

Abstract
Aim:
Regulatory and health technology assessment (HTA) agencies have increasingly published frameworks, guidelines, and recommendations for the use of real-world evidence (RWE) in healthcare decision-making. Variations in the scope and content of these documents, with updates running in parallel, may create challenges for their implementation especially during the market authorization and reimbursement phases of a medicine's life cycle. This environmental scan aimed to comprehensively identify and summarize the guidance documents for RWE developed by most well-established regulatory and reimbursement agencies, as well as other organizations focused on healthcare decision-making, and present their similarities and differences.
Methods:
RWE guidance documents, including white papers from regulatory and HTA agencies, were reviewed in March 2024. Data on scope and recommendations from each body were extracted by two reviewers and similarities and differences were summarized across four topics: study planning, choosing fit-for-purpose data, study conduct, and reporting. Post-authorization or non-pharmacological guidance was excluded.
Results:
Forty-six documents were identified across multiple agencies; US FDA produced the most RWE-related guidance. All agencies addressed specific and often similar methodological issues related to study design, data fitness-for-purpose, reliability, and reproducibility, although inconsistency in terminologies on these topics was noted. Two HTA bodies (National Institute for Health and Care Excellence [NICE] and Canada's Drug Agency) each centralized all related RWE guidance under a unified framework. RWE quality tools and checklists were not consistently named and some differences in preferences were noted. European Medicines Agency, NICE, Haute Autorité de Santé, and the Institute for Quality and Efficiency in Health Care included specific recommendations on the use of analytical approaches to address RWE complexities and increase trust in its findings.
Conclusion:
Similarities in agencies' expectations on RWE studies design, quality elements, and reporting will facilitate evidence generation strategy and activities for manufacturers facing multiple, including global, regulatory and reimbursement submissions and re-submissions. A strong preference by decision-making bodies for local real-world data generation may hinder opportunities for data sharing and outputs from international federated data networks. Closer collaboration between decision-making agencies towards a harmonized RWE roadmap, which can be centrally preserved in a living mode, will provide manufacturers and researchers clarity on minimum acceptance requirements and expectations, especially as novel methodologies for RWE generation are rapidly emerging.
Keywords: drug development, frameworks, HTA, health technology assessment, real-world data, real-world evidence, regulatory, reimbursement
Plain language summary
What is the article about?
Real-world data (RWD) is collected outside clinical trial settings, from sources such as patient medical records from routine clinical practice. The knowledge gained from analyses of RWD is called real-world evidence (RWE) and it can be used alongside clinical trials to assess how effective and safe treatments perform in the real world. Organizations that assess the value of medicines have provided and continue to provide guidance on how to generate and use RWE for healthcare decisions, such as market authorizations and reimbursement. In this article we aimed to identify and summarize the guidance on RWE developed by regulatory and reimbursement agencies, as well as other organizations focused on healthcare decision-making, and present similarities and differences.
What are the results?
We looked at guidance documents from regulatory agencies (such as FDA and EMA), from organizations that assess the value of healthcare technologies compared with standard of care for pricing and reimbursement decisions (such as NICE), and from organizations focused on healthcare decision making (such as ISPOR, ISPE). Overall, the documents covered similar topics when talking about issues of data quality and of transparency in results reporting, but the documents differed in the level of detail and specificity in their recommendations. In preparation for regulatory and health technology assessment submissions, this maze of RWE frameworks and guidance can be unclear and cumbersome to navigate for manufacturers and researchers.
What do the results of the study mean?
The findings revealed a duplication of effort during the development of guidance documents and the lack of a uniform, clear set of guidelines and expectations. We believe more collaboration between the organizations is needed to improve clarity and efficiency of everyone involved. Ideally, a central resource with up-to-date information and standardized guidance and approaches should be established.
Shareable abstract
Confused by RWD/RWE guidance for healthcare decision-making? Review and comparison of guidance documents by regulatory and reimbursement agencies, as well as research groups, revealed the need for a harmonized roadmap and standards across agencies! #RWD #RWE #DrugDevelopment #EvidenceGeneration #Healthcare #Regularory #HTA
Data-driven healthcare decision-making is evolving at an exponential pace. Recent methodological developments have re-shaped the criteria for the types of data and evidence considered acceptable to substantiate the value of health technologies in regulatory and reimbursement submissions. Data from randomized controlled trials (RCT) still hold an undisputed place at the top of the evidence hierarchy to demonstrate the clinical value of a new product. However, there is an increased recognition that real-world data (RWD) and real-world evidence (RWE) have a unique role in providing valuable insight on the effectiveness and safety of treatments, and this realization is transforming the evidence generation landscape during the lifecycle of a healthcare technology.
The US FDA has defined RWD as data relating to patient health status and/or the delivery of healthcare routinely collected from a variety of sources (typically outside the clinical trial setting) and RWE as the clinical evidence about the usage and potential benefits or risks of a medical product derived from analysis of RWD. These definitions are now well established and have been adopted by most decision-makers and organizations conducting research on this topic [1].
The changing healthcare ecosystem creates the fruitful ground for the wider consideration of RWD and RWE in decision-making: unprecedented access to a large amount of patient data via digital technologies, the increased shift toward patient-centric healthcare, the need for early access of promising technologies especially in rare/very rare diseases, and the wider recognition that RCTs are not always sufficient sources to answer questions about patient generalizability and equity [2–4]. The transformative power of RWD and RWE alters the entire healthcare environment from drug development and clinical trial development to regulatory and reimbursement decision-making, and value-based care. Every stakeholder (industry, regulatory, reimbursement, market access, post-launch) holds a unique perspective of the benefits and risks when considering RWD.
Decision-makers, however, have continued to express their lack of trust in and skepticism of RWD/RWE to support effectiveness and safety claims of healthcare technologies; issues of data quality and validity combined with access and safety have contributed to delays in the recognition of the potential of RWE by authorities worldwide. Not only is no progress being made on alleviating these concerns, in fact, in some cases they are now seen to be regressing [5].
Against this background, agencies worldwide have created specific centers (e.g., the European Medicines Agency [EMA] Coordination Centre for the Data Analysis and Real-World Interrogation Network [DARWIN EU®], the US FDA's Advancing RWE Program) and alliances (e.g., RWE Collaborative through the Duke-Margolis Institute for Health Policy, Real-World Evidence Alliance) to monitor trends in RWD/RWE topics and methodologies and have started developing standards and frameworks to guide and standardize the integration of RWE in manufacturer submissions [6,7].
Healthcare decision-makers are attempting to keep pace with the general trend for wider use/acceptance of RWD/RWE by publishing guidance or position documents to clarify how their processes have been adjusted to accommodate the “disrupted” nature of this type of evidence in their assessments; this is especially true when RWD/RWE is integrated to support comparative effectiveness assessments. However, in a very short period, the landscape of RWE guidance/frameworks has gone from barren to overcrowded, with multiple publications around this topic from different organizations around the globe. The scope and content of these guidance documents and frameworks vary considerably, with several updates running in parallel. In the meantime, international organizations, such as the Duke-Margolis Center for Health Policy, The Professional Society for Health Economics and Outcomes Research (ISPOR) and International Society for Pharmacoepidemiology (ISPE), have invested in creating their own task forces, best practices, specific toolkits and checklists to promote the understanding and standardizing of RWE methods and data quality issues among their communities. This abundance of RWE guidance publications has added a layer of complexity on how manufacturers and researchers prepare most effectively for regulatory and health technology assessment (HTA)/reimbursement submissions, which are sometimes at a global level. In addition, manufacturers and researchers are constantly tasked with navigating a continually changing stream of RWD/RWE terminology, methodologies and guidance.
Against this background, this research aimed to identify existing RWD-/RWE-related guidance documents across decision-making (regulatory and HTA) agencies, international organizations and research institutes across the globe and pinpoint similarities and differences among a selected list of agencies.
Methods
The websites of the most well-established regulatory agencies, including the US FDA, EMA, the UK's Medicines and Healthcare products Regulatory Agency (MHRA), Health Canada, Taiwan FDA, China National Medical Products Administration, Japan's Pharmaceuticals and Medical Devices Agency, Singapore Health Sciences Authority and HTA/HTA-supporting agencies including Canada's Drug Agency (CDA-AMC), National Institute for Health and Care Excellence (NICE), Institute for Quality and Efficiency in Health Care (IQWiG), Haute Autorité de Santé (HAS), European Network for HTA (EUnetHTA) and the Center for Drug Evaluation Taiwan, were manually searched in March 2024 (independently by two reviewers) for RWE methodological guidance documents and frameworks, including white papers, quality standards and policies framing official positions of these agencies around RWD and RWE methods and topics. The individual websites searched, and search strategies are presented in Supplementary Table 1. A separate review was conducted for RWE-related documents endorsed by international, local, and academic organizations, as well as working groups focused on healthcare decision-making, including the US Institute for Clinical and Economic Review, ISPOR, ISPE, HTA International and Duke-Margolis Health Policy Center. A snowball search through organizational websites or reference tracking was also performed to identify any additional relevant documents. Documents were excluded if they were not published in English, were not endorsed or represented by the organizations listed above, or were related to digital technologies, medical devices, artificial intelligence, or disease-specific RWE topics. For pragmatic reasons and to keep the scope of this review manageable to allow comprehensive synthesis of findings, we excluded guidance related to pragmatic trials and post-marketing authorization guidance as RWD/RWE is routinely used for these assessments. A pre-designed form was designed for the purposes of this review to capture information per guidance on the scope and content of recommendations, including general information about the type of organization and date of publication. Extraction was independently conducted by two reviewers and a quality check was performed by a more senior researcher. Results were narratively synthesized to identify similarities and differences in guidance (including proposed tools/checklists) across agencies by mapping this information across four pre-defined phases in RWE studies: study planning, choosing fit-for-purpose data, study conduct and study reporting. Recommended tools or checklists were separately presented.
Results
Forty-six documents on RWD/RWE guidance were identified from regulatory and HTA agencies worldwide (Figure 1 & Supplementary Table 2). All, except for two documents (NICE Technical Support Document 17, 2015 and FDA, 2017), were published after 2018 [8,9]. All guidance documents represented a single agency or an institutional organization, except for some ISPOR/ISPE joint initiatives and for CDA-AMC, which aimed to harmonize RWE principles for both Canadian HTA agencies and regulators.
Figure 1. . Overview of identified real-world evidence guidance by geography.
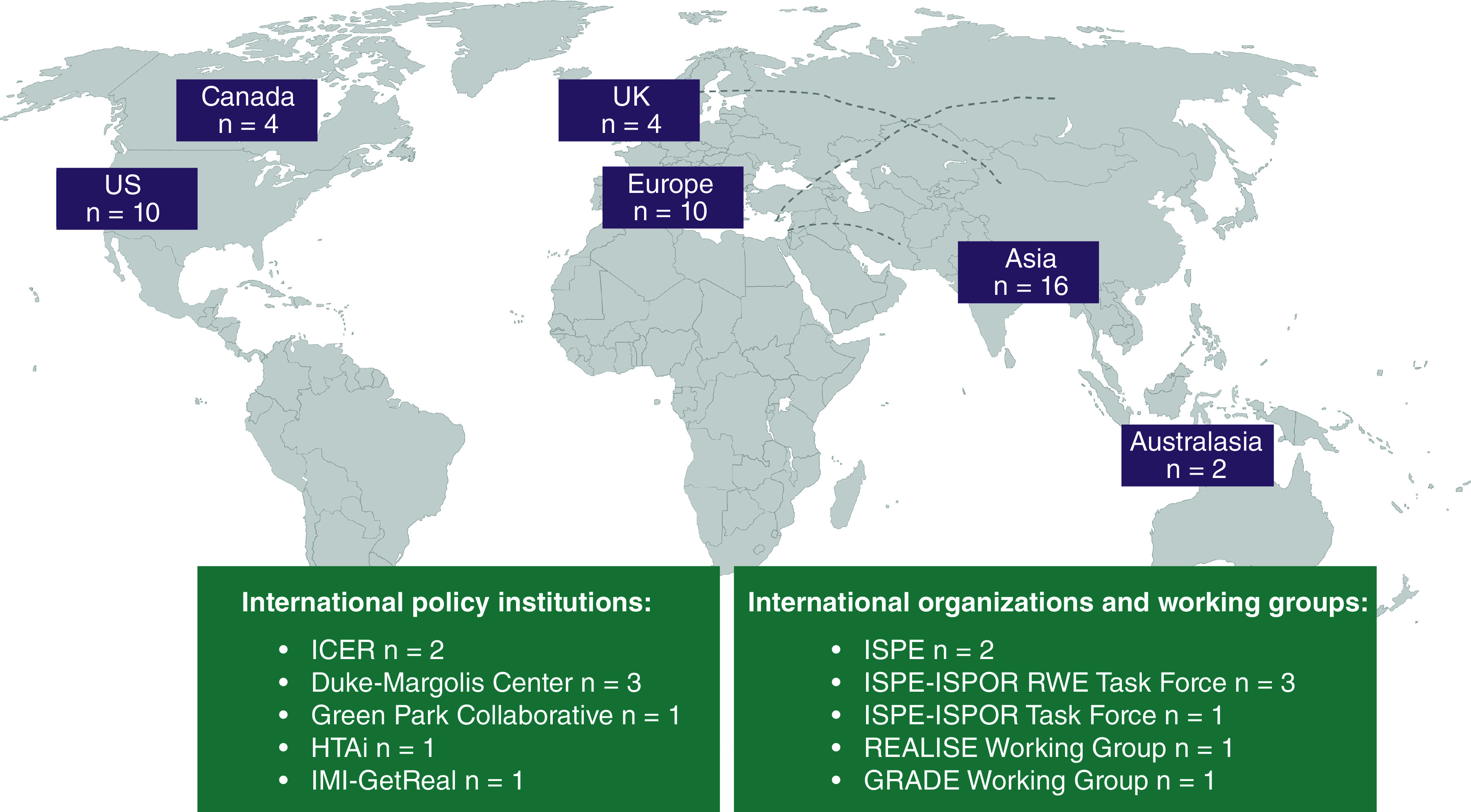
Purple rectangles represent counts for RWE publications from regulatory and HTA agencies.
GRADE: Grading of Recommendation Assessment, Development and Evaluation; HTAi: Health Technology Assessment international; ICER: Institute for Clinical and Economic Review; IMI: Innovative Medicines Initiative; ISPOR: The Professional Society for Health Economics and Outcomes Research; ISPE: International Society for Pharmacoepidemiology; REALISE: REAL World Data In Asia for Health Technology Assessment in Reimbursement; RWE: Real-world evidence.
In general, documents from regulatory agencies were referred to as explicit guidance to be followed by manufacturers when considering RWD/RWE in their submissions. This differs from HTA guidance documents, which were referred to as good practice recommendations or practical benchmarks related to methodological aspects of RWD/RWE that are not binding and do not set minimum requirements. The scope for each of the identified documents across regulatory and HTA agencies is summarized in Figure 2.
Figure 2. . Overview of identified real-world evidence guidance by drug development phase.
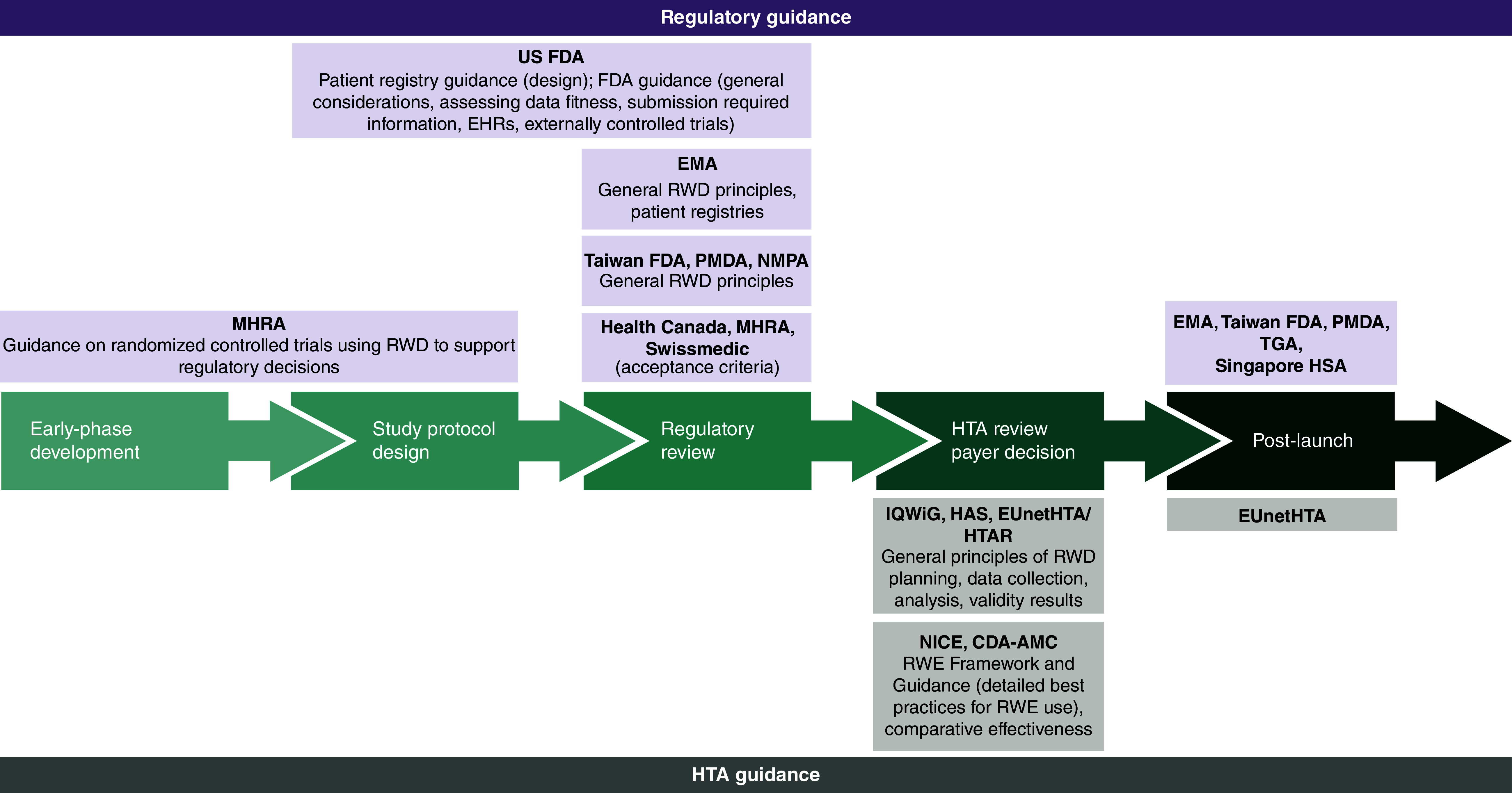
CDA-AMC: Canada's Drug Agency; EHR: Electronic health record; EMA: European Medicines Agency; EUnetHTA: European Network for Health Technology Assessment; FDA: Food and Drug Administration; HAS: Haute Autorité de Santé; HSA: Health Sciences Authority; HTA: Health technology assessment; HTAR: Health Technology Assessment Regulation; IQWiG: Institute for Quality and Efficiency in HealthCare; MHRA: Medicines and Healthcare products Regulatory Agency; NICE: National Institute for Health and Care Excellence; NMPA: National Medical Products Administration; PMDA: Pharmaceuticals and Medical Devices Agency; RWD: Real-world data; RWE: Real-world evidence; TGA: Therapeutic Goods Administration.
Among the included frameworks/guidance documents, only the US FDA specifically included RWE as part of a legislation act (21st Century Cures Act) which mandated the FDA to set up the RWE Program in 2018. As a result, FDA had the highest number of RWD/RWE guidance documents (n = 10).
In Europe, the first RWE framework and guidance by the EMA were in 2019 and 2020 with the Operational, Technical and Methodological Framework (OPTIMAL) and the Regulatory Science to 2025 strategic document, respectively [10–16]. Since then, several RWE-related guidance documents were published by the EMA on topics such as patient registries [11], electronic health records (EHR) [17] and external control arms [18]. The MHRA in the UK published guidance for RWD only when produced within the remit of a clinical trial development program (e.g., in hybrid trials when some of the data are RWD, and some are collected specifically for the clinical trial and not through RWD) [19,20]. NICE and CDA-AMC frameworks each provided one centralized document presenting good practice standards for all phases of RWE development from study planning and design to data reporting [3,4]. HAS RWE methodological guide aims to provide practical benchmarks relating to specific aspects of real-world/observational studies such as data quality, use of pre-existing data and integration of patient-reported outcomes during the product's assessment [21], whereas the IQWiG rapid report considers the requirements for the individual steps in the generation and analysis of routine practice data for benefit assessments (EHRs, registries, claims data from health insurance funds) [22]. EUnetHTA RWE guidance focused on validity concerns in RWD analyses reporting and in RWE uses in indirect treatment comparisons alongside with RCTs [23,24], whereas the latest EU HTA Regulation (2024) strongly recommends the limited RWE use in indirect treatment comparisons and only dependent on the availability of individual patient data [24].
Sixteen additional RWD/RWE guidance documents were retrieved through a separate search of websites from international policy institutions, organizations and working groups (Figure 1). More details regarding these documents are provided in Supplementary Table 3. Lastly, 28 publications on various RWE topics were identified as endorsed by ISPE (Supplementary Table 4). Some of these documents were also summarized as background materials during the development of HAS [21], CDA-AMC [3] and NICE [4] RWE-related guidance.
We pre-selected the following regulatory and HTA agencies across North America (FDA, CDA-AMC), England (MHRA, NICE) and Europe (HAS, IQWiG, EUnetHTA) to construct a roadmap of similarities and differences in exact guidance and RWE minimum acceptance criteria. Details from each of the guidance documents were dual extracted and systematically arranged across the well-defined phases in RWE studies (study planning, choosing fit-for-purpose data, study conduct, and study reporting). A summary of all recommendations by phase was produced and mapped by agency to allow a qualitative assessment of similarities and differences across bodies (Figure 3). The recommended tools or checklists by each document were also recorded (Table 1); these are described in more detail in Supplementary Table 5.
Figure 3. . Overview of real-world evidence guidance across pre-selected regulatory and health technology assessment agencies.
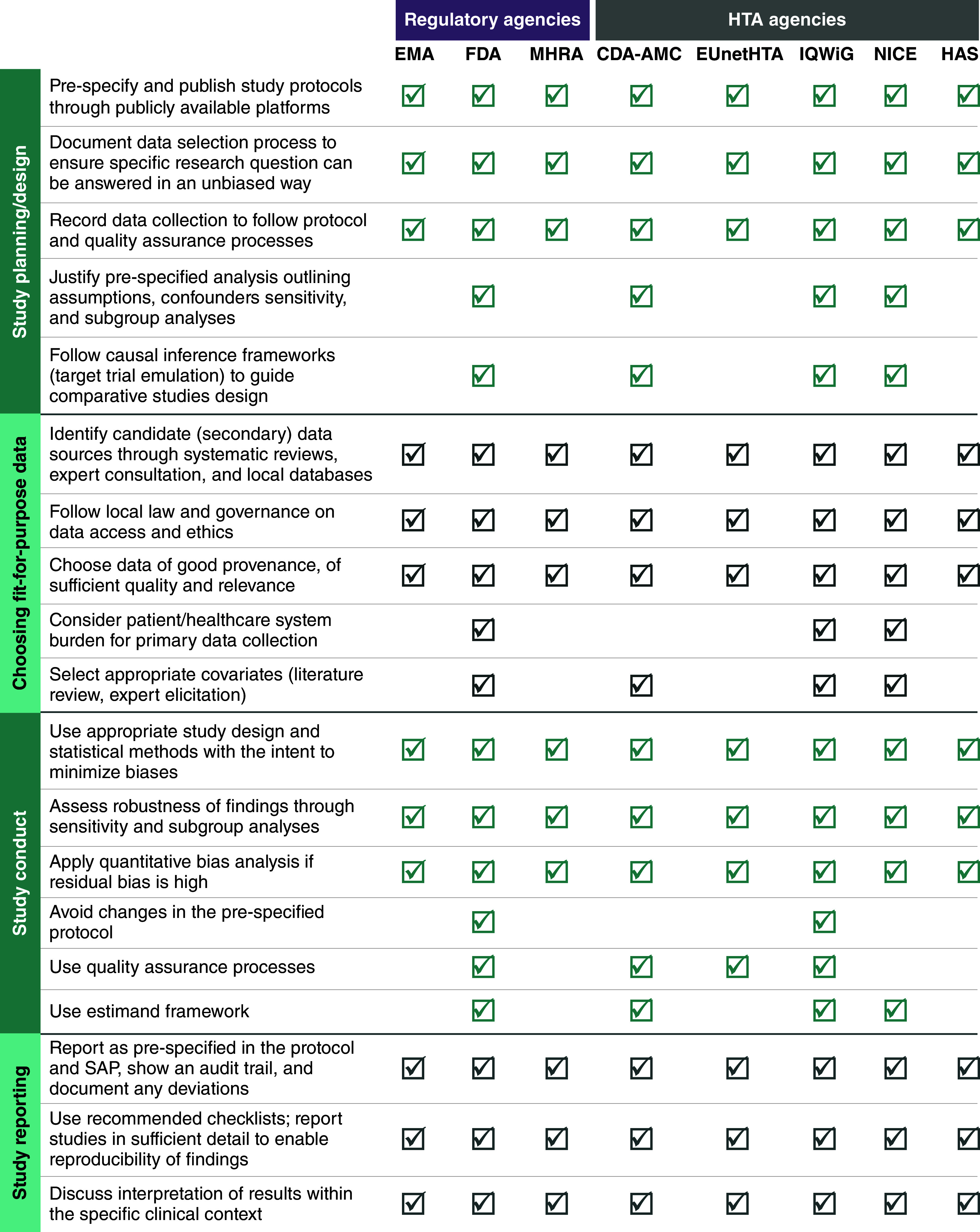
CADTH (Canadian Agency for Drugs and Technologies in Health) is now operating under the CDA-AMC name.
EMA: European Medicines Agency; EUnetHTA: European Network for Health Technology Assessment; FDA: Food and Drug Administration; HAS: Haute Autorité de Santé; HTA: Health technology assessment; IQWiG: Institute for Quality and Efficiency in HealthCare; MHRA: Medicines and Healthcare products Regulatory Agency; NICE: National Institute for Health and Care Excellence; SAP: Statistical analysis plan.
Table 1. . Recommended tools by regulatory and health technology assessment agencies for each phase of real-world evidence studies.
| Guidance agency | Study planning | Choosing fit-for-purpose data | Study conduct | Study reporting |
|---|---|---|---|---|
| FDA | • Checklist for drafting results reports (STROBE) | |||
| EMA | • ENCePP Checklist for study protocols • ENCePP Methodological Standards • Protocol template for evaluation of RWE studies (HARPER) |
• ENCePP Methodological Standards | • ENCePP Code of Conduct Checklist • ENCePP Methodological Standards |
• ENCePP Code of Conduct Checklist • ENCePP Methodological Standards |
| MHRA | • ISPE Guidelines for good database searches | |||
| NICE | • Protocol template for evaluation of RWE studies (HARPER) • Methods reporting template (Appendix 2 of NICE real-world evidence framework) • Database with high-quality core outcome sets (COMET) • Core outcome set study design (COS-STAD) and protocol development (COS-STAP) • ENCePP Checklist for study protocols • ENCePP Methodological Standards • Study design diagram templates for longitudinal studies (REPEAT) |
• Assessment of data suitability (DataSAT) • Checklist for the conduct of studies on routinely collected databases (RECORD) • ENCePP Methodological Standards |
• Checklist for the conduct of studies on routinely collected databases (RECORD, RECORD-PE) • ENCePP Code of Conduct Checklist • ENCePP Methodological Standards • Assess risk of bias in non-randomized studies of interventions (ROBINS-I) |
• Checklist for drafting results reports (STROBE) • Planning and reporting of real-world studies (StaRT-RWE) • Checklist for the conduct of studies on routinely collected databases (RECORD, RECORD-PE) • Core outcome set study reporting (COS-STAR) • ENCePP Code of Conduct Checklist • ENCePP Methodological Standards |
| EUnetHTA | • AHRQ Developing a protocol for observational comparative effectiveness research (checklists) | • Assess methodological quality of case series studies (JBI critical appraisal tool) • Assess the methodological quality of non-randomized surgical studies, comparative or non-comparative (MINORS) |
• Assess risk of bias in non-randomized studies (ROBINS-I, RoBANS, ROBIS, ACROBAT-NRSI) • Rate quality of evidence and strength of recommendations (GRADE) |
• Evaluate the quality of data collection systems (REQueST) • Classification of study design in systematic reviews (DAMI) |
| CDA-AMC | • Recommendation checklist in Appendix 3 of CADTH guidance for reporting of real-world evidence • Protocol template for evaluation of RWE studies (HARPER) • ENCePP Checklist for study protocols • ENCePP Methodological Standards • Planning and reporting of real-world studies (StaRT-RWE) |
• Recommendation checklist in Appendix 3 of CADTH guidance for reporting of RWE • Evaluate the quality of data collection systems (REQueST) • ENCePP Methodological Standards • Checklist for the conduct of studies on routinely collected databases (RECORD, RECORD-PE) |
• Recommendation checklist in Appendix 3 of CADTH guidance for reporting of RWE • Quality assurance for search strategies (PRESS) • ENCePP Code of Conduct Checklist • ENCePP Methodological Standards • Checklist for the conduct of studies on routinely collected databases (RECORD, RECORD-PE) • Evaluation of research (CASP) • Assessment of Real-World Observational Studies (ArRoWS) • Assess quality of non-randomized studies in meta-analyses (NOS) • Assess risk of bias in non-randomized studies of interventions (ROBINS-I) • Rate quality of observational studies of comparative effectiveness (GRACE) |
• Recommendation checklist in Appendix 3 of CADTH guidance for reporting of RWE • Evaluate the quality of data collection systems (REQueST) • ENCePP Code of Conduct Checklist • ENCePP Methodological Standards • Checklist for drafting results reports (STROBE) • Planning and reporting of real-world studies (StaRT-RWE) |
| IQWiG | • Checklists of criteria for data quality (Tables 2, 7, 8, 14 in Guidance ‘Concepts for the generation of routine practice data and their analysis for the benefit assessment of drugs according to §35a Social Code Book V (SGB V)’) | • Checklist for the conduct of studies on routinely collected databases (RECORD, RECORD-PE) | • Checklist for the conduct of studies on routinely collected databases (RECORD, RECORD-PE) • Confidence in findings from qualitative evidence (CERQuaL) • Quality assurance for search strategies (PRESS) • Assess risk of bias in systematic reviews (ROBIS) • Assess risk of bias in non-randomized studies of interventions (ROBINS-I) |
• Checklist for the conduct of studies on routinely collected databases (RECORD, RECORD-PE) • Confidence in findings from qualitative evidence (CERQuaL) • Reporting in systematic reviews and meta-analyses (PRISMA) |
| HAS | • ENCePP Checklist for study protocols • ENCePP Methodological Standards • AHRQ Developing a protocol for observational comparative effectiveness research (checklists) • Planning and reporting of real-world studies (StaRT-RWE) |
• ENCePP Methodological Standards • Checklist for the conduct of studies on routinely collected databases (RECORD, RECORD-PE) • Evaluate the quality of data collection systems (REQueST) |
• ENCePP Code of Conduct Checklist • ENCePP Methodological Standards • ISPOR-ISPE good practices for RWD studies of treatment and/or comparative effectiveness • AHRQ Developing a protocol for observational comparative effectiveness research (checklists) • Checklist for the conduct of studies on routinely collected databases (RECORD, RECORD-PE) |
• ENCePP Code of Conduct Checklist • ENCePP Methodological Standards • Checklist for drafting results reports (STROBE, STROBE-ME) • AHRQ Developing a protocol for observational comparative effectiveness research (checklists) • Drafting of observational study meta-analyses (MOOSE) • Evaluation of systematic reviews of literature containing randomized and non-randomized studies (AMSTAR 2) • Planning and reporting of real-world studies (StaRT-RWE) |
ACROBAT-NRSI: A Cochrane Risk Of Bias Assessment Tool for Non-Randomized Study of Intervention; AHRQ: Agency for Healthcare Research and Quality; AMSTAR 2: Assessing the Methodological Quality of Systematic Reviews, version 2; ArRoWS: Assessment of Real-World Observational Studies; CASP: Critical Appraisal Skill Programme; CDA-AMC, Canada's Drug Agency; CERQual: Confidence in the Evidence from Reviews of Qualitative research; COMET: Core Outcome Measure in Effectiveness Trials; COS-STAD: Core Outcome Set – Study Design; COS-STAP: Core Outcome Set – Protocol Development; COS-STAR: Core Outcome Set – Reporting; DAMI: Design Algorithm for Medical literature on Intervention; DataSAT: Data Suitability Assessment Tool; EMA: European Medicines Agency; ENCePP: European Network of Centre for Pharmacoepidemiology and Pharmacovigilance; EUnetHTA: European Network for Health Technology Assessment; FDA: Food and Drug Administration; GRACE: Good Research for Comparative Effectiveness; GRADE: Grading of Recommendations Assessment, Development and Evaluation; HARPER: HARmonized Protocol template to Enhance Reproducibility; HAS: Haute Autorité de Santé; IQWiG: Institute for Quality and Efficiency in HealthCare; ISPOR: The Professional Society for Health Economics and Outcomes Research; ISPE: International Society for Pharmacoepidemiology; JBI: Joanna Briggs Institute; MHRA: Medicines and Healthcare products Regulatory Agency; MINORS: Methodological Index for NOn-Randomized Studies; MOOSE: Meta-analysis of Observational Studies in Epidemiology; NICE: National Institute for Health and Care Excellence; NOS: Newcastle-Ottawa Scale; PRESS: Peer Review of Electronic Search Strategies; PRISMA: Preferred Reporting Items for Systematic Reviews and Meta-Analyses; RECORD: REporting of studies Conducted using Observational Routinely collected health Data; RECORD-PE: REporting of studies Conducted using Observational Routinely collected health Data statement for PharmacoEpidemiology; REPEAT: Reproducible Evidence: Practices to Enhance and Achieve Transparency; REQueST: Registry Evaluation and Quality Standards Tool; RoBANS: Risk of Bias Assessment for Non-randomized Studies; ROBINS-I: Risk Of Bias in non-randomized Studies – of Interventions; ROBIS: Risk of Bias in Systematic Reviews; RWD: Real-world data; RWE: Real-world evidence; STaRT-RWE: Structured template for planning and reporting on the implementation of real-world studies; STROBE: STrengthening the Reporting of OBservational studies in Epidemiology; STROBE-ME: STrengthening the Reporting of OBservational studies in Epidemiology – Molecular Epidemiology.
In a few instances, due to variation in the level of detail/full explanation provided in the recommendations across the reviewed documents, some of the assessments with regard to the presence or absence of a recommendation in Figure 3 was largely dependent on the explicit reference to these points. Furthermore, in some guidance documents referred to as providing specific recommendations for RWE study reporting (e.g., IQWiG [22]), their content was re-structured for the purposes of compiling their guidance into the four pre-defined phases (Figure 3).
Study planning/design
The importance of RWE study planning and design was covered by all the identified guidance documents and frameworks developed by the pre-selected agencies. RWD can be based either through new data collection (“primary” data) or using existing databases (“secondary” data). All HTA agencies promote the use of national (local) databases for secondary use when basic quality criteria are met and promote setting up disease-specific patient registries, especially for rare diseases. Across the documents, common principles in this phase were:
Clear justification of the research question (according to population, intervention, comparison, outcomes structure); regulatory bodies prioritized confirmation of research objectives through robust evidence, while HTA agencies emphasized the role of RWE in addressing knowledge gaps using RWE.
Detailed recording of all aspects of study design in a pre-designed study protocol is a must.
The application of appropriate measures to ensure the highest data quality (e.g., study design to minimize biases, confounders availability, reduce data missingness through data linkage) and representativeness are selected.
The HARmonized Protocol Template to Enhance Reproducibility (HARPER), an output of a joint ISPE/ISPOR task force effort, is recommended by most agencies (EMA, NICE, CDA-AMC) [25]. EMA published a freely accessible RWD catalog through an online platform that allows searches of available RWD sources and associated studies to allow their integration in decision-making (https://catalogues.ema.europa.eu/search). Some agencies (FDA, CDA-AMC, NICE, IQWiG) require transparency in the description of planned quantitative analyses, including the use of bias minimization and/or quantification methods (conduct sensitivity and subgroup analyses) and the need for detailed recording for any protocol amendments. Regulatory guidance by FDA and EMA emphasized the need of tailored statistical analysis based on RWD nuances, whereas NICE and IQWiG probed into specific acceptable analytical approaches such as direct adjustment and propensity score matching. The publication of a study's protocol in international platforms (European Network of Centres for Pharmacoepidemiology and Pharmacovigilance, ClinicalTrials.gov, RWE Transparency Initiative [26]) is encouraged as an effort to increase transparency. Additionally, NICE, CDA-AMC, IQWiG and HAS specifically include recommendations on named tools and templates to be used during RWE study design (Table 1 & Supplementary Table 4). Only HAS points out that the presence of a multidisciplinary scientific committee is required for the validation of an RWE study protocol development; the multidisciplinary scientific committee should be composed of scientific experts, patients and user association representatives. For RWE studies aiming to answer causal research questions, some agencies (FDA, NICE, CDA-AMC, IQWiG) recommend methodologies such as the target trial emulation framework [27] in which every element in the RWE study design (patient eligibility criteria, intervention, comparison, outcomes, follow-up, statistical analysis) can be implemented in an analog way to an RCT with the aim to avoid critical sources of biases (e.g., selection and immortal time bias) and allow trustworthy estimate of causal effects. In addition, CDA-AMC and NICE explicitly suggest the use of causal diagrams to reflect the structure and the assumptions in the causal relationships between interventions, outcomes and confounders (e.g., using causal diagrams known as directed acrylic graphs [28]).
Choosing fit-for purpose data
Guidance on the importance of choosing fit-for-purpose data for the specific decision problem under consideration is considered critical and was included in all documents and frameworks by the pre-selected regulatory and HTA agencies (Figure 3); close adherence to local laws and governance around data access is critical. However, some variance in the level of detail, terminologies and concepts (“labelling”) around fitness-for-purpose of data was evident across these documents. For example, the FDA guidance emphasizes describing fit-for-purpose data in relation to data reliability and relevance whereas the EMA documents also include data extensiveness, coherence and timeliness. The NICE RWE framework [4] refers to the issue of data “quality” to describe the overall umbrella term covering topics related to reliability (completeness and accuracy) whereas CDA-AMC includes additional issues related to data access and version of a database [3]. Figure 4 tries to unravel the complexity in definitions and concept expressions used across agencies and captures the interconnectedness of terminologies used under the concept of fitness-for-purpose data. The NICE RWE Framework also recommends the use of an in-house developed tool, the Data Suitability Assessment Tool (DataSAT) (Appendix 1), which is designed to methodically assess the suitability of data using consistent criteria, as well as referring to related published guidance from ISPE on this topic (Supplementary Table 4).
Figure 4. . Maze of fitness-for-purpose concepts and their interconnections.
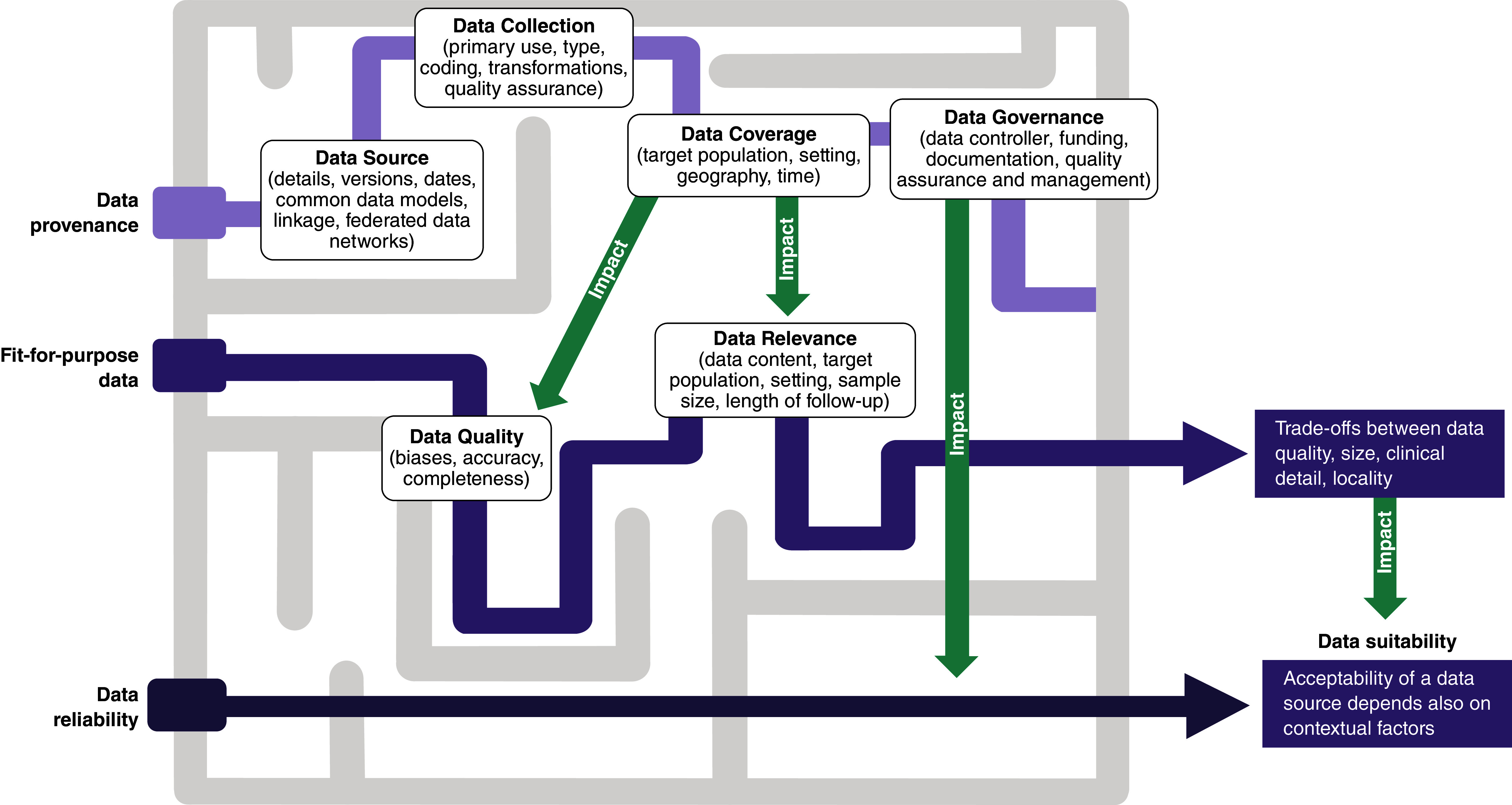
The darkness of the color of the lines represents the number of intermediate concepts connecting one of the three core concepts at the left (data provenance, fit-for-purpose data, data reliability) to data suitability at the right.
Study conduct
The similarities between RWE considerations to be followed in study planning/design and study conduct were evident. These regarded data transformation (e.g., data linkage), organization (e.g., use of common data model, selection of confounders) and applying appropriate statistical analyses based on a priori protocols (research questions) and ensuring transparency in the rationale for the selected methods. The quantification of the role and sources of potential biases in RWD sources either by undertaking additional analyses (such as sensitivity and subgroup analysis) to ensure robustness of results, as well as applying quality assurance steps designed to minimize human errors were recommended by all agencies. Quantitative bias analysis (QBA) is recommended by the NICE RWE Framework and CDA-AMC guide as a type of sensitivity analyses to estimate the direction, magnitude and uncertainty of study results due to RWE biases that cannot only identify potential sources of systematic errors but also provide ranges of potential impacts of biases on study results (e.g., particularly valuable in studies using RWE external controls [29]). These two HTA bodies also provided specific methods guidance on how to reliably combine evidence from RWD sources and RCTs to answer causal research questions (e.g., combining RWE with RCTs in comparative effectiveness research [30]). IQWiG does not support a common data pool when analyzing several registries. Instead, it suggested conducting identically designed studies within each registry, followed by a meta-analysis to synthesize the evidence.
Overall, regulatory documents generally provided more detailed guidance than others. Given the distinct evidentiary requirements of regulatory and HTA bodies, there were differences in the emphasis on data quality and methodological rigor for data collection. And for data linkage, regulatory bodies focused more on scientific validity and regulatory compliance aspects, while HTA bodies spotlight practical considerations for healthcare decision-making.
Study reporting
In terms of study reporting, all documents emphasized the importance of providing a clear and reproducible record of how RWD were selected, accessed, processed and analyzed with any deviations from the original study protocol explicitly stated (internal validity considerations). All decision-makers agreed that a clear reporting structure of RWE studies would eliminate data misinterpretation during regulatory and HTA assessments and facilitate reproducibility and validity assessment during evidence synthesis. The Structured template for planning and reporting on the implementation of real-world studies (STaRT RWE) tool, a structured template for planning and reporting on the implementation of RWE studies of the safety and effectiveness of treatments was recommended as a tool to guide the design and conduct of reproducible RWE studies [31]. EUnetHTA developed a de novo tool called Registry Evaluation and Quality Standards Tool (REQueST) to evaluate specific data collected from patient registries which was also adopted by other HTA agencies (NICE, CDA-AMC, HAS, IQWiG) [32]. Efforts to provide meaningful interpretations of results in the context of the decision problem, especially in relation to generalizability of study results (external validity considerations) to reflect the targeted population, while discussing limitations was considered a key reporting element.
Discussion
The increased attention to the wider potential of RWD/RWE use in estimating the effectiveness and safety of a treatment in healthcare decision-making (beyond the description of disease burden, epidemiology and providing inputs to economic models) has resulted in a surge of guidance, recommendations and best practices documents published since 2018. Advanced RWE methods continuously grow in popularity through the implementation of real cases (e.g., RCT duplicate, DARWIN case studies [14,33]) and wider communication of their findings. Most importantly, the introduction of RWE guidance and lately, of RWE frameworks across the globe, aims to provide a structure under which high-quality, reliable RWE can be generated and advanced analytics could be formally applied in regulatory and HTA decision-making.
Previously, other researchers conducted similar exercises; however, our environmental scan had a wider scope than previous reviews which aimed to summarize RWE guidance either only from regulatory authorities or on specific aspects from all decision-makers which become quickly outdated given the dynamic, rapidly moving field of RWD/RWE in healthcare policy [34–36]. The cut-off point for our review was less than six months; we detailed similarities and differences in the recommended RWE quality tools and checklists, something not comprehensively covered by previous research and the results can be used as a one-stop blueprint RWD/RWE guide and as a basis for future RWE updates (in living mode). This may facilitate discussions around harmonization efforts in this sphere but also as an internal training tool for manufacturers and researchers working in this area. After our review was completed, similar activities were undertaken by a European public-private partnership (Integration of Heterogeneous Data and Evidence toward Regulatory and HTA Acceptance) which produced a scoping review of HTA and regulatory RWE documents to inform policy recommendations with similar concluding remarks to the findings of this review but also covering topics (e.g., governance and ethics) which were beyond the scope of our environmental scan [37].
All authorities set up guidance and best practices that emphasize the need for best available, high-quality RWD to inform technologies submissions and clear reporting of their findings. The categorization of “acceptable criteria” was strong and consistent around RWD study planning and design (defined as robustness, reliability and of good provenance) and reporting (defined as transparency) with some variation in definitions and details provided, as previously explained, given the different scope and remit of regulatory and HTA agencies. The most noticeable disagreement between the reviewed documents seems to exist in the provision of detail regarding the use and implementation of RWE analytical methods beyond the need for adjusting for confounders and the selection of standardized tools or templates to allow detailed reporting of considerations applied during study conduct and reporting. This disconnect in the documents may be explained by the high RWE heterogeneity in terms of data availability (sources) and feasibility of rigorous collection methods which would require case-specific approaches; therefore, some guidance would be more general in some instances to allow greater flexibility in its use for a variety of RWE applications. However, this may leave a maze of options for manufacturers to navigate and cover while preparing for regulatory and HTA submissions across the globe.
As expected, the topic of RWE use in comparative effectiveness research was exclusively covered in HTA guidance given the focus on estimating comparative estimates of effectiveness and safety for a new healthcare technology versus existing standard of care. Recommendations on the analytic approaches around RWE use in comparative treatment estimation were presented in all the reviewed documents from HTA agencies with varying levels of detail. For example, EU Heatlh Technology Assessment Regulation (HTAR) and EUnetHTA guidance provided specific reflections on the use of RWE in direct and indirect treatment comparisons such as issues around effect modification (e.g., how to reliably identify and list a priori all potential treatment effect modifiers), careful considerations of validity issues and use of standardized tools. The NICE framework, on the other hand, recommends the use of specific analytical frameworks to build up a comparator arm (external control) to estimate treatment effects against a single-arm trial or to add controls in RCTs, transport treatment effects from other countries, explore heterogeneity in intervention effects from under-presented populations, and techniques (informing priors, increasing power or filling evidence gaps) for evidence synthesis across RCTs and observational studies producing RWE.
Noticeably, there were some redundancies in what appeared to be duplicated efforts during the development of these guidance documents. For example, environmental scans of RWE-related guidance (CDA-AMC, NICE, HAS) that informed final guidance or the development of specific checklists (e.g., CDA-AMC Recommended Checklist, Appendix 3) which overlap with existing risk-of-bias tools (e.g., Risk Of Bias In Non-randomized Studies – of Interventions) and other checklists developed by international organizations (e.g., StaRT-RWE template) were noted. Similarly, attempts at building consensus around RWE standardized use and assessment themselves are occurring in duplicate, as evidenced by some example cases such as the EMA's Methodology Working Party (https://www.ema.europa.eu/en/committees/working-parties-other-groups/chmp/methodology-working-party) and various workshops [38,39] that have published recommendations on the use of RWE without specific plans for how these recommendations can be implemented in real practice. This lack of guidance standardization could lead to wastage of valuable resources funded by taxpayers and creates an environmental question of what the best resource-use strategy is when it comes to guidelines development. Recent recommendations [38] have highlighted the need for clear guidance on data quality standards for HTA agencies but do so by stating that the onus should be on the individual HTA agencies, which serves to only entrench the siloed nature of guidance recommendations, and is likely to create confusion, and impose resource strains for manufacturers and other stakeholders working on regulatory and HTA submissions across the globe using RWE. Furthermore, given the exponential increase in RWE analytical methodologies and related publications, and as more RWE detailed taskforces are initiated by international organizations, some of the formal guidance may become quickly outdated. All the reviewed guidance, except for the NICE RWE framework and CDA-AMC guidance, did not consider setting up the RWE guidance in a living mode (i.e., to be regularly updated) which may restrict their relevance as the RWE field rapidly evolves. International efforts such as those by the Duke-Margolis International Harmonization of Real-World Evidence Standards Dashboard (https://healthpolicy.duke.edu/projects/international-harmonization-real-world-evidence-standards-dashboard) will help researchers and other stakeholders to track in real time relevant regulatory guidance and frameworks including international documents. Recommendations on the use of RWE need to be converted to actionable plans for their consistent implementation across disease areas and case studies will be key in providing additional insights.
Conclusion
There is some consensus among the frameworks and methodological documents on RWE by regulatory and HTA agencies, as well as organizations focused on healthcare decision-making, regarding the importance of using the best available, high-quality RWD to inform decision-making; and, in general, the documents reflected a high level of agreement on the common principles of study planning/design and study reporting. There was unanimous agreement that RWD and RWE need to be robust, reliable and of good provenance, with transparency of reporting elements noted as a core element to increase trust in the findings. However, differences were observed in the provision of detail regarding the use and implementation of analytical methods for the RWD analysis, which will likely require manufacturers to navigate a maze of options while preparing for multiple regulatory and HTA submissions globally. Furthermore, there is a duplication of effort during the development of RWE guidance documents from regulatory and HTA agencies, and other organizations focusing on healthcare decision-making, as well as a lack of a uniform, clear set of guidelines and expectations by these agencies. Harmonized expectations on RWE studies design, quality elements, and reporting will facilitate evidence generation strategy and activities for manufacturers facing multiple, including global, regulatory and reimbursement submissions and re-submissions. Closer collaboration between decision-making agencies towards a harmonized RWE roadmap, which can be centrally preserved in a living mode, will provide manufacturers and researchers clarity on minimum acceptance requirements and expectations, especially as novel methodologies for RWE generation are rapidly emerging.
Summary points
There is increasing awareness that real-world data (RWD) and real-world evidence (RWE) have a unique role in providing valuable insight on the effectiveness and safety of treatments, which is transforming how evidence is generated throughout the lifecycle of a healthcare technology.
Changes are occurring in the attitudes that have traditionally limited trust in RWE study design and RWE approaches and how these can be used to inform healthcare decision-making.
The number of RWE guidance documents from different organizations around the globe has increased dramatically in a short time, with considerable variation in scope and content.
A search of websites of relevant regulatory and health technology assessment (HTA) agencies worldwide returned 46 RWE-related documents.
There was consensus among the frameworks and methodological documents on the importance of using the best available, high-quality RWD to inform decision-making; in general, the documents reflected a high level of agreement on the common principles of study planning/design and study reporting.
There was unanimous agreement that RWD and RWE need to be robust, reliable and of good provenance, with transparency of reporting elements noted as a core element to increase trust in the findings.
Many differences were seen in the provision of detail regarding the use and implementation of analytical methods for the RWD analysis, which will likely require manufacturers to navigate a maze of options while preparing for multiple regulatory and HTA submissions globally.
The review revealed a duplication of effort during the development of these guidance documents.
The failure to standardize RWE guidance could result in a costly, inefficient use of resources.
As a next step, harmonization of good standard practices and actionable plans that result in the consistent implementation of RWE use in healthcare decision-making is needed across agencies and disease areas.
Supplementary Material
Acknowledgments
The authors thank Colleen Dumont of Cytel, Inc., for writing and editorial support of this manuscript.
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: https://bpl-prod.literatumonline.com/doi/10.57264/cer-2024-0061
Author contributions
The authors declare that this is an original work. All authors contributed equally.
Financial disclosure
The review of materials and editorial assistance for this manuscript was funded by Takeda Pharmaceuticals America, Inc. G Sarri is an employee of Cytel, Inc. LG Hernandez is an employee of and owns stock of Takeda. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Competing interests disclosure
The authors have no competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Writing disclosure
Editorial assistance for this manuscript was funded by Takeda Pharmaceuticals America, Inc.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.US Food & Drug Administration. Real-World Evidence. https://www.fda.gov/science-research/science-and-research-special-topics/real-world-evidence
- 2.Nishioka K, Makimura T, Ishiguro A et al. Evolving acceptance and use of RWE for regulatory decision making on the benefit/risk assessment of a drug in Japan. Clin. Pharmacol. Ther. 111(1), 35–43 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Canadian Agency for Drugs and Technologies in Health. Guidance for Reporting Real-World Evidence 2023. https://www.cadth.ca/sites/default/files/RWE/MG0020/MG0020-RWE-Guidance-Report-Secured.pdf ; •• This framework aims to promote standardization in the reporting of real-world evidence (RWE) studies to inform reimbursement decision-making in Canada.
- 4.National Institute for Health and Care Excellence. NICE real-world evidence framework 2022. https://www.nice.org.uk/corporate/ecd9/resources/nice-realworld-evidence-framework-pdf-1124020816837#:~:text=The%20framework%20aims%20to%20improve,the%20section%20on%20NICE%20guidance [DOI] [PMC free article] [PubMed]; •• This framework describes best practices for planning, conducting and reporting RWE studies with the aim to increase the quality and transparency of this type of evidence.
- 5.International Coalition of Medicines Regulatory Authorities. ICMRA statement on international collaboration to enable real-world evidence (RWE) for regulatory decision-making 2022. https://www.icmra.info/drupal/sites/default/files/2022-07/icmra_statement_on_rwe.pdf
- 6.Duke-Margolis Institute for Health Policy. Real-World Evidence Collaborative. https://healthpolicy.duke.edu/projects/real-world-evidence-collaborative
- 7.Real-world Evidence Alliance. https://rwealliance.org/
- 8.US Food & Drug Administration. Guidance document. Use of Real-World Evidence to Support Regulatory Decision-Making for Medical Devices. Guidance for Industry and Food and Drug Administration Staff 2017. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-real-world-evidence-support-regulatory-decision-making-medical-devices
- 9.Faria R, Hernandez Alava M, Manca A et al. NICE DSU Technical Support Document 17: the use of observational data to inform estimates of treatment effectiveness in technology appraisal: Methods for comparative individual patient data 2015. http://www.nicedsu.org.uk
- 10.European Medicines Agency. European medicines agencies network strategy to 2025. https://www.ema.europa.eu/en/documents/report/european-union-medicines-agencies-network-strategy-2025-protecting-public-health-time-rapid-change_en.pdf ; •• Outlines areas of strategic focus to handle the rapidly changing environment surrounding how to ensure patient access to safe, effective medicines in the European Union.
- 11.European Medicines Agency. Guideline on registry-based studies 2021. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-registry-based-studies_en.pdf-0
- 12.European Commission. European Health Data Space. https://health.ec.europa.eu/ehealth-digital-health-and-care/european-health-data-space_en
- 13.European Medicines Agency. Technical workshop on real-world metadata for regulatory purposes. https://www.ema.europa.eu/en/events/technical-workshop-real-world-metadata-regulatory-purposes
- 14.European Medicines Agency. Data Analysis and Real World Interrogation Network (DARWIN EU). https://www.ema.europa.eu/en/about-us/how-we-work/big-data/data-analysis-real-world-interrogation-network-darwin-eu
- 15.European Medicines Agency. Learnings initiative webinar for optimal use of big data for regulatory purpose. https://www.ema.europa.eu/en/events/learnings-initiative-webinar-optimal-use-big-data-regulatory-purpose
- 16.European Commission. Horizon Europe Work Programme 2021-2022 2022. https://ec.europa.eu/info/funding-tenders/opportunities/docs/2021-2027/horizon/wp-call/2021-2022/wp-4-health_horizon-2021-2022_en.pdf
- 17.US Food & Drug Administration. Real-World Data: Assessing Electronic Health Records and Medical Claims Data To Support Regulatory Decision-Making for Drug and Biological Products. Draft Guidance for Industry 2021. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/real-world-data-assessing-electronic-health-records-and-medical-claims-data-support-regulatory [DOI] [PMC free article] [PubMed]; • This guidance, issued as part of the 21st Century Cures Act, aims to provide relevant stakeholders with considerations on an evidence base that relies on electronic health records or medical claims data in clinical studies.
- 18.US Food & Drug Administration. Considerations for the Design and Conduct of Externally Controlled Trials for Drug and Biological Products. Guidance document 2023. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-design-and-conduct-externally-controlled-trials-drug-and-biological-products
- 19.Medicines & Healthcare Products Regulatory Agency. MHRA guidance on the use of real-world data in clinical studies to support regulatory decisions 2021. https://www.gov.uk/government/publications/mhra-guidance-on-the-use-of-real-world-data-in-clinical-studies-to-support-regulatory-decisions/mhra-guidance-on-the-use-of-real-world-data-in-clinical-studies-to-support-regulatory-decisions
- 20.Medicines & Healthcare Products Regulatory Agency. Guidance on randomised controlled trials using real-world data to support regulatory decisions (2021). https://www.gov.uk/government/publications/mhra-guidance-on-the-use-of-real-world-data-in-clinical-studies-to-support-regulatory-decisions/mhra-guideline-on-randomised-controlled-trials-using-real-world-data-to-support-regulatory-decisions#:~:text=This%20guideline%20is%20aimed%20at,RWD%20for%20supporting%20regulatory%20decisions
- 21.Haute Autorité de Santé. Real-world studies for the assessment of medicinal products and medical devices (2021). https://www.has-sante.fr/jcms/p_3284524/en/real-world-studies-for-the-assessment-of-medicinal-products-and-medical-devices
- 22.Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG). Concepts for the generation of routine practice data and their analysis for the benefit assessment of drugs according to §35a Social Code Book V (SGB V) 2020. https://www.iqwig.de/download/a19-43_routine-practice-data-for-the-benefit-assessment-of-drugs_extract-of-rapid-report_v1-0.pdf ; • Outlines and assesses concepts related to data collected outside a clinical trial setting, specifically in regard to suitability, quality, methodology and reporting.
- 23.European Network for Health Technology Assessment (EUnetHTA). Individual Practical Guideline Document: D4.3.1: Direct and Indirect Comparisons 2022. https://www.eunethta.eu/wp-content/uploads/2022/12/EUnetHTA-21-D4.3.1-Direct-and-indirect-comparisons-v1.0.pdf
- 24.HTA CG. Methodological Guideline for Quantitative Evidence Synthesis: Direct and Indirect Comparisons 2024. https://health.ec.europa.eu/document/download/4ec8288e-6d15-49c5-a490-d8ad7748578f_en?filename=hta_methodological-guideline_direct-indirect-comparisons_en.pdf
- 25.Wang SV, Pottegård A, Crown W et al. HARmonized Protocol Template to Enhance Reproducibility of hypothesis evaluating real-world evidence studies on treatment effects: a good practices report of a joint ISPE/ISPOR task force. Pharmacoepidemiol. Drug Saf. 32(1), 44–55 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Provides a template to guide the development of clear, reproducible RWE study protocols.
- 26.Orsini LS, Berger M, Crown W et al. Improving transparency to build trust in real-world secondary data studies for hypothesis testing-why, what, and how: recommendations and a road map from the Real-World Evidence Transparency Initiative. Value Health 23(9), 1128–1136 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Hernán MA, Robins JM. Using big data to emulate a target trial when a randomized trial is not available. Am. J. Epidemiol. 183(8), 758–764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shrier I, Platt RW. Reducing bias through directed acyclic graphs. BMC Med. Res. Methodol. 8, 70 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lash TL, Fox MP, MacLehose RF et al. Good practices for quantitative bias analysis. Int. J. Epidemiol. 43(6), 1969–1985 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Sarri G, Patorno E, Yuan H et al. Framework for the synthesis of non-randomised studies and randomised controlled trials: a guidance on conducting a systematic review and meta-analysis for healthcare decision making. BMJ Evid. Based Med. 27(2), 109–119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang SV, Pinheiro S, Hua W et al. STaRT-RWE: structured template for planning and reporting on the implementation of real world evidence studies. BMJ 372, m4856 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.European Network for Health Technology Assessment (EUnetHTA). Vision paper on the sustainable availability of the proposed Registry Evaluation and Quality Standards Tool (REQueST). https://www.eunethta.eu/wp-content/uploads/2019/10/EUnetHTAJA3_Vision_paper-on-REQueST-tool.pdf
- 33.Franklin JM, Patorno E, Desai RJ et al. Emulating randomized clinical trials with nonrandomized real-world evidence studies: first results from the RCT DUPLICATE Initiative. Circulation 143(10), 1002–1013 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burns L, Roux NL, Kalesnik-Orszulak R et al. Real-world evidence for regulatory decision-making: guidance from around the world. Clin. Ther. 44(3), 420–437 (2022). [DOI] [PubMed] [Google Scholar]
- 35.Jaksa A, Wu J, Jónsson P et al. Organized structure of real-world evidence best practices: moving from fragmented recommendations to comprehensive guidance. J. Comp. Eff. Res. 10(9), 711–731 (2021). [DOI] [PubMed] [Google Scholar]
- 36.Beyrer J, Abedtash H, Hornbuckle K et al. A review of stakeholder recommendations for defining fit-for-purpose real-world evidence algorithms. J. Comp. Eff. Res. 11(7), 499–511 (2022). [DOI] [PubMed] [Google Scholar]
- 37.IDERHA. Public Consultation. https://www.iderha.org/outreach/public-consultation
- 38.Kent S, Burn E, Dawoud D et al. Common problems, common data model solutions: evidence generation for health technology assessment. Pharmacoeconomics 39(3), 275–285 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Claire R, Elvidge J, Hanif S et al. Advancing the use of real world evidence in health technology assessment: insights from a multi-stakeholder workshop. Front. Pharmacol. 14, 1289365 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.