

Abstract
The stress kinase MAPK13 (aka p38δ-MAPK) is an attractive entry point for therapeutic intervention because it regulates the structural remodeling that can develop after epithelial barrier injury in the lung and likely other tissue sites. However, a selective, safe, and effective MAPK13 inhibitor is not yet available for experimental or clinical application. Here we identify a first-in-kind MAPK13 inhibitor using structure-based drug design combined with a screening funnel for cell safety and molecular specificity. This inhibitor (designated NuP-4) down-regulates basal-epithelial stem cell reprogramming, structural remodeling, and pathophysiology equivalently to Mapk13 gene-knockout in mouse and mouse organoid models of post-viral lung disease. This therapeutic benefit persists after stopping treatment as a sign of disease modification and attenuates key aspects of inflammation and remodeling as an indication of disease reversal. Similarly, NuP-4 treatment can directly control cytokine-stimulated growth, immune activation, and mucinous differentiation in human basal-cell organoids. The data thereby provide a new tool and potential fix for long-term stem cell reprogramming after viral injury and related conditions that require MAPK13 induction-activation.
Keywords: mitogen-activated protein kinase (MAPK), p38-MAPK, epithelial stem cell, inflammatory memory, respiratory viral infection, asthma, chronic obstructive pulmonary disease (COPD), long-term Covid
Introduction
The family of mitogen-activated protein kinases (MAPKs), and in particular the subgroup of p38-kinases (MAPK11-14 aka p38α-δ-MAPK), are implicated in a broad set of experimental models of disease and the corresponding human diseases (1, 2). These studies led to a corresponding array of drug development programs directed at MAPK targets, particularly MAPK14 (aka p38α-MAPK) as the first and best studied member of the MAPK family (3–12). Despite this effort, no inhibitors for MAPK14 have been approved for clinical application. The basis for failure in clinical trials range from suboptimal efficacy and safety to difficulties in target validation, biomarker application, and disease modification. Nonetheless, MAPK14 and related kinases continue to be an attractive target for ongoing drug discovery and development programs.
In thar context, the relatively undrugged MAPK13 (aka p38δ-MAPK) is implicated in diseases that include diabetes, sepsis, neurodegeneration, and carcinoma (13–18). Given this diversity, we questioned whether MAPK13 might represent a more fundamental control over cellular programming. Thus, our earlier studies identified MAPK13 as a therapeutic target in chronic lung disease. This idea derived from a MAPK13 requirement for mucus production in human basal-epithelial stem cell (basal-ESC) models and a corresponding upregulation of MAPK13 expression and activation in lung tissue samples from COPD (19). In follow up, recent work demonstrates a MAPK13 requirement in vivo based on attenuation of basal-ESC reprogramming and post-viral lung disease (PVLD) in Mapk13 gene-knockout mice (20). Similarly, MAPK13 induction-activation was localized to basal-ESCs in lung samples from asthma and COPD. In addition, MAPK13 gene-deficiency attenuated basal-ESC growth in mouse and human cell models as evidence of a fundamental role for MAPK13 in epithelial barrier function (20). These findings provided a pathway to practical application since epithelial stem cell-orchestrated inflammation and mucus production are linked to morbidity and mortality in chronic lung diseases such as asthma and COPD (19, 21, 22).
The present study therefore addresses the goal of discovering and developing a highly selective MAPK13 inhibitor to address the issue of barrier injury and consequent disease. Previous studies by our group could not address this issue given the broader activity of first- and second-generation inhibitors and the absence of studies in a model of basal-ESC reprogramming that might exhibit long-term inflammation and structural remodeling (19, 23). In contrast, the present study arrives at a third-generation MAPK13 inhibitor that is remarkably selective compared to previous compounds from our lab and others (11, 12, 19, 23). In addition, the proof-of-concept is developed in a more suitable model for basal-ESC reprogramming for long-term growth, immune activation, and mucinous differentiation after barrier injury. Together, the present experimental approaches provide a previously unavailable tool for MAPK13 blockade and proof-of-concept for down-regulating this target as a disease-modifying strategy.
Results
Structure-based drug design yields a selective MAPK13 inhibitor
Given the possible benefits of MAPK13 blockade, we aimed to develop a small-molecule inhibitor that was selective for MAPK13 versus the previous focus on MAPK14 or MAPK13-14. Accordingly, we extended a drug-design strategy to fully modify a MAPK14-inhibitor parent compound (BIRB-796, NuP-43 in our chemical library) to better fit into the left-hand hinge region, ATP-binding pocket, and right-hand allosteric pocket of MAPK13 as defined in our high-resolution X-ray crystal structure (19, 23–25). These chemical analogs (n=520) as recently published (26) entered a screening funnel that included a primary screen for suitable chemical properties (molecular weight, Lipinski and Veber criteria, partition coefficient, and topological polar surface area) followed by a secondary partial screen for inhibition of MAPK13 and MAPK14 enzyme activities, a tertiary screen for cell toxicity based on transepithelial electrical resistance (TEER) as a readout for monolayer integrity in primary-culture human tracheobronchial epithelial cells (hTECs), and a final full screen of all candidates for MAPK13 enzyme inhibition (Figure 1A). This process arrived at a lead candidate drug designated NuP-4 with a 122-fold increase in MAPK13 inhibition and at least a 250-fold decrease in MAPK14 inhibition compared to the parent compound NuP-43 (Figure 1B). This performance improves on first-generation compounds with a modest (3-7-fold) increase in MAPK13 inhibitory activity (19) and second-generation compounds, including NuP-3 that improved MAPK13 inhibition (130-fold) compared to the parent compound NuP-43 (23). Further, NuP-4 demonstrated high selectivity for MAPK13 in a 425-kinase inhibitor screen compared to parent compound NuP-43 or MAPK13-14 inhibitor NuP-3 (Figure 1C and Supplemental Figure 1).
Fig. 1. Structure-based drug design yields a lead candidate MAPK13 inhibitor (NuP-4).
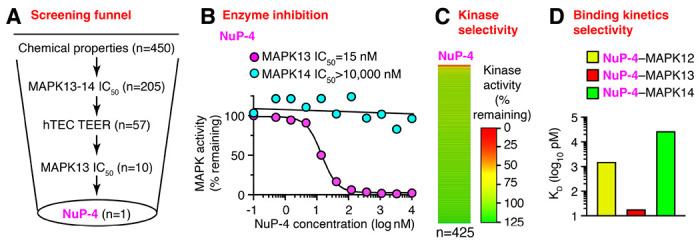
A, Screening funnel steps for chemical compound library (n=520) using chemical properties, MAPK13-14 enzyme assays, trans-epithelial electrical resistance (TEER) for human tracheobronchial epithelial cell (hTEC) monolayer integrity for cell toxicity, and additional MAPK13 assays. B, Enzyme inhibition activities for NuP-4 based on MAPK13 and MAPK14 phosphorylation of kinase substrate. C, Heat map for enzyme inhibition activities for NuP-4 based on a 425-kinase panel. D, Values for KD derived from biolayer interferometry (BLI) analysis of NuP-4 binding for MAPK13 versus MAPK12 or MAPK14.
By design, NuP-4 (like other chemical analogs in our series) maintains a DFG-out binding mode that provides favorable slow dissociation kinetics and consequent high potency and long duration of action of a Type II kinase inhibitor (19, 27, 28) that fits with modeling of this process (29, 30). These biding kinetics were confirmed using biolayer interferometry (Supplemental Fig. 2). Notably, we also found 200-fold higher binding (lower KD) for NuP-4 to MAPK13 compared to even its closest homologue MAPK12 based on a remarkably long Koff rate (Figure 1D). This data demonstrates high selectivity and potency of NuP-4, which fits with our analysis of MAPK13 structure (19, 24) and function. This profile also predicts low off-target toxicity. Indeed, dose-range finding showed no adverse effects or significant abnormalities in clinical laboratory values in toxicology studies done in rat (Supplemental Fig. 3A–E) or dog (Supplemental Fig. 4A–D).
Mapk13 inhibition blocks PVLD
We next determined whether NuP-4 treatment might provide MAPK13 blockade and therapeutic benefit in vivo. For this goal, we took advantage of a mouse model of PVLD that that develops after a natural pathogen Sendai virus (SeV) and depends on basal-ESC reprogramming (31, 32) that might in turn be linked to Mapk13 function (20, 23). For drug delivery in this model, NuP-4 was formulated as the corresponding salt (designated NuP-4A) in excipient 2-hydroxypropyl-β-cyclodextrin (Cdx) to optimize compound solubility suitable for intravenous (i.v.) or intraperitoneal (i.p.) injection dosing. Using this formulation, pharmacokinetic (PK) analysis showed that NuP-4A levels were consistently increased in target lung tissue compared to plasma when given by i.v. or i.p. injection to mice (Figure 2A). For example, NuP-4A achieves 9-32-fold higher area-under-the-curve (AUC) in lung tissue compared to plasma consistent with high uptake and target affinity in lung. These lung tissue levels of NuP-4A were maintained for at least 8 h at a concentration (>100 nM) that was effective for fully blocking MAPK13 enzyme activity (Figure 1B). In that context, we proceeded with a dose of 4 mg/kg given i.p. twice per day, and to approximate clinical treatment conditions, we initiated treatment at 5 d after infection and assessed animals at 49 d when PVLD phenotype is maximal (31, 33–39).
Fig. 2. NuP-4A benefit persists after stopping treatment to correct PVLD.

A, Levels of NuP-4A in lung tissue and plasma after a single dose (4 mg/kg i.p.) in mice (n=3). AUC, area under curve. B, Protocol scheme for NuP-4A or vehicle treatment twice daily (bid) for 5-21 d with final samples at 49 d after SeV infection. C, Body weights for conditions in (B). D, Immunostaining for Mapk13 and Krt5 with DAPI counterstaining in lung sections at 49 d after SeV or PBS and with NuP-4A or vehicle treatment for conditions in (B). E, Quantitation of immunostaining for (D). F, Protocol scheme for basal cell isolation, quantitation, and 3D organoid culture. G, Cell numbers from flow cytometry of basal epithelial cells for conditions in (F). H, Organoid numbers from 3D cultures of basal cells for conditions in (F). I, Immunostaining for Krt5 with DAPI counterstaining in lung sections for conditions in (A). J, PAS-hematoxylin staining of lung sections for conditions in (A). K, Quantitation of staining for (I,J). L, Immunostaining for IL-33 and Sftpc with DAPI counterstaining of lung sections for conditions in (B). M, Quantitation of immunostaining for (L). For (C-M), data are representative of three separate experiments with n=5-7 animals per condition in each experiment (mean ± s.e.m.). *P <0.05 using ANOVA and Tukey correction.
Under this protocol (as diagrammed in Figure 2B), we found that acute illness and associated weight loss were unchanged in mice treated with NuP-4A (Figure 2C). In contrast, the usual progression to PVLD was markedly attenuated in NuP-4A-treated mice. Thus, the typical increase in nuclear-Mapk13+ basal cells in remodeling regions was significantly diminished with NuP-4A-treatment compared to vehicle control at 49 d after SeV infection (Figure 2D,E). Relatedly, treatment resulted in decreased levels of EpCAM+Aqp3+ basal cell counts in lung tracked with flow cytometry (Figure 2F,G) and corresponding decreases in basal-ESC-derived organoid formation in 3D culture conditions (Figure 2H). Similarly, attenuation of Krt5+ basal cell hyperplasia was detected by immunostaining of lung sections (Figure 2I). In concert with control of basal cell overgrowth, the levels of periodic acid Schiff (PAS) and hematoxylin tissue staining (reflecting mucus production and cellularity, respectively) in lung sections were also attenuated in NuP-4A-treated mice at 49 d after infection (Figure 2J). These treatment-related improvements in Krt5 and PAS staining were confirmed as significant based on quantitative morphology (Figure 2K). In contrast to down-regulation of the basal cell reprogramming and remodeling events, we found no significant change in Sftpc+IL-33+ AT2 cell level with NuP-4A treatment compared to vehicle control (Figure 2L,M), providing evidence of basal-cell selectivity for NuP-4A treatment effect.
In concert with histopathology findings, treatment with NuP-4 also resulted in down-regulation of mRNA biomarkers linked to chronic lung disease. Thus, treatment resulted in significant decreases in basal-ESC growth marked by Krt5, Aqp3, and Trp63; immune activation marked by Serpinb2, Ltf, Cxcl17, and Nos2; type 2 inflammation marked by Il13, Arg1, and Trem2; type1/2 inflammation marked by Il6; and mucinous differentiation marked by Muc5ac and Clca1(Figure 3A–F). No significant decreases were found for mucinous differentiation when marked by Muc5b (Figure 3E), alarmin signal marked by Il33 mRNA (Figure 3G), or conventional type 1 inflammation marked by Ifng, Tnfa, and Il1b mRNA (Fig. 3H). We also found that NuP-4 treatment resulted in correction of clinical disease phenotypes: immune activation based on Nos2+ immunostaining of lung sections (Figure 4A,B); macrophage infiltration based on F4/80+ immunostaining of these sections (Figure 4C,D); mucinous differentiation based on immunostaining for Muc5ac and Muc5b (Figure 4E,F); and hypoxemia based on SpO2 measurements (Figure 4G). Each of these findings supported a beneficial effect for Mapk13 inhibitor treatment that was similar to the case for Mapk13 gene deficiency. Further, PVLD did not recur after stopping treatment, suggesting that basal cell control might confer a disease-modifying benefit.
Fig. 3. NuP-4A effect on biomarkers to track PVLD.

A-G, Lung levels of mRNA biomarkers for indicated disease endpoints for NuP-4A treatment for 5-21 d with final samples at 49 d after SeV infection as shown in Fig. 2B. Data are representative of three separate experiments with n=8 animals per condition in each experiment (mean ± s.e.m.). *P <0.05 using ANOVA and Tukey correction.
Fig. 4. NuP-4A effect on phenotypes that track PVLD.

A, Immunostaining for Nos2 with DAPI counterstaining of lung sections from mice for NuP-4A treatment for 5-21 d with final samples at 49 d after SeV or PBS as shown in Fig. 2B. B, Quantitation of staining in (A). C, Immunostaining for F4/80+ macrophages and DAPI counterstaining for conditions in (A). D, Quantitation of staining in (C). E, Immunostaining for Muc5ac and Muc5b with DAPI counterstaining for conditions in (A). F, Quantitation of staining in (E). G, Oximeter levels for blood oxygen saturation (SpO2) for conditions in (A). Data are representative of three separate experiments with n=5-14 animals per condition in each experiment (mean ± s.e.m.). *P <0.05 using ANOVA and Tukey correction.
We extended this analysis of NuP-4A efficacy to provide additional preclinical guidance. First, we determined whether NuP-4A might be safe and effective even when delivered during infectious illness. For this approach, NuP-4A treatment was started 2 d before infection. In these experiments, we also we compared NuP-4A to a combined MAPK13-14 inhibitor (NuP-3A) (23) and Mapk13−/− mice at 21 d after infection when changes in disease biomarkers become significant (as diagrammed in Supplemental Figure 5A). Under this protocol, acute weight loss and lung levels for viral RNA and titer were not significantly different among NuP-4A-treated, NuP-3A-treated, Mapk13−/− mice, and control vehicle-treated mice (Supplemental Figure 5B–D). In addition, NuP-4A treatment was as effective as NuP-3A or Mapk13-deficiency in attenuating basal cell hyperplasia, mucinous differentiation, and cellularity in lung sections (Supplemental Figure 5E–G). Similarly, mRNA biomarkers showed down-regulation of basal-ESC growth, immune activation, type 2 inflammation, and mucinous differentiation, and Mapk13 induction, (Supplemental Figure 6A–H). Second, we assessed whether NuP-4A treatment could reverse PVLD, providing a basis for wider clinical application when basal-ESC growth might be less prominent but immune activation and mucinous differentiation might still be ongoing (31). Indeed, we found that NuP-4A treatment delivered at 33-48 d after SeV infection (as diagrammed and monitored in Supplemental Figure 7A,B) was less effective in attenuating basal cell hyperplasia but still significantly decreased mucus production based on results from Mapk13+, Krt5+, and PAS-hematoxylin+ staining (Supplemental Figures 7C–E) and mRNA biomarker expression (Supplemental Figure 8A–H). Similarly, NuP-4A treatment significantly decreased macrophage infiltration and mucinous differentiation based on immunostaining of lung sections but did not affect Nos2+ immunostaining given its localization to basal and basal-lineage cells at 49 d after infection (Supplemental Fig. 9A–F). These findings suggest special sensitivity of IL-13 and mucus production to NuP-4 treatment.
MAPK13 activation and function in human basal cells
To better define NuP-4 mechanism, especially in humans, we extended our approach to studies of human cell models. In that regard, we recognized that MAPK13 expression and activation can be up-regulated and localized to basal-epithelial cells in the lung, particularly in tissue samples from diseases (notably asthma and COPD) linked to basal-ESC reprogramming (19, 20). Further, these findings fit with MAPK13 function in regulating growth and mucinous differentiation based on studies of MAPK13 gene knockdown in human basal-ESC cultures (19, 20). Accordingly, we performed additional studies of NuP-4 effects in hTEC cultures. To assess basal-ESC growth, hTECs were maintained under submerged 2D culture conditions that preserve basal-ESCs (34). Under these conditions (as diagrammed in Figure 5A), NuP-4 treatment inhibited basal-ESC growth (Figure 5B). Drug effectiveness (IC50=30 nM) was close to the value for enzyme-based MAPK13 inhibition (IC50=15 nM) based on generic conditions for substrate phosphorylation (Figure 1B). These inhibitory effects are at least 1000-fold lower than levels that produce LDH-based cell toxicity for NuP-4 (Supplemental Figure 10A,B). Moreover, the effect of NuP-4 on basal cell growth was not found with a potent and selective MAPK14 inhibitor NuP-401 (VX-745) (40) under these conditions (Figure 5B). We also took advantage of our discovery that GPNMB provides a vital signal for basal-ESC growth in mouse models of PVLD and ex vivo models of mouse and human organoid formation (32). Here we found that NuP-4 treatment also attenuated media-stimulated and GPNMB-enhanced organoid formation in 3D culture conditions (Figure 5C,D). This inhibition was found at similar potency (IC50=10 nM) to growth in submerged cultures. Further, NuP-4 treatment (at the same potency) also blocked baseline and GPNMB-stimulated immune activation marked by CXCL17 chemokine and IL33 alarmin gene expression tracked with corresponding mRNA levels (Figure 5E). In contrast to effects on growth and immune activation, NuP-4 was even more effective in blocking IL-13-stimulated differentiation into mucous cells in IL-13-stimulated air-liquid interface (ALI) cultures that promote this process. Here efficacy (IC50=0.02 nM) was equivalent to binding-kinetics KD for NuP-4 specific to MAPK13 (Figure 1D and Supplemental Figure 2), providing a structural mechanism for the observed MAPK13 function. Together, these results provide initial evidence that MAPK13 might be overexpressed, activated, and susceptible to therapeutic blockade in human basal cells as predicted by our mouse models.
Fig. 5. NuP-4 controls basal-ESC growth and differentiation.

A, Protocol scheme for human tracheobronchial epithelial cell (hTEC) study using submerged culture conditions. B, Cell number levels for conditions in (A). C, Protocol for hTEC-derived organoids for compound effect on cell growth and immune activation. D, Levels of organoids for conditions in (C). E, Levels of CXCL17 and IL33 mRNA for conditions in (C). F, Protocol for hTEC study under air-liquid interface (ALI) culture conditions for effect of NuP-4 on MUC5AC mRNA level with and without IL-13 stimulation. G, Levels of target mRNA for conditions in (F). For (B,D,E,G), values represent mean ± s.e.m. for a single subject representative of 3 subjects. *P <0.05 using ANOVA and Tukey correction.
Discussion
This study engages drug discovery and development to identify a highly selective, effective, and safe MAPK13 inhibitor that can be applied to the specific problem of chronic lung disease and the general challenge of tissue repair and remodeling after injury. Key findings include: (1) drug design-based generation and formulation of a first-in-kind, small-molecule MAPK13 inhibitor (designated NuP-4); (2) demonstration that this drug candidate can deliver long-term correction of basal-ESC reprogramming and chronic lung disease after viral injury in mouse and mouse-cell models; and (3) corresponding MAPK13 inhibitor control of the same basal-ESC endpoints (excess growth, immune activation, and mucinous differentiation) in human-cell models. Together, the data provide a practical strategy for modifying this type of disease (as diagrammed in Figure 6) and perhaps other diseases that already implicate a similar mechanism. Here we highlight the present findings in the context of recent work that demonstrates the value of MAPK13 blockade as a therapeutic target based on gene-knockout/knockdown (20). Thus, we highlight three major points of the present data.
Fig. 6. Model for basal-ESC reprogramming towards excess growth, immune activation, and mucus production as targets for MAPK13 blockade in airway disease.
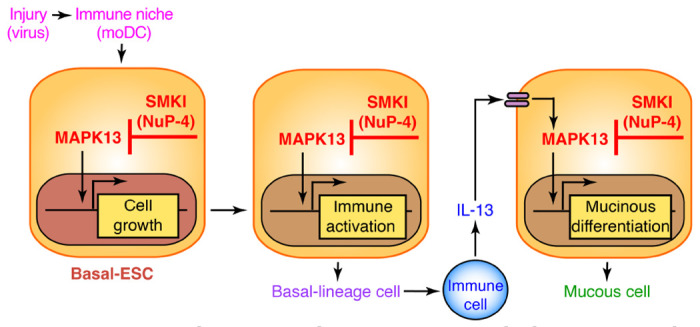
Sequential steps include initial barrier injury (e.g., viral infection); immune niche support (e.g., moDC recruitment and activation); basal-ESC growth, differentiation, and immune activation (e.g., chemokine production); immune cell recruitment and production of cytokines (e.g., IL-13), and further basal-lineage cell differentiation towards mucus production in mucous cells. SMKI, small-molecule kinase inhibitor (e.g., NuP-4).
First, in relation to drug discovery, it has been difficult to drug MAPKs selectively, effectively, and safely in general but particularly challenging in the case of MAPK13 (41). Previous discovery efforts generally based on broad kinase-inhibitor screens did not deliver a suitable drug candidate (42–44), perhaps revealing structural limitations in access to the working parts of the drug target. Cell-based screening were similarly unsuccessful, potentially due to the usually low levels of MAPK13 expression under homeostatic conditions and the challenges with artificial overexpression of MAPK13 (45). Given these problems, we took advantage of structural data for the molecular target (MAPK13) and pathobiological data for the cellular target of interest (basal-ESCs) in combination with experimental and clinical disease models. This combined approach led to a candidate drug with remarkable increases in potency, selectivity, binding characteristics, and pharmacological properties all validated with the MAPK13 target. These advantages translated to a therapeutic benefit that is equivalent to MAPK13 deficiency in animal and cell models. This new compound might thereby prove useful to other disease models and diseases wherein MAPK13 control is implicated but still based primarily on gene-knockout/knockdown strategies (15, 20, 46–48).
Second, in relation to drug mechanism, it has also been historically difficult to achieve efficacy and avoid toxicity. In general, a solution required a greater margin of safety for dosing, with lower dosing favoring safety. Here we find that NuP-4 maintains at least two levels of inhibitory activities: the first linked to cell growth and immune activation that is sensitive to nM levels of compound for inhibition of target. phosphorylation/activation; and the second connected to mucinous differentiation that is sensitive to only pM levels of compound for inhibition of target binding even without phosphorylation. Both inhibitory effects demonstrate a high level of compound selectivity for the MAPK13 target, but the binding data shows remarkable specificity even compared to the most closely related MAPK12 target. This data thereby provides a previously unappreciated opportunity for molecular specificity and thereby efficacy with safety. The full structural basis for this NuP-4-MAPK13 interaction needs further definition with NuP-4 analogs, but the finding already translates to practical application to downstream substrates. There is already and will likely be more downstream MAPK13 substrates that translate to distinct signaling functions (41, 43). The present data suggests that these substrates might engage with varying affinities to account for the differential potency for NuP-4 inhibition of MAPK13-dependent cell growth, immune activation, and differentiation. Here we find that MAPK13 blockade can also interrupt both baseline and GPNMB-stimulated growth and differentiation. The possible intersection of GPNMB-CD44 and MAPK13 signals should also serve to define the basis for selective stem cell control with improvements in efficacy and safety. It will also be critical to identify the native substrates for upstream activation of MAPK13, presumably mediated at least in part by MAPK-MAP2K interaction similar to MAPK14 (49).
Third, in relation to this issue of target selectivity, the present data for NuP-4 also bolsters the proposal that MAPK13 function is linked specifically to the basal-ESC behavior. In particular, NuP-4 action can be localized to MAPK13 in basal-ESCs in situ and ex vivo, consistent with studies of MAPK13 gene-knockout/knockdown (19, 20). Of note, NuP-4 treatment exhibit blockade of basal-cell growth without significantly changing AT2-cell growth in vivo as a measure of functional sensitivity. These findings provide additional specificity for drug action based on cellular target to compliment the selectivity for molecular target noted in the above discussion. Whether there might be additional MAPK13 action in other cell types still needs to be further defined, and studies of conditional gene-knockouts and inhaled drug delivery are ongoing to address this issue. However, even at present, this cell-type information represents a shift from the historical focus on immune-cell directed MAPK14 inhibitors for treatment of inflammation in general and lung disease in particular (3–6, 8, 11, 50–55). That approach resulted in MAPK14-specific inhibitors that were highly effective in blocking conventional cytokine (e.g., TNF-α and IL-1β) signaling (56) as a pathway to therapeutic benefit (57). Indeed, our initial approach aimed at development of a combined MAPK13-14 inhibitor designated NuP-3 (23), recognizing as shown here that there would be a loss of drug specificity. Further, even the most advanced versions of MAPK14 inhibitors have proved to be ineffective in clinical trials of patients with lung disease, e.g., COPD (8). The data here provide a basis for this failure in efficacy (and off-target actions) by implicating the dominant action of MAPK13 in the disease process. As predicted from our earlier work (19, 20, 23), MAPK13 activation appears to be required for type-2 cytokine (particularly IL-13 and IL-4) signaling towards airway inflammation and excess mucus production. This paradigm predicted the success for IL-13 blockade in subsets of asthma and COPD (58). The present data reinforces this more specific therapeutic strategy, however, adding the additional element of a stem cell target for long-term correction of disease phenotypes and perhaps inflammatory memory.
Finally, the present data provides a small-molecule MAPK13 inhibitor that demonstrates effectiveness in models of MAPK13-dependent disease. The findings thereby offer a new tool for studies of MAPK13 function and further solidify the value of MAPK13 as a therapeutic target in the pathway to chronic lung disease. However, these results also highlight shared pathogenesis and therapeutic effect across the spectrum of epithelial barrier injury and repair. In the case of respiratory viruses, our lab and others provide evidence that a similar basal and immune cell response develops across different respiratory viruses (including EV-D68, IAV, RSV, HRV) and triggers (including allergens and pollutants) in animal models and humans (32, 36, 39, 59–62). These findings suggest a pathway to a broader and more lasting strategy to inhaled agents that are responsible for respiratory failure in the intermediate-term and initiation, progression, and exacerbation of chronic respiratory diseases in the long-term. The present data for disease correction after stopping treatment and disease reversal with late-stage treatment further suggest the potential for disease modification. This represents an advancement over current drug and biologic treatments (63) by introducing a practical and precise (biomarker-guided) means to correct basal-ESC reprogramming and post-viral lung disease as a guide to testing in related diseases in the respiratory tract and other sites.
Materials and Methods
Compound generation, analysis, and formulation
MAPK inhibitor candidates were developed as a chemical analog series (n=520 compounds) from parent compound NuP-43 (BIRB-796) with modifications based on X-ray crystal structures for MAPK13 (unphosphorylated and phosphorylated forms) and co-crystal structure for the MAPK13-NuP-3 complex (19, 23–25). The entire series of analogs was subjected to a screening funnel that included assessments of chemical properties, MAPK13 and MAPK14 enzyme inhibition assays, and hTEC TEER values. Individual kinase inhibition assays were performed using the HotSpot assay platform (Reaction Biology, Malvern, PA) as described previously (23, 42) using MAP2K6 (MKK6) as upstream kinase activator and myelin basic protein as downstream kinase substrate. The same approach was used to test NuP-43, NuP-3, and NuP-4 for inhibitory activities in a comprehensive human kinase panel (Reaction Biology, Malvern, PA). Binding kinetics for NuP-4 interaction with MAPK12, 13, and 14 were assessed with biolayer interferometry (BLI) as described previously (23).
Lead candidate compound (NuP-4) was formulated as the HBr salt (designated NuP-4A) to improve solubility for all studies of parenteral administration. For pharmacokinetic (PK) analysis, compound levels were determined in plasma and lung tissue samples from mice at a single dose of 2-8 mg/kg and 4 mg/ml in 20% (wt/vol) 2-hydroxypropyl-β-cyclodextrin (Cdx) given by intravenous (i.v.) or intraperitoneal (i.p.) injection. Compound levels were determined using a Shimadzu DGU-20A5R(C) HPLC and LCMS-8060 LC/MS/MS instrument or a Prominence Degasser DGU-20A5T(C) HPLC and an AB Sciex Triple Quad 5500 LC/MS/MS instrument. For mouse model experiments, compound was also dissolved in Cdx (20% wt/vol) at a compound concentration of 4 mg/ml and stored at 4 °C for each experiment. Compounds were reconstituted in DMSO vehicle (≤1:1000 vol/vol) for cell model experiments. For each preparation, compound stability and purity were verified at >99% using an Agilent 1100 Series HPLC and LC/MSD system and a Varian 400-MHz NMR spectrometer.
Toxicology
For rat (Sprague-Dawley) and dog (Beagle) toxicology studies, NuP-4 was tested in maximum tolerated dose (MTD) and dose-range finding (DRF) protocols using intravenous injection of NuP-4A at 4 mg/ml in 20% 2-hydroxypropyl-β-cyclodextrin (Cdx) solution (tail vein for rats and leg vein for dogs). Maximal doses were limited by feasibility based on NuP-4A solubility and injection volume. Levels of NuP-4 for lung tissue and plasma samples were determined using a bioanalytical assay with LC/MS/MS as described above.
Mouse models
Male and female wild-type (WT) C57BL/6J mice (000664) mice were obtained from Jackson Laboratory. The Mapk13 gene knockout mice (Mapk13−/−) were generated in the C57BL/6J background using CRISPR/Cas9 technology as described in our recent report (20). All mice were maintained and co-housed in a barrier facility using cages fitted with micro-isolator lids. Animal husbandry and experimental procedures were approved by the Animal Studies Committees of Washington University School of Medicine in accordance with the guidelines from the National Institutes of Health. Sendai virus (SeV, Sendai/52 Fushimi strain, ATCC VR-105) was obtained from ATCC and prepared and titered by plaque-forming assay and qPCR assay as described previously (33). Mice were infected with SeV (2.6 x 105 PFU) as described previously (36). Virus or an equivalent amount of UV-inactivated virus or PBS alone was delivered intranasally in 30 μl of PBS under ketamine/xylazine anesthesia at 6-9 wk of age. Results from male and female mice were pooled since no significant differences were found between sexes as reported initially (64) and confirmed recently (36) and in the present experiments (data not shown). Viral titers for stock solutions and lung infections were monitored by quantitative PCR (qPCR) assay using primers for SeV-NP RNA as defined previously and in Supplemental Table 1 using SeV-NP-expressing plasmids as an internal standard (36)
Tissue histology and staining
Lung tissue was fixed with 10% formalin, embedded in paraffin, cut into 5-μm sections and adhered to charged slides. Sections were stained with PAS and hematoxylin as described previously (31, 36). For immunostaining, sections were deparaffinized in Fisherbrand® CitriSolv® (Fisher), hydrated, and heat-treated with antigen unmasking solution (Vector Laboratories, Inc). Immunostaining was performed with the commercially available primary antibodies as detailed in Supplemental Table 2. Primary antibodies were detected with secondary Abs labeled with Alexa Fluor 488 (Thermo Fisher Scientific) or Alexa Fluor 594 (Thermo Fisher Scientific) followed by DAPI counterstaining. Slides were imaged by light microscopy using a Leica DM5000 B and by immunofluorescent microscopy using an Olympus BX51, and staining was quantified in whole lung sections using a NanoZoomer S60 slide scanner (Hamamatsu) and ImageJ software as described previously (31, 36).
Flow cytometry and FACS
Single cell suspensions were generated from minced lung tissue that was subjected to collagenase (Liberase TM Research Grade, Roche), hyaluronidase (Sigma), DNAse I (Sigma), and Dispase II (Roche) digestion for 45 min at 37 °C and then treated with ACK buffer (Lonza) to remove red blood cells. Following FcR blockade, lung cell suspensions were incubated with labeled antibodies and were sorted using a Sony SY3200 Synergy high-speed cell sorter. The following antibodies were used: anti-mouse CD31 (clone MEC 13.3; BD Biosciences), anti-mouse CD45 (clone 30-F11; BD Biosciences), anti-mouse EpCAM (clone G8.8; BioLegend), anti-Aqp3 (Abcam ab125219) and anti-Ki-67 (clone SolA15, eBiosciences). Anti-Aqp3 antibody was labeled using the Zenon antibody labeling kit (Molecular Probes). FACS results were plotted and analyzed using FlowJo software (TreeStar).
Mouse epithelial cell culture
For mouse epithelial cell cultures, CD45−CD31−EpCAM+Aqp3+ lung epithelial cells from FACS (as described above) were resuspended in SAGM (Lonza) mixed 1:1 with growth-factor reduced Matrigel (BD Biosciences) and plated at 3 x 103 cells per 100 μL in 24-well Transwells (Corning). For the first 5 d, SAGM was added with 10 μM Rock inhibitor (Sigma) but without FBS. After 5 d, SAGM was added with 10 % FBS but without Rock inhibitor. The medium was changed every 2-3 d. Organoids were cultured for 14 d to monitor stem cell growth (using organoid levels) as described previously (32). For compound treatment experiments, the same protocol was used, and NuP-4 (10-1000 nM) or vehicle control was added at each medium change.
RNA analysis
RNA was purified from homogenized lung tissue using Trizol (Invitrogen) or from isolated cells with the RNeasy mini kit (Qiagen) and was used to generate cDNA with the High-Capacity cDNA Archive kit (Life Technologies). We quantified target mRNA and viral RNA levels using real-time qPCR assay with specific fluorogenic probe-primer combinations and Fast Universal PCR Master Mix systems (Applied Biosystems) with mouse-specific forward and reverse primers and probes as described previously (37) and in Supplemental Table 3. For the Mapk13 assay, primers and probe were designed to amplify WT but not Mapk13 indel sequence in Mapk13−/− mice. All samples were assayed using the 7300HT or QuantStudio 6 Fast Real-Time PCR System and analyzed using Fast System Software (Applied Biosystems). All real-time PCR data was normalized to the level of Gapdh mRNA. Values were expressed as fold-change based on the delta-delta Ct method as described previously (65).
Lung function tests
To test lung function, we assessed blood oxygen saturation (SpO2) and airway reactivity to methacholine (MCh). For SpO2 monitoring, we used a MouseSTAT® pulse oximeter (Kent Scientific, Torrington, CT) applied to the paw skin under isoflurane anesthesia as described previously (31). To decrease variability in readings for this procedure, anesthetized mice were placed on the RightTemp warming pad, and the SpO2 reading was recorded when the Oxiwave signal was stable.
Human epithelial cell culture
For non-disease control samples, lung tissue was obtained from our Advanced Lung Disease Tissue Registry that contains whole lung explants harvested from consented patients in our lung transplant program; lungs that were not usable for transplantation from the local Organ Procurement Organization, Mid-America Transplant; and lungs from a tissue procurement service (IIAM, Edison, NJ) as described previously (19, 20, 39, 66, 67). Human studies were conducted with protocols approved by the Washington University (St. Louis, MO) Institutional Review Board and USAMRDC Office of Research Protection. For the present experiments, non-disease control samples were used to isolate human tracheobronchial epithelial cells (hTECs) using enzymatic digestion and then studied to monitor cell growth, immune activation, mucinous differentiation, and toxicity. To monitor cell growth, hTECs were cultured under submerged conditions as described previously (34), and growth was monitored using the CyQUANT cell proliferation assay (ThermoFisher). In addition, hTECs were also cultured under 3D-Matrigel conditions with and without GPNMB (100 ng/ml) to permit organoid formation as described previously (32). To assess immune activation, hTECs were cultured under the same 3D conditions and assessed for CXCL17 and IL33 mRNA using real-time qPCR assay. To monitor mucinous differentiation, hTECs were cultured under ALI conditions as described previously (19, 23). In this case, cells were cultured in 24-well Transwell plates (6.5-mm diameter inserts, 0.4 μm pore size) from Corning (Corning, NY) with 2% NuSerum medium (68) supplemented with Primocin (50 μg/ml, (InvivoGen, San Diego, CA), and retinoic acid (1 x 10−8 M, Sigma, St. Louis, MO) with or without human IL-13 (10 ng/ml, Peprotech, Rocky Hill, NJ) under submerged conditions for 7 d and then ALI conditions for 21 d. Cells were cultured in the presence or absence of inhibitors or vehicle that were added 2 d before addition of IL-13 and were re-added with each medium change/IL-13 treatment (twice per week). Transepithelial electrical resistance (TEER) was monitored for monolayer integrity as described previously (19, 69). Compound effect on mucus production was based on target mRNA levels that were determined using real-time qPCR assay with primers and probes as listed in Supplemental Table 4. To assess cell toxicity, hTECs were cultured under submerged conditions to achieve confluence and then ALI conditions to assess toxicity to compound treatment. Toxicity was determined using cell supernatant levels of LDH release measured with the CyQUANT LDH cytotoxicity assay (ThermoFisher).). For all cell culture experiments, compounds were prepared and stored as 10 mM in DMSO stock solution and for each experiment diluted at least 10,000-fold in cell culture media for cell treatment.
Statistical analysis
All data are presented as mean and s.e.m. and are representative of at least three experiments with at least 5 data points per experiment. For cell and molecular data, mixed-model repeated measures analysis of variance with Tukey correction for multiple comparisons were used to assess statistical significance between means. In all cases, significance threshold was set at P < 0.05.
Supplementary Material
New and noteworthy.
This study identifies a small-molecule inhibitor for MAPK13 with efficacy and safety in models of the long-term disease that features epithelial stem cell reprogramming towards inflammation and structural remodeling. The present model has direct implications for respiratory disease triggered by viral infection and other inhaled toxins, but the tissue distribution of MAPK13 implies related actions at other epithelial barrier sites. The findings also refine a hypothesis for therapeutic intervention based on proper adjustment of MAPK13 function with a selective kinase inhibitor.
Acknowledgments
We thank the Pulmonary Morphology Core and Division of Comparative Medicine for technical support and the NuPeak advisory team (Drs. Broschat, Montgomery, Redmann, Reed, and Reiss) for expert input on drug development.
Funding
This work was supported by grants from the National Institutes of Health (National Heart, Lung, and Blood Institute UH2-HL123429, R35-HL145242, and STTR R41-HL149523, National Institute of Allergy and Infectious Diseases R01-AI130591, Department of Defense TTDA W81XWH2010603 and W81XWH2210281, and Harrington Discovery Institute.
Abbreviations used in this article:
- basal-ESC
basal-epithelial stem cell
- CLCA1
chloride channel accessory 1
- COPD
chronic obstructive pulmonary disease
- Covid-19
coronavirus disease of 2019
- hTEC
human tracheobronchial epithelial cell
- MAPK
mitogen-activated protein kinase
- MUC5AC
mucin 5AC
- MUC5B
mucin 5B
- SeV
Sendai virus
Footnotes
Disclosures
MJH is the Founder of NuPeak Therapeutics, Inc. KW, YZ, AGR, and MJH are inventors on a patent for MAPK inhibitors and methods of use thereof. MJH, KW, and YZ are inventors on a provisional patent for Methods of use for GPNMB-CD44 blockade in chronic respiratory disease.
References
- 1.Canovas B, and Nebreda AR. Diversity and versatility of p38 kinase signaling in health and disease. Nat Rev Mol Cell Biol. 2021;22:346–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cuenda A, and Sanz-Ezquerro JJ. p38gamma and p38delta: From Spectators to Key Physiological Players. Trends in biochemical sciences. 2017;42(6):431–42. [DOI] [PubMed] [Google Scholar]
- 3.Underwood DC, Osborn RR, and Kotzer CJ. SB 239063, a potent p38 MAP kinase inhibitor, reduces inflammatory cytokine production, airways eosinophil infiltration, and persistence. J Pharmacol Exp Ther. 2000;293:281–8. [PubMed] [Google Scholar]
- 4.Duan W, Chan JH, McKay K, Crosby JR, Choo HH, Leung BP, Karras JG, and Wong WSF. Inhaled p38alpha mitogen-activated protein kinase antisense oligonucleotide attenuates asthma in mice. Am J Respir Crit Care Med. 2005;171:571–8. [DOI] [PubMed] [Google Scholar]
- 5.Medicherla S, Fitzgerald MF, Spicer D, Woodman P, Ma JY, Kapoun AM, Chakravarthy S, Dugar S, Protter AA, and Higgins LS. p38a-selective mitogen-activated protein kinase inhibitor SD-282 reduces inflammation in a subchronic model of tobacco smoke-induced airway inflammation. J Pharmacol Exp Ther. 2008;324:921–9. [DOI] [PubMed] [Google Scholar]
- 6.Renda T, Baraldo S, Pelaia G, Bazzan E, Turato G, Papi A, Maestrelli P, Maselli R, Vatrella A, Fabbri LM, Zuin R, Marsico SA, and Saetta M. Increased activation of p38 MAPK in COPD. Eur Respir J. 2008;31:62–9. [DOI] [PubMed] [Google Scholar]
- 7.Millan DS, Bunnage ME, Burrows JL, Butcher KJ, Dodd PG, Evans TJ, Fairman DA, Hughes SJ, Kilty IC, Lemaitre A, Lewthwaite RA, Mahnke A, Mathias JP, Philip J, Smith RT, Stefaniak MH, Yeadon M, and Phillips C. Design and synthesis of inhaled p38 inhibitors for the treatment of chronic obstructive pulmonary disease. J Med Chem. 2011;54:7797–814. [DOI] [PubMed] [Google Scholar]
- 8.MacNee W, Allan RJ, Jones I, Cristina De Salvo M, and Tan LF. Efficacy and safety of the oral p38 inhibitor PH-797804 in chronic obstructive pulmonary disease: a randomised clinical trial. Thorax. 2013;68:738–45. [DOI] [PubMed] [Google Scholar]
- 9.Watz H, Barnacle H, Hartley BF, and Chan R. Efficacy and safety of the p38 MAPK inhibitor losmapimod for patients with chronic obstructive pulmonary disease: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2014;2:63–72. [DOI] [PubMed] [Google Scholar]
- 10.Patel NR, Cunoosamy DM, Fageras M, Taib Z, Asimus S, Hegelund-Myrback T, Lundin S, Pardali K, Kurian N, Ersdal E, Kristensson C, Korsback K, Palmer R, Brown MN, Greenaway S, Siew L, Clarke GW, Rennard SI, Make BJ, Wise RA, and Jansson P. The developmen tof AZD7624 for prevention of exacerbations in COPD: a randomized controlled trial. Int J COPD. 2018;13:1009–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haller V, Nahidino P, Forster M, and Laufer SA. An updated patent review of p38 MAPK kinase inhibitors (2014-2019). Expert Opin Ther Pat. 2020;30:453–66. [DOI] [PubMed] [Google Scholar]
- 12.Pelaia C, Vatre A., Gallelli L, Lombardo N, Sciacqua A, Savino R, and Pelaia G. Role of p38 mitogen-activated protein kinase in asthma and COPD: pathogenic aspects and potential targeted therapies. Drug Design, Development and Therapy. 2021;15:1275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goedert M, Hasegawa M, Jakes R, Lawler S, Cuenda A, and Cohen P. Phosphorylation of microtube-associated protein tau by stress-activated protein kinases. FEBS Lett. 1997;409:57–62. [DOI] [PubMed] [Google Scholar]
- 14.Sumara G, Formentini I, Collins S, Sumara I, Windak R, Bodenmiller B, Ramracheya R, Caille D, Jiang H, Platt KA, Meda P, Aebersold R, Rorsman P, and Ricci R. Regulation of PKD by the MAPK p38δ in insulin secretion and glucose homeostasis. Cell. 2009;136:235–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ittner A, Block H, Reichel CA, Varjosalo M, Gehart H, Sumara G, Gstaiger M, Krombach F, Zarbock A, and Ricci R. Regulation of PTEN activity by p38d-PKD1 signaling in neutrophils confers inflammatory responses in the lung. J Exp Med. 2012;209:2229–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schindler E, Hindes A, Gribben E, Burns C, Yin Y, Lin M, Owen R, Longmore G, Kissling G, Arthur J, and Efimova T. p38delta Mitogen-activated protein kinase is essential for skin tumor development in mice. Cancer Res. 2009;69:4648–55. [DOI] [PubMed] [Google Scholar]
- 17.Risco A, Del Fresno C, Mambol A, Alsina-Beauchamp D, MacKenzie KF, Yang H-T, Barber DF, Morcelle C, Arthur JSC, Ley SC, Ardavin C, and Cuenda A. p38g and p38d kinases regulate the Toll-like receptor 4 (TLR4)-induced cytokine production by controlling ERK1/2 protein kinase pathway activation. Proc Natl Acad Sci U S A. 2012;109:11200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomas-Loba A, Manieri E, Gonzalez-Teran B, Mora A, Leiva-Vega L, Santamans AM, Romero-Becerra R, Rodriguez E, Pintor-Chocano A, Feixas F, Lopez JA, Caballero B, Trakala M, Blanco O, Torres JL, Hernandez-Cosido L, Montalvo-Romeral V, Matesanz N, Roche-Molina M, BJ A., Mischo H, Leon M, Caballero A, Miranda-Saavedra D, Ruiz-Cabello J, Nevzorova YA, Cubero FJ, Bravo J, Vazquez J, Malumbres M, Marcos M, Osuna S, and Sabio G. p38g is essential for cell cycle progression and liver tumoriogenesis. Nature. 2019;568:557–60. [DOI] [PubMed] [Google Scholar]
- 19.Alevy Y, Patel AC, Romero AG, Patel DA, Tucker J, Roswit WT, Miller CA, Heier RF, Byers DE, Brett TJ, and Holtzman MJ. IL-13–induced airway mucus production is attenuated by MAPK13 inhibition. J Clin Invest. 2012;122:4555–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu K, Zhang Y, Mao D, Iberg C, Yin-Declue H, Sun K, Keeler S, Wikfors H, Young D, Yantis J, Austin S, Byers D, Brody S, Crouch E, Romero A, and Holtzman M. MAPK13 controls structural remodeling and disease after epithelial injury. bioRxiv. 2024; 10.1101/2024.05.31.596863. [DOI] [Google Scholar]
- 21.Radicioni G, Ceppe A, Ford AA, Alexis NE, Barr RG, Bleecker ER, Christenson SA, Cooper CB, Han MK, Hansel NN, Hastie AT, Hoffman EA, Kanner RE, Martinez FJ, Ozkan E, Paine R 3rd, Woodruff PG, O’Neal WK, Boucher RC, and Kesimer M. Airway mucin MUC5AC and MUC5B concentrations and the initiation and progression of chronic obstructive pulmonary disease: an analysis of the SPIROMICS cohort. Lancet Respir Med. 2021;9(11):1241–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang M, Elicker BM, Henry T, Gierada DS, Schiebler ML, Huang BK, Peters MC, Castro M, Hoffman EA, Fain SB, Ash SY, Choi JY, Hall C, Phillips BR, Mauger DT, Denlinger LC, Jarjour NN, Israel E, Phipatanakul W, Levy BD, Wenzel SE, Bleecker ER, Woodruff PG, Fahy JV, Dunican EM, and Program-3 NSAR. Mucus plugs persist in asthma, and changes in mucus plugs associate with changes in airflow over time. Am J Respir Crit Care Med. 2022;205:1036–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keeler SP, Wu K, Zhang Y, Mao D, Li M, Iberg CA, Austin SP, Glaser SA, Yantis J, Podgorny S, Brody SL, Chartock JR, Han Z, Byers DE, Romero AG, and Holtzman MJ. A potent MAPK13-14 inhibitor prevents airway inflammation and mucus production. Am J Physiol Lung Cell Mol Physiol. 2023;325:L726–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yurtsever Z, Schaeffer SM, Romero AG, Holtzman MJ, and Brett TJ. The crystal structure of phosphorylated MAPK13 reveals common structural features and diffeerences in p38 MAPK family activation. Acta Crystallogr D Biol Crystallogr. 2015;71:790–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yurtsever Z, Patel DA, Kober DL, Su A, Miller CA, Romero AG, Holtzman MJ, and Brett TJ. First comprehensive structure and biophysical analysis of MAPK13 inhibitors targeting DFG-in and DFG-out binding modes. Biochim Biophys Acta. 2016;1860:2335–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holtzman MJ, Romero AG, Gerovac BJ, Han Z, Keeler SP, and Wu K. In: USPTO ed. United States Patent Application Publication. US: Washington University; 2023. [Google Scholar]
- 27.Pargellis C, Tong L, Churchill L, Cirillo PF, Gilmore T, Graham AG, Grob PM, Hickey ER, Moss N, Pav S, and Regan J. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Biol. 2002;9:268–72. [DOI] [PubMed] [Google Scholar]
- 28.Regan J, Pargellis CA, Cirillo PF, Gilmore T, Hickey ER, Peet GW, Proto A, Swinamer A, and Moss N. The kinetics of binding to p38 MAP kinase by analogues of BIRB 796. Bioorg Med Chem. 2003;13:3101–4. [DOI] [PubMed] [Google Scholar]
- 29.Casasnovas R, Limongelli V, Tiwary P, Carloni P, and Parrinello M. Unbinding kinetics of a p38 MAP kinase type II inhibitor from metadynamics simulations. J Am Chem Soc. 2017;139:4780–8. [DOI] [PubMed] [Google Scholar]
- 30.Huang Y. Multiscale computational study of ligand binding pathways: case of p38 MAP kinase and its inhibitors. Biophysical J. 2021;120:3881–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu K, Kamimoto K, Zhang Y, Yang K, Keeler SP, Gerovac BJ, Agapov EV, Austin SP, Yantis J, Gissy KA, Byers DE, Alexander-Brett J, Hoffmann CM, Wallace M, Hughes ME, Morris SA, and Holtzman MJ. Basal-epithelial stem cells cross an alarmin checkpoint for post-viral lung disease. J Clin Invest. 2021;131:e149336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu K, Zhang Y, Yin-Declue H, Sun K, Mao D, Austin S, Crouch E, Brody S, Byers D, Hoffmann C, Hughes M, and Holtzman M. A correctable immune niche for basal-epithelial stem cell reprogramming and post-viral lung diseases. J Clin Invest. 2024;134:e183092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, Benoit LA, Byers DE, Alevy Y, Tucker J, Swanson S, Tidwell R, Tyner JW, Morton JD, Castro M, Polineni D, Patterson GA, Schwendener RA, Allard JD, Peltz G, and Holtzman MJ. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med. 2008;14:633–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Byers DE, Alexander-Brett J, Patel AC, Agapov E, Dang-Vu G, Jin X, Wu K, You Y, Alevy YG, Girard J-P, Stappenbeck TS, Patterson GA, Pierce RA, Brody SL, and Holtzman MJ. Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J Clin Invest. 2013;123:3967–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu K, Byers DE, Jin X, Agapov E, Alexander-Brett J, Patel AC, Cella M, Gilfilan S, Colonna M, Kober DL, Brett TJ, and Holtzman MJ. TREM-2 promotes macrophage survival and lung disease after respiratory viral infection. J Exp Med. 2015;212:681–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Mao D, Keeler SP, Wang X, Wu K, Gerovac BJ, Shornick LP, Agapov E, and Holtzman MJ. Respiratory enterovirus (like parainfluenza virus) can cause chronic lung disease if protection by airway epithelial STAT1 is lost. J Immunol. 2019;202:2332–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu K, Wang X, Keeler SP, Gerovac BJ, Agapov E, Byers DE, Gilfillan S, Colonna M, Zhang Y, and Holtzman MJ. Group 2 innate lymphoid cells must partner with the myeloid-macrophage lineage for long-term postviral lung disease. J Immunol. 2020;205:1084–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Wu K, Keeler SP, Mao D, Agapov EV, Zhang Y, and Holtzman MJ. TLR3-activated monocyte-derived dendritic cells trigger progression from acute viral infection to chronic disease in the lung. J Immunol. 2021;206:1297–314 (selected for Top-Reads p. 115). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu K, Zhang Y, Yin Declue H, Austin SR, Byers DE, Crouch EC, and Holtzman MJ. Lung remodeling regions in long-term coronavirus disease 2019 feature basal epithelial cell reprogramming. Am J Pathol 2023;193:680–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duffy JP, Harrington EM, Salituro FG, Cochran JE, Green J, Gao H, Bemis GW, Evindar G, Galulio VP, Ford PJ, Germann UA, Wilson KP, Bellon SF, Chen G, Taslimi P, Jones P, Huang C, Pazhanisamy S, Wang YM, Murcko MA, and Su MS. The discovery of VX-745: a novel and selective p38a kinase inhibitor. ACS Med Chem Lett. 2011;2:758–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holtzman M, Zhang Y, Wu K, and Romero AG. Mitogen-activated protein kinase-guided drug discovery for post-viral and related types of lung disease. Eur Respir Rev. 2024;33:230220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anastassiadis T, Deacon SW, Devarajan K, Ma H, and Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1039–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig P-A, Reinecke M, Ruprecht B, Petzoldt S, Meng C, Zecha J, Reiter K, Qiao H, Helm D, Koch H, Schoof M, Canevari G, Casale E, Depaolini SR, Feuchtinger A, Wu Z, Schmidt T, Rueckertr L, Becker W, Huenges J, Garz A-K, Gohlke B-O, Zolg DP, Kayser G, Vooder T, Preissner R, Hahne H, Tonisson N, Kramer K, Gotze K, Bassermann F, Schlegl J, Ehrlich H-C, Aiche S, Walch A, Greif PA, Schneider S, Felder ER, Ruland J, Medard G, Jeremias I, Spiekermann K, and Kuster B. The target landscape of clinical kinase drugs. Science. 2017;358:1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cohen P, Cross D, and Janne PA. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat Rev Drug Discov. 2021;20:551–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Varjosalo M, Keskitalo S, Van Drogen A, Nurkkala H, Vichalkovski A, Aebersold R, and Gstaiger M. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013;3(1306-1320). [DOI] [PubMed] [Google Scholar]
- 46.Alsina-Beauchamp D, Escos A, Fajardo P, Gonzalez-Romero D, Diaz-Mora E, R A., Martin-Serrano MA, dF C., Dominguez-Andres J, Aparicio N, Zur R, Shpiro N, Brown GD, Ardavin C, Netea MC, Alemany S, Sanz-Ezquerro JJ, and Cuenda A. Myeloid cell deficiency of p38g/p38d protects against candidiasis and regulates antifungal immunity. EMBO Mol Med. 2018;10:e8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Escos A, Martin-Gomez J, Gonzalez-Romero D, Diaz-Mora E, Francisco-Velilla R, Santiago C, Cueza J, Dominguez-Zorita S, Martinez-Salas E, Sonenberg N, Sanz-Ezquerro J, Jafanejad S, and Cuenda A. TPL2 kinase expression is regulated by the p38g/p38d-dependent association with aconitase-1 with TPL2 mRNA. Proc Natl Acad Sci U S A. 2022;119:e2204752119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Escos A, Diaz-Mora E, Pattison M, Fajardo P, Gonzalez-Romero D, Risco A, Martin-Gomez J, Bonnell E, Sonenberg N, Jafanejad SM, Sanz-Ezquerro JJ, Ley SC, and Cuenda A. p38g and p38d modulate innate immune response by regulating MEF2D activation. eLife. 2023;12:e86200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Juyoux P, Galdadas J, Gobbo D, von Velson J, Pelosse M, Tully M, Vadas O, Gervasio FL, Pellegrini E, and Bowler MW. Architecture of the MKK6-p38a complex defines the basis of MAPK specifcity and activation. Science. 2023;381:1217–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawai T, and Akira S. Pathogen recognition with Toll-like receptors. Curr Opin Immunol. 2005;17(4):338–44. [DOI] [PubMed] [Google Scholar]
- 51.Ha U, LJ H., Jono H, Koga T, Srivastava A, Malley R, Pages G, Pouyssegur J, and Li J-D. A novel role for IkB kinase (IKK) a and IKKb in ERK-dependent up-regulation of MUC5AC mucin transcription by Streptococcus pneumoniae. J Immunol. 2007;178:1736–47. [DOI] [PubMed] [Google Scholar]
- 52.Fujisawa T, Velichko S, Thai P, Hung LY, Huang F, and Wu R. Regulation of airway MUC5AC expression by IL-1beta and IL-17A; the NF-kappaB paradigm. J Immunol. 2009;183(10):6236–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Na HG, Kim Y-D, and Bae CH. High concentration of insulin induces MUC5AC expression via phosphoinositide 3 kinase/AKT and mitogen-activated protein kinase signaling pathways in human airway epithelial cells. Am J Rhinol Allergy. 2018. [DOI] [PubMed] [Google Scholar]
- 54.Xu H, Sun Q, Lu L, Luo F, Zhou L, Liu J, Cao L, Wang Q, Xue J, Yang Q, Yang P, Lu J, Xiang Q, and Liu Q. MicroRNA-218 acts by repressing TNFR1-mediated activation of NF-kB, which is involved in MUC5AC hyper-production and inflammation in smoking-induced bronchiolitis of COPD. Toxicol Lett. 2017;280:171–80. [DOI] [PubMed] [Google Scholar]
- 55.Wu S, Li H, Yu L, Wang N, Li X, and Chen W. IL-1b upregulates Muc5ac expression via NF-kB-induced HIF-1a in asthma. Immunol Lett. 2017;192:20–6. [DOI] [PubMed] [Google Scholar]
- 56.Selness SR, Devraj RV, Devadas B, Walker JK, Boehm TL, Durley RC, Shieh H, Xing L, Rucker PV, Jerome KD, and Benson AG. Discovery of PH-797804, a highly selective and potent inhibitor of p38 MAP kinase. Bioorg Med Chem Lett. 2011;21:4066–71. [DOI] [PubMed] [Google Scholar]
- 57.McQualter JL, Yuen K, Williams B, and Bertoncello I. Evidence of an epithelial stem/progenitor cell hierarchy in the adult mouse lung. Proc Natl Acad Sci U S A. 2010;107(4):1414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bhatt SP, Rabe KF, Hanania NA, Vogelmeier CF, Cole J, Bafadhel M, Christenson SA, Papi A, Singh D, Laws E, Mannent LP, Patel N, Staudinger HW, Yancopoulos GD, Mortensen ER, Akinlade B, Maloney J, Lu X, Bauer D, Bansal A, Robinson LB, and Abdulai RM. Dupilumab for COPD with type 2 inflammation indicated by eosinophil counts. N Engl J Med. 2023;389:205–14. [DOI] [PubMed] [Google Scholar]
- 59.Sigurs N, Aljassim F, Kjellman B, Robinson PD, Sigurbergsson F, Bjarnason R, and Gustafsson PM. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax. 2010;65(12):1045–52. [DOI] [PubMed] [Google Scholar]
- 60.Holtzman MJ, Byers DE, Alexander-Brett J, and Wang X. The role of airway epithelial cells and innate immune cells in chronic respiratory disease. Nat Rev Immunol. 2014;14:686–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mallia P, Message SD, Gielen V, Contoli M, Gray K, Kebadze T, Aniscenko J, Laza-Stanca V, Edwards MR, Slater L, Papi A, Stanciu LA, Kon OM, Johnson M, and Johnston SL. Experimental rhinovirus infection as a human model of chronic obstructive pulmonary disease exacerbation. Am J Respir Crit Care Med. 2011;183:734–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Keeler SP, Agapov EV, Hinojosa ME, Letvin AN, Wu K, and Holtzman MJ. Influenza A virus infection causes chronic lung disease linked to sites of active viral RNA remnants. J Immunol. 2018;201:2354–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schleich F, Bougard N, Moermans C, Sabbe M, and Louis R. Cytokine-targeted therapies for asthma and COPD. Eur Respir Rev. 2023;32:220193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van Nunen MCJ, and van der Veen J. Experimental infection with Sendai virus in mice. Arch Gesamte Virusforsch. 1967;22:388–97. [DOI] [PubMed] [Google Scholar]
- 65.Livak KJ, and Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 66.Deslee G, Woods J, Moore C, Conradi S, Gierada D, Atkinson J, Battaile J, Liu L, Patterson A, Adair-Kirk T, Holtzman M, and Pierce R. Oxidative damage to nucleic acids in severe emphysema. Chest. 2009;135:965–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Byers DE, Wu K, Dang-Vu G, Jin X, Agapov E, Zhang X, Battaile JT, Schechtman KB, Yusen R, Pierce RA, and Holtzman MJ. Triggering receptor expressed on myeloid cells-2 (TREM-2) expression tracks with M2-like macrophage activity and disease severity in COPD. Chest. 2018;153:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lechner JF, and LaVeck MA. A serum-free method for culturing normal human bronchial epithelial cells at clonal density. J Tissue Culture Meth. 1985;9:43–8. [Google Scholar]
- 69.Patel DA, Patel AC, Nolan WC, Zhang Y, and Holtzman MJ. High throughput screening for small molecule enhancers of the interferon signaling pathway to drive next-generation antiviral drug discovery. PloS ONE. 2012;7:e36594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.