

Abstract
Nuclear exclusion and cytoplasmic accumulation of the RNA-binding protein TDP43 are characteristic of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). Despite this, the origin and ultrastructure of cytosolic TDP43 deposits remain unknown. Accumulating evidence suggests that abnormal RNA homeostasis can drive pathological TDP43 mislocalization, enhancing RNA misprocessing due to loss of nuclear TDP43 and engendering a cycle that ends in cell death. Here, we show that adding small monovalent oligonucleotides successfully recapitulates pathological TDP43 mislocalization and aggregation in iPSC-derived neurons (iNeurons). By employing a multimodal in situ cryo-correlative light and electron microscopy pipeline, we examine how RNA influences the localization and aggregation of TDP43 in near-native conditions. We find that mislocalized TDP43 forms ordered fibrils within lysosomes and autophagosomes in iNeurons as well as in patient tissue, and provide the first high-resolution snapshots of TDP43 aggregates in situ. In so doing, we provide a cellular model for studying initial pathogenic events underlying ALS, FTLD, and related TDP43-proteinopathies.
Trans-active response DNA/RNA binding protein of 43 kDa (TDP43) is a ubiquitously expressed nuclear protein responsible for RNA splicing, transport, and stability that is integrally linked with amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD)(1–4). Nearly 95% of individuals with ALS and ~50% of FTLD patients exhibit nuclear exclusion and cytosolic aggregation of TDP43 within affected neurons and glia(5). TDP43 mislocalization and cytosolic aggregation are not limited to ALS and FTLD; TDP43 pathology is increasingly recognized as a common event in elderly patients with slowly progressive cognitive decline, termed limbic predominant age-related TDP43 encephalopathy (LATE-NC)(6). Once excluded from the nucleus, TDP43 primarily accumulates within hyperphosphorylated and ubiquitinated aggregates in the cytoplasm(7, 8). Within cortical regions, these deposits contain amyloidogenic C-terminal TDP43 fragments(9–11), while spinal cord inclusions are rich in full-length TDP43(12). These observations suggest that the TDP43 aggregate composition, the mechanisms underlying their formation, and downstream consequences may differ depending on the brain region involved.
Despite its prevalence and increasingly recognized connection with disease pathogenesis, the origin of TDP43 mislocalization and the architecture of native TDP43 inclusions within intact neurons remain mysterious. Engineered mutations disrupting the nuclear localization signal of TDP43(13–15), application of exogenous stressors(16, 17), or the use of artificial aggregation-promoting domains can all elicit TDP43 mislocalization(18), yet these circumstances are disconnected from the pathophysiology of the disease. More recent discoveries suggest that RNA is one of the most significant determinants of TDP43 localization(19–21). In light of the extensive and widespread abnormalities in RNA processing identified in ALS/FTLD patient material and models(22–24), this observation raises the intriguing possibility that RNA dysregulation may underlie TDP43 mislocalization and aggregation in these conditions. Even so, all previous studies were performed over short time scales (~hours) and in transformed cell lines.
Here, we show that prolonged application of RNA oligonucleotides leads to TDP43 mislocalization and the formation of cytosolic TDP43 inclusions in human neurons that resemble ubiquitinated and phosphorylated pathological TDP43 aggregates characteristic of human disease. Notably, cytosolic TDP43 aggregates accumulate primarily within lysosomes and autophagosomes, suggesting that these organelles are critical for the formation and/or clearance of mislocalized TDP43. Combining cryogenic-correlative light and electron microscopy (cryo-CLEM) and cryo-electron tomography (cryo-ET), we demonstrate that lysosomal and autophagosomal TDP43 adopts filamentous morphologies mirroring those isolated from ALS/FTLD patients with TDP43 pathology (FTLD-TDP)(25, 26). Based on these observations, we propose that RNA dyshomeostasis, in conjunction with ineffective autophagy or lysosomal turnover — processes often impaired in aging(27–34) — contribute to nuclear TDP43 mislocalization and the accumulation of pathogenic fibrillar aggregates characteristic of TDP43 proteinopathies such as ALS and FTLD-TDP.
Results
Establishing an in vitro neuronal model to recapitulate TDP43 pathology
Short oligonucleotides (oligos) bearing GU motifs recognized by TDP43 effectively promote TDP43 nuclear efflux in transformed cell lines(19–21). To test whether this is true in mature neurons, we applied fluorescently-labeled short monovalent GU-rich RNA (5’-Cy5-GUGUGUGUGUGU-3’, Cy5-(GU)6 hereafter) to rodent primary mixed cortical neurons. We assessed TDP43 localization and oligo uptake by immunofluorescence (Fig. S1A). Cy5-(GU)6 oligos elicited a clear time-dependent reduction in the nuclear/cytoplasmic ratio of TDP43, modeled through generalized estimating equations (Fig. S1B–D) (35). In contrast to cultured cells, where TDP43 mislocalization was highly prevalent and rapid (i.e., occurring within 6h of oligo treatment) (19–21), maximal displacement of TDP43 in primary neurons required up to 24h, with only ~20% of treated neurons showing a >50% reduction in the nuclear/cytoplasmic ratio of TDP43 (Fig. S1B–E).
Given the species-specific differences in TDP43 substrates(36–39) and the presence of distinct TDP43 isoforms in rodent vs. human brains(40), we next confirmed these findings in human induced pluripotent stem cell (iPSC) derived neurons (iNeurons). A customized cassette encoding the master transcription factors NGN1 and NGN2, under the control of a doxycycline (dox)-inducible promoter, was integrated into the CLYBL safe harbor locus of iPSCs, facilitating robust and rapid differentiation of iPSCs into glutamatergic, forebrain-like iNeurons (Fig. 1A)(23, 41–46). These cells were treated with (GU)6 oligos prior to the assessment of TDP43 localization by immunostaining and fluorescence microscopy. We observed punctate cytoplasmic accumulations of TDP43 in iNeurons as early as 4h after (GU)6 application (Fig. S2A–D), with more pronounced puncta after prolonged (24h) treatment (Fig. 1B–E), confirming RNA-induced TDP43 nuclear egress and cytoplasmic deposition in human neurons.
Fig. 1. TDP43 mislocalizes to autophagosomes, lysosomes, and stress granules upon treatment with GU-rich oligonucleotides.

(A) Schematic of iPSC differentiation into forebrain-like neurons and subsequent (GU)6 oligonucleotide treatment. (B-E) Representative confocal microscopy images with xz and yz orthogonal views of (GU)6-treated DIV10 iNeurons fixed and immunostained for MAP2 (gray), TDP43 (magenta), and (B) LC3B, (C) p62, (D) LAMP2, or (E) G3BP1 (cyan). Scale bar, 5μm. Insets provide ~4.8x magnification; scale bar, 1μm. White arrowheads in xz and yz orthogonal views indicate TDP43 puncta present in inset.
Loss of nuclear TDP43 results in characteristic splicing changes, including the abnormal inclusion of unannotated and non-conserved (cryptic) exons. We, therefore, assessed whether treatment with (GU)6 oligos was associated with cryptic splicing and downregulation of UNC13A, a TDP43 substrate that is consistently misspliced in ALS and FTLD-TDP(47, 48). Cryptic UNC13A products were dramatically upregulated in U2OS cells treated with (GU)6 for 24h, consistent with reductions in nuclear TDP43 and the formation of cytosolic TDP43 puncta in these cells (Fig. S3A, C, D). Significant differences in cryptic UNC13A products could not be confirmed in (GU)6-treated iNeurons by qRT-PCR, likely due to the relatively low prevalence of TDP43 mislocalization in treated neurons (Fig. S1E, Fig. S3B) in combination with the instability of UNC13A cryptic splice products(47). However, we detected significant reductions in canonical UNC13A transcripts by hybridization chain reaction fluorescence in situ hybridization (HCR-FISH) in treated iNeurons, suggesting that GU-rich oligos induce TDP43 loss of function (Fig. S3E, F).
To determine the composition of cytosolic TDP43 puncta induced by (GU)6 application, we systematically examined oligo-treated iNeurons for markers of TDP43 post-translational modifications, as well as cytosolic membrane-bound and membrane-less organelles through immunostaining. Similar to pathological TDP43 aggregates(3, 7, 8, 49), cytoplasmic TDP43 puncta are phosphorylated (S409/S410) and ubiquitinated (Fig. S2E, F). Strikingly, cytosolic TDP43 puncta also overlapped with markers of autophagosomes (LC3B, p62, Fig. 1B, C), lysosomes (LAMP2, Fig. 1D), and stress granules (G3BP1, Fig. 1E). We also found evidence for the recruitment of Galectin-3, indicating membrane rupture in a fraction of lysosomes (Fig. S4A) and ESCRT machinery (CHMP4B, Fig. S4B). Taken together, these findings suggest that RNA-induced perturbation of TDP43 localization in iNeurons recapitulates key features of ALS/FTLD-TDP pathology, including TDP43 nuclear egress, UNC13A downregulation, and the formation of ubiquitinated and phosphorylated cytosolic TDP43 deposits(7, 47, 48, 50).
Mislocalized TDP43 accumulates within lysosomes and autophagosomes
To verify that TDP43 accumulates within autophagosomes and lysosomes upon oligo treatment, we utilized an engineered line of HEK293T cells in which the endogenous MAP1LC3B gene was fused to an open reading frame encoding the photoconvertible fluorescent protein Dendra2(43). These Dendra2-LC3B HEK293T cells were treated with (GU)6 for 4h. To isolate Dendra2-positive autophagosome-rich and lysosomal-rich fractions (hereafter autophagosomes and lysosomes, respectively), we lysed Dendra2-LC3B HEK293T treated cells, isolated the autophagic membrane fraction, and separated organelles by density gradient centrifugation (Fig. S5A). In accordance with our prior results in iNeurons, we observed a significant shift in the distribution of TDP43 upon (GU)6 treatment towards lysosomal and autophagosomal fractions (Fig. 2B). The application of Torin1, a potent mTOR inhibitor that effectively induces bulk macroautophagy, also shifts TDP43 into autophagosomal and lysosomal fractions, albeit to a lesser extent than (GU)6 (Fig. 2B). We next confirmed that the shift of TDP43 towards lysosomal and autophagosomal fractions was due to the sequence specificity of RNA. We observed that treating HEK293T cells with A13 oligos did not yield any significant migration of TDP43 into autophagosomes and lysosomes in our fractionation experiments (Fig. 2B).
Fig. 2. GU-rich oligonucleotides induce the formation of TDP43 fibrils in autophagosomes and lysosomes.

(A) Schematic of Nycodenz density gradient fractionation protocol to isolate autophagosomes (AP) and lysosomes (Lyso) from HEK293T cells. (B) (i) Representative immunoblots of fractions 2–12 and membrane fraction input (in) isolated from oligonucleotide-treated Dendra2-LC3B HEK293T cells, detected with anti-TDP43, -LAMP1, and -LC3B antibodies. n=3. Quantification of TDP43 in fractions 4 (ii) and 6 (iii) (mean ± SEM, ANOVA, Tukey’s test, *p < 0.05). (C) Anti-TDP43 immunogold labeling on an ultrathin section of (GU)6 treated Dendra2-LC3B HEK293T cells (left). n=6. Quantification of TDP43 positive vesicles in oligonucleotide-treated Dendra2-LC3B HEK293T cells (right). (Welch’s t-test, **p < 0.005). Scale bar, 100nm. (D) Representative negative stain EM images of immunogold labeled detergent insoluble TDP43 fibrils from Dendra2-LC3B HEK293T cells using anti-TDP43 antibodies. Scale bar, 50nm. (E) Correlative cryo-ET of lysosome enriched fraction from (GU)6-treated Dendra2-LC3B HEK293T cells. Cryo-EM micrograph of lysosome overlaid with the Dendra2-LC3B cryo-fluorescence signal (left). Slice through the tomogram of the lysosome shown in the left panel (middle). White arrowheads highlight TDP43 fibrils. TDP43 fibrils and lysosome membranes in left/middle panels are segmented (right). Color scheme: red - TDP43; blue - lysosome membrane. Scale bars, 200nm.
Given this result, we next asked if the accumulation of TDP43 within autophagosomes was secondary to a global induction of autophagy upon (GU)6 application. To answer this question, we used an engineered iPSC line in which HaloTag was fused to the endogenous MAP1LC3B gene (encoding the autophagy marker and substrate LC3B) on one allele. These lines provide an accessible and quantitative means of measuring autophagy flux through the cleavage of HaloTag from LC3 within mature autophagosomes(51). We observed a single band for HaloTag-LC3B, and no significant change in the levels of LC3B-II, TDP43, or p62 in treated HaloTag-LC3B iNeurons, arguing against induction of macroautophagy via application of oligos (Fig. S6A–D).
Next, we prepared ultrathin sections of high-pressure frozen and freeze-substituted Dendra2-LC3B HEK293T cells, allowing us to examine RNA-induced TDP43 mislocalization by immuno-electron microscopy (immunoEM). Gold-labeled anti-TDP43 antibodies reaffirmed the presence of TDP43 within single membrane-bound endolysosomes (Fig. 2C, Fig. S7). Membrane-bound vesicles exhibited significantly more gold beads in (GU)6-treated cells in comparison to untreated (control) cells (Fig. 2C), indicative of greater endolysosomal TDP43 immunoreactivity following RNA application.
We also isolated the sarkosyl-insoluble fractions from control and (GU)6-treated Dendra2-LC3B HEK293T cells to probe whether (GU)6 oligos led to the formation of amyloidogenic aggregates. Negative stain transmission electron microscopy (TEM) of these samples revealed the presence of fibrils only in the (GU)6-treated samples, reminiscent of TDP43 filaments obtained from postmortem brain tissue(25, 26). Immunogold labeling with TDP43-specific antibodies once again confirmed that these fibrils are comprised of TDP43 (Fig. 2D). To probe if fibrils are already present in isolated organelles and to verify that they are not an artifact of sarkosyl extraction, we performed cryo-correlative light and electron microscopy (cryo-CLEM) on plunge-frozen lysosomal and autophagosomal fractions isolated from Dendra2-LC3B HEK293T cells treated with (GU)6 oligos (Fig. S5B). In these samples, Dendra2-LC3B fluorescence enabled the precise localization of autophagosomes and lysosomes. Subsequent cryo-electron tomography (cryo-ET) tilt-series data collection at these areas, followed by tomogram reconstruction, revealed multiple filaments ensconced within membrane-bound vesicles (white arrows, Fig. 2E, Movie 1). These filaments displayed a mean width of 4.9 ± 0.79 nm, mirroring the amyloidogenic C-terminus of TDP43 obtained by limited proteolysis of the protein(52) (Fig. S10A). Three-dimensional segmentation of the tomograms revealed that a majority of these filaments are stacked and rarely appear as individual fibrils (Fig. 2E).
Movie 1. Cryo-ET of isolated lysosome from HEK293T cells.
Sequential sections of a tomogram of lysosomes obtained from fraction 4 of gradient fractionation reveal ensconced TDP43 filaments. Accompanies Fig. 2.
A custom cryo-CLEM workflow confirms lysosomal and autophagosomal TDP43 mislocalization in situ
To obtain high-resolution insights into the native architecture of mislocalized TDP43 in human neurons, we established an in situ cryo-correlative light and electron microscopy (cryo-CLEM) workflow and adapted it for use with iNeurons (Fig. 3). Initial experiments took advantage of iNeurons derived from human iPSCs that express native LC3B fused with an enhanced green fluorescent protein (EGFP), obtained from the Allen Cell Collection(42). mEGFP-LC3B iPSCs were differentiated into iNeurons as described (Fig. 1A), but on micropatterned gold electron microscopy grids (Fig. 3A). Next, we applied Cy3 labeled (GU)6, plunge-froze the cells after 24h, and collected cryo-confocal Z-stacks of candidate iNeurons (Fig. 3B). Autogrids containing cells were transferred to a dual beam cryo-focused ion beam scanning electron (cryo-FIB/SEM) microscope, and cryo-confocal images were used to precisely target the region of interest (Fig. 3B, C). Following cryo-focused ion beam (cryo-FIB) milling, the grids were assessed by cryo-TEM, allowing for a second correlation of the maximum intensity projection image obtained from cryo-confocal Z-stack with the medium magnification cryo-TEM image (6500x, Fig. 3D). Tilt-series data collection followed by tomogram reconstruction on areas guided by the fluorescence signal revealed the presence of long filaments within lysosomes and autophagosomes selectively in Cy3-(GU)6-treated cells (Fig. 3D, white arrowheads). These filaments were identical, in shape and width, to TDP43-immunopositive structures observed in Dendra2-LC3B HEK293T cells, confirming lysosomal localization of fibrillar TDP43 upon treatment with (GU)6 oligos (Fig. S10A).
Fig. 3. Cryo-correlative workflow to study aggregates and probe proteinopathies in situ.

Representative images appear below each step of the workflow, with a schematic on the left. (A) Micropatterning and vitrification: doxycycline-inducible mEGFP-LC3B iPSCs are differentiated to iNeurons on micropatterned EM grids. Scale bar, 40μm. (B) Cryo-confocal microscopy: Cryo-confocal z-stacks with mEGFP fluorescence (green) and reflected light (grey) are collected of mEGFP-LC3B iNeurons (green) on grid squares for precise correlative cryo-FIB/SEM milling. Scale bar, 20μm. (C) Correlative cryo-focused ion beam milling (cryo-FIB): Cryo-confocal maximum intensity projection (MIP) of mEGFP-LC3B (green) overlaid on cryo-FIB image (grey) of mEGFP-LC3B iNeuron before milling (top). 3D-correlation toolbox (3DCT) was used to correlate the cryo-confocal and cryo-FIB data. Scale bar, 20μm. Bottom: Final lamella from cryo-FIB (grey) with MIP of mEGFP-LC3B (green) correlated fluorescence. Scale bar, 5μm. (D) In situ cryo-CLEM: Correlation of MIP of mEGFP-LC3B (green) and 6500x cryo-EM montage of cryo-lamella guides tilt-series positioning. Tilt-series were collected at the region of cryo-lamella highlighted with an orange box. Scale bar, 2μm. Slice through a tomogram of lysosome with TDP43 fibrils (bottom). White arrowheads indicate individual TDP43 fibrils. Scale bar, 200 nm. (E) Analysis: 3D segmentation corresponding to the volume represented in panel (D). Scale bar, 200 nm.
In situ TDP43 architecture at molecular resolution
To verify TDP43 accumulation within lysosomes of (GU)6-treated neurons, we employed CRISPR/Cas9 to insert the HaloTag open reading frame just downstream of the endogenous TARDBP start codon in human iPSCs, thereby generating a fusion of HaloTag with the N-terminus of native TDP43 (Fig. S8A). Correct positioning of HaloTag was confirmed by Sanger sequencing, and an appropriate shift in the molecular weight of TDP43 was observed on immunoblotting (Fig. S8B). After integrating the NGN1/NGN2 cassette, HaloTag-TDP43 iPSCs were differentiated into iNeurons and labeled using cell-permeable and photostable HaloTag-compatible dye (JF635)(53). We then applied Cy3-labeled (GU)6 oligos for 24h and employed our cryo-CLEM workflow to visualize mislocalized, JF635-positive HaloTag-TDP43 puncta within the cytoplasm in greater detail (Fig. S8C–E). Some of these HaloTag-TDP43 foci appeared to have very little organized ultrastructure, instead forming amorphous accumulations that were notable for their spherical appearance and the exclusion of ribosomes and other cellular organelles (Fig. 4A, Movie 2). Fluorescence signals from Cy3-(GU)6 often overlapped with these spherical puncta, suggesting that these structures may represent phase-separated ribonucleoprotein condensates. Notably, such ‘ribosome-excluded zones’ frequently localize to the surface of endolysosomes, where they adopt the curvature of the adjacent membrane (Fig. 4A, Movie 2).
Fig. 4. In situ structural analysis of mislocalized TDP43 in (GU)6-treated human iNeurons.

Slice through tomograms and corresponding segmentation of the volumes (images with solid black outlines on the left) highlighting TDP43 inclusion architecture in DIV10 iNeurons treated with (GU)6 RNA for 24h, within an amorphous organelle excluded zone (A), within autophagosomes (B), or lysosomes (C), respectively. Cellular features are labeled and colored as: ribosome (grey) TDP43 (red), endoplasmic reticulum (ER, pink), mitochondria (mito, yellow), microtubules (MT, green), lysosome (lyso, blue), autophagosome (AP, purple), degraded membranes (lilac), and endosome/multivesicular bodies (MVB, sky blue). Scale bars, 200nm. TDP43 shows amorphous condensate-like morphology in the cytosol (A) but fibril-like morphology in autophagosomes (B) and lysosomes (C). The two right panels in (A-C, dotted black outline) are enlarged views of the slices through tomogram and segmentation on the left, highlighting TDP43 specifically. White arrowheads indicate individual TDP43 fibrils. Scale bars, 200nm. (D) Slice through a tomogram of the autophagosome with TDP43 fibrils (left panel). TDP43 forms sheets (cyan arrowheads) and individual filaments (red arrowheads). 3D segmentation of TDP43 sheets (cyan) and single filaments (red) en face and rotated 90° around the y-axis (right panel). Scale bar, 50nm. (E) Fit of TDP43 Type B variant 1 (PDB: 7PY2) atomic model to the subtomogram averaged map of TDP43 fibrils in iNeurons. Scale bar, 5 nm.
Movie 2. In situ cryo-ET of TDP43 condensate.
Sequential sections of a tomogram of cryo-FIB milled human neurons reveal amorphous TDP43 in the cytosol. Accompanies Fig. 4.
We also observed several larger cytoplasmic TDP43 foci located within double membrane-bound structures reminiscent of autophagosomes (Fig. 4B, Movie 3, Fig. S8C–E). In some cases, multiple sheets of TDP43 fibrils span the entirety of autophagosomes. As in our cryo-ET analysis of isolated organelles, most of the filaments form stacked sheets (Fig. 2E, Fig. 4D). TDP43-containing autophagosomes rarely exhibited other cellular debris or material, implying selective rather than bulk (macro) autophagy of TDP43 fibrils.
Movie 3. In situ cryo-ET of TDP43 engulfed within autophagosome.
Sequential sections of a tomogram of cryo-FIB milled human neurons reveal TDP43 filaments engulfed by an autophagosome. Accompanies Fig. 4.
The large majority of mislocalized TDP43 was fibrillar rather than amorphous and present within single membrane-bound endolysosomes (Fig. 4C, Movie 4). Within these structures, the density of TDP43 filaments was less than that found in double membrane-bound autophagosomes (Fig. 4B). In fact, we rarely found TDP43 filaments outside of acidified membrane-enclosed organelles such as lysosomes and autophagosomes, suggesting that fibril formation may be enhanced by changes in pH, molecular crowding, and/or partial cleavage of TDP43 within these organelles.
Movie 4. In situ cryo-ET of TDP43 engulfed within the lysosome.
Sequential sections of a tomogram of cryo-FIB milled human neurons reveal lysosomal engulfment of TDP43 filaments. Accompanies Fig. 4.
We computationally extracted these filaments and performed subtomogram averaging (Fig. S9), obtaining a low-resolution Coulombe potential map of TDP43 within lysosomes (Fig. S9B). Our analysis revealed that the length and width of the TDP43 filaments within lysosomes in HEK293T cells and iNeurons are remarkably similar (Fig. S10A, B). PDB models of insoluble TDP43 fibrils from both FTLD-TDP type A and ALS/FTLD-TDP type B(25, 26), consisting of C-terminal residues 272–360 (PDB ID: 8CG3), 281–360 (PDB ID: 8CGH), 281–360 (PDB ID: 8CGG) and 282–360 (PDB ID: 7PY2), fit very well to maps of fibrillar TDP43 from iNeurons, with ALS/FTLD-TDP type B showing an overall better fit, suggesting misfolding and reorganization of the TDP43 C-terminus within individual filaments (Fig. 4E, Fig. S10C), similar to what is observed in post-mortem material.
TDP43 mislocalizes to autophagosomes and lysosomes in ALS patient tissue
Our observations of TDP43 fibrils within lysosomes and autophagosomes suggested that the earliest stages of pathological TDP43 deposition may occur within these organelles. To pursue this possibility, we performed dual immunohistochemistry for TDP43 and LAMP1 or LC3B in ALS patient spinal cord sections. Unaffected spinal neurons within the anterior horn displayed nuclear TDP43 and the expected distribution of lysosomal (LAMP1) and autophagosomal (LC3B) signals within the cytosol (Fig. 5A, B). However, spinal neurons with pathological nuclear exclusion of TDP43 instead displayed marked overlap of cytosolic TDP43 puncta with lysosomes and less so autophagosomes, confirmed by color deconvolution of dual stained tissue (Fig. 5C). As with unaffected neurons, we did not observe colocalization between TDP43 and lysosomes or autophagosomes in spinal neurons from control subjects (Fig. S11A, B). The overlap between TDP43 and these organelles was further confirmed by immunofluorescence microscopy. We observed clear colocalization of TDP43 with lysosomal (LAMP1) and autophagosomal (LC3B) markers in affected neurons displaying TDP43 mislocalization, but not unaffected cells or those from controls (Fig. 5D, E, Fig. S11C, D). To verify the lysosomal accumulation of mislocalized TDP43 and the formation of fibrillar TDP43 within these organelles, we adapted the method we previously used for isolating lysosomes and autophagosomes from cultured cells and used it to fractionate ALS/FTLD-TDP prefrontal cortex (Fig. S5A). In lysosome-rich fractions, we detected full-length phosphorylated TDP43 (S409/S410) as well as truncated species of ~35 and 25kDa (Fig. S11E). Cryo-ET analysis of these lysosomes revealed filamentous inclusions within membrane-bound structures that were remarkably similar to what we observed in iNeurons (Fig. 5F, Movie 5). The widths of filaments in patient-derived lysosomes displayed a bimodal distribution, with thinner filaments having a mean width of 4.4 ± 0.46 nm and thicker filaments with a mean width of 6.7 ± 0.7 nm (Fig. S11F, G). Together, these findings validate and extend our observations from (GU)6-treated iNeurons, suggesting that RNA dysregulation may similarly foster TDP43 mislocalization and aggregation in disease.
Fig. 5. TDP43 mislocalizes to lysosomes and autophagosomes in patient tissue.
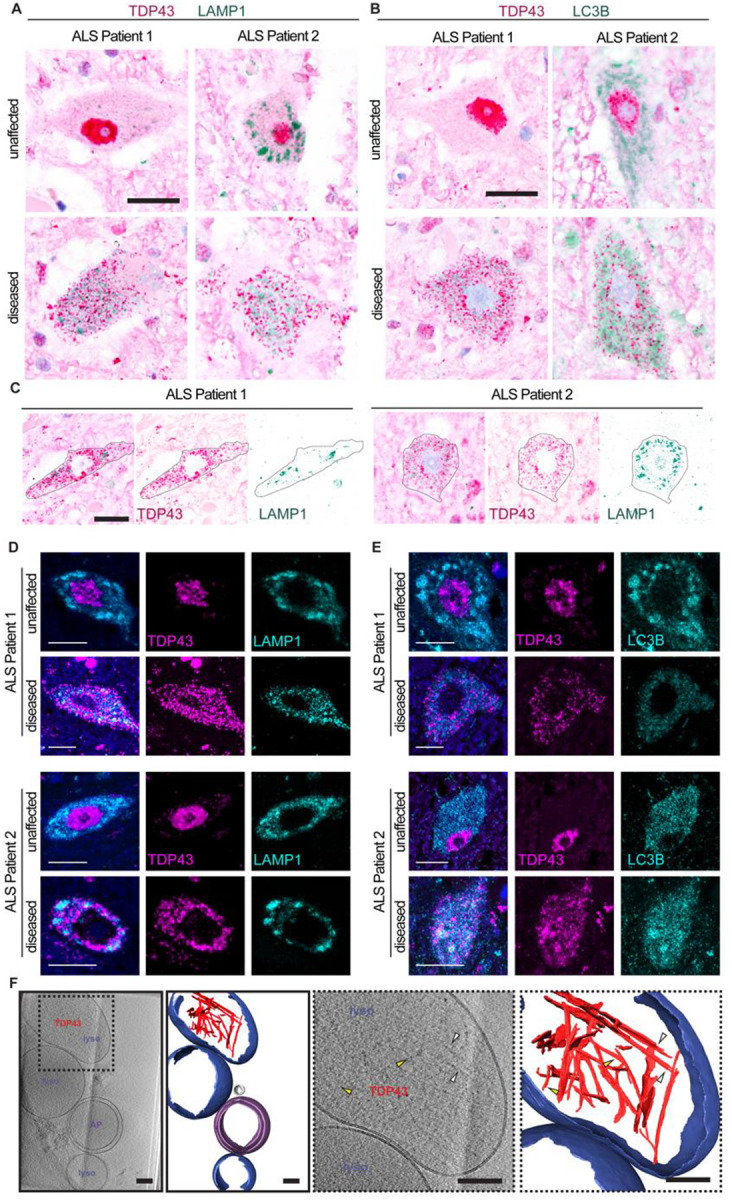
(A-B) Immunohistochemistry for TDP43 (magenta) and (A) LAMP1 or (B) LC3B (green) in spinal cord of two patients with sporadic ALS show unaffected (top row) and diseased (bottom row) neurons. (C) Diseased neurons (outlined in black) in deconvolved immunohistochemistry images for TDP43 (magenta) and LAMP1 (green) in the spinal cord of two patients with sporadic ALS show colocalization of TDP43 and LAMP1. Scale bars, 20μm. (D-E) Immunofluorescence for TDP43 (magenta) and (D) LAMP1 or (E) LC3B (cyan) in the spinal cord of two patients with sporadic ALS show unaffected (top row) and diseased (bottom row) neurons. Diseased neurons show colocalization of LAMP1 or LC3B (cyan) with mislocalized, cytosolic TDP43 (magenta). Scale bars, 10μm. (F) Slice through tomogram and corresponding segmentation of the volume (images with solid black outlines on the left) highlighting TDP43 (red) within lysosomes (blue) isolated from ALS/FTLD patient postmortem brain tissue. Cellular features are labeled and colored as lysosome (lyso, blue), autophagosome (AP, purple), and vesicle (grey). The two right panels (dotted black outline) are enlarged views of the slices through tomogram and segmentation on the left, highlighting TDP43 specifically. Yellow arrowheads highlight thinner filaments and white arrowheads highlight thicker filaments. Scale bars, 100nm.
Movie 5. Cryo-ET of isolated lysosomes from ALS/FTLD patient brain tissue.
Sequential sections of lysosomes obtained from fraction 1 of gradient fractionation reveal ensconced TDP43 filaments. Accompanies Fig. 5.
Discussion
TDP43 recognizes almost ⅓ of transcribed genes(54, 55) and is crucial for diverse aspects of RNA metabolism. Loss of this protein from the nucleus and its cytosolic accumulation are closely tied with neurodegeneration in cellular and animal ALS/FTLD-TDP models, as well as in human postmortem samples(14, 38, 56, 57). Indeed, TDP43 knockdown leads to dramatic and widespread changes in RNA splicing and stability(22, 36, 37, 47, 48, 58), implying that RNA misprocessing in ALS/FTLD-TDP is downstream of TDP43 mislocalization. Our data, together with recent studies illustrating the significance of RNA for TDP43 localization(19–21), are consistent with an alternative possibility: RNA dyshomeostasis may precede and drive TDP43 mislocalization, eliciting a self-perpetuating positive feedback loop that further facilitates and enhances RNA misprocessing. The end result is a spiral of RNA dysfunction and TDP43 mislocalization that expedites neuronal toxicity and degeneration in disease.
RNA misprocessing may originate from genetic and/or acquired means. For instance, mutations in genes encoding RNA binding proteins such as FUS, hnRNPA2B1, MATR3, and TDP43 itself lead to ALS and FTLD-TDP(59–65). Co-transcriptional RNA modifications, including methylation at the N6 position of adenosine (m6A), may also affect TDP43 binding affinity and/or the fate of transcribed RNA(23, 66–69). Abnormal patterns and extent of RNA methylation are characteristic of not just ALS and FTLD-TDP but also other conditions, such as Alzheimer’s disease, that display TDP43 pathology(23, 24, 70, 71). RNA-based sequestration of TDP43 into cytosolic granules is also observed during skeletal muscle regeneration in mice and humans(72). These myo-granules are rich in fibrillar TDP43, suggesting common misfolding events upon cytosolic TDP43 mislocalization. Here, we used TDP43 substrate, GU-rich RNA oligonucleotides, to initiate the efflux of TDP43 from the nucleus. Mislocalized TDP43 overlaps with bona fide markers of autophagosomes, lysosomes, stress granules, and ESCRT machinery (Fig. 1, Fig. S4), and biochemical gradient centrifugation experiments reveal the presence of full-length TDP43 within lysosomes and autophagosomes (Fig. 2). This is consistent with the appearance of full-length TDP43 within pathological aggregates in ALS spinal motor neurons(12), the overlap between TDP43 inclusions and lysosomal/autophagosomal markers in post-mortem ALS samples (Fig. 5A, B, and Fig. S11E) and the appearance of fibrillar material in purified lysosomes from ALS/FTLD-TDP brain (Fig. 5F).
Structural studies on purified protein fragments or fibrils extracted from patient samples are invaluable for deriving small molecule ligands or tracers specific for protein inclusions; nevertheless, these investigations are limited in their ability to uncover molecular mechanisms of TDP43 aggregation. To address this, we established an in situ cryo-CLEM platform (Fig. 3) that provides the first visual snapshots of endogenous TDP43 mislocalization and aggregation within human neurons. Application of short GU-rich oligos leads to the appearance of amorphous ‘condensate-like’ TDP43 inclusions within the cytosol (Fig. 4A, Movie 2), whose material properties may depend on chaperones (i.e., HSPB1)(73) and/or RNA length, size, modifications, and motif valency. Cytosolic TDP43 condensates were often found in association with lysosomal membranes, but within these organelles, TDP43 adopted filamentous and sheet-like structures (Fig. 4C, Movie 4). The acidified environment of lysosomes may promote the demixing of TDP43 from condensates, leading to its misfolding and filament formation(74–76). The appearance of TDP43 within autophagosomes argues against chaperone-mediated autophagy in the clearance of cytosolic TDP43(77) but would be consistent with selective autophagy that would require an as-yet-unknown adapter. Prior studies identified a single LC3-interacting region (LIR) containing protein in association with TDP43—TATA-box binding protein (TBP)(78) - but further studies are required to determine whether this factor is required for the delivery of cytosolic TDP43 to lysosomes and/or autophagosomes.
The TDP43 C-terminal low complexity domain (LCD) is critical for forming pathological amyloid-like aggregates in vitro. Within this region, amino acid stretches 286–331 in primary cortical neurons(79), and 341–367 in neuronal cell lines are sufficient for fibril formation(9, 10). Here, we show that introducing GU-rich oligos reproduces TDP43 mislocalization and aggregation within human neurons and other commonly established model cell lines. Mislocalized TDP43 forms fibril-like structures reminiscent of patient-derived fibrils, mainly in autophagosomes and lysosomes, underscoring the importance of these acidified organelles in the formation and/or clearance of nascent TDP43 aggregates (Fig. 4, Movie 3–4). The presence of the N-terminus of TDP43 and whether the GU-rich RNA is bound to the two RRM domains of TDP43 within these organelles remains to be investigated. We believe these features are dynamic and lost during averaging, as observed in TDP43 cryo-EM maps(25, 26). Similarly, we could not detect specific structures assumed by RNA in our reconstruction.
Our data are consistent with disease-associated mutations in autophagy-related genes (i.e., UBQLN2, C9ORF72, OPTN, SQSTM1)(80–84), age-related declines in the efficiency of neuronal autophagy, and the broad neuroprotection afforded by autophagy-inducing strategies in ALS/FTLD-TDP models(43, 56, 85, 86). Based on this, we propose a model for TDP43 mislocalization, clearance, and its aggregation in disease (Fig. 6). External stressors (i.e., injury) or internal events (i.e., RNA dyshomeostasis) may trigger nuclear TDP43 egress. In the cytosol, TDP43 undergoes RNA-dependent liquid-like condensation, as observed for other RNA-binding proteins in similar environments(87). At this stage, at least two scenarios are possible. If the stressors are self-limited and autophagy is functional, mislocalized TDP43 is degraded via autophagy, and nuclear TDP43 levels are restored by new protein synthesis. Alternatively, if the stressors persist, both existing and newly synthesized pools of TDP43 are mislocalized and targeted to autophagy, leading to TDP43 accumulation within autophagosomes and lysosomes (Fig. 6D). Eventually, TDP43 forms fibrils in these acidified organelles, and it can no longer repress cryptic exons due to its continued mislocalization from the nucleus. These events culminate in neurotoxicity and subsequent neurodegeneration.
Fig. 6. An autophagy-centric model of TDP43 pathology.

(A) TDP43 (red) primarily localizes to the nucleus in healthy neurons. RNA misprocessing, genetic mutations, or environmental factors may trigger TDP43 nuclear egress and cytosolic accumulation. (B) Increasing amounts of mislocalized TDP43 leads to the accumulation of mislocalized TDP43 in autophagosomes and lysosomes. (C) Increased burden of mislocalized TDP43 leads to the loss of TDP43 splicing activity and cytosolic TDP43 fibrilization, as seen in ALS, FTD, and other TDP43 proteinopathies. (D) TDP43, LC3B, and LAMP1 immunohistochemistry (left) and immunofluorescence (right) of ALS patient tissue. Scale bars, 20μm (left) and 5μm (right). (E) Restoring TDP43 nuclear localization has pharmacotherapeutic potential to prevent TDP43 pathology.
Taken together, we establish a model of TDP43 mislocalization and cytoplasmic aggregation, a signature pathologic event in ALS/FTLD-TDP. Using this model, we provide the first visual snapshots of TDP43 aggregates within neuronal lysosomes and autophagosomes in situ, highlighting the critical connection between autophagy and the pathogenesis of ALS/FTLD-TDP. We expect this platform to be invaluable for investigating not just the pathogenesis and origins of TDP43 pathology but also for developing the means to detect and even reverse TDP43 mislocalization in ALS/FTLD-TDP and related TDP43-proteinopathies.
Supplementary Material
Acknowledgments:
We thank all members of the Barmada, Baldridge, and Mosalaganti laboratories for their advice and suggestions. We thank Dr. Jing Liang and the Microscopy Core of the University of Michigan Biomedical Research Core Facilities for help with immunogold labeling experiments. We thank Dr. Alister Burt for suggestions on subtomogram averaging of TDP43 filaments. We thank Dr. Vinson Lam for his assistance with data collection and segmentation analysis.
Funding:
This work was supported by:
National Institutes of Health DP2GM150019-01 (SM)
National Institutes of Health R01NS097542, R01NS113943 and 1R56NS128110-01 (SJB)
National Institutes of Health F31NS134123-01 (MD)
National Institutes of Health AWD012778 (EP)
National Institutes of Health R35GM128592 (RDB)
National Institutes of Health T32GM007544 (JER)
National Institutes of Health T32GM141840 to (MF)
National Institutes of Health P30AG072931 to the University of Michigan Brain Bank and Alzheimer’s Disease Research Center
National Institutes of Health S10OD030275 and the Arnold and Mabel Beckmann Foundation award to the University of Michigan Cryo-EM facility
University of Michigan Pioneer Postdoctoral Fellowship (ALE)
University of Michigan Life Sciences Institute
Klatskin Sutker Discovery Fund (SM)
The funders had no role in the study design, data collection, and analysis or the content and publication of this manuscript.
Competing interests:
S.J.B. serves on the advisory board for Neurocures, Inc., Symbiosis, Eikonizo Therapeutics, Ninesquare Therapeutics, the Live Like Lou Foundation, and the Robert Packard Center for ALS Research. S.J.B. has received research funding from Denali Therapeutics, Biogen, Inc., Lysoway Therapeutics, Amylyx Therapeutics, Acelot Therapeutics, Meira GTX, Inc., Prevail Therapeutics, Eikonizo Therapeutics, and Ninesquare Therapeutics.
Funding Statement
This work was supported by:
National Institutes of Health DP2GM150019-01 (SM)
National Institutes of Health R01NS097542, R01NS113943 and 1R56NS128110-01 (SJB)
National Institutes of Health F31NS134123-01 (MD)
National Institutes of Health AWD012778 (EP)
National Institutes of Health R35GM128592 (RDB)
National Institutes of Health T32GM007544 (JER)
National Institutes of Health T32GM141840 to (MF)
National Institutes of Health P30AG072931 to the University of Michigan Brain Bank and Alzheimer’s Disease Research Center
National Institutes of Health S10OD030275 and the Arnold and Mabel Beckmann Foundation award to the University of Michigan Cryo-EM facility
University of Michigan Pioneer Postdoctoral Fellowship (ALE)
University of Michigan Life Sciences Institute
Klatskin Sutker Discovery Fund (SM)
The funders had no role in the study design, data collection, and analysis or the content and publication of this manuscript.
Footnotes
CRediT roles:
Conceptualization: SJB, SM
Methodology: ALE, MLC, MGF, DA, MB, JER, TSB, TH, EMHT
Investigation: ALE, MLC, MGF, DA, MB, JER, RS, ESP, DT, TSB, MD, XL, TH
Visualization: ALE, MLC, MGF, DA, RS, ESP
Funding acquisition: RDB, SJB, SM
Project administration: SJB, SM
Supervision: ALE, RDB, SJB, SM
Writing – original draft: SM
Writing – review & editing: ALE, RDB, SJB, SM
Data and materials availability:
All correspondence regarding the materials and data presented in this paper should be addressed to sbarmada@med.umich.edu or mosalaga@umich.edu. The in situ subtomogram averaging map of the TDP43 is deposited in Electron Microscopy DataBank with the accession code EMD-45215. All electron microscopy data analysis was performed using existing software.
References and Notes:
- 1.Barmada S. J., Linking RNA Dysfunction and Neurodegeneration in Amyotrophic Lateral Sclerosis. Neurotherapeutics 12, 340–351 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weskamp K., Barmada S. J., TDP43 and RNA instability in amyotrophic lateral sclerosis. Brain Research 1693, 67–74 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neumann M., Molecular Neuropathology of TDP-43 Proteinopathies. Int J Mol Sci 10, 232–246 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baralle M., Buratti E., Baralle F. E., The role of TDP-43 in the pathogenesis of ALS and FTLD. Biochem Soc Trans 41, 1536–1540 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Barmada Sami J., Finkbeiner Steven, Pathogenic TARDBP Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia: Disease-Associated Pathways. Reviews in the Neurosciences 21, 251–272 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Nelson P. T., Dickson D. W., Trojanowski J. Q., Jack C. R., Boyle P. A., Arfanakis K., Rademakers R., Alafuzoff I., Attems J., Brayne C., Coyle-Gilchrist I. T. S., Chui H. C., Fardo D. W., Flanagan M. E., Halliday G., Hokkanen S. R. K., Hunter S., Jicha G. A., Katsumata Y., Kawas C. H., Keene C. D., Kovacs G. G., Kukull W. A., Levey A. I., Makkinejad N., Montine T. J., Murayama S., Murray M. E., Nag S., Rissman R. A., Seeley W. W., Sperling R. A., White C. L., Yu L., Schneider J. A., Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 142, 1503–1527 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., Bruce J., Schuck T., Grossman M., Clark C. M., McCluskey L. F., Miller B. L., Masliah E., Mackenzie I. R., Feldman H., Feiden W., Kretzschmar H. A., Trojanowski J. Q., Lee V. M. Y., Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., Mann D., Tsuchiya K., Yoshida M., Hashizume Y., Oda T., TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochemical and Biophysical Research Communications 351, 602–611 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Guenther E. L., Cao Q., Trinh H., Lu J., Sawaya M. R., Cascio D., Boyer D. R., Rodriguez J. A., Hughes M. P., Eisenberg D. S., Atomic structures of TDP-43 LCD segments and insights into reversible or pathogenic aggregation. Nature Structural and Molecular Biology 25, 463–471 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao Q., Boyer D. R., Sawaya M. R., Ge P., Eisenberg D. S., Cryo-EM structures of four polymorphic TDP-43 amyloid cores. Nature Structural and Molecular Biology 26, 619–627 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berning B. A., Walker A. K., The Pathobiology of TDP-43 C-Terminal Fragments in ALS and FTLD. Front. Neurosci. 13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Igaz L. M., Kwong L. K., Xu Y., Truax A. C., Uryu K., Neumann M., Clark C. M., Elman L. B., Miller B. L., Grossman M., McCluskey L. F., Trojanowski J. Q., Lee V. M.-Y., Enrichment of C-Terminal Fragments in TAR DNA-Binding Protein-43 Cytoplasmic Inclusions in Brain but not in Spinal Cord of Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. The American Journal of Pathology 173, 182–194 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winton M. J., Igaz L. M., Wong M. M., Kwong L. K., Trojanowski J. Q., Lee V. M.-Y., Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem 283, 13302–13309 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barmada S. J., Skibinski G., Korb E., Rao E. J., Wu J. Y., Finkbeiner S., Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci 30, 639–649 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walker A. K., Spiller K. J., Ge G., Zheng A., Xu Y., Zhou M., Tripathy K., Kwong L. K., Trojanowski J. Q., Lee V. M.-Y., Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol 130, 643–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDonald K. K., Aulas A., Destroismaisons L., Pickles S., Beleac E., Camu W., Rouleau G. A., Vande Velde C., TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet 20, 1400–1410 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Liu-Yesucevitz L., Bilgutay A., Zhang Y.-J., Vanderweyde T., Citro A., Mehta T., Zaarur N., McKee A., Bowser R., Sherman M., Petrucelli L., Wolozin B., Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 5, e13250 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mann J. R., Gleixner A. M., Mauna J. C., Gomes E., DeChellis-Marks M. R., Needham P. G., Copley K. E., Hurtle B., Portz B., Pyles N. J., Guo L., Calder C. B., Wills Z. P., Pandey U. B., Kofler J. K., Brodsky J. L., Thathiah A., Shorter J., Donnelly C. J., RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron 102, 321–338.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duan L., Zaepfel B. L., Aksenova V., Dasso M., Rothstein J. D., Kalab P., Hayes L. R., Nuclear RNA binding regulates TDP-43 nuclear localization and passive nuclear export. Cell Rep 40, 111106 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.dos Passos P. M., Hemamali E. H., Mamede L. D., Hayes L. R., Ayala Y. M., RNA-mediated ribonucleoprotein assembly controls TDP-43 nuclear retention. PLOS Biology 22, e3002527 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X., Das T., Chao T. F., Trinh V., Carmen-Orozco R. P., Ling J. P., Kalab P., Hayes L. R., Multivalent GU-rich oligonucleotides sequester TDP-43 in the nucleus by inducing high molecular weight RNP complexes. iScience 27, 110109 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tank E. M., Figueroa-Romero C., Hinder L. M., Bedi K., Archbold H. C., Li X., Weskamp K., Safren N., Paez-Colasante X., Pacut C., Thumma S., Paulsen M. T., Guo K., Hur J., Ljungman M., Feldman E. L., Barmada S. J., Abnormal RNA stability in amyotrophic lateral sclerosis. Nat Commun 9, 2845 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McMillan M., Gomez N., Hsieh C., Bekier M., Li X., Miguez R., Tank E. M. H., Barmada S. J., RNA methylation influences TDP43 binding and disease pathogenesis in models of amyotrophic lateral sclerosis and frontotemporal dementia. Mol Cell 83, 219–236.e7 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y., Dou X., Liu J., Xiao Y., Zhang Z., Hayes L., Wu R., Fu X., Ye Y., Yang B., Ostrow L. W., He C., Sun S., Globally reduced N6-methyladenosine (m6A) in C9ORF72-ALS/FTD dysregulates RNA metabolism and contributes to neurodegeneration. Nat Neurosci 26, 1328–1338 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arseni D., Hasegawa M., Murzin A. G., Kametani F., Arai M., Yoshida M., Ryskeldi-Falcon B., Structure of pathological TDP-43 filaments from ALS with FTLD. Nature 601, 139–143 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arseni D., Chen R., Murzin A. G., Peak-Chew S. Y., Garringer H. J., Newell K. L., Kametani F., Robinson A. C., Vidal R., Ghetti B., Hasegawa M., Ryskeldi-Falcon B., TDP-43 forms amyloid filaments with a distinct fold in type A FTLD-TDP. Nature 620, 898–903 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finkbeiner S., The Autophagy Lysosomal Pathway and Neurodegeneration. Cold Spring Harb Perspect Biol 12, a033993 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nixon R. A., The aging lysosome: an essential catalyst for late-onset neurodegenerative diseases. Biochim Biophys Acta Proteins Proteom 1868, 140443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malik B. R., Maddison D. C., Smith G. A., Peters O. M., Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Molecular Brain 12, 100 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rubinsztein D. C., Mariño G., Kroemer G., Autophagy and Aging. Cell 146, 682–695 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Menzies F. M., Fleming A., Caricasole A., Bento C. F., Andrews S. P., Ashkenazi A., Füllgrabe J., Jackson A., Jimenez Sanchez M., Karabiyik C., Licitra F., Lopez Ramirez A., Pavel M., Puri C., Renna M., Ricketts T., Schlotawa L., Vicinanza M., Won H., Zhu Y., Skidmore J., Rubinsztein D. C., Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 93, 1015–1034 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Carmona-Gutierrez D., Hughes A. L., Madeo F., Ruckenstuhl C., The crucial impact of lysosomes in aging and longevity. Ageing Research Reviews 32, 2–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan J. X., Finkel T., Lysosomes in senescence and aging. EMBO reports n/a, e57265 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rajawat Y. S., Hilioti Z., Bossis I., Aging: Central role for autophagy and the lysosomal degradative system. Ageing Research Reviews 8, 199–213 (2009). [DOI] [PubMed] [Google Scholar]
- 35.LIANG K.-Y., ZEGER S. L., Longitudinal data analysis using generalized linear models. Biometrika 73, 13–22 (1986). [Google Scholar]
- 36.Klim J. R., Williams L. A., Limone F., Guerra San Juan I., Davis-Dusenbery B. N., Mordes D. A., Burberry A., Steinbaugh M. J., Gamage K. K., Kirchner R., Moccia R., Cassel S. H., Chen K., Wainger B. J., Woolf C. J., Eggan K., ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci 22, 167–179 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melamed Z., López-Erauskin J., Baughn M. W., Zhang O., Drenner K., Sun Y., Freyermuth F., McMahon M. A., Beccari M. S., Artates J. W., Ohkubo T., Rodriguez M., Lin N., Wu D., Bennett C. F., Rigo F., Da Cruz S., Ravits J., Lagier-Tourenne C., Cleveland D. W., Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci 22, 180–190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ling J. P., Pletnikova O., Troncoso J. C., Wong P. C., TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–655 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Humphrey J., Emmett W., Fratta P., Isaacs A. M., Plagnol V., Quantitative analysis of cryptic splicing associated with TDP-43 depletion. BMC Medical Genomics 10, 38 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.D’Alton S., Altshuler M., Lewis J., Studies of alternative isoforms provide insight into TDP-43 autoregulation and pathogenesis. RNA 21, 1419–1432 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weskamp K., Tank E. M., Miguez R., McBride J. P., Gómez N. B., White M., Lin Z., Gonzalez C. M., Serio A., Sreedharan J., Barmada S. J., Shortened TDP43 isoforms upregulated by neuronal hyperactivity drive TDP43 pathology in ALS. Journal of Clinical Investigation 130, 1139–1155 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chua J. P., Bedi K., Paulsen M. T., Ljungman M., Tank E. M. H., Kim E. S., McBride J. P., Colón-Mercado J. M., Ward M. E., Weisman L. S., Barmada S. J., Myotubularin-related phosphatase 5 is a critical determinant of autophagy in neurons. Curr Biol 32, 2581–2595.e6 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Safren N., Tank E. M., Malik A. M., Chua J. P., Santoro N., Barmada S. J., Development of a specific live-cell assay for native autophagic flux. J Biol Chem 297, 101003 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fernandopulle M. S., Prestil R., Grunseich C., Wang C., Gan L., Ward M. E., Transcription Factor–Mediated Differentiation of Human iPSCs into Neurons. Current Protocols in Cell Biology 79 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Busskamp V., Lewis N. E., Guye P., Ng A. H. M., Shipman S. L., Byrne S. M., Sanjana N. E., Murn J., Li Y., Li S., Stadler M., Weiss R., Church G. M., Rapid neurogenesis through transcriptional activation in human stem cells. Mol Syst Biol 10, 760 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lam R. S., Töpfer F. M., Wood P. G., Busskamp V., Bamberg E., Functional Maturation of Human Stem Cell-Derived Neurons in Long-Term Cultures. PLOS ONE 12, e0169506 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown A.-L., Wilkins O. G., Keuss M. J., Hill S. E., Zanovello M., Lee W. C., Bampton A., Lee F. C. Y., Masino L., Qi Y. A., Bryce-Smith S., Gatt A., Hallegger M., Fagegaltier D., Phatnani H., Newcombe J., Gustavsson E. K., Seddighi S., Reyes J. F., Coon S. L., Ramos D., Schiavo G., Fisher E. M. C., Raj T., Secrier M., Lashley T., Ule J., Buratti E., Humphrey J., Ward M. E., Fratta P., TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 603, 131–137 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma X. R., Prudencio M., Koike Y., Vatsavayai S. C., Kim G., Harbinski F., Briner A., Rodriguez C. M., Guo C., Akiyama T., Schmidt H. B., Cummings B. B., Wyatt D. W., Kurylo K., Miller G., Mekhoubad S., Sallee N., Mekonnen G., Ganser L., Rubien J. D., Jansen-West K., Cook C. N., Pickles S., Oskarsson B., Graff-Radford N. R., Boeve B. F., Knopman D. S., Petersen R. C., Dickson D. W., Shorter J., Myong S., Green E. M., Seeley W. W., Petrucelli L., Gitler A. D., TDP-43 represses cryptic exon inclusion in the FTD–ALS gene UNC13A. Nature 603, 124–130 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Otero P. C., Todd T. W., Shao W., Jones C. J., Huang K., Daughrity L. M., Yue M., Sheth U., Gendron T. F., Prudencio M., Oskarsson B., Dickson D. W., Petrucelli L., Zhang Y.-J., Generation and characterization of monoclonal antibodies against pathologically phosphorylated TDP-43. PLOS ONE 19, e0298080 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prudencio M., Humphrey J., Pickles S., Brown A.-L., Hill S. E., Kachergus J. M., Shi J., Heckman M. G., Spiegel M. R., Cook C., Song Y., Yue M., Daughrity L. M., Carlomagno Y., Jansen-West K., de Castro C. F., DeTure M., Koga S., Wang Y.-C., Sivakumar P., Bodo C., Candalija A., Talbot K., Selvaraj B. T., Burr K., Chandran S., Newcombe J., Lashley T., Hubbard I., Catalano D., Kim D., Propp N., Fennessey S., NYGC ALS Consortium, Fagegaltier D., Phatnani H., Secrier M., Fisher E. M., Oskarsson B., van Blitterswijk M., Rademakers R., Graff-Radford N. R., Boeve B. F., Knopman D. S., Petersen R. C., Josephs K. A., Thompson E. A., Raj T., Ward M., Dickson D. W., Gendron T. F., Fratta P., Petrucelli L., Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J Clin Invest 130, 6080–6092 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yim W. W.-Y., Yamamoto H., Mizushima N., A pulse-chasable reporter processing assay for mammalian autophagic flux with HaloTag. Elife 11, e78923 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumar S. T., Nazarov S., Porta S., Maharjan N., Cendrowska U., Kabani M., Finamore F., Xu Y., Lee V. M.-Y., Lashuel H. A., Seeding the aggregation of TDP-43 requires post-fibrillization proteolytic cleavage. Nat Neurosci 26, 983–996 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grimm J. B., English B. P., Choi H., Muthusamy A. K., Mehl B. P., Dong P., Brown T. A., Lippincott-Schwartz J., Liu Z., Lionnet T., Lavis L. D., Bright photoactivatable fluorophores for single-molecule imaging. Nat Methods 13, 985–988 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Polymenidou M., Lagier-Tourenne C., Hutt K. R., Huelga S. C., Moran J., Liang T. Y., Ling S.-C., Sun E., Wancewicz E., Mazur C., Kordasiewicz H., Sedaghat Y., Donohue J. P., Shiue L., Bennett C. F., Yeo G. W., Cleveland D. W., Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14, 459–468 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tollervey J. R., Curk T., Rogelj B., Briese M., Cereda M., Kayikci M., König J., Hortobágyi T., Nishimura A. L., Zupunski V., Patani R., Chandran S., Rot G., Zupan B., Shaw C. E., Ule J., Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 14, 452–458 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barmada S. J., Serio A., Arjun A., Bilican B., Daub A., Ando D. M., Tsvetkov A., Pleiss M., Li X., Peisach D., Shaw C., Chandran S., Finkbeiner S., Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol 10, 677–685 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sakae N., Bieniek K. F., Zhang Y.-J., Ross K., Gendron T. F., Murray M. E., Rademakers R., Petrucelli L., Dickson D. W., Poly-GR dipeptide repeat polymers correlate with neurodegeneration and Clinicopathological subtypes in C9ORF72-related brain disease. Acta Neuropathol Commun 6, 63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seddighi S., Qi Y. A., Brown A.-L., Wilkins O. G., Bereda C., Belair C., Zhang Y.-J., Prudencio M., Keuss M. J., Khandeshi A., Pickles S., Kargbo-Hill S. E., Hawrot J., Ramos D. M., Yuan H., Roberts J., Sacramento E. K., Shah S. I., Nalls M. A., Colón-Mercado J. M., Reyes J. F., Ryan V. H., Nelson M. P., Cook C. N., Li Z., Screven L., Kwan J. Y., Mehta P. R., Zanovello M., Hallegger M., Shantaraman A., Ping L., Koike Y., Oskarsson B., Staff N. P., Duong D. M., Ahmed A., Secrier M., Ule J., Jacobson S., Reich D. S., Rohrer J. D., Malaspina A., Dickson D. W., Glass J. D., Ori A., Seyfried N. T., Maragkakis M., Petrucelli L., Fratta P., Ward M. E., Mis-spliced transcripts generate de novo proteins in TDP-43-related ALS/FTD. Sci Transl Med 16, eadg7162 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kwiatkowski T. J., Bosco D. A., Leclerc A. L., Tamrazian E., Vanderburg C. R., Russ C., Davis A., Gilchrist J., Kasarskis E. J., Munsat T., Valdmanis P., Rouleau G. A., Hosler B. A., Cortelli P., de Jong P. J., Yoshinaga Y., Haines J. L., Pericak-Vance M. A., Yan J., Ticozzi N., Siddique T., McKenna-Yasek D., Sapp P. C., Horvitz H. R., Landers J. E., Brown R. H., Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208 (2009). [DOI] [PubMed] [Google Scholar]
- 60.Vance C., Rogelj B., Hortobágyi T., De Vos K. J., Nishimura A. L., Sreedharan J., Hu X., Smith B., Ruddy D., Wright P., Ganesalingam J., Williams K. L., Tripathi V., Al-Saraj S., Al-Chalabi A., Leigh P. N., Blair I. P., Nicholson G., de Belleroche J., Gallo J.-M., Miller C. C., Shaw C. E., Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim H. J., Kim N. C., Wang Y.-D., Scarborough E. A., Moore J., Diaz Z., MacLea K. S., Freibaum B., Li S., Molliex A., Kanagaraj A. P., Carter R., Boylan K. B., Wojtas A. M., Rademakers R., Pinkus J. L., Greenberg S. A., Trojanowski J. Q., Traynor B. J., Smith B. N., Topp S., Gkazi A.-S., Miller J., Shaw C. E., Kottlors M., Kirschner J., Pestronk A., Li Y. R., Ford A. F., Gitler A. D., Benatar M., King O. D., Kimonis V. E., Ross E. D., Weihl C. C., Shorter J., Taylor J. P., Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson J. O., Pioro E. P., Boehringer A., Chia R., Feit H., Renton A. E., Pliner H. A., Abramzon Y., Marangi G., Winborn B. J., Gibbs J. R., Nalls M. A., Morgan S., Shoai M., Hardy J., Pittman A., Orrell R. W., Malaspina A., Sidle K. C., Fratta P., Harms M. B., Baloh R. H., Pestronk A., Weihl C. C., Rogaeva E., Zinman L., Drory V. E., Borghero G., Mora G., Calvo A., Rothstein J. D., ITALSGEN, Drepper C., Sendtner M., Singleton A. B., Taylor J. P., Cookson M. R., Restagno G., Sabatelli M., Bowser R., Chiò A., Traynor B. J., Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 17, 664–666 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sreedharan J., Blair I. P., Tripathi V. B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J. C., Williams K. L., Buratti E., Baralle F., de Belleroche J., Mitchell J. D., Leigh P. N., Al-Chalabi A., Miller C. C., Nicholson G., Shaw C. E., TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 319, 1668–1672 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kabashi E., Valdmanis P. N., Dion P., Spiegelman D., McConkey B. J., Velde C. V., Bouchard J.-P., Lacomblez L., Pochigaeva K., Salachas F., Pradat P.-F., Camu W., Meininger V., Dupre N., Rouleau G. A., TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40, 572–574 (2008). [DOI] [PubMed] [Google Scholar]
- 65.Van Deerlin V. M., Sleiman P. M. A., Martinez-Lage M., Chen-Plotkin A., Wang L.-S., Graff-Radford N. R., Dickson D. W., Rademakers R., Boeve B. F., Grossman M., Arnold S. E., Mann D. M. A., Pickering-Brown S. M., Seelaar H., Heutink P., van Swieten J. C., Murrell J. R., Ghetti B., Spina S., Grafman J., Hodges J., Spillantini M. G., Gilman S., Lieberman A. P., Kaye J. A., Woltjer R. L., Bigio E. H., Mesulam M., Al-Sarraj S., Troakes C., Rosenberg R. N., White C. L., Ferrer I., Lladó A., Neumann M., Kretzschmar H. A., Hulette C. M., Welsh-Bohmer K. A., Miller B. L., Alzualde A., Lopez de Munain A., McKee A. C., Gearing M., Levey A. I., Lah J. J., Hardy J., Rohrer J. D., Lashley T., Mackenzie I. R. A., Feldman H. H., Hamilton R. L., Dekosky S. T., van der Zee J., Kumar-Singh S., Van Broeckhoven C., Mayeux R., Vonsattel J. P. G., Troncoso J. C., Kril J. J., Kwok J. B. J., Halliday G. M., Bird T. D., Ince P. G., Shaw P. J., Cairns N. J., Morris J. C., McLean C. A., DeCarli C., Ellis W. G., Freeman S. H., Frosch M. P., Growdon J. H., Perl D. P., Sano M., Bennett D. A., Schneider J. A., Beach T. G., Reiman E. M., Woodruff B. K., Cummings J., Vinters H. V., Miller C. A., Chui H. C., Alafuzoff I., Hartikainen P., Seilhean D., Galasko D., Masliah E., Cotman C. W., Tuñón M. T., Martínez M. C. C., Munoz D. G., Carroll S. L., Marson D., Riederer P. F., Bogdanovic N., Schellenberg G. D., Hakonarson H., Trojanowski J. Q., Lee V. M.-Y., Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 42, 234–239 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Coots R. A., Liu X.-M., Mao Y., Dong L., Zhou J., Wan J., Zhang X., Qian S.-B., m6A Facilitates eIF4F-Independent mRNA Translation. Mol Cell 68, 504–514.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roundtree I. A., Luo G.-Z., Zhang Z., Wang X., Zhou T., Cui Y., Sha J., Huang X., Guerrero L., Xie P., He E., Shen B., He C., YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. eLife 6, e31311 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang X., Lu Z., Gomez A., Hon G. C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G., Ren B., Pan T., He C., N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sendinc E., Shi Y., RNA m6A methylation across the transcriptome. Mol Cell 83, 428–441 (2023). [DOI] [PubMed] [Google Scholar]
- 70.Deng J., Chen X., Chen A., Zheng X., m6A RNA methylation in brain injury and neurodegenerative disease. Front Neurol 13, 995747 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jiang L., Lin W., Zhang C., Ash P. E. A., Verma M., Kwan J., van Vliet E., Yang Z., Cruz A. L., Boudeau S., Maziuk B. F., Lei S., Song J., Alvarez V. E., Hovde S., Abisambra J. F., Kuo M.-H., Kanaan N., Murray M. E., Crary J. F., Zhao J., Cheng J.-X., Petrucelli L., Li H., Emili A., Wolozin B., Interaction of tau with HNRNPA2B1 and N6-methyladenosine RNA mediates the progression of tauopathy. Mol Cell 81, 4209–4227.e12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vogler T. O., Wheeler J. R., Nguyen E. D., Hughes M. P., Britson K. A., Lester E., Rao B., Betta N. D., Whitney O. N., Ewachiw T. E., Gomes E., Shorter J., Lloyd T. E., Eisenberg D. S., Taylor J. P., Johnson A. M., Olwin B. B., Parker R., TDP-43 and RNA form amyloid-like myo-granules in regenerating muscle. Nature 563, 508–513 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lu S., Hu J., Arogundade O. A., Goginashvili A., Vazquez-Sanchez S., Diedrich J. K., Gu J., Blum J., Oung S., Ye Q., Yu H., Ravits J., Liu C., Yates J. R., Cleveland D. W., Heat-shock chaperone HSPB1 regulates cytoplasmic TDP-43 phase separation and liquid-to-gel transition. Nat Cell Biol 24, 1378–1393 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Patni D., Jha S. K., Thermodynamic modulation of folding and aggregation energy landscape by DNA binding of functional domains of TDP-43. Biochim Biophys Acta Proteins Proteom 1871, 140916 (2023). [DOI] [PubMed] [Google Scholar]
- 75.Gu J., Zhou X., Sutherland L., Kato M., Jaczynska K., Rizo J., McKnight S. L., Oxidative regulation of TDP-43 self-association by a β-to-α conformational switch. Proc Natl Acad Sci U S A 120, e2311416120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Al Ojaimi Y., Slek C., Osman S., Alarcan H., Marouillat S., Corcia P., Vourc’h P., Lanznaster D., Blasco H., The effect of pH alterations on TDP-43 in a cellular model of amyotrophic lateral sclerosis. Biochem Biophys Rep 38, 101664 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ormeño F., Hormazabal J., Moreno J., Riquelme F., Rios J., Criollo A., Albornoz A., Alfaro I. E., Budini M., Chaperone Mediated Autophagy Degrades TDP-43 Protein and Is Affected by TDP-43 Aggregation. Front Mol Neurosci 13, 19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cho N. H., Cheveralls K. C., Brunner A.-D., Kim K., Michaelis A. C., Raghavan P., Kobayashi H., Savy L., Li J. Y., Canaj H., Kim J. Y. S., Stewart E. M., Gnann C., McCarthy F., Cabrera J. P., Brunetti R. M., Chhun B. B., Dingle G., Hein M. Y., Huang B., Mehta S. B., Weissman J. S., Gómez-Sjöberg R., Itzhak D. N., Royer L. A., Mann M., Leonetti M. D., OpenCell: proteome-scale endogenous tagging enables the cartography of human cellular organization. Science 375, eabi6983 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guo W., Chen Y., Zhou X., Kar A., Ray P., Chen X., Rao E. J., Yang M., Ye H., Zhu L., Liu J., Xu M., Yang Y., Wang C., Zhang D., Bigio E. H., Mesulam M., Shen Y., Xu Q., Fushimi K., Wu J. Y., An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat Struct Mol Biol 18, 822–830 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yin P., Bai D., Deng F., Zhang C., Jia Q., Zhu L., Chen L., Li B., Guo X., Ye J., Tan Z., Wang L., Li S., Li X.-J., SQSTM1-mediated clearance of cytoplasmic mutant TARDBP/TDP-43 in the monkey brain. Autophagy 18, 1955–1968 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Renton A. E., Chiò A., Traynor B. J., State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17, 17–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maruyama H., Morino H., Ito H., Izumi Y., Kato H., Watanabe Y., Kinoshita Y., Kamada M., Nodera H., Suzuki H., Komure O., Matsuura S., Kobatake K., Morimoto N., Abe K., Suzuki N., Aoki M., Kawata A., Hirai T., Kato T., Ogasawara K., Hirano A., Takumi T., Kusaka H., Hagiwara K., Kaji R., Kawakami H., Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226 (2010). [DOI] [PubMed] [Google Scholar]
- 83.DeJesus-Hernandez M., Mackenzie I. R., Boeve B. F., Boxer A. L., Baker M., Rutherford N. J., Nicholson A. M., Finch N. A., Flynn H., Adamson J., Kouri N., Wojtas A., Sengdy P., Hsiung G.-Y. R., Karydas A., Seeley W. W., Josephs K. A., Coppola G., Geschwind D. H., Wszolek Z. K., Feldman H., Knopman D. S., Petersen R. C., Miller B. L., Dickson D. W., Boylan K. B., Graff-Radford N. R., Rademakers R., Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Renton A. E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J. R., Schymick J. C., Laaksovirta H., van Swieten J. C., Myllykangas L., Kalimo H., Paetau A., Abramzon Y., Remes A. M., Kaganovich A., Scholz S. W., Duckworth J., Ding J., Harmer D. W., Hernandez D. G., Johnson J. O., Mok K., Ryten M., Trabzuni D., Guerreiro R. J., Orrell R. W., Neal J., Murray A., Pearson J., Jansen I. E., Sondervan D., Seelaar H., Blake D., Young K., Halliwell N., Callister J. B., Toulson G., Richardson A., Gerhard A., Snowden J., Mann D., Neary D., Nalls M. A., Peuralinna T., Jansson L., Isoviita V.-M., Kaivorinne A.-L., Hölttä-Vuori M., Ikonen E., Sulkava R., Benatar M., Wuu J., Chiò A., Restagno G., Borghero G., Sabatelli M., Heckerman D., Rogaeva E., Zinman L., Rothstein J. D., Sendtner M., Drepper C., Eichler E. E., Alkan C., Abdullaev Z., Pack S. D., Dutra A., Pak E., Hardy J., Singleton A., Williams N. M., Heutink P., Pickering-Brown S., Morris H. R., Tienari P. J., Traynor B. J., A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 72, 257–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chua J. P., De Calbiac H., Kabashi E., Barmada S. J., Autophagy and ALS: mechanistic insights and therapeutic implications. Autophagy 18, 254–282 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kumar S., Phaneuf D., Cordeau P., Boutej H., Kriz J., Julien J.-P., Induction of autophagy mitigates TDP-43 pathology and translational repression of neurofilament mRNAs in mouse models of ALS/FTD. Molecular Neurodegeneration 16, 1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Maharana S., Wang J., Papadopoulos D. K., Richter D., Pozniakovsky A., Poser I., Bickle M., Rizk S., Guillén-Boixet J., Franzmann T. M., Jahnel M., Marrone L., Chang Y.-T., Sterneckert J., Tomancak P., Hyman A. A., Alberti S., RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 360, 918–921 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Goedhart J., SuperPlotsOfData—a web app for the transparent display and quantitative comparison of continuous data from different conditions. MBoC 32, 470–474 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Livak K. J., Schmittgen T. D., Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
- 90.Choi H. M. T., Schwarzkopf M., Fornace M. E., Acharya A., Artavanis G., Stegmaier J., Cunha A., Pierce N. A., Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145, dev165753 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.-Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A., Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hagen W. J. H., Wan W., Briggs J. A. G., Implementation of a cryo-electron tomography tilt-scheme optimized for high resolution subtomogram averaging. Journal of Structural Biology 197, 191–198 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mastronarde D. N., Automated electron microscope tomography using robust prediction of specimen movements. Journal of Structural Biology 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 94.Mastronarde D. N., Held S. R., Automated tilt series alignment and tomographic reconstruction in IMOD. Journal of Structural Biology 197, 102–113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Toro-Nahuelpan M., Zagoriy I., Senger F., Blanchoin L., Théry M., Mahamid J., Tailoring cryo-electron microscopy grids by photo-micropatterning for in-cell structural studies. Nat Methods 17, 50–54 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arnold J., Mahamid J., Lucic V., de Marco A., Fernandez J.-J., Laugks T., Mayer T., Hyman A. A., Baumeister W., Plitzko J. M., Site-Specific Cryo-focused Ion Beam Sample Preparation Guided by 3D Correlative Microscopy. Biophysical Journal 110, 860–869 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wolff G., Limpens R. W. A. L., Zheng S., Snijder E. J., Agard D. A., Koster A. J., Bárcena M., Mind the gap: Micro-expansion joints drastically decrease the bending of FIB-milled cryo-lamellae. Journal of Structural Biology 208 (2019). [DOI] [PubMed] [Google Scholar]
- 98.Tegunov D., Cramer P., Real-time cryo-electron microscopy data preprocessing with Warp. Nature Methods, doi: 10.1038/s41592-019-0580-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Castaño-Díez D., Kudryashev M., Arheit M., Stahlberg H., Dynamo: A flexible, user-friendly development tool for subtomogram averaging of cryo-EM data in high-performance computing environments. Journal of Structural Biology 178, 139–151 (2012). [DOI] [PubMed] [Google Scholar]
- 100.Burt A., Gaifas L., Dendooven T., Gutsche I., A flexible framework for multi-particle refinement in cryo-electron tomography. PLOS Biology 19, e3001319 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bharat T. A. M., Scheres S. H. W., Resolving macromolecular structures from electron cryo-Tomography data using subtomogram averaging in RELION. Nature Protocols 11, 2054–2065 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Goddard T. D., Huang C. C., Meng E. C., Pettersen E. F., Couch G. S., Morris J. H., Ferrin T. E., UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Science 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lamm L., Zufferey S., Righetto R. D., Wietrzynski W., Yamauchi K. A., Burt A., Liu Y., Zhang H., Martinez-Sanchez A., Ziegler S., Isensee F., Schnabel J. A., Engel B. D., Peng T., MemBrain v2: an end-to-end tool for the analysis of membranes in cryo-electron tomography. bioRxiv [Preprint] (2024). 10.1101/2024.01.05.574336. [DOI] [Google Scholar]
- 104.Martinez-Sanchez A., Garcia I., Asano S., Lucic V., Fernandez J.-J., Robust membrane detection based on tensor voting for electron tomography. Journal of Structural Biology 186, 49–61 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All correspondence regarding the materials and data presented in this paper should be addressed to sbarmada@med.umich.edu or mosalaga@umich.edu. The in situ subtomogram averaging map of the TDP43 is deposited in Electron Microscopy DataBank with the accession code EMD-45215. All electron microscopy data analysis was performed using existing software.