

ABSTRACT
Background
Contiguous gene deletion in the short arm of chromosome 4 is linked to various neurodevelopmental disorders.
Methods
In this study, we conducted peripheral blood chromosome G‐banding karyotyping and whole‐exome sequencing (WES) on a proband presenting with anal atresia, global developmental delay, lymphocytosis, and other multisystem anomalies. Additionally, chromosome G‐banding karyotyping was also carried out on the proband's parents and brother.
Results
The 7‐month‐old proband was found to have a 26.738 Mb 4p15.33‐p14 deletion as identified by chromosome G‐banding karyotyping and WES.
Conclusion
We identified a patient with proximal 4p deletion syndrome by karyotype and WES analysis, which might explain some of his phenotypes. Our research enhances clinicians' knowledge of this rare condition, and offers valuable genetic counseling to the affected family. Further research is necessary to identify the causative gene or critical region associated with proximal 4p deletion syndrome.
Keywords: chromosome 4p, deletion, developmental delay, whole‐exome sequencing
Karyotypic analysis of chromosome G‐banding in proband: 46, XY, del (4) (p15.33‐ p14).

1. Introduction
Chromosome 4 is the fourth largest chromosome in human cells, with a short arm spanning approximately 50 Mb and housing 379 genes (Hannes and Vermeesch 2008). The deletion syndrome affecting the short arm of chromosome 4 (4p syndrome) involves deletions in the 4p11‐p16 region, categorized as either short arm terminal deletions or proximal interstitial deletions. Concurrent deletions from the 4p16.3 region to the end of 4p are associated with Wolf‐Hirschhorn syndrome (WHS [MIM: 194190]). Compared to WHS, proximal 4p interstitial deletions are rarer. Deletions in the 4p15 region are centrally located, with the smallest deletion region encompassing 4p15.2‐p15.32 (Basinko et al. 2008). In this study, we investigated a male child with global developmental delay, anal atresia and lymphocytosis as the main symptoms, and a genetic analysis was done on him.
2. Subjects and Methods
2.1. Subjects
The clinical data of a patient with global development delay and family were collected. Whole‐blood samples were obtained from the proband, his parents, brother, and their maternal grandmother and stored at −20°C. The study was approved by the Institutional Ethics Committee of the Sichuan Provincial Maternity and Child Health Care Hospital (protocol code: 202300911‐225 and date: September 11, 2023).
2.2. Methods
Chromosomal karyotyping and WES analysis were employed.
2.2.1. Sampling
The advantages and limitations of chromosomal karyotyping and WES were explained to the parents of the patient. After obtaining their informed consent, 2 tubes of venous blood were drawn from the patient and the parents. The parental specimens were used for verification.
2.2.2. Chromosomal Karyotype Analysis
The 0.5 mL of peripheral blood was collected and subjected to routine lymphocyte culture (BIOSCIENCES medium, Israel) for 72 h under sterile conditions. The harvested cells were then sectioned, banded with Giemsa staining, and counted using the MetaSystems Ikaros Chromosome Automated Scanning and Analysis System (ZEISS, Germany). Twenty split phases were counted, and five karyotypes were analyzed and described according to the International System for Human Cytogenetic or Cytogenomic Nomenclature 2020 (ISCN2020) standard.
2.2.3. Whole Exome Sequencing
Genomic DNA was isolated from peripheral blood leukocytes using the QIAamp DNA Blood Mini Kit (Düsseldorf, Germany) and sent to Saifu Decoding (Beijing) Genetic Science and Technology Co. The raw data obtained had a size greater than 10G and a Q30 score of at least 80%. For the bioinformatic analysis and variant screening process, the raw data was first converted from .bcl files to .fastq files using bcl2fastq. Then, the reads were aligned to the human reference genome GRCh38/hg38 using BWA, Samtools, and Picard software. The resulting .bam files were locally realigned using the GATK software, followed by duplicate sequence removal and variant detection. The variant file (.vcf) was annotated with Annovar. Pathogenic variants sites were identified based on the following criteria: (1) screening for variants in exonic regions and nonsynonymous variants sites, (2) checking the carrier rate in databases such as ExAC_EAS, ExAC_ALL, 1000Genomes, and gnomAD to ensure they are not commonly found in normal human populations or have a carrier rate of less than 5%, and (3) evaluating the pathogenicity of variant loci using databases like dbSNP, OMIM, HGMD, and ClinVar. (4) Protein function prediction was conducted to assess the impact of gene variants using multiple software tools including SIFT, Polyphen2, LRT, MutationTaster, and FATHMM. The detection followed the ACMG classification guidelines and considered the clinical phenotype of the patients.
3. Results
3.1. Phenotype of the Patient
A 7‐month‐old boy was hospitalized with a diagnosis of “Global developmental delay.” He is the second child of young healthy non‐consanguineous parents whose first son was healthy. The proband was delivered via caesarean section at 38 + 1 weeks due to uterine scarring. At birth, he weighed 2910 g (<25th percentile), a birth length of 49.5 cm (50th percentile) and a birth head circumference of 33.5 cm (50th percentile). On the first day of life, the patient experienced post‐feeding vomiting and was diagnosed with anal atresia. A bowel ultrasound and abdominal x‐ray indicated a low intestinal obstruction. Emergency anoplasty was performed, and the patient was later discharged from the pediatric surgery department. Intraoperatively, the pathological biopsy revealed only smooth muscle tissue without any interosseous ganglion (Figure 1A). At 4‐months‐old, the patient was diagnosed with “pneumonia” and hospitalized at another hospital. Blood routine analysis showed high levels of leukocytes, mainly elevated lymphocytes. The leukocyte count ranged from 18.98 to 28.88 × 109/L (reference range: 5.0–14.2 × 109/L), while the lymphocyte count ranged from 11.17 to 20.15 × 109/L (reference range: 2.8–10 × 109/L). Additionally, the haemoglobin levels were between 83 and 110 g/L (reference range: 103–138 g/L). After receiving anti‐infective treatment, the patient's cough and other symptoms improved, but the leukocyte count remained elevated for 3 months. At the age of 7 months, during hospitalization, a physical examination revealed the following: head circumference 40.5 cm (<3th percentile), weight 7.2 kg (<25th percentile), length 67 cm (<25th percentile), hypertelorism, depressed nasal ridge, thick eyebrow, hypotonia and patent ductus arteriosus. The patient's bone marrow aspiration revealed 5% naive lymphocytes, although bone marrow flow cytometry was not completed at that time. (Figure 1B).
FIGURE 1.
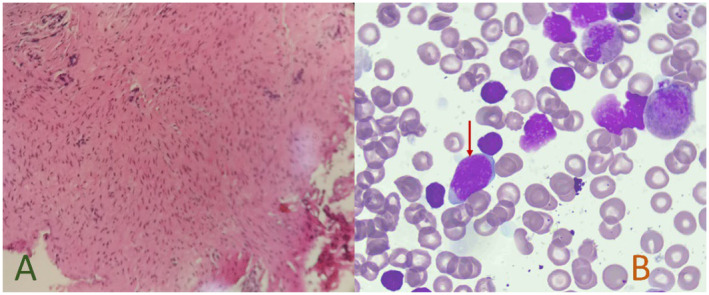
(A) H&E staining, pathological report of the end of the rectum showing smooth muscle tissue in the figure, magnification ×100; (B) Wright–Giemsa staining, bone marrow cell morphology. Red arrows show naive lymphocytes, magnification ×100.
At 8 months of age, the child exhibited delayed gross motor development and language development. A child health care physician conducted a professional evaluation on him, the Gesell developmental schedule indicated a developmental age of 3.1 months and a developmental quotient of 37, indicating severe developmental delay.
3.2. Genetic Results
Karyotypic analysis of chromosome G‐banding in proband: 46,XY,del(4)(p15.33p14) (Figure 2A). Karyotypic analyses of the parental and older brother's chromosomes were normal. A comprehensive comparative analysis of WES reads revealed a deletion of 26.738 Mb of heterozygosity, specifically identified as seq[hg38]del(4)(p15.33p14) chr4:g.11,399,082_38,137,335del in the samples. No additional single nucleotide variations were detected in this specimen (Figure 2B). The ClinGen (https://www.ncbi.nlm.nih.gov/projects/dbvar/clingen) dosage sensitivity evaluation system was used to assess genes located within the deletion region of 11,399,082–38,137,335 (Thaxton et al. 2021).
FIGURE 2.
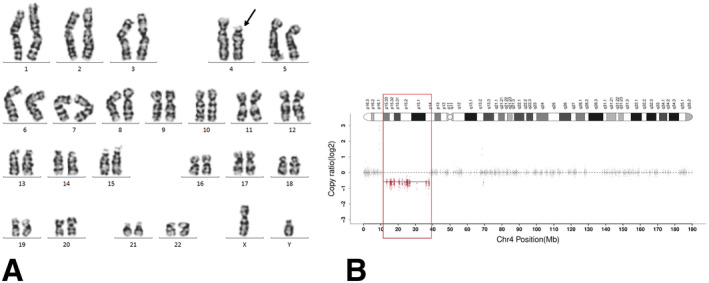
Genetic results, (A) karyotypic analysis of chromosome G‐banding, black arrow indicating the site of deletion; (B) Whole exome sequencing (WES), red part indicating the region of deletion.
The UCSC Genome Browser (https://genome.ucsc.edu/) revealed a total of 45 protein‐coding genes at the deletion region, with 11 genes phenotypically associated with the OMIM database (https://www.omim.org) (Table 1). Our molecular analyses show 10 genes in this deleted region for our proband, BOD1L1, FBXL5, LDB2, etc, have a pLI score of more than 0.9 (https://gnomad.broadinstitute.org), and thus likely to have phenotypic effects (Table 2).
TABLE 1.
The genotypes and phenotypes associated with this patient's deletion gene region are documented in the OMIM database.
| Gene | OMIM | Cytogenetic location | Genomic coordinates (GRCh38) | Inheritance | Phenotype | Dysfunction of gene product | pLI score |
|---|---|---|---|---|---|---|---|
| RAB28 | 612,994 | 4p15.33 | 4:13,367,724‐13,484,340 | AR | Cone‐rod dystrophy 18 | Retina | 0.02 |
| NKX3‐2 | 602,183 | 4p15.33 | 4:13,540,830‐13,547,744 | AR | Spondylo‐megaepiphyseal‐metaphyseal dysplasia | Chromosome rearrangement | 0.64 |
| CC2D2A | 612,013 | 4p15.32 | 4:15,469,865‐15,601,557 | AR | COACH syndrome 2, Joubert syndrome‐ 9, Meckel syndrome‐ 6 | Meckel diverticulum | 0.00 |
| PROM1 | 604,365 | 4p15.32 | 4:15,968,228‐16,084,023 | AD, AR, AD, AR, AD | Cone‐rod dystrophy 12, Macular dystrophy, Retinal, Retinitis pigmentosa 41, Stargardt disease 4 | Retina; leukaemia | 0.00 |
| TAPT1 | 612,758 | 4p15.32 | 4:16,160,505‐16,227,390 | AR | Osteochondrodysplasia, complex lethal, Symoens‐Barnes‐Gistelinck type | Ciliopathy | 0.03 |
| QDPR | 612,676 | 4p15.32 | 4:17,486,395‐17,512,090 | AR | BH4‐deficient, C | Hyperphenylalaninemia | 0.00 |
| CLRN2 | 618,988 | 4p15.32 | 4:17,515,165‐17,527,104 | AR | Deafness, autosomal recessive 117 | Ciliopathy | 0.00 |
| SEPSECS | 613,009 | 4p15.2 | 4:25,120,014‐25,160,582 | AR | Pontocerebellar hypoplasia type 2D | Late‐onset cerebellar atrophy | 0.00 |
| SLC34A2 | 604,217 | 4p15.2 | 4:25,655,851‐25,678,748 | AR | Pulmonary alveolar microlithiasis | pH‐sensitive sodium‐dependent phosphate transporter. | 0.00 |
| RBPJ | 147,183 | 4p15.2 | 4:26,105,449‐26,435,131 | AD | Adams‐Oliver syndrome 3 | Tumor of the kidney | 1.00 |
| DTHD1 | 616,979 | 4p14 | 4:36,281,616‐36,347,511 | NA | Variant of unknown signifcance | Glaucoma | 0.00 |
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; NA, not applicable; pLI, probability of loss‐of‐function intolerance.
TABLE 2.
Genes have pLI scores more than 0.9 in the presented case.
| Gene | OMIM | Gene Function | pLI score |
|---|---|---|---|
| BOD1L1 | 616,746 | Component of the fork protection pathway | 1 |
| FBXL5 | 605,655 | Function in phosphorylation‐dependent ubiquitination | 1 |
| LCORL | 611,799 | Mediates methylation of (H3K27) | 0.92 |
| LDB2 | 603,450 | As adapter molecules to allow assembly of transcriptional regulatory complexes | 0.95 |
| SLIT2 | 603,746 | Conserved roles in axon guidance and neuronal migration | 1 |
| DHX15 | 603,403 | Nuclear ATP‐dependent helicase, implicated in pre‐mRNA splicing | 1 |
| PPARGC1A | 604,517 | Coactivator of nuclear receptors and other transcription factors, involved in energy metabolism | 1 |
| STIM2 | 610,841 | Regulate calcium concentrations in the cytosol and endoplasmic reticulum | 0.96 |
| RBPJ | 147,183 | Transcriptional regulator important in the Notch signaling pathway | 1 |
| PCDH7 | 602,988 | Thought to function in cell–cell recognition and adhesion | 1 |
Abbreviations: H3K27, histone H3 (see 602,810) lys27; pLI, probability of loss‐of‐function intolerance.
The observed depletion region does not encompass the known causative gene or critical region for the established HI genomic region. We did not find enough evidence to classify certain genes and regions in 4p15.33‐p14 as potentially disease‐causing due to haploinsufficiency. A detailed evaluation of genomic content was conducted using cases from published literature and public databases (Maldziene et al. 2017; Di et al. 2023; Chen et al. 2013; Liang et al. 2016; Park et al. 2020). According to the ACMG 2019 guidelines, this depletion region, with assigned point values resulting in a final point value >0.99, is considered “pathogenic” (Riggs et al. 2020).
4. Discussion
In 1965 Cooper and Hirschhorn reported the first case of a human patient with a deletion at the end of the short arm of chromosome 4 (4p15.1‐pter), leading to the identification of Wolf‐Hirschhorn Syndrome (WHS) (Battaglia, Carey, and South 2015). Individuals with WHS typically present with distinct craniofacial abnormalities, including a high forehead, broad nose, and cleft palate, often likened to a ‘Greek warrior helmet’. These facial features can be detected prenatally through ultrasound (Maymon et al. 2004; Friebe‐Hoffmann et al. 2016). Cytogenetically, terminal deletions of chromosome 4 are known to be the primary cause of WHS, with an incidence rate ranging from 1 in 25,000 to 50,000 (Battaglia, Carey, and South 2015). The DECIPHER database (https://www.deciphergenomics.org), has documented a total of 120 patients with 4p deletions (Figure 3).
FIGURE 3.
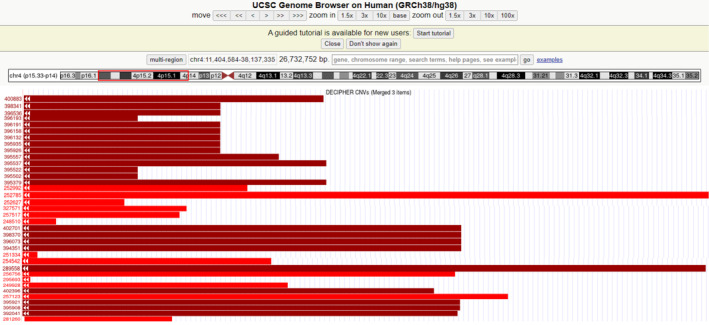
Genomic locations of 36 deletions based on the results obtained in the DECIPHER databases. Patient 252,785 and Patient 289,558 deletions in locations close to our patient to our case. The annotation is based on GRCh38/hg38.
In contrast to WHS, the 4p proximal interstitial deletion (4p12‐p16) is even rarer. The first documented case of this deletion on chromosome 4's short arm dates back to 1977, showing varying deletion regions (Di et al. 2023). To date, approximately 35 cases of chromosome 4 interstitial deletion have been documented globally. Between 2005 and 2017, an increasing number of instances of proximal 4p deletion syndrome were identified. Most of these cases were diagnosed using cytogenetic methods alone, without molecular genetic analysis, and did not undergo a detailed examination of the genes involved (Basinko et al. 2008; Maldziene et al. 2017; Chen et al. 2013; Liang et al. 2016; South et al. 2005; Moller et al. 2007; Makrythanasis et al. 2012). Compared to other reported cases, our patient exhibits a larger deletion region. However, there is limited understanding of the genes that influence the 4p14p16.1 deletion phenotype. The genetic cause of our patient was only identified post‐delivery rather than through prenatal diagnosis. Initially, due to financial constraints and the cost of analysis, the patient's family opted for chromosome and WES analysis. When we recommended further copy number variation (CNV)‐seq verification of the abnormal results, the patient's parents declined this examination due to concerns about repeated blood draws.
The RAB28 gene encodes a protein that may regulate intracellular transport and is associated with hereditary retinopathy. The DTHD1 gene is involved in the apoptotic pathway, while the NKX3‐2 gene plays a role in chondrogenic differentiation and regulation of signaling pathways, such as MAPK and BMP, and is associated with skeletal and tumoral disorders. The CC2D2A gene is linked to primary cilia formation, the CP110‐CEP290‐CC2D2A signaling pathway, and ciliopathies. Additionally, the QDPR gene catalyzes the reduction of quinonoid dihydrobiopterin, which is highly expressed in the brain. It is well established that Bi‐allelic Pathogenic variants cause hyperphenylalaninemia (BH4‐deficient, type C) (Zhu et al. 2024). The functions of the CLRN2 gene remain unclear, but it is thought to be involved in maintaining the structure and function of stereocilia. Mouse studies have linked it to deafness. The SLC34A2 gene codes for a protein that transports phosphate in a sodium‐dependent manner, playing a role in calcium and phosphorus metabolism. The TAPT1 gene influences primary cilia formation and variants it can disrupt the Golgi apparatus, impacting axial bone development in mice. The RBPJ gene is part of the RBPJ/DAPK3/UBE3A signaling pathway, which modulates the Notch signaling pathway and has been associated with epilepsy and neurodevelopmental disorders. Variants in the SEPSECS gene have been linked to deafness and neurodevelopmental issues. Conversely, the PROM1 gene, also known as CD133, has been implicated in various cancers and is found on cancer stem cells in mixed‐lineage leukemia (MLL) and B cell acute lymphoblastic leukemia (B‐ALL) (Glumac and LeBeau 2018; Li et al. 2018; Godfrey et al. 2020).
Our patient was in close proximity to the site of deletion of case 2 reported by Chitayat et al. (1995). However, our patient did not exhibit the severe skeletal deformities mentioned in the literature. There is a 25‐year age difference between the two patients, and many phenotypes may not yet manifest, necessitating long‐term follow‐up observation.
Lymphocytosis persisted in our patient for over 4 months, necessitating differentiation from lymphoproliferative disorders. Based on a comprehensive medical history and physical examination, the hematologist excluded recent infections such as Epstein–Barr virus (EBV), cytomegalovirus (CMV), human immunodeficiency virus (HIV‐1), Bordetella pertussis, among others (Devi et al. 2022). It is noted that immature peripheral lymphocytes in infants or young children and hematogone in the bone marrow can resemble cells in lymphocytic leukemia, which can be differentiated by flow cytometry. Unfortunately, the patient's family declined this essential examination. Noninfectious reactive lymphocytosis is frequently triggered by drug hypersensitivity and stress, both of which are not applicable to this patient. Although a bone marrow examination at 7 months of age revealed 5% naive lymphocytes, there was insufficient evidence to indicate hematologic malignancy (e.g., leukemia, etc.). It has been noted that individuals with WHS may be at risk of developing combined malignancies like neuroblastoma. The reasons behind the abnormally high peripheral blood lymphocytes in our patient remain unknown, warranting further evaluation by a hematologist using techniques such as flow cytometry and cytogenetics.
Most individuals with proximal 4p interstitial deletion have de novo variants, and familial inheritance is rare (Di et al. 2023). Unlike the craniofacial anomalies seen in WHS, the craniofacial anomalies observed in these patients are typically not prominent. Detecting these anomalies solely through prenatal ultrasound imaging can be challenging. A review of the literature identified a single case in which thickening of the nuchal fold (NF) was observed on prenatal ultrasound, and subsequently confirmed to be 46,XX,del(4)(p15.1p15.32) through FISH analysis. The region of the deletion was found to be 14.5 Mb. (South et al. 2005). The pathogenic variant on proximal 4p deletion syndrome, which may indicate a true contiguous gene syndrome requiring further research for confirmation, has not yet been identified.
5. Conclusions
In this case study, a patient with de novo proximal 4p deletion syndrome was diagnosed using chromosomal karyotype analysis and WES. Furthermore, it remains unclear whether 4p deletion syndrome correlates with anal atresia and persistent lymphocyte elevation in our study; thus, additional evidence is required to support this hypothesis. This case underscores the variability of 4p deletion syndrome and emphasizes the necessity for more clinical data to improve our understanding of genotype–phenotype correlation.
Author Contributions
P.Y., X.F. and Z.S. collected and integrated data, and wrote the article. X.F. and Z.S. participated in the study's design and coordination. L.H., C.C., J.N. and P.G. interpreted the results of the bone marrow cell smear. S.L. explained the pathology report. Z.L., W.J. and C.A. explained the chromosome and W.E.S. results. Z.S. and Y.L. participated in the evaluation of the patient. All authors reviewed the article critically for intellectual content and agreed to the published version of the manuscript.
Ethics Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Ethics Committee of Sichuan Provincial Hospital for Women and Children (protocol code 202300911‐225 and date of 2023. 09. 11). No financial or non‐financial benefits have been received or will be received from any party related directly or indirectly to the subject of this article.
Consent
Informed consent was obtained from all subjects involved in the study. Written informed consent was obtained from the patient's parents for their anonymized information to be published in this article.
Acknowledgements
We are grateful to the family for their willing participation and cooperation.
Funding: This work was supported by Chengdu Science and Technology Bureau (2021‐YF05‐01658‐SN).
Fu Xiong and Shuyao Zhu contributed equally to this work.
Contributor Information
Fu Xiong, Email: xiongfu606@163.com.
Shuyao Zhu, Email: 330986673@qq.com.
Data Availability Statement
The data that support the findings of this study are openly available in ClinVar at https://www.ncbi.nlm.nih.gov/clinvar/variation/2445987/, reference number VCV002445987.1.
References
- Basinko, A. , Douet‐Guilbert N., Parent P., et al. 2008. “Familial Interstitial Deletion of the Short arm of Chromosome 4 (p15.33‐p16.3) Characterized by Molecular Cytogenetic Analysis.” American Journal of Medical Genetics. Part A 146A, no. 7: 899–903. [DOI] [PubMed] [Google Scholar]
- Battaglia, A. , Carey J. C., and South S. T.. 2015. “Wolf‐Hirschhorn Syndrome: A Review and Update.” American Journal of Medical Genetics. Part C, Seminars in Medical Genetics 169, no. 3: 216–223. [DOI] [PubMed] [Google Scholar]
- Chen, C. P. , Lee M. J., Chern S. R., et al. 2013. “Prenatal Diagnosis and Molecular Cytogenetic Characterization of a de Novo Proximal Interstitial Deletion of Chromosome 4p (4p15.2→p14).” Gene 529, no. 2: 351–356. [DOI] [PubMed] [Google Scholar]
- Chitayat, D. , Ruvalcaba R. H., Babul R., et al. 1995. “Syndrome of Proximal Interstitial Deletion 4p15: Report of Three Cases and Review of the Literature.” American Journal of Medical Genetics 55, no. 2: 147–154. [DOI] [PubMed] [Google Scholar]
- Devi, A. , Thielemans L., Ladikou E. E., Nandra T. K., and Chevassut T.. 2022. “Lymphocytosis and Chronic Lymphocytic Leukaemia: Investigation and Management.” Clinical Medicine 22, no. 3: 225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di, J. , Yenwongfai L., Rieger H. T., Zhang S., and Wei S.. 2023. “Familial 4p Interstitial Deletion Provides New Insights and Candidate Genes Underlying This Rare Condition.” Genes (Basel) 14, no. 3: 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe‐Hoffmann, U. , Reister F., Gaspar H., Hummler H., Lindner W., and Lato K.. 2016. “The Wolf‐Hirschhorn Syndrome.” Zeitschrift für Geburtshilfe und Neonatologie 220, no. 5: 195–199. [DOI] [PubMed] [Google Scholar]
- Glumac, P. M. , and LeBeau A. M.. 2018. “The Role of CD133 in Cancer: A Concise Review.” Clinical and Translational Medicine 7, no. 1: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey, L. , Crump N. T., O'Byrne S., et al. 2020. “H3K79me2/3 Controls Enhancer–Promoter Interactions and Activation of the Pan‐Cancer Stem Cell Marker PROM1/CD133 in MLL‐AF4 Leukemia Cells.” Leukemia 35, no. 1: 90–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannes, F. , and Vermeesch J. R.. 2008. “Benign and Pathogenic Copy Number Variation on the Short arm of Chromosome 4.” Cytogenetic and Genome Research 123, no. 1–4: 88–93. [DOI] [PubMed] [Google Scholar]
- Li, D. , Hu Y., Jin Z., et al. 2018. “TanCAR T Cells Targeting CD19 and CD133 Efficiently Eliminate MLL Leukemic Cells.” Leukemia 32, no. 9: 2012–2016. [DOI] [PubMed] [Google Scholar]
- Liang, L. , Xie Y., Shen Y., Yin Q., and Yuan H.. 2016. “A Rare de Novo Interstitial Duplication at 4p15.2 in a boy With Severe Congenital Heart Defects, Limb Anomalies, Hypogonadism, and Global Developmental Delay.” Cytogenetic and Genome Research 150, no. 2: 112–117. [DOI] [PubMed] [Google Scholar]
- Makrythanasis, P. , Gimelli S., Bena F., et al. 2012. “Homozygous Deletion of a Gene‐Free Region of 4p15 in a Child With Multiple Anomalies: Could Biallelic Loss of Conserved, Non‐Coding Elements Lead to a Phenotype?” European Journal of Medical Genetics 55, no. 1: 63–66. [DOI] [PubMed] [Google Scholar]
- Maldziene, Z. , Preiksaitiene E., Ignotiene S., Kapitanova N., Utkus A., and Kucinskas V.. 2017. “A de Novo Pericentric Inversion in Chromosome 4 Associated With Disruption of PITX2 and a Microdeletion in 4p15.2 in a Patient With Axenfeld‐Rieger Syndrome and Developmental Delay.” Cytogenetic and Genome Research 151, no. 1: 5–9. [DOI] [PubMed] [Google Scholar]
- Maymon, R. , Strauss S., Herman A., Wiener Y., Heyman E., and Goldman M.. 2004. “The Prenatal Scan Pitfall for the Diagnosis of Renal Mass: Case Report.” Prenatal Diagnosis 24, no. 11: 932–934. [DOI] [PubMed] [Google Scholar]
- Moller, R. S. , Hansen C. P., Jackson G. D., et al. 2007. “Interstitial Deletion of Chromosome 4p Associated With Mild Mental Retardation, Epilepsy and Polymicrogyria of the Left Temporal Lobe.” Clinical Genetics 72, no. 6: 593–598. [DOI] [PubMed] [Google Scholar]
- Park, S. , Jeon B. R., Lee Y. K., Ki C.‐S., and Jang M.‐A.. 2020. “The First Korean Case of De Novo Proximal 4p Deletion Syndrome in a Child With Developmental Delay.” Annals of Laboratory Medicine 40, no. 5: 435–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs, E. R. , Andersen E. F., Cherry A. M., et al. 2020. “Technical Standards for the Interpretation and Reporting of Constitutional Copy‐Number Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen).” Genetics in Medicine 22, no. 2: 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- South, S. T. , Corson V. L., McMichael J. L., Blakemore K. J., and Stetten G.. 2005. “Prenatal Detection of an Interstitial Deletion in 4p15 in a Fetus With an Increased Nuchal Skin Fold Measurement.” Fetal Diagnosis and Therapy 20, no. 1: 58–63. [DOI] [PubMed] [Google Scholar]
- Thaxton, C. , Good M. E., DiStefano M. T., et al. 2021. “Utilizing ClinGen Gene‐Disease Validity and Dosage Sensitivity Curations to Inform Variant Classification.” Human Mutation 43, no. 8: 1031–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, S. , Hu Q., Yang Y., et al. 2024. “Genotype Characterization of Tetrahydrobiopterin Deficiency in Two Tibetan Children.” Heliyon 10, no. 5: e27050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are openly available in ClinVar at https://www.ncbi.nlm.nih.gov/clinvar/variation/2445987/, reference number VCV002445987.1.