 per HL-60 group, n = 4 in PBS group). (B) Survival of mice bearing HL-60 cells that received either 1 × 106 1H7.int or 1 × 106 FMC63 CAR T cells 1 week after receiving 1 × 106 HL-60 cells (n = 5 per group). (C) Survival of mice bearing HL-60 cells that received either 2 × 106 1H7.int, 2 × 106 1H7.sh, or 2 × 106 FMC63 CAR T cells 2 weeks after receiving 2 × 106 HL-60 cells (n = 5 per group). (D) Survival of mice bearing KG-1a cells having received either 2 × 106 CAR T cells or vehicle control 2 weeks following transfer of 2 × 106 KG-1a cells (n = 5 in KG-1a group, n = 3 in PBS group). (E) CD33 expression on GFP+ KG-1a AML cells in peripheral blood as measured by flow cytometry in mice that received 2 × 106 1H7 CAR T cells or (F) PBS control. (G) CD8+ CD33PAN (1H7.int) or CD33V-set (A33.int) CAR T cells were co-cultured with MOLM-14 cells at various E:T cell ratios as indicated and assessed for cytotoxicity by chromium-51 release 4 h later. Shown are mean ± SEM from technical triplicates. ∗∗p < 0.01; ∗∗∗∗p < 0.0001; ns (not significant) by two-way ANOVA with post hoc Tukey correction. (H) Survival of NSG mice injected with 5 × 105 firefly luciferase-expressing MOLM-14 that then received CD33PAN (1H7.int) or CD33V-set (A33.int) 5 × 106 CAR T cells in a 1:1 ratio of CD4:CD8 T cells 1 week later by tail vein injection (n = 5 per group). Shown are mean ± SEM. ∗∗p < 0.01; ns (not significant) by log rank test.
Antigenic heterogeneity presents a major challenge to therapeutic success in AML.18,19 Thus, we sought to assess the efficacy of CD33PAN CAR T cells to clear CD33+ cells in mice bearing KG-1a cells, which, in vivo, lack CD33 on a subset (16%) of cells. In this study, NSG mice received 2 × 106 combined CD4+ and CD8+ 1H7 CAR T cells in a 1:1 ratio 2 weeks after tail vein injection of KG-1a cells, and were then bled weekly to serially assess CD33 expression on circulating human leukemia cells. A lower dose of CAR T cells was used in this experiment to circumvent potential xenogeneic graft-versus-host disease (GVHD) in this model of less proliferative AML. Supporting the notion of potent target-specific activity of CD33PAN CAR T cells against AML cells expressing CD33, clearance of CD33+ cells was seen in the peripheral blood of mice that received 1H7 CAR T cells but not in the blood of control mice, with mice eventually dying with an increasing burden of CD33− leukemia cells (Figures 5D–5F; CD33− cells gated by fluorescence-minus-one [FMO] as per Figure S9). We then chose to compare CD33PAN (1H7) CAR T cells to CD33V-set (A33) CAR T cells (both bearing intermediate spacers in their CARs) in a model of aggressive AML. The 1H7 CD8+ CAR T cells showed increased in vitro cytotoxicity against MOLM-14 cells when compared to A33 CD8+ CAR T cells at most E:T ratios (Figure 5G). We explored this difference further in NSG mice xenotransplanted with MOLM-14 cells, which have previously been shown to reproducibly lead to animals’ deaths from leukemia within a median of 3 weeks.35 NSG mice were injected with 5 × 105 leukemia cells via tail vein, and 1 week later, mice received 5 × 106 1H7 or A33 CAR T cells in a CD4:CD8 ratio of 1:1 or PBS control and were then sequentially imaged for bioluminescent expression to dynamically assess tumor burdens.35 As shown in Figure 5H, 1H7 CAR T cells prolonged survival relative to A33 CAR T cells, with mice eventually succumbing to high tumor burden (Figure S8E). In all cases, 1H7 CAR T cells extended the survival of mice bearing xenografts without evidence of weight loss at later time points when compared with control or A33 CAR T cell-treated mice (Figure S10).
CD33PAN CAR T cells show superior cytotoxicity compared to CD33V-set CAR T cells without evidence of exhaustion
The data from the mouse model with MOLM-14 cells depicted in Figure 5H were consistent with the hypothesis that the shorter membrane-to-membrane synaptic distance leads to enhanced cytotoxicity of CD33PAN compared to CD33V-set CAR T cells. To test this notion in more detail, we conducted a series of comparative in vitro studies between CD33PAN and CD33V-set CAR T cells. In these studies, done with CD8+ cells, 1H7 CD33PAN CAR T cells displayed greater cytotoxicity than My96 CD33V-set CAR T cells against several human acute leukemia cell lines spanning a range of CD33 expression (Figures 6A and 6B). My96 transgene expression was higher in these CD8+ CAR T cells compared to 1H7 CAR T cells (Figure S11). Importantly, CD33PAN CAR T cells showed no increase in coincident expression of exhaustion markers (PD-1, TIM-3, LAG-3; Figure 6C) relative to CD33V-set CAR T cells, neither without CD33 nor when stimulated with CD33+ cells for 3 days.
Figure 6.

Comparative analysis of in vitro cytotoxicity of 1H7 CD33PAN and My96 CD33V-set CAR T cells against target cells expressing various levels of CD33
(A) Quantibrite-PE assessment of cell surface expression of CD33 on human leukemia cell lines. Shown are mean ± SEM from technical triplicates. (B) CD8+ CD33PAN (1H7) or CD8+ CD33V-set (My96) CAR T cells were co-cultured with either ML-1 cells with CRISPR-Cas9-mediated deletion of the endogenous CD33 locus (CD33KO) or parental CD33+ AML cell lines as indicated and assessed for cytotoxicity by chromium-51 release assay 4 h later. Shown are mean ± SEM from technical triplicates of one representative out of three separate experiments. (C) Percentage of CAR T cells positive for PD-1, LAG-3, and TIM-3 following 3 days of co-culture of CD8+ CD33PAN (1H7) or CD33V-set (My96) CAR T cells with cell lines or media as indicated. Shown are concatenated values from technical triplicates. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; ns (not significant) by two-way ANOVA with post hoc Tukey correction).
To further study the cytotoxic properties of CD33PAN CAR T cells, we generated CAR constructs like the optimized 1H7 construct using scFvs from two additional CD33PAN mAbs that we previously raised (CD33-9G2 and CD33-3A5). As shown in Figures 7A–7C, 9G2 and 1H7 CAR T cells expressed similar numbers of transduction marker molecules/cells and showed equivalent cytotoxicity in CD4+ and CD8+ T cell fractions, whereas reduced cytotoxicity (and reduced CAR-containing transgene expression) was observed with 3A5 CD8+ CAR T cells and, to a lesser extent, with CD4+ CAR T cells.
Figure 7.

Comparative analysis of in vitro cytotoxicity of CD33PAN CAR T cells
(A) CD8+ or (B) CD4+ CD33PAN (1H7, 9G2, and 3A5) CAR T cells were co-cultured with ML-1 cells with CRISPR-Cas9-mediated deletion of the endogenous CD33 locus (CD33KO) or parental CD33+ AML cells and assessed for cytotoxicity by flow cytometry after overnight co-culture. Percent cell death was calculated by subtracting percent cell death from background cell death in non-T cell-containing wells. (C) Surface tCD19 expression as measured by Quantibrite-PE. Shown are mean ± SEM from technical triplicates of one representative out of two separate experiments. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; ns (not significant) by two-way ANOVA with post hoc Tukey correction.
CD33PAN CAR T cells show superior in vitro activity against primary human AML cells compared to CD33V-set CAR T cells
In a final series of studies, we assessed the anti-leukemic activity of CD33PAN CAR T cells against primary human AML cells. As first step, we quantified surface expression of CD33 with both a CD33V-set-binding mAb (P67.6) and a CD33PAN-binding mAb (9G2) in a set of 10 biospecimens from patients with relapsed/refractory AML cells on bulk cells (gated on CD45 vs. side scatter), leukemic progenitor/stem cells (gated on CD34+CD38−), and other leukemic cells (gated on CD34+CD38+; see Figure S12 for gating strategy). As shown in Figure 8A, P67.6 and 9G2 detected similar numbers of CD33 molecules on the surface of bulk cells and CD34+/CD38+ blasts. However, a higher number of CD33 molecules was measured with 9G2 relative to P67.6 on CD34+/CD38− cells. In a different set of 14 samples from patients with identifiable AML blasts spanning a range of CD33 expression levels (Figure 8B), we then assessed the relative cytotoxic properties of CD33V-set (My96) and CD33PAN (1H7 and 9G2) CAR T cells. Given our results above showing decreasing membrane-membrane distance improved cytotoxicity in CD4+ CAR T cells (Figure 2), we assessed both CD8+ and CD4+ CAR T cells in these experiments with primary human AML samples. As summarized in Figures 8C and 8D, all three CAR T cell products induced CD33-specific cytotoxicity in an E:T cell ratio-dependent manner compared to non-transduced T cells and non-targeting CAR T cells (13R4). CD33PAN CAR T cells showed statistically significantly greater cytotoxicity than CD33V-set (My96) CAR T cells at higher E:T cell ratios in both CD8+ and CD4+ T cell fractions.
Figure 8.

Targeting of primary human AML cells with CD33PAN and CD33V-set CAR T cells
(A) Bone marrow specimens from 10 patients with relapsed/refractory AML were stained with PE-conjugated CD33V-set (P67.6) and CD33PAN antibody (9G2). Cells were gated on all AML cells (CD45 vs. SCC), AML blasts (CD34+/CD38+), and AML progenitor/stem cells (CD34+/CD38−). Peripheral blood AML samples from a separate cohort of patients was assessed for expression of CD33 by P67.6 (B) and cell death when co-cultured with CD8+ (C) and CD4+ (D) engineered T cells expressing CD33PAN CAR (9G2 and 1H7), CD33V-set CAR (My96), or a non-binding CAR (13R4) or untransduced T cells. Percent cell death was calculated by subtracting percent cell death from background cell death in non-T cell-containing wells. Bar graphs show mean ± SEM. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 by paired two-way ANOVA with Tukey post hoc correction.
Discussion
Limited clinical efficacy of current CD33 CAR T cells prompted us to identify novel ways of how such adoptive cellular therapies could be optimized. We primarily focused on the location of the binding epitope on CD33 because we previously found that engaging CD33 close to the membrane enhances the efficacy of CD33/CD3 BiAbs,31 consistent with data from other cell surface antigens showing enhanced activity of mAbs, BiAbs, or CAR T cells with membrane proximal targeting.24,25,26,27,28,29 Our results support three main conclusions: (1) decreasing the distance from the T cell to the leukemia cell membrane by targeting the CD33 membrane proximally and by shortening the spacer between the scFv and the transmembrane domain of the CAR enhances the cytolytic activity and TNF-α production of CAR T cells; (2) spacer-optimized CD33PAN CAR T cells (i.e., cells targeting the membrane-proximal C2-set domain of CD33) are highly potent in vivo and in vitro against CD33+ human acute leukemia cells, including cells with a very limited display of CD33; and (3) CD33PAN CAR T cells have superior in vitro and in vivo efficacy compared to CD33V-set CAR T cells. Together, these data provide a compelling rationale to study CD33PAN CAR T cells further toward possible clinical testing. Such testing will need to include careful examination of the relative effects of CD33PAN CAR T cells toward normal and neoplastic CD33+ cells.
In our studies with conventional CD33V-set CAR T cells, we determined the optimal distance from the T cell membrane to the leukemia cell membrane via two strategies. In the first strategy, we engineered CARs using different spacer lengths separating the scFv from the transmembrane domain of the CAR construct to identify the spacer that provides optimal CAR T cell activity. Relative to T cells expressing a CD33V-set-directed CAR with a long spacer, those with a short spacer showed enhanced cytolytic activity. Thus, shortening the membrane-membrane distance via a short spacer is one strategy to maximize CD33 CAR T cell efficacy, suggesting that there is no significant steric hindrance—in other words, the target antigen on CD33 remains accessible to engineered T cells despite limited flexibility within the extracellular region of the CAR.
In the second strategy, we diminished the distance between the binding epitope on CD33 and the membrane of the leukemia cell by using an artificial CD33 protein in which the V-set domain was brought adjacent to the cell membrane. We did not find any evidence of steric hindrance when short-spacer CD33V-set CAR T cells were used against human leukemia cells expressing the targeted domain of CD33 immediately adjacent to the cell membrane. In fact, such CAR T cells yielded greater cytotoxicity and produced more TNF-α with these target cells than cells expressing full-length CD33, again indicating minimizing the cell-cell distance enhances the efficacy of CD33 CAR T cells. Increased TNF-α production may be an important feature of efficacy-optimized CAR T cells, considering TNF-α expression has been associated with enhanced responses with CD19 CAR T cell therapy.43 In our studies, we observed improved cytotoxicity when membrane-membrane distances were minimized, not only with CD8+ but also with CD4+ CAR T cells. The latter is significant in light of data associating cytotoxic CD4+ CAR T cells with prolonged remissions in patients following CAR T cell therapy.44 Thus, enhancing cytotoxicity of CD4+ CAR T cells by optimizing the membrane-membrane synaptic distance might increase the likelihood of success with CD33 CAR T cells.
Developing CAR T cells recognizing the membrane-proximal C2-set domain of CD33 logically follows our findings. Our experiments with CD33PAN CAR T cells demonstrate the effectiveness of this approach against multiple human leukemia cell lines and primary human AML cell samples displaying a range of CD33 expression levels. Because the C2-set domain of CD33 is expressed in all known CD33 variants,5,33 targeting the C2-set domain of CD33 via CD33PAN-directed CARs offers the advantage over CD33V-set CAR T cells of targeting all known isoforms of CD33, potentially limiting CD33 escape variants. Our immunophenotyping studies of a limited series of biospecimens from patients with AML also suggested the possibility that some AML cell fractions might indeed display CD33 molecules expressing only the C2-set, not the V-set domain—such cells would only be targetable with CD33PAN but not CD33V-set CAR T cells—although further studies in larger series of samples will be necessary to test this idea further. Notably, even in the setting of CD33PAN CAR T cells, short spacer CAR constructs yielded the greatest cytolytic activity.
As a central finding of our studies, careful comparative analyses between spacer-optimized CD33PAN and spacer-optimized CD33V-set CAR T cells showed greater cytolytic activity of CD33PAN CAR T cells in vitro against human leukemia cells expressing low, intermediate, or high levels of CD33, as well as against primary human AML cells. CD33PAN CAR T cells also resulted in better leukemia control and longer survival of mice bearing human AML cell xenografts relative to CD33V-set CAR T cells. In a model of human AML, in which a subset of cells lacked CD33, CD33PAN CAR T cells improved the survival of mice, which ultimately succumbed to CD33− leukemia, highlighting the pressure of these engineered cells to force target antigen-negative escape. Importantly, while CD33PAN CAR T cells showed better in vitro and in vivo efficacy than CD33V-set CAR T cells, we did not observe increased expression of PD-1, LAG-3, and/or TIM-3—markers of exhausted CAR T cells45—on CD33PAN CAR T cells.
Together, by showing that decreased membrane-membrane distance improves CAR T cell efficacy, our findings highlight the importance of epitope selection for optimized CD33 CAR T cell therapy. Demonstration of improved efficacy of CAR T cells when directed against membrane-proximal epitopes of CD33 charts a new course for highly effective CD33-targeted immunotherapy that is based on binding sequences from C2-set domain-recognizing CD33PAN mAbs.
Materials and methods
Transgene expression of CD33
pRRLsin.cPPT.MSCV lentivirus constructs containing an internal ribosome entry site-EGFP cassette for expression of full-length CD33 (CD33FL) or CD33 lacking the exon 3/4-encoded C2-set domain (CD33ΔE3-4; Figure 1A) have been described.31,33,34,46,47
Parental and engineered human leukemia cell lines
Human myeloid K562, KG-1a, ML-1, and OCI-AML3 cells and human lymphoid RS4;11 cells were cultured as described.31,34,48 MOLM-14 cells were cultured in RPMI-1640 with 10% fetal bovine serum (FBS; HyClone, Thermo Fisher Scientific, Waltham, MA). Lentivirally transduced sublines were generated at MOIs of 0.25–50, and EGFP+ cells were isolated by fluorescence-activated cell sorting (FACS).31,33,34,46,47,49 Cell lines were routinely tested for mycoplasma contamination and authenticated using standard short tandem repeat combined DNA index system typing.
Primary AML patient specimens
Frozen aliquots of primary AML patient specimens were obtained from an institutional repository under protocol 10,142 approved by the Fred Hutchinson Cancer Center (FHCC) institutional review board and cultured as described.31 All patients provided written informed consent for sample collection and use for research.
CAR T cell vectors
An epHIV7 lentiviral backbone encoding the CD28-transmembrane domain, the CD3zeta and 4-1BB intracellular signaling domains, and tCD19 as a transduction marker was used for all CAR constructs (Figure 1B).50,51 Due to non-specific staining, we were unable to directly quantify CD33 CAR expression; however, we have previously shown transduction marker expression to correlate with CAR expression with this CAR construct.52 Codon optimized nucleic acid sequences for scFvs of CD33 mAbs in heavy-chain/Gly-Ser linker/light-chain orientation were integrated together with the following spacers: short, hinge region of human immunoglobulin G4 (IgG4) (12 amino acids [aa]); intermediate, short spacer plus the CH3 domain of human IgG4 (combined, 119 aa); and long, intermediate spacer plus the CH2 domain of human IgG4 (combined, 228 aa). For V-set domain targeting of CD33, two CAR constructs were built from published scFvs: My96 (US patent 9,777,061, sequence ID: 146), and A33 (US patent 10,933,132, sequence ID: 227). For C2-set domain targeting, scFvs were generated from three CD33PAN mAbs we previously raised (CD33-1H7, CD33-9G2, and CD33-3A5).31
CAR T cell generation
CAR T cells were generated through the transduction of negatively selected CD8+ or CD4+ T cells from healthy donors as described.50 tCD19-expressing CD8+ and CD4+ cells were expanded in IL-7 and IL-15 (10 ng/mL; Peprotech, Rocky Hill, NJ) each for 12–14 days, with media and cytokine changes every other day.
Genetic deletion of CD33
CRISPR-Cas9 editing of CD33 was carried out as described.31,34,53 CD33− single cells were isolated via FACS and genomic DNA from individual clones sequenced to confirm disruption or frameshift mutation at all CD33 alleles.
Quantification of CD33 expression
Expression of CD33 and variants was quantified flow cytometrically using a directly labeled CD33 mAb (clone P67.6; BD Biosciences, San Jose, CA) or unlabeled P67.6 followed by APC-conjugated or phycoerythrin (PE)-conjugated goat anti-mouse Ig (Thermo Fisher Scientific). In some experiments, expression of CD33 was quantitated with Quantibrite-PE beads (BD Biosciences). To identify non-viable cells, samples were stained with DAPI or live-dead fixable violet stain (Thermo Fisher Scientific). Around 10,000–20,000 events were acquired flow cytometrically. CD33 expression on cryopreserved bone marrow aspirates and peripheral blood was measured with PE-conjugated mAbs P67.6 (BD Biosciences) and in-house PE-conjugated 9G2.
Quantification of in vitro cytokine expression/secretion and CAR T cell-induced cytotoxicity
Cytokine production was measured by co-culturing CAR T cells with GFP+ target cells or target cells labeled with CellVue Jade or Burgundy (Polysciences, Warrington, PA). After 12–20 h, cells were cultured with monensin (BD Biosciences) and a CD107a mAb for 4 h, followed by staining with live-dead fixable blue stain (Thermo Fisher Scientific) and mAbs against CD4, CD8, and CD3, fixation/permeabilization with the FoxP3 intracellular staining or Cytofix/Cytoperm Fixation/Permeabilization Kit (both BD Biosciences), and staining for TNF-α, IFN-γ, IL-2, and granzyme B. The percentage of marker-positive cells was calculated relative to an FMO control. Expression of markers and absolute target cell counts were flow cytometrically determined, collecting at least 10,000 events. Cell surface expression of PD-1, LAG-3, and TIM-3 was quantified by flow cytometry, with the percentage of marker-positive cells calculated relative to an FMO control. Secretion of soluble effector molecules (TNF-α, IFN-γ, IL-2, perforin, granzyme A, and granzyme B) was measured in duplicate with a custom LegendPlex Cytokine Bead Array (BioLegend, San Diego, CA). In early experiments, CAR T cell cytotoxicity was assessed following incubation with chromium-51-labeled target cells for 4 h as described.51 CAR T cell cytotoxicity by chromium-51 release assay was quantitated using the formula: (scintillation count experimental condition − scintillation count spontaneous wells)/(scintillation count maximum lysis by detergent − scintillation count spontaneous wells). Because of reagent unavailability related to the COVID-19 pandemic, CAR T cell cytotoxicity was quantified flow cytometrically 18–24 h after co-culture with tumor targets in later experiments, with cytotoxicity calculated relative to an antigen-negative, parental cell line or a no-T cell control, with dead cell identification by live-dead fixable blue or violet stain.54,55
In vivo testing of CAR T cells
All animal experiments were approved by the FHCC Institutional Animal Care and Use Committee (IACUC). NSG mice, 6–8 weeks old, randomly assigned to individual treatment groups, were intravenously injected with GFP-expressing and firefly luciferase-expressing AML cell lines. Between 1 and 2 weeks later, animals were injected with CAR T cells and monitored for tumor growth and survival as described.56 To quantitate circulating AML cells, mice were bled weekly by alternating retro-orbital bleeds, followed by ACK lysis and flow cytometric quantitation of CD33+ and CD33− populations within mCD45−/huCD45+ GFP+ cells. To minimize potential confounding by xenogeneic GVHD, animals were followed for only 100 days after tumor transfer. Following local IACUC requirements, mice were euthanized if they lost >20% body weight, showed abnormal behavior or appearance, or developed tumors that compromised normal function.
Statistical analysis
All in vitro and in vivo data were analyzed with Prism version 9.5.1 (GraphPad, San Diego, CA). All flow cytometry data were analyzed with FlowJo version 10 (BD Biosciences). Hypothesis tests of significance and significance levels were performed as indicated.
Data and code availability
For original data and reagents, please contact the corresponding author (rwalter@fredhutch.org).
Acknowledgments
The research reported in this publication was supported by the National Institutes of Health/National Cancer Institute (NIH/NCI; R21-CA245594 and R50-CA274319), as well as the FHCC Vector Core, which is funded by NIH/National Institute of Diabetes and Digestive and Kidney Diseases Cooperative Center of Excellence in Hematology grant (U54-DK106829). S.F. acknowledges the support of the Haematology Society of Australia and a New Zealand Clinical Fellowship Grant. E.L.K. acknowledges the support of the Paul Calabresi Career Development Award for Clinical Oncology in Pediatric and Medical Hematology/Oncology (NIH/NCI; K12-CA076930). R.B.W. acknowledges support from the José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research. We thank the Comparative Medicine facility for assistance in animal experiments and Dr. Derek L. Stirewalt for access to human AML samples.
Author contributions
S.F. conceptualized and designed the study, performed the research, analyzed and interpreted the data, and wrote the manuscript. S.Y.T.L., E.L.K., T.-D.P., M.C.L.-H., D.R.K., and J.H. performed the research and revised the manuscript. G.S.L. generated critical reagents, analyzed and interpreted the data, and revised the manuscript. H.-P.K. assisted in conceptualizing the study, interpreting the data, and revising the manuscript. C.J.T. conceptualized and designed the study, participated in data analysis and interpretation, and revised the manuscript. R.B.W. conceptualized and designed this study, participated in data analysis and interpretation, and wrote the manuscript. All authors gave final approval to submit for publication.
Declaration of interests
S.F., G.S.L., C.J.T., and R.B.W. filed a patent application related to CD33 antibodies and CD33-directed CAR T cell therapy. S.F. has filed patents on optimizing CAR T cell cytotoxicity, received research laboratory grants from Bristol Myers Squibb, and is a consultant for Prescient Therapeutics. E.L.K. received research support from Juno Therapeutics. H.-P.K. received support as the inaugural recipient of the José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research and the Stephanus Family Endowed Chair for Cell and Gene Therapy, and is or was a consultant to and has or had ownership interests with Rocket Pharma, Homology Medicines, VOR, and Ensoma. C.J.T. received research funding from Juno Therapeutics/BMS and Nektar Therapeutics; serves on advisory boards for Caribou Biosciences, T-CURX, Myeloid Therapeutics, ArsenalBio, Cargo Therapeutics, and Celgene/BMS Cell Therapy; has served on ad hoc advisory boards/consulting (last 12 months) for Nektar Therapeutics, Century Therapeutics, Legend Biotech, Allogene, Sobi, Syncopation Life Sciences, Prescient Therapeutics, Orna Therapeutics, and IGM Biosciences; and holds stock options in Eureka Therapeutics, Caribou Biosciences, Myeloid Therapeutics, ArsenalBio, and Cargo Therapeutics. R.B.W. received laboratory research grants and/or clinical trial support from Aptevo, Celgene/Bristol Myers Squibb, ImmunoGen, Janssen, Jazz, Kite, Kura, Pfizer, and VOR, and has been a consultant to Wugen.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omton.2024.200854.
Supplemental information
References
- 1.Taussig D.C., Pearce D.J., Simpson C., Rohatiner A.Z., Lister T.A., Kelly G., Luongo J.L., Danet-Desnoyers G.A.H., Bonnet D. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106:4086–4092. doi: 10.1182/blood-2005-03-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walter R.B., Appelbaum F.R., Estey E.H., Bernstein I.D. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood. 2012;119:6198–6208. doi: 10.1182/blood-2011-11-325050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cowan A.J., Laszlo G.S., Estey E.H., Walter R.B. Antibody-based therapy of acute myeloid leukemia with gemtuzumab ozogamicin. Front. Biosci. 2013;18:1311–1334. doi: 10.2741/4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laszlo G.S., Estey E.H., Walter R.B. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014;28:143–153. doi: 10.1016/j.blre.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Godwin C.D., Gale R.P., Walter R.B. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia. 2017;31:1855–1868. doi: 10.1038/leu.2017.187. [DOI] [PubMed] [Google Scholar]
- 6.Knapp D.J.H.F., Hammond C.A., Hui T., van Loenhout M.T.J., Wang F., Aghaeepour N., Miller P.H., Moksa M., Rabu G.M., Beer P.A., et al. Single-cell analysis identifies a CD33(+) subset of human cord blood cells with high regenerative potential. Nat. Cell Biol. 2018;20:710–720. doi: 10.1038/s41556-018-0104-5. [DOI] [PubMed] [Google Scholar]
- 7.Hauswirth A.W., Florian S., Printz D., Sotlar K., Krauth M.T., Fritsch G., Schernthaner G.H., Wacheck V., Selzer E., Sperr W.R., Valent P. Expression of the target receptor CD33 in CD34+/CD38-/CD123+ AML stem cells. Eur. J. Clin. Invest. 2007;37:73–82. doi: 10.1111/j.1365-2362.2007.01746.x. [DOI] [PubMed] [Google Scholar]
- 8.Pollard J.A., Alonzo T.A., Loken M., Gerbing R.B., Ho P.A., Bernstein I.D., Raimondi S.C., Hirsch B., Franklin J., Walter R.B., et al. Correlation of CD33 expression level with disease characteristics and response to gemtuzumab ozogamicin containing chemotherapy in childhood AML. Blood. 2012;119:3705–3711. doi: 10.1182/blood-2011-12-398370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krupka C., Kufer P., Kischel R., Zugmaier G., Bögeholz J., Köhnke T., Lichtenegger F.S., Schneider S., Metzeler K.H., Fiegl M., et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell-engaging antibody AMG 330. Blood. 2014;123:356–365. doi: 10.1182/blood-2013-08-523548. [DOI] [PubMed] [Google Scholar]
- 10.Perna F., Berman S.H., Soni R.K., Mansilla-Soto J., Eyquem J., Hamieh M., Hendrickson R.C., Brennan C.W., Sadelain M. Integrating proteomics and transcriptomics for systematic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell. 2017;32:506–519.e5. doi: 10.1016/j.ccell.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haubner S., Perna F., Köhnke T., Schmidt C., Berman S., Augsberger C., Schnorfeil F.M., Krupka C., Lichtenegger F.S., Liu X., et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia. 2019;33:64–74. doi: 10.1038/s41375-018-0180-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duan S., Paulson J.C. Siglecs as immune cell checkpoints in disease. Annu. Rev. Immunol. 2020;38:365–395. doi: 10.1146/annurev-immunol-102419-035900. [DOI] [PubMed] [Google Scholar]
- 13.Willier S., Rothämel P., Hastreiter M., Wilhelm J., Stenger D., Blaeschke F., Rohlfs M., Kaeuferle T., Schmid I., Albert M.H., et al. CLEC12A and CD33 coexpression as a preferential target for pediatric AML combinatorial immunotherapy. Blood. 2021;137:1037–1049. doi: 10.1182/blood.2020006921. [DOI] [PubMed] [Google Scholar]
- 14.Hills R.K., Castaigne S., Appelbaum F.R., Delaunay J., Petersdorf S., Othus M., Estey E.H., Dombret H., Chevret S., Ifrah N., et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15:986–996. doi: 10.1016/S1470-2045(14)70281-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walter R.B. Expanding use of CD33-directed immunotherapy. Expet Opin. Biol. Ther. 2020;20:955–958. doi: 10.1080/14712598.2020.1788540. [DOI] [PubMed] [Google Scholar]
- 16.Wang Q.S., Wang Y., Lv H.Y., Han Q.W., Fan H., Guo B., Wang L.L., Han W.D. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol. Ther. 2015;23:184–191. doi: 10.1038/mt.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walter R.B. Investigational CD33-targeted therapeutics for acute myeloid leukemia. Expet Opin. Invest. Drugs. 2018;27:339–348. doi: 10.1080/13543784.2018.1452911. [DOI] [PubMed] [Google Scholar]
- 18.Acharya U.H., Walter R.B. Chimeric antigen receptor (CAR)-modified immune effector cell therapy for acute myeloid leukemia (AML) Cancers. 2020;12:3617. doi: 10.3390/cancers12123617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fiorenza S., Turtle C.J. CAR-T cell therapy for acute myeloid leukemia: preclinical rationale, current clinical progress, and barriers to success. BioDrugs. 2021;35:281–302. doi: 10.1007/s40259-021-00477-8. [DOI] [PubMed] [Google Scholar]
- 20.Tambaro F.P., Singh H., Jones E., Rytting M., Mahadeo K.M., Thompson P., Daver N., DiNardo C., Kadia T., Garcia-Manero G., et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia. 2021;35:3282–3286. doi: 10.1038/s41375-021-01232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maucher M., Srour M., Danhof S., Einsele H., Hudecek M., Yakoub-Agha I. Current limitations and perspectives of chimeric antigen receptor-T-cells in acute myeloid leukemia. Cancers. 2021;13:6157. doi: 10.3390/cancers13246157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koedam J., Wermke M., Ehninger A., Cartellieri M., Ehninger G. Chimeric antigen receptor T-cell therapy in acute myeloid leukemia. Curr. Opin. Hematol. 2022;29:74–83. doi: 10.1097/MOH.0000000000000703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pasvolsky O., Daher M., Alatrash G., Marin D., Daver N., Ravandi F., Rezvani K., Shpall E., Kebriaei P. CARving the path to allogeneic CAR T cell therapy in acute myeloid leukemia. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bluemel C., Hausmann S., Fluhr P., Sriskandarajah M., Stallcup W.B., Baeuerle P.A., Kufer P. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol. Immunother. 2010;59:1197–1209. doi: 10.1007/s00262-010-0844-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin T.S. Ofatumumab: a novel monoclonal anti-CD20 antibody. Pharmgenomics Pers. Med. 2010;3:51–59. doi: 10.2147/pgpm.s6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haso W., Lee D.W., Shah N.N., Stetler-Stevenson M., Yuan C.M., Pastan I.H., Dimitrov D.S., Morgan R.A., FitzGerald D.J., Barrett D.M., et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cleary K.L.S., Chan H.T.C., James S., Glennie M.J., Cragg M.S. Antibody distance from the cell membrane regulates antibody effector mechanisms. J. Immunol. 2017;198:3999–4011. doi: 10.4049/jimmunol.1601473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J., Stagg N.J., Johnston J., Harris M.J., Menzies S.A., DiCara D., Clark V., Hristopoulos M., Cook R., Slaga D., et al. Membrane-proximal epitope facilitates efficient T cell synapse formation by anti-FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell. 2017;31:383–395. doi: 10.1016/j.ccell.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao Q., Zhang X., Tu L., Cao J., Hinrichs C.S., Su X. Size-dependent activation of CAR-T cells. Sci. Immunol. 2022;7 doi: 10.1126/sciimmunol.abl3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dagher O.K., Posey A.D., Jr. Forks in the road for CAR T and CAR NK cell cancer therapies. Nat. Immunol. 2023;24:1994–2007. doi: 10.1038/s41590-023-01659-y. [DOI] [PubMed] [Google Scholar]
- 31.Godwin C.D., Laszlo G.S., Fiorenza S., Garling E.E., Phi T.D., Bates O.M., Correnti C.E., Hoffstrom B.G., Lunn M.C., Humbert O., et al. Targeting the membrane-proximal C2-set domain of CD33 for improved CD33-directed immunotherapy. Leukemia. 2021;35:2496–2507. doi: 10.1038/s41375-021-01160-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sterner R.C., Sterner R.M. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11:69. doi: 10.1038/s41408-021-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laszlo G.S., Harrington K.H., Gudgeon C.J., Beddoe M.E., Fitzgibbon M.P., Ries R.E., Lamba J.K., McIntosh M.W., Meshinchi S., Walter R.B. Expression and functional characterization of CD33 transcript variants in human acute myeloid leukemia. Oncotarget. 2016;7:43281–43294. doi: 10.18632/oncotarget.9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Godwin C.D., Laszlo G.S., Wood B.L., Correnti C.E., Bates O.M., Garling E.E., Mao Z.J., Beddoe M.E., Lunn M.C., Humbert O., et al. The CD33 splice isoform lacking exon 2 as therapeutic target in human acute myeloid leukemia. Leukemia. 2020;34:2479–2483. doi: 10.1038/s41375-020-0755-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saland E., Boutzen H., Castellano R., Pouyet L., Griessinger E., Larrue C., de Toni F., Scotland S., David M., Danet-Desnoyers G., et al. A robust and rapid xenograft model to assess efficacy of chemotherapeutic agents for human acute myeloid leukemia. Blood Cancer J. 2015;5:e297. doi: 10.1038/bcj.2015.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugita M., Yamazaki T., Alhomoud M., Martinet J., Latouche J.B., Golden E., Boyer O., Van Besien K., Formenti S.C., Galluzzi L., Guzman M.L. Radiation therapy improves CAR T cell activity in acute lymphoblastic leukemia. Cell Death Dis. 2023;14:305. doi: 10.1038/s41419-023-05829-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider D., Xiong Y., Hu P., Wu D., Chen W., Ying T., Zhu Z., Dimitrov D.S., Dropulic B., Orentas R.J. A unique human immunoglobulin heavy chain variable domain-only CD33 CAR for the treatment of acute myeloid leukemia. Front. Oncol. 2018;8:539. doi: 10.3389/fonc.2018.00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borot F., Wang H., Ma Y., Jafarov T., Raza A., Ali A.M., Mukherjee S. Gene-edited stem cells enable CD33-directed immune therapy for myeloid malignancies. Proc. Natl. Acad. Sci. USA. 2019;116:11978–11987. doi: 10.1073/pnas.1819992116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baroni M.L., Sanchez Martinez D., Gutierrez Aguera F., Roca Ho H., Castella M., Zanetti S.R., Velasco Hernandez T., Diaz de la Guardia R., Castaño J., Anguita E., et al. 41BB-based and CD28-based CD123-redirected T-cells ablate human normal hematopoiesis in vivo. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2020-000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jetani H., Navarro-Bailón A., Maucher M., Frenz S., Verbruggen C., Yeguas A., Vidriales M.B., González M., Rial Saborido J., Kraus S., et al. Siglec-6 is a novel target for CAR T-cell therapy in acute myeloid leukemia. Blood. 2021;138:1830–1842. doi: 10.1182/blood.2020009192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qin H., Yang L., Chukinas J.A., Shah N., Tarun S., Pouzolles M., Chien C.D., Niswander L.M., Welch A.R., Taylor N., et al. Systematic preclinical evaluation of CD33-directed chimeric antigen receptor T cell immunotherapy for acute myeloid leukemia defines optimized construct design. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2021-003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uhlen M., Karlsson M.J., Zhong W., Tebani A., Pou C., Mikes J., Lakshmikanth T., Forsström B., Edfors F., Odeberg J., et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science. 2019;366 doi: 10.1126/science.aax9198. [DOI] [PubMed] [Google Scholar]
- 43.Finney O.C., Brakke H.M., Rawlings-Rhea S., Hicks R., Doolittle D., Lopez M., Futrell R.B., Orentas R.J., Li D., Gardner R.A., Jensen M.C. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J. Clin. Invest. 2019;129:2123–2132. doi: 10.1172/JCI125423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Melenhorst J.J., Chen G.M., Wang M., Porter D.L., Chen C., Collins M.A., Gao P., Bandyopadhyay S., Sun H., Zhao Z., et al. Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature. 2022;602:503–509. doi: 10.1038/s41586-021-04390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Long A.H., Haso W.M., Shern J.F., Wanhainen K.M., Murgai M., Ingaramo M., Smith J.P., Walker A.J., Kohler M.E., Venkateshwara V.R., et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015;21:581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walter R.B., Raden B.W., Kamikura D.M., Cooper J.A., Bernstein I.D. Influence of CD33 expression levels and ITIM-dependent internalization on gemtuzumab ozogamicin-induced cytotoxicity. Blood. 2005;105:1295–1302. doi: 10.1182/blood-2004-07-2784. [DOI] [PubMed] [Google Scholar]
- 47.Laszlo G.S., Gudgeon C.J., Harrington K.H., Dell'Aringa J., Newhall K.J., Means G.D., Sinclair A.M., Kischel R., Frankel S.R., Walter R.B. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood. 2014;123:554–561. doi: 10.1182/blood-2013-09-527044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laszlo G.S., Orozco J.J., Kehret A.R., Lunn M.C., Huo J., Hamlin D.K., Scott Wilbur D., Dexter S.L., Comstock M.L., O'Steen S., et al. Development of [(211)At]astatine-based anti-CD123 radioimmunotherapy for acute leukemias and other CD123+ malignancies. Leukemia. 2022;36:1485–1491. doi: 10.1038/s41375-022-01580-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laszlo G.S., Gudgeon C.J., Harrington K.H., Walter R.B. T-cell ligands modulate the cytolytic activity of the CD33/CD3 BiTE antibody construct, AMG 330. Blood Cancer J. 2015;5 doi: 10.1038/bcj.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turtle C.J., Hanafi L.A., Berger C., Hudecek M., Pender B., Robinson E., Hawkins R., Chaney C., Cherian S., Chen X., et al. Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci. Transl. Med. 2016;8 doi: 10.1126/scitranslmed.aaf8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berger C., Jensen M.C., Lansdorp P.M., Gough M., Elliott C., Riddell S.R. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Invest. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kirchmeier D.R., Sheih A., Fiorenza S., Hirayama A.V., Chou C., Kimble E., Gauthier J., Williams A., Price J., Correnti C., Turtle C.J. Recombinant CD19 glycomutant accurately and reproducibly detects CD19-directed CAR-T cells by flow cytometry [review] Blood. 2021;138:1724. [Google Scholar]
- 53.Humbert O., Laszlo G.S., Sichel S., Ironside C., Haworth K.G., Bates O.M., Beddoe M.E., Carrillo R.R., Kiem H.P., Walter R.B. Engineering resistance to CD33-targeted immunotherapy in normal hematopoiesis by CRISPR/Cas9-deletion of CD33 exon 2. Leukemia. 2019;33:762–808. doi: 10.1038/s41375-018-0277-8. [DOI] [PubMed] [Google Scholar]
- 54.Kandarian F., Sunga G.M., Arango-Saenz D., Rossetti M. A flow cytometry-based cytotoxicity assay for the assessment of human NK cell activity. J. Vis. Exp. 2017 doi: 10.3791/56191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu X., Zhang Y., Li Y., Schmidt-Wolf I.G.H. Improvements in flow cytometry-based cytotoxicity assay. Cytometry A. 2021;99:680–688. doi: 10.1002/cyto.a.24242. [DOI] [PubMed] [Google Scholar]
- 56.Sommermeyer D., Hill T., Shamah S.M., Salter A.I., Chen Y., Mohler K.M., Riddell S.R. Fully human CD19-specific chimeric antigen receptors for T-cell therapy. Leukemia. 2017;31:2191–2199. doi: 10.1038/leu.2017.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For original data and reagents, please contact the corresponding author (rwalter@fredhutch.org).