

Abstract
PURPOSE
Our study aimed to explore real-world treatment scenarios for children and adolescents with neurotrophic tropomyosin receptor kinase (NTRK)–fused tumors, emphasizing access, responses, side effects, and outcomes.
PATIENTS AND METHODS
Pooled clinical data from 17 pediatric cases (11 soft-tissue sarcomas, five brain tumors, and one neuroblastoma) treated with larotrectinib and radiologic images for 14 patients were centrally reviewed. Testing for gene fusions was prompted by poor response to treatment, tumor progression, or aggressiveness.
RESULTS
Six different NTRK fusion subtypes were detected, and various payment sources for testing and medication were reported. Radiologic review revealed objective tumor responses (OR) in 11 of 14 patients: Complete responses: two; partial responses: nine; and stable disease: three cases. Grades 1 or 2 Common Terminology Criteria for Adverse Events adverse effects were reported in five patients. Regarding the entire cohort's clinical information, 15 of 17 patients remain alive (median observation time: 25 months): four with no evidence of disease and 11 alive with disease (10 without progression). One patient developed resistance to the NTRK inhibitor and died from disease progression while another patient died due to an unrelated cause.
CONCLUSION
This real-world study confirms favorable agnostic tumor OR rates to larotrectinib in children with NTRK-fused tumors. Better coordination to facilitate access to medication remains a challenge, particularly in middle-income countries like Brazil.
Study explores real-world treatment of neurotrophic tropomyosin receptor kinase–fused cancer in a Latin American pediatric population.
INTRODUCTION
Molecular evaluation of pediatric tumors has allowed a better understanding of the genetic mechanisms driving childhood cancer.1,2 Gene fusions involving the neurotrophic tropomyosin receptor kinase (NTRK) family are of particular interest.3 The NTRK1/NTRK2/NTRK3 genes present an intracellular domain with tyrosine-dependent kinase activity connected through the transmembrane structure to an extracellular domain made of two immunoglobulin-like high-affinity receptors and three leucine-rich motifs.4 Under normal conditions, it determines the activation of key downstream intracellular pathways, leading to cell survival, proliferation, and differentiation.5,6 Each neurotrophin has specificity for a particular TRK6 (Fig 1A). Numerous described fusions involve the NTRK gene's 3ʹ end (containing the tyrosine kinase domain) juxtaposed with an unrelated gene at 5ʹ end, forming in-frame chimeric receptors, retaining the kinase domain. This results in constitutively activated receptors, disrupting downstream pathways7,8 (Figs 1B and 1C). Additionally, fusion partners often harbor a dimerization/oligomerization domain, such as coiled coil or zinc finger domains, further contributing to receptor activation.9
FIG 1.
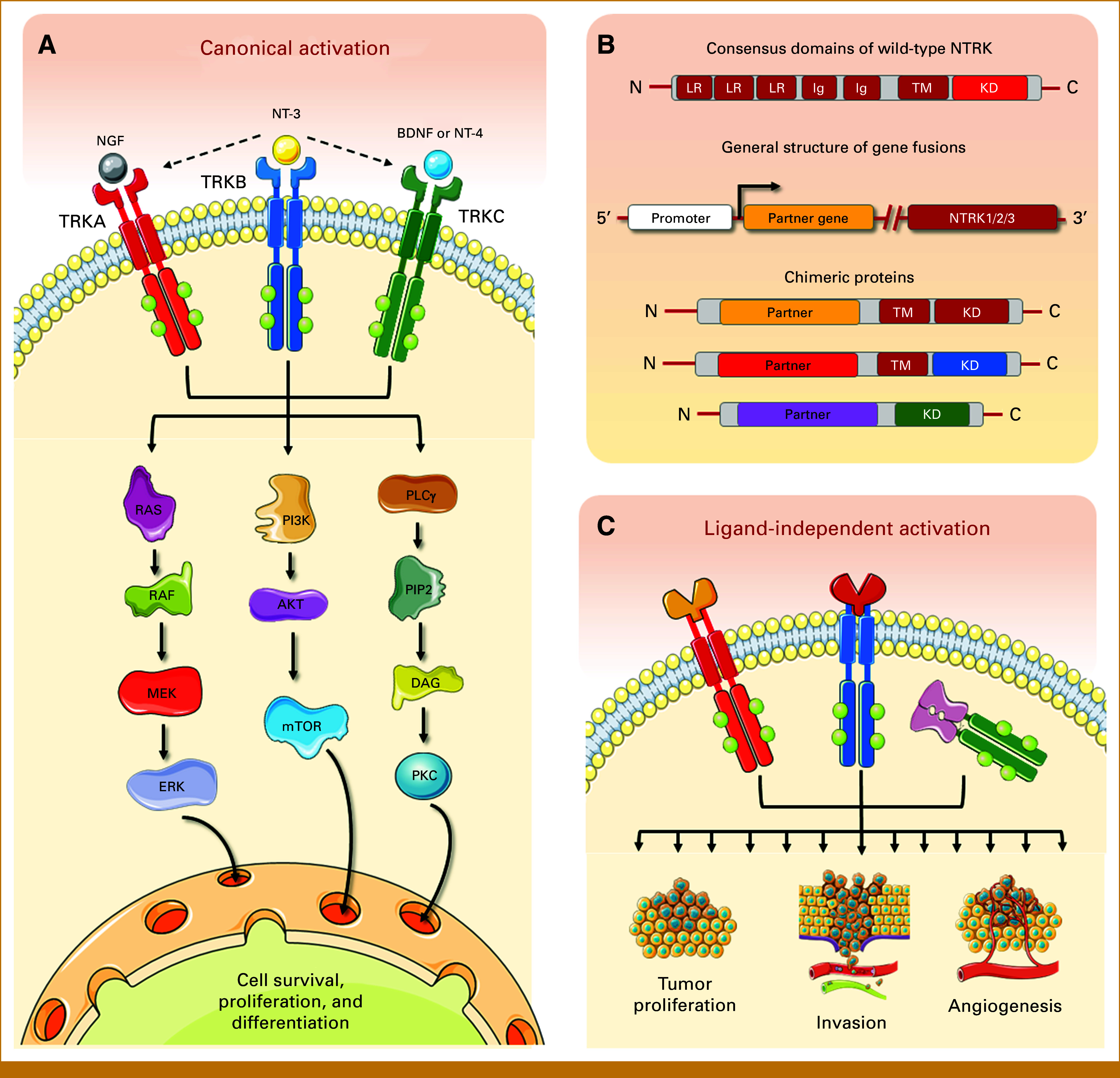
(A) NTRK1, NTRK2, and NTRK3 genes encode tyrosine kinase receptors that under normal conditions respond to the binding of neutrophins (NGF, BDNF, NT-3 or NT-4) and activate key downstream intracellular pathways associated with cell survival, proliferation, and differentiation; (B) the receptors have a consensus structures consisting of an intracellular domain with tyrosine-KD connected through the TM structure to an extracellular domain made of two Ig-like high-affinity receptors and three LR motifs. Gene fusions with a myriad of unrelated partner genes lead to the overexpression of varied chimeric proteins that retain constitutive activation in a ligand-independent manner. In some cases, the TM domain can also be lost, resulting in intracellular kinases (ie, ETV6-NTRK3); (C) dysregulated constitutive activation leads to cell transformation, uncontrolled growth, and tumor progression. The figure was composed with the aid of illustrations from the SMART-servier Medical Art. BDNF, brain-derived neurotrophic factor; Ig, immunoglobulin; KD, kinase domain; LR, leucine-rich; NGF, nerve growth factor; NT-3, neurotrophin-3; NT-4, neurotrophin-4; NTRK, neurotrophic tropomyosin receptor kinase; TM, transmembrane.
CONTEXT
Key Objective
This research, led by the Brazilian Committee of Precision Medicine in Pediatric Oncology-the Brazilian Society of Pediatric Oncology, aims to explore real-world treatment for children and adolescents with neurotrophic tropomyosin receptor kinase (NTRK)–fused tumors.
Knowledge Generated
Seventeen cases with various tumor types and NTRK fusions received larotrectinib. Tumor response and adverse effects were consistent with previous reports. Secondary resistance to treatment was observed in one case. Fifteen of 17 patients remain alive with a median observation time of 25 months. Payment sources varied, with time to access treatment ranging from less than a month to 1 year from initial prescription of the medication.
Relevance
This is the first study to evaluate a pediatric cohort of Latin American patients, in a real-world setting, treated with an NRTK inhibitor. Although our data confirm the good tumor response rates, timely and continuous access to this type of treatment remains a challenge for middle-income countries such as Brazil.
Although the overall frequency of these fusions is low among cancers in general,10,11 they are enriched in different subsets of neoplasms such as infantile fibrosarcomas (IFs), mammary analog secretory carcinoma of the salivary gland, and mesoblastic nephroma.12 Of note, patients with NTRK-fused tumors often experience clinical benefits and high response rates (>75%) when receiving first-generation oral NTRK inhibitors such as larotrectinib or entrectinib.13 Moreover, these responses are observed regardless of tumor histology, which places these agents as tissue-agnostic therapeutic treatment.13
Initial data on the safety and efficacy of larotrectinib described high tumor responses.14 Doz et al15 confirmed the high rates of progression-free survival (56%) and overall survival (85%). Clinical data updates have corroborated the excellent clinical and sustained responses to NTRK inhibitor.16 Similar encouraging results were observed using entrectinib for treating NTRK- or ROS-fused tumors.17 While pivotal clinical trial data are crucial for confirming an agent's efficacy and biosafety profile, the controlled environment of such studies may not fully reflect the diverse clinical variables observed in real-world settings. Additionally, data on the efficacy of NTRK inhibitors are largely underreported in Latin American populations.18 Hence, this multicenter study aims to compile Brazilian cases of children and adolescents diagnosed with NTRK-positive tumors and treated with oral NTRK inhibitors in real-world settings.
PATIENTS AND METHODS
This was a retrospective and descriptive clinical study. Invitation to participate was emailed to all pediatric oncologists in Brazil affiliated with the Brazilian Society of Pediatric Oncology. Pediatric patients (age 0-17 years) who received an NTRK inhibitor from January 1, 2018, to June 1, 2023, were eligible. All study participants, or their legal guardian, provided informed written consent before study enrollment. All patients were treated in tertiary care center. The following inclusion criteria were considered: (1) diagnostic confirmation of an NTRK1, NTRK2, or NTRK3 fusion in tumor, detected by a next-generation sequencing (NGS) method; (2) treatment with an oral NTRK inhibitor for a minimum period of 3 months; and (3) availability of clinical data in medical records regarding access to therapy, response, adverse effects (AEs) and discontinuations, and clinical outcomes. Exclusion criteria were refusal to participate by patient and/or legal guardians or nonsignature of the informed consent form. This multicentric study was approved by the Research Ethics Committee (CAAE 66469323.6.1001.5440).
Clinical and epidemiological data were extracted from electronic health records (EHRs), and radiologic data were exported in anonymized DICOM files. Radiologic examinations were centrally reviewed to verify the degree of response to therapy. The RECIST v1.1 or Response Assessment in Neuro-Oncology v2.0 criteria were used to define response to treatment.19,20 Raw image data were independently analyzed by two experienced radiologists (A.A.C. and V.S.Y.D.). Images (magnetic resonance imaging [MRI] and/or computed tomography [CT]) before treatment initiation with NTRK inhibitor, after at least 3 months of treatment, and at last follow-up were reviewed. The results were categorized as complete response (CR): disappearance of all target lesions; partial response (PR): ≥30% decrease in the sum of the longest diameters (SLD) of all target lesions; progression disease (PD): ≥20% increase in SLD of the target lesions, or the appearance of new unequivocal metastatic lesions, and stable disease (SD) as neither PR nor PD, compared with the examination immediately pretherapy with the NTRK inhibitor.
Data on AEs of NTRK inhibitor therapy were collected. AEs were graded according to the Common Terminology Criteria for Adverse Events (CTCAE), version 5.0.21 The main characteristics of the data set for this descriptive statistics analysis were reported in mean, median, frequency, and variance. This article followed major recommendations of the ESMO Guidance for Reporting Oncology real-World evidence.22
RESULTS
Clinical Data of the Cohort
Twenty-four cases from 14 pediatric oncology centers were identified; seven cases were not eligible for accrual because of diagnosis of an NTRK-fused tumors without receiving treatment with NTRK inhibitor (three patients); legal guardians did not consent to participate (two cases); medical center refused to participate; and not enough time on oral NTRK inhibitor (one case each). The final cohort was composed of 17 patients (11 male and six female), with a median age at diagnosis of 10.9 months (varying from 0.1 to 159.6). Histologic subgroups of tumors harboring NTRK fusions were as follows: 11 soft-tissue sarcomas, five CNS glial/glioneuronal tumors, and one neuroblastoma. Tumors were localized in 16 cases and metastatic in one case (an IF—lung metastasis). Table 1 summarizes the main demographic, clinical, pathologic, and genetic data of the cohort.
TABLE 1.
Summary of the Demographic and Pathologic Data, Type of NTRK-Rearrangement, and Centrally Reviewed Images on 17 Cases of Pediatric Tumors
| Case No. | Sex | Age at Diagnosis, Months | Pathologic Diagnosis | Presence of Metastasis at Diagnosis | Type of Fusion Detected by NGS | Images Centrally Reviewed? |
|---|---|---|---|---|---|---|
| 1 | M | 53.9 | WHO grade IV analogous malignant glioneuronal tumor | No | ETV6-NTRK3 | Yes |
| 2 | F | 13.3 | Infantile fibrosarcoma | No | TPM3-NTRK1 | No |
| 3 | M | 10.9 | Pediatric high-grade glioma | No | TPR-NTRK1 | Yes |
| 4 | F | 8.5 | Grade II diffuse astrocytoma | No | STRN-NTRK2 | Yes |
| 5 | M | 14.3 | Infantile fibrosarcoma | No | ETV6-NTRK3 | Yes |
| 6 | F | 34.4 | Peripheral nerve sheath sarcoma | No | TPM3-NTRK1 | Yes |
| 7 | M | 159.5 | Diffuse glioneuronal tumor with similar oligodendroglial features and nuclear clusters | No | ETV6-NTRK3 | Yes |
| 8 | M | 3.3 | Infantile fibrosarcoma | Yes | TPM3-NTRK1 | No |
| 9 | M | 7.4 | Infantile fibrosarcoma | No | ETV6-NTRK3 | Yes |
| 10 | F | 8.8 | Neuroblastoma | No | SCAPER-NTRK3 | Yes |
| 11 | F | 39.7 | Low-grade soft tissue sarcoma, NOE | No | TPM3-NTRK1 | Yes |
| 12 | F | 8.6 | Infantile fibrosarcoma | No | TPM3-NTRK1 | Yes |
| 13 | M | 0.1 | Infantile fibrosarcoma | No | ETV6-NTRK3 | Yes |
| 14 | M | 1.5 | Infantile fibrosarcoma | No | ETV6-NTRK3 | Yes |
| 15 | M | 40.1 | Mesenchymal spindle cell sarcoma/malignant peripheral nerve sheath tumor | No | TPM3-NTRK1 | Yes |
| 16 | M | 3.2 | Infantile fibrosarcoma | No | TPR-NTRK1 | Yes |
| 17 | M | 90.8 | Low-grade glioma | No | NOTCH2NL-NTRK1 | No |
Abbreviations: F, female; M, male; NGS, next-generation sequencing; NOE, not otherwise specified; NTRK, neurotrophic tropomyosin receptor kinase.
Prior treatment modalities for the cohort included surgery (three cases), chemotherapy (nine cases), and radiotherapy (three cases); in two cases an NTRK inhibitor was offered up-front. Specifically for sarcomas, the majority presented with advanced disease at the time of diagnosis, as classified by the Intergroup Rhabdomyosarcoma Study (IRS) staging system: IRS-I (n = 1), IRS-II (n = 3), IRS-III (n = 5), and IRS-IV (n = 1). The tumor stage was not described in one case. Some of these cases were published as case reports elsewhere.18,23,24
Motivation to Test, Access to NGS Examinations, and Types of NTRK Fusions
Physicians directly caring for patients were surveyed about their reasons for ordering an NGS panel. In seven cases, poor response to initial treatment, tumor progression, or aggressiveness were primary reasons. For six cases, the test was ordered because of tumor type and potential association with NTRK fusions. In two cases, NGS panels intended to aid diagnosis while in one case each, testing was conducted because it was readily available or because of depletion of therapeutic options. NTRK fusions were detected in all 17 cases through DNA- or RNA-based NGS panels. Pharmaceutical industry sponsored testing for eight patients. In four cases, testing was conducted in-house (Archer FusionPlex solid tumor panel), provided complimentary by the treating institution (Barretos Cancer Hospital). In three cases, families covered the molecular test costs, and in two cases, research funding supported testing expenses.
ETV6-NTRK3 was the most frequent transcript observed (four sarcomas; two glioneuronal tumors), followed by TPM3-NTRK1 fusion (four sarcoma cases). Rarer fusions involving NTRK1 and two and three gene partners were also identified: TPR-NTRK1, NOTCH2NL-NTRK1, STRN-NTRK2 (all CNS gliomas), and SCAPER-NTRK3 (neuroblastoma). All physicians affirmed that molecular information aided in patient management. Physicians' perspectives on the timing to access and start medication varied: Nine felt the NTRK inhibitor was timely prescribed and started while seven believed it was delayed. In one case, treatment started sooner than deemed necessary by the attending physician. Appendix Table A1 outlines how NGS results and medication access influenced clinical care, as perceived by pediatric oncologists.
Access to Treatment and Doses of the NTRK Inhibitor
All 17 patients in this cohort were treated with larotrectinib. This choice was not an inclusion restriction for the study, but rather a decision made by the treating physician. The time between the prescription of the NTRK inhibitor medication and the actual start of treatment was categorized as follows: within 30 days of the prescription; between 1 and 3 months; between 3 and 6 months; and between 6 months and 1 year. Five patients started the medication within the prescribed month; seven between 1 and 2 months, four patients between 3 and 6 months, and one patient waited between 6 months and 1 year for the medication. Payment sources included health insurance (six), the patient's institution covering medication costs (five), participation in expanded access programs (two), family-funded payment (one), and miscellaneous payment sources (two cases). Larotrectinib administration included oral solution (16 cases) and hard capsules (one case). All prescribed doses were 100 mg/m2 twice daily; for patients with a body surface area of at least 1 m2, the dose was also 100 mg twice daily.
Treatment Evaluation and Outcome
Fourteen cases had images available for central radiologic review. The concordance between the two independent radiologists on response categories (PD, SD, PR, or CR) for all cases was 100%. Objective responses (CR + PR) were observed in 11 of 14 patients. The best tumor responses after treatment with larotrectinib alone were CR (two cases), PR (nine cases), and SD (three cases). One additional case (6) achieved CR with additional surgical resection and radiotherapy following the NTRK inhibitor. Figure 2A shows different treatment schedules received for each patient throughout a timeline, and Figure 2B shows the best tumor response compared with baseline, tumor type, NTRK fusion type, last follow-up outcome, and observation time since the initiation of larotrectinib in months.
FIG 2.
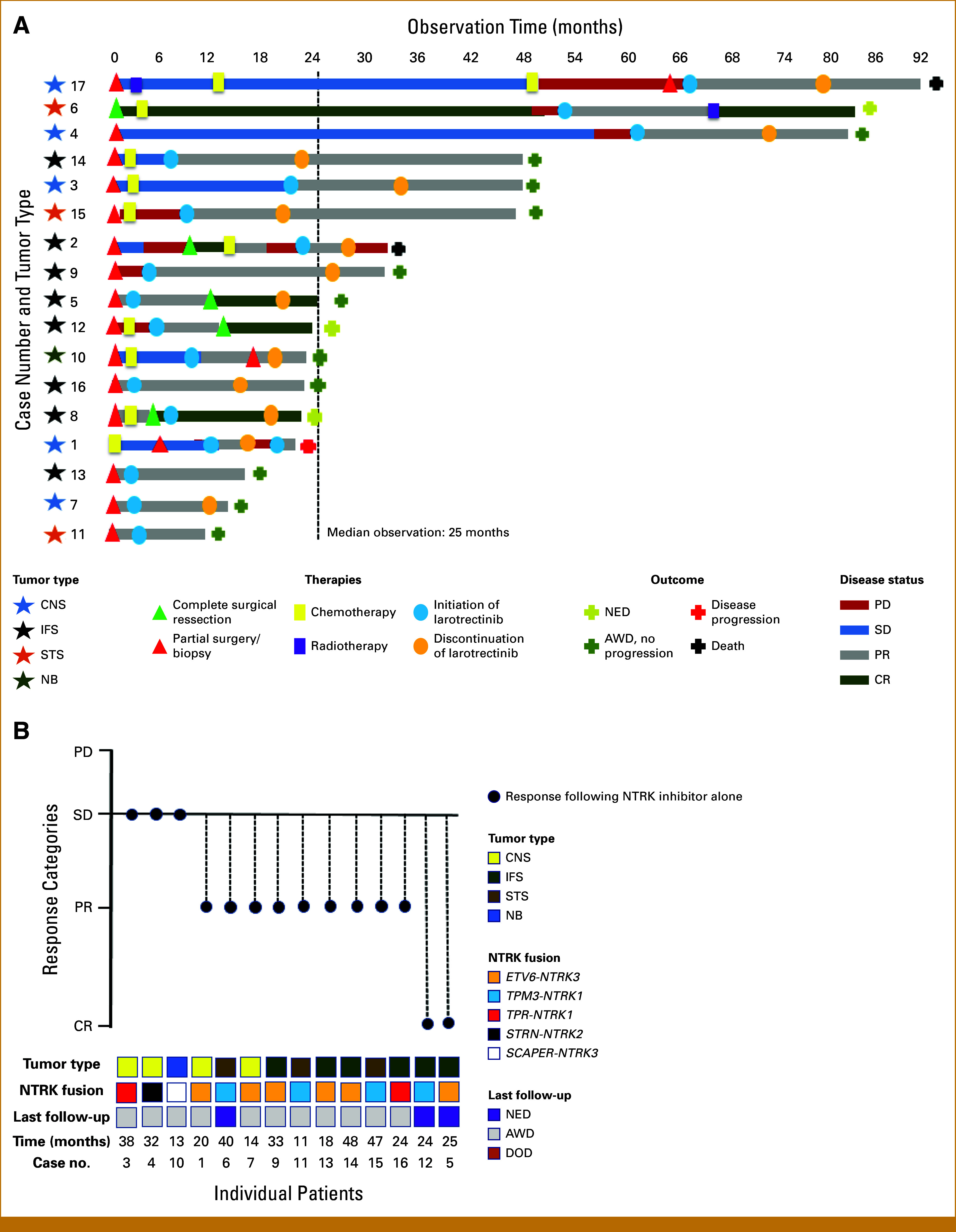
(A) Swimmer plot demonstrating different treatment modalities (surgery; chemotherapy, radiotherapy) and treatment with larotrectinib, along with best tumor response achieved by each treatment approach. (B) Best response to the NTRK inhibitor (larotrectinib) for each individual patient that had their tumor images centrally and independently reviewed. The response is associated with tumor type, NTRK fusion subtype, patients' clinical status at the last follow-up, and the observation time since the start of therapy with larotrectinib in months. CR, complete response; IFS, infantile fibrosarcoma; NBL, neuroblastoma; NTRK, neurotrophic tropomyosin receptor kinase; PD, progressive disease; PR, partial response; SD, stable disease; STS, other soft tissue sarcomas.
Fifteen of 17 patients remain alive, with a mean observation time of 25 months (11-48 months): four with no evidence of disease, 11 patients are alive with disease, in PR (10 without progression). Although images were not available for central review, case 2 experienced tumor progression after an initial period of tumor response (PR) after treatment with larotrectinib. This patient died of disease progression. One additional patient (17) died due to complications not related to the tumor (pneumonia), in a state of PR.
One case deserves special attention regarding response to therapy. An 8-month-old girl was diagnosed with neuroblastoma (NB) (10), with a primary tumor in the mediastinum and metastasis to the skin and bones by metaiodobenzylguanidine (MIBG) scintigraphy (Fig 3); MYCN status of the tumor was inconclusive. The patient was initially treated with conventional chemotherapy for high-risk NB. After 1 year of treatment, although metastatic lesions resolved at MIBG scintigraphy, she persisted with the mediastinal uptaking lesion. A second-look surgery (partial resection) was performed and confirmed a residual poorly differentiated NB specimen at mediastinum. NGS analysis showed a SCAPER-NTRK3 fusion. At this point, the patient started to receive larotrectinib. After 3 months on the medication, a new re-evaluation by MRI showed SD. A new biopsy was performed and revealed a completely mature NB tissue (ganglioneuroma). The patient continued to receive larotrectinib for 13 months, and new MIBG scintigraphy did not show uptake in the primary lesion or metastatic sites.
FIG 3.
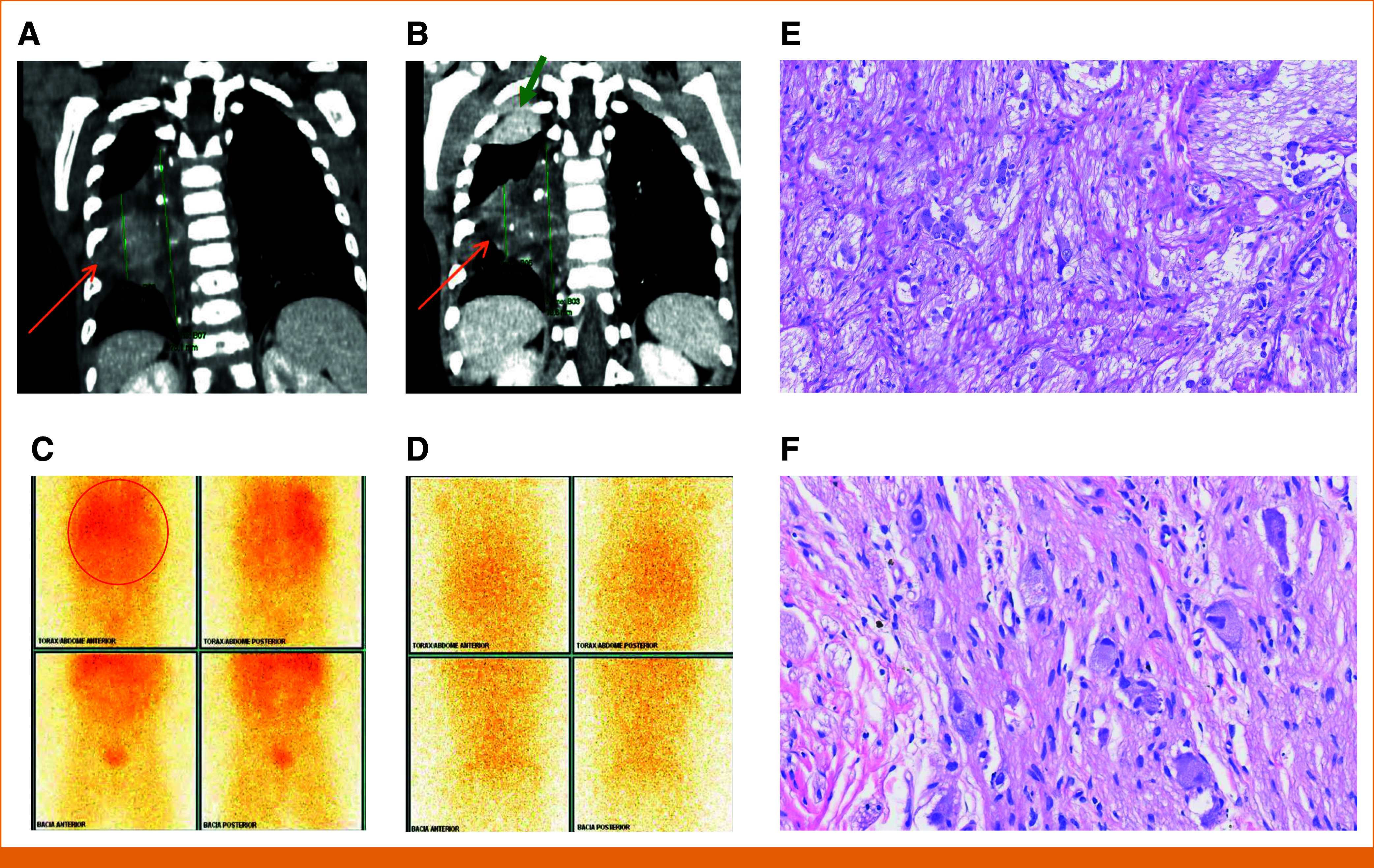
The chest CT scan (soft tissue window, coronal reformatting) shows a heterogeneous right paravertebral mass in the mid thoracic segment, with (A) hypoattenuating areas and avid uptake on the (C) PET-MIBG study. (B) After treatment, there was a slight reduction in size (being characterized as a stable disease by RECIST) and the onset of small foci of calcification (orange arrows) and (D) no more PET uptake. (B) Also, an atelectatic opacity was observed at the apex of the right lung, probably related to decubitus (green arrow). The histopathological study of specimens before and after larotrectinib treatment shows (E) a poorly differentiated NB specimen and (F) a completely mature NB tissue compatible with ganglioneuroma. CT, computed tomography; MIBG, metaiodobenzylguanidine; NB, XXX; PET, positron emission tomography.
Second-Look Surgeries
Second-look surgeries after the use of NTRK inhibitors varied at the physician's discretion, and 10 patients did not undergo a new surgical procedure. Patients 3, 4, and 7 (CNS tumors) were not reoperated because of difficult tumor locations in eloquent areas (frontal hemispheric, thalamus, and pineal, respectively) of the brain. Interestingly, patients 4 and 7 remain alive with no evidence of progression for 12 and 7 months, respectively, after larotrectinib discontinuation, with only stable minor alterations on MRI. Patient 3 achieved CR with larotrectinib alone. Regarding IFs cases: 5 and 8 achieved CR with larotrectinib, with no need for further surgical procedures; for cases 9 and 16, second-look surgery was not indicated because of tumor location (face, with deep vascular involvement, and skull, respectively)—both cases remain in PR without tumor progression after larotrectinib discontinuation (7 and 10 months, respectively); case 12 had second-look surgery with 100% necrosis of tumor specimens after larotrectinib, and cases 13 and 14 are waiting for second-look surgical procedures. For soft tissue sarcomas (STS), case 6 experienced sequelae related to primary surgery, and radiation therapy was offered for local tumor control; the tumor involved the brachial plexus for case 15, which contraindicated second-look surgery; case 11 achieved PR and awaits a second-look surgical procedure. Specifically for the NB case, only partial resection was achieved at the second-look surgery; this patient discontinued larotrectinib and remains in PR without progression. Information on second-look surgery was unavailable for patients 15 and 17 (CNS and STS, respectively).
Adverse Events and Treatment Interruptions
Five patients experienced AEs with larotrectinib therapy. Reported side effects according to CTCAE included hepatic toxicity (grade 2) in two cases, weight gain (grade 2) in two cases, and fatigue (grade 2) and somnolence (grade 1) in one case. These side effects did not necessitate drug interruption or dose reduction. Of the 15 surviving patients in this cohort, four were still using an NTRK inhibitor (cases 6, 11, 12, and 13) when the data for this study were compiled. In two cases, the medication was discontinued after the patient achieved complete remission (5 and 10). In the remaining nine patients, the discontinuation of larotrectinib occurred for various reasons: in six cases, discontinuation occurred because of the decision of the medical team responsible for the patient's care and in two cases because of difficulties in obtaining the medication continuously and regularly by the family; in one case, the reason for discontinuation was not reported. In one of these cases (1), the medication was resumed after a brief suspension period, and an excellent clinical response was observed again (1). The median duration time of medication use among patients who discontinued larotrectinib was 12 months (ranging from 11 to 25 months).
DISCUSSION
In this retrospective series, we explored clinical aspects regarding treatment of NTRK-fused pediatric cancer within a real-world Brazilian experience. The most common types of tumors observed in this cohort are in accordance with those reported in this setting,25 except for a neuroblastoma case, which, to our knowledge, has not been described to date. Regarding fusion types, ETV6-NTRK3 and TPM3-NTRK1 were the most frequent fusions (10/17 cases) detected. Of note, our cohort identified some infrequent translocations involving rarer NTRK1/3 partners. Two cases of TPR-NTRK1 fusions (high-grade glioma—3 and IF—16) were observed. The TPR-NTRK1 fusion was described in only 0.04% of all samples tested by the American Association for Cancer Research GENIE project26; this fusion was previously described in sarcoma cases, but not in brain tumors. The NOTCH2NL-NTRK1 (17) fusion was reported twice in the literature: one case of a patient with lung adenocarcinoma with neuroendocrine differentiation where this transcript emerged after osimertinib treatment27 and one case of metastatic squamous non–small cell lung cancer that was primarily resistant to larotrectinib.28 Both cases harboring NOTCH2NL-NTRK1 fusion were observed in adults, and this fusion has not been previously reported neither in children nor in brain cancer. The STRN-NTRK2 (4) was previously reported in pediatric sarcoma,29 malignant glioneuronal tumor of the brain,30 papillary thyroid cancer in adults,31 and lung adenocarcinoma.32 Finally, the SCAPER-NTRK3 (neuroblastoma—10) was previously described only once, in a very rare case of epithelioid melanocytoma in a child.33
The largest populational study evaluated 295,676 cases for NTRK1/2/3 fusion positivity and observed a prevalence of 1.34% and 0.28% of NTRK-fused tumors in the pediatric and adult population, respectively.11 NTRK fusion frequency varies across tumors, grouped as follows: (1) rare tumors where NTRK fusions are defining features (frequencies often >90%); (2) relatively common tumors with intermediate NTRK fusion rates (5%-25%); and (3) tumors where NTRK fusions are rare (<5%, usually <1%).34 A recent tumor genomic profiling of approximately 1,200 pediatric patients found NTRK fusions in 2.22% of all tumors and 3.08% of solid tumors. These fusions were observed more often in childhood tumors than in adult tumors, indicating a broader panel of fusion partners and a wider range of pediatric tumors than previously recognized.25 Moreover, fusions presented a certain tissue tropism: pediatric thyroid tumors predominantly involved NTRK1 and NTRK3, whereas CNS tumors primarily have fusions associated with NTRK2.25,35,36 In this regard, as more tumors are being studied for fusions, higher the number of partners and different breakpoints. Indeed, the webserver for fusion integrative analysis FPIA37 shows that for each NTRK gene, considering 33 cancer types from The Cancer Genome Atlas Program data, breaks occur at different exonic, splicing (within 2-bp of a splicing junction), non-coding RNA (overlaps a transcript without coding annotation in the gene definition), intronic, and intergenic locations (Fig 4A). The literature revealed 127 different fusion partners5,25,34,35,38,39 from which 90 (70.9%) were associated with adult tumor histologies, 22 (17.3%) were restricted to pediatric tumors, and 15 (11.8%) are shared by both settings (Fig 4B). When examining the recombinational background of each fusion partner, 78% of them translocate with all NTRK members in adult tumors. However, this promiscuity decreases to 44% in pediatric tumors. Additionally, 17.6% of fusion partners are exclusively associated with NTRK1, 20.6% with NTRK2, and 11.8% with NTRK3 (Fig 5).
FIG 4.

(A) The analysis of gene breakpoints for each member of the NTRK family through the FPIA webserver37 showed varied break locations. Data was compiled from samples from 9,966 adult tumors (33 different tumor types) present at the TCGA consortium; (B) in the literature, 127 different translocation partners were found. From these, <30% have been described in pediatric tumors; only 22 are restricted to this setting. Venn diagram and comparing lists were analyzed with the aid of the Venny 2.1 program. NTRK, neurotrophic tropomyosin receptor kinase; TCGA, The Cancer Genome Atlas Program.
FIG 5.

The recombinational background of each fusion partner shows that in adult tumors the majority translocates with the three NTRK members while in the pediatric setting, it is reduced to 44%, with more exclusively associated partners. Chord diagram generated through the SRplot platform. NTRK, neurotrophic tropomyosin receptor kinase.
Response rates observed in our study were similar to the literature.14,15 One case exhibited resistance to therapy. Acquired resistance to NTRK inhibitors, though rare, is well-documented, often linked to specific NTRK gene mutations or off-target alterations.40 Second-generation NTRK inhibitors (repotrectinib, selitrectinib) aim to address tumor resistance, yet resistance to these newer inhibitors may also emerge.41
Several different algorithms have been published to guide NTRK testing both in pediatric and adult populations.42-45 Motivation to test cases in our cohort was related to different clinical aspects such as poor treatment response, tumor progression, and less frequently, tumor type and NTRK fusion prevalence. Time to get access to medication is a problematic issue in the context of rapidly growing pediatric cancer at difficult-to-treat locations (ie, CNS tumors and deep-located mesenchymal cancer). Public governmental initiatives to offer pharmaceutical benefits46 for reimbursement of NTRK testing, and medicine for patients diagnosed with specific tumors at specific ages are being implemented in Australia and New Zealand. These ongoing efforts aim to establish cost-effective approaches and expedite access to testing and medication, although they are still being evaluated. Doses effectively used in our cohort did not differ from the recommended doses of larotrectinib for pediatric population.47
We observed AEs in five of 17 patients (29%). Grades 1 or 2 AEs retrieved were hepatic, weight gain, fatigue, and somnolence. Side effects reported did not lead to drug interruption or dose reduction. Our observations are in accordance with those previously reported in other pediatric populations receiving the same NTRK inhibitor. Pooled data from different phase 1/2 studies on larotrectinib in patients with solid tumors harboring NTRK fusions gathered data on safety information for 260 patients treated with larotrectinib. The authors found AEs to be infrequent, occurring at any grade in at least 15% of patients, and in grades 3 or 4 in 13% and <1%, respectively.16
Our study has some limitations. It is a retrospective, nonrandomized study that retrieved information on EHRs to produce real-world evidence on the outcome of children and adolescents treated with NTRK inhibitors, which can make it difficult to interpret direct treatment effects. There was no specific recommendation for monitoring AEs and serious AEs (SAEs) for this retrospective study, and most physicians used their own institutional recommendations and/or available guidelines, and AEs/SAEs may have been underreported. All patients were treated outside controlled clinical trials and disease assessments and intervals between MRIs or CT were scheduled at the discretion of physicians. This circumstance does not allow us to assess the time to the best response to NTRK inhibitors in this cohort. Moreover, the tumor classification was not centralized, and distinct methodologies were used for tumor histological classification.
In conclusion, this real-world study was the first to assess the outcome of children and adolescents with NTRK-fused tumors treated with larotrectinib in a Latin American population. Our findings confirmed the good tolerability and tumor responses in a tissue agnostic scenario. Yet, aspects related to drug access remain a major challenge, pointing to the need of a strong coordination between public health, medical insurance, and community to ease access to medication. Additionally, many questions and challenges persist regarding the use of NTRK inhibitors in real-world settings. Systematic follow-up and updates on these cases, focusing on clinical outcomes and pharmacoeconomics, would help pediatric oncologists in their decision-making process, particularly in middle-income countries like Brazil.
ACKNOWLEDGMENT
We are grateful to all participants of the Brazilian Committee of Precision Medicine in Pediatric Oncology (BC-PMPO). We also thank the Brazilian Society of Pediatric Oncology for their technical support for this study.
APPENDIX
TABLE A1.
Case Number, Diagnosis, Age at Diagnosis, Pediatric Oncologists' Perceptions of NTRK-Fused Tumor Results, Impact on Clinical Management, Time to Start Larotrectinib, and Current Medication Usage by Patients (data censored on September 1, 2023)
| No. | Pathologic Diagnosis | Age at Diagnosis, Months | How NGS Results and Access to NTRK Inhibitors Helped Pediatric Oncologists to Better Assist Their Patients, According to Their Own Clinical Perception? | Time to Start NTRK Inhibitor Therapy (Too Early/Proper/Late) | Is the Patient Still on Therapy With a NTRK Inhibitor? |
|---|---|---|---|---|---|
| 1 | WHO grade IV analogous malignant glioneuronal tumor | 53.9 | Treatment with conventional chemotherapy was not effective | Proper | No |
| 2 | Infantile fibrosarcoma | 13.3 | The result helped in the therapeutic planning and targeted therapy with good clinical results | Late | No |
| 3 | Pediatric high-grade glioma | 10.9 | The result allowed us to request target drug (larotrectinib); radiation therapy was avoided for this child, with good clinical outcome | Proper | No |
| 4 | Grade II diffuse astrocytoma | 8.5 | The result allowed us to prescribe larotrectinib, with a very good clinical outcome | Late | No |
| 5 | Infantile fibrosarcoma | 14.3 | We were allowed to prescribe NTRK inhibitor and avoid other cytotoxic treatments | Proper | No |
| 6 | Peripheral nerve sheath sarcoma | 34.4 | The patient had already used chemotherapy and radiotherapy, without remission of the disease. Patient already had sequelae related to surgical procedures—no additional treatment options | Late | Yes |
| 7 | Diffuse glioneuronal tumor with similar oligodendroglial features and nuclear clusters | 159.5 | Targeted therapy was able to be offered, after conventional treatments have shown little success | Too early | No |
| 8 | Infantile fibrosarcoma | 3.3 | Patient with poor response to conventional chemotherapy | Proper | No |
| 9 | Infantile fibrosarcoma | 7.4 | Oral treatment, with lesser toxicity, outpatient, making it possible not to use traditional cytotoxic chemotherapy, dispensing the use of long-term catheters | Late | No |
| 10 | Neuroblastoma | 8.8 | Primary posterior mediastinal neuroblastoma with spinal canal invasion. Surgery difficult to perform (high morbidity). Change of treatment (conventional intravenous chemotherapy) to specific oral medication | Proper | No |
| 11 | Low-grade soft tissue sarcoma, NOE | 39.7 | Patient did not respond to conventional chemotherapy and showed a lot of toxicity; treatment with larotrectinib offered excellent quality of life and partial clinical response on imaging | Proper | Yes |
| 12 | Infantile fibrosarcoma | 8.6 | The tumor did not shrink with any conventional chemotherapy as per the 2005 EpSSG NRTS Protocol for localized non-rabdo sarcomas. In the second cycle of more intensive chemotherapy, the patient had an anaphylactic reaction | Proper | No |
| 13 | Infantile fibrosarcoma | 0.1 | Microscopic margins compromised by the tumor (difficult tumor location at abdomen); decision about offering neoadjuvant therapy (in infants) with available target therapy (as opposed to offering chemotherapy) was preferred | Proper | Yes |
| 14 | Infantile fibrosarcoma | 1.5 | Large tumor, inoperable because of its location on the face, poorly responsive to chemotherapy | Late | No |
| 15 | Mesenchymal spindle cell sarcoma/malignant peripheral nerve sheath tumor | 40.1 | Patient initially received intensive chemotherapy, without response and with many adverse effects | Late | Yes |
| 16 | Infantile fibrosarcoma | 3.2 | Helped us to manage a tumor weighting 477 g in child weighing 4.8 kg; surgical margins compromised; therapy used as adjunctive therapy | Proper | No |
| 17 | Low-grade glioma | 90.8 | Exhaustion of therapeutic resources; the result allowed us to offer novel therapy to this case | Late | No |
NOTE. Observations: These are qualitative and descriptive data about the personal impression of the physician responsible for the patient's treatment regarding the time window between the prescription of the NTRK inhibitor medication and its actual availability for patient use (early, proper, or late). Data from column 4 were free translations of reports from the attending physicians regarding their own perception of how NGS results and access to the NTRK inhibitor may have affected the health care of their patients.
Abbreviations: NGS, next-generation sequencing; NOE, not otherwise specified; NTRK, neurotrophic tropomyosin receptor kinase.
Footnotes
C.S.C.V. and M.S.B. contributed equally to this work.
AUTHOR CONTRIBUTIONS
Conception and design: Carolina Sgarioni Camargo Vince, Maria Sol Brassesco, Bruna Minniti Mançano, Cassia Silvestre Mariano, Carla Donato Macedo, Luciana Nunes Silva, Flavia Delgado Martins, Mara Albonei Dudeque Pianovski, Paulo Vidal Campregher, Neviçolino Pereira Carvalho Filho, Elvis Terci Valera
Administrative support: Mara Albonei Dudeque Pianovski, Elvis Terci Valera
Provision of study materials or patients: Bruna Minniti Mançano, Lauro Jose Gregianin, Edna Kakitani Carbone, Roberta Zeppini Menezes da Silva, Cassia Silvestre Mariano, Juliana França da Mata, Marcelo Oliveira Silva, Eliana Maria Monteiro Caran, Gildene Alves da Costa, Tereza Cristina Esteves, Luciana Nunes Silva, Lilian Maria Cristófani, Marcelo Milone Silva, Rui Manuel Reis, Elvis Terci Valera
Collection and assembly of data: Carolina Sgarioni Camargo Vince, Bruna Minniti Mançano, Lauro Jose Gregianin, Edna Kakitani Carbone, Adham do Amaral e Castro, Viviane Sayuri Yamachira Dwan, Roberta Zeppini Menezes da Silva, Juliana França da Mata, Marcelo Oliveira Silva, Eliana Maria Monteiro Caran, Carla Donato Macedo, Gildene Alves da Costa, Tereza Cristina Esteves, Luciana Nunes Silva, Sima Esther Ferman, Lilian Maria Cristófani, Vicente Odone-Filho, Marcelo Milone Silva, Rui Manuel Reis, Mayara Satsuki Kunii, Karla Emilia de Sá Rodrigues, Elvis Terci Valera
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Edna Kakitani Carbone
Speakers' Bureau: Recordati
Research Funding: Amgen—Local PI, Pfizer
Juliana França da Mata
Travel, Accommodations, Expenses: Alexion Pharmaceuticals
Luciana Nunes Silva
Honoraria: Recordati Rare Diseases
Travel, Accommodations, Expenses: Alexion Pharmaceuticals
Mara Albonei Dudeque Pianovski
Consulting or Advisory Role: Zodiac Pharma
Paulo Vidal Campregher
Stock and Other Ownership Interests: Pfizer
Honoraria: Astellas Pharma, Pfizer, SERVIER, Janssen, AstraZeneca
Mayara Satsuki Kunii
Expert Testimony: Bayer
Neviçolino Pereira Carvalho Filho
Speakers' Bureau: Recordati
Travel, Accommodations, Expenses: RECORDATI
Elvis Terci Valera
Consulting or Advisory Role: Pfizer, Bayer
No other potential conflicts of interest were reported.
REFERENCES
- 1.Flaherty KT, Gray RJ, Chen AP, et al. : Molecular landscape and actionable alterations in a genomically guided cancer clinical trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH). J Clin Oncol 38:3883-3894, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rahal Z, Abdulhai F, Kadara H, et al. : Genomics of adult and pediatric solid tumors. Am J Cancer Res 8:1356-1386, 2018 [PMC free article] [PubMed] [Google Scholar]
- 3.Zito Marino F, Pagliuca F, Ronchi A, et al. : NTRK fusions, from the diagnostic algorithm to innovative treatment in the era of precision medicine. Int J Mol Sci 21:3718, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lemmon MA, Schlessinger J: Cell signaling by receptor tyrosine kinases. Cell 141:1117-1134, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aepala MR, Peiris MN, Jiang Z, et al. : Nefarious NTRK oncogenic fusions in pediatric sarcomas: Too many to Trk. Cytokine Growth Factor Rev 68:93-106, 2022 [DOI] [PubMed] [Google Scholar]
- 6.Amatu A, Sartore-Bianchi A, Bencardino K, et al. : Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann Oncol 30:viii5-viii15, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saliba J, Church AJ, Rao S, et al. : Standardized evidence-based approach for assessment of oncogenic and clinical significance of NTRK fusions. Cancer Genet 264–265:50-59, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manea CA, Badiu DC, Ploscaru IC, et al. : A review of NTRK fusions in cancer. Ann Med Surg (Lond) 79:103893, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hechtman JF: NTRK insights: Best practices for pathologists. Mod Pathol 35:298-305, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cocco E, Scaltriti M, Drilon A: NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol 15:731-747, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Westphalen CB, Krebs MG, Le Tourneau C, et al. : Genomic context of NTRK1/2/3 fusion-positive tumours from a large real-world population. NPJ Precis Oncol 5:69, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albert CM, Davis JL, Federman N, et al. : TRK fusion cancers in children: A clinical review and recommendations for screening. J Clin Oncol 37:513-524, 2019 [DOI] [PubMed] [Google Scholar]
- 13.Kummar S, Lassen UN: TRK inhibition: A new tumor-agnostic treatment strategy. Target Oncol 13:545-556, 2018 [DOI] [PubMed] [Google Scholar]
- 14.Drilon A, Laetsch TW, Kummar S, et al. : Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med 378:731-739, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doz F, van Tilburg CM, Geoerger B, et al. : Efficacy and safety of larotrectinib in TRK fusion-positive primary central nervous system tumors. Neuro Oncol 24:997-1007, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong DS, DuBois SG, Kummar S, et al. : Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol 21:531-540, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desai AV, Robinson GW, Gauvain K, et al. : Entrectinib in children and young adults with solid or primary CNS tumors harboring NTRK, ROS1, or ALK aberrations (STARTRK-NG). Neuro Oncol 24:1776-1789, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mançano BM, Dos Reis MB, Moreno DA, et al. : A unique case report of infant-type hemispheric glioma (gliosarcoma subtype) with TPR-NTRK1 fusion treated with larotrectinib. Pathobiology 89:178-185, 2022 [DOI] [PubMed] [Google Scholar]
- 19.Therasse P, Eisenhauer EA, Verweij J: RECIST revisited: A review of validation studies on tumour assessment. Eur J Cancer 42:1031-1039, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Wen PY, Macdonald DR, Reardon DA, et al. : Updated response assessment criteria for high-grade gliomas: Response assessment in neuro-oncology working group. J Clin Oncol 28:1963-1972, 2010 [DOI] [PubMed] [Google Scholar]
- 21.Freites-Martinez A, Santana N, Arias-Santiago S, et al. : Using the common terminology criteria for adverse events (CTCAE - Version 5.0) to evaluate the severity of adverse events of anticancer therapies. Actas Dermosifiliogr (Engl Ed) 112:90-92, 2021 [DOI] [PubMed] [Google Scholar]
- 22.Castelo-Branco L, Pellat A, Martins-Branco D, et al. : ESMO Guidance for Reporting Oncology real-World evidence (GROW). Ann Oncol 34:1097-1112, 2023 [DOI] [PubMed] [Google Scholar]
- 23.D’Almeida Costa F, Alves de Castro JV, Domenici Kulikowski L, et al. : Pineal region high grade neuroepithelial tumors with NTRK fusions belonging to the novel methylation class “diffuse high grade glioma, IDH-wildtype, subtype E” (HGG_E)—A distinct clinicopathological and molecular presentation. Abstracts of the 20th International Congress of Neuropathology Berlin, Germany, September 13-16, 2023. Brain Pathol 33:e13194, 2023 [Google Scholar]
- 24.Romagnol FT, Caran EM, Romagnol FT, et al. : Targeted therapy using the selective tropomyosin kinase receptor inhibitor larotrectinib in an infant with infantile fibrosarcoma with a TPM3–NTRK1 gene fusion with lung and central nervous system metastases: Case report. Clin Oncol Res 4:1-6, 2021 [Google Scholar]
- 25.Zhao X, Kotch C, Fox E, et al. : NTRK fusions identified in pediatric tumors: The frequency, fusion partners, and clinical outcome. JCO Precis Oncol 1:204-214, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.AACR Project GENIE Consortium. AACR Project GENIE: Powering precision medicine through an international consortium. Cancer Discov 7:818-831, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin G, Liu Y, Li H, et al. : Emergence of NOTCH2-NTRK1 after osimertinib in a patient with lung adenocarcinoma with neuroendocrine differentiation. Clin Lung Cancer 22:e712-e715, 2021 [DOI] [PubMed] [Google Scholar]
- 28.Boulanger MC, Temel JS, Mino-Kenudson M, et al. : Primary resistance to larotrectinib in a patient with squamous NSCLC with subclonal NTRK1 fusion: Case report. JTO Clin Res Rep 4:100501, 2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu LW, Pavlock T, Patterson A, et al. : Durable clinical response to larotrectinib in an adolescent patient with an undifferentiated sarcoma harboring an STRN-NTRK2 fusion. JCO Precis Oncol 2:1-8, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyer J, Birzu C, Bielle F, et al. : Dramatic response of STRN-NTRK-fused malignant glioneuronal tumor to larotrectinib in adult. Neuro Oncol 23:1200-1202, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bastos AU, de Jesus AC, Cerutti JM: ETV6-NTRK3 and STRN-ALK kinase fusions are recurrent events in papillary thyroid cancer of adult population. Eur J Endocrinol 178:83-91, 2018 [DOI] [PubMed] [Google Scholar]
- 32.Solomon JP, Linkov I, Rosado A, et al. : NTRK fusion detection across multiple assays and 33,997 cases: Diagnostic implications and pitfalls. Mod Pathol 33:38-46, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedman BJ, Hernandez S, Fidai C, et al. : A pediatric case of pigmented epithelioid melanocytoma with chromosomal copy number alterations in 15q and 17q and a novel NTRK3-SCAPER gene fusion. J Cutan Pathol 47:70-75, 2020 [DOI] [PubMed] [Google Scholar]
- 34.Conde E, Hernandez S, Alonso M, et al. : Pan-TRK immunohistochemistry to optimize the detection of NTRK fusions: Removing the hay when looking for the needle. Mod Pathol 36:100346, 2023 [DOI] [PubMed] [Google Scholar]
- 35.Casado-Medrano V, O'Neill A, Halada S, et al. : NTRK-fusions in pediatric thyroid tumors: Current state and future perspectives. Cancer Genet 264–265:23-28, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moch H, Cathomas G, Frattini M, et al. : Use of diagnostic algorithms for NTRK fusion-positive tumors in pathology institutes in Switzerland. healthbook TIMES Oncol Hematol 7:14-21, 2021 [Google Scholar]
- 37.Huang L, Zhu H, Luo Z, et al. : FPIA: A database for gene fusion profiling and interactive analyses. Int J Cancer 150:1504-1511, 2022 [DOI] [PubMed] [Google Scholar]
- 38.Amatu A, Sartore-Bianchi A, Siena S: NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 1:e000023, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gatalica Z, Xiu J, Swensen J, et al. : Molecular characterization of cancers with NTRK gene fusions. Mod Pathol 32:147-153, 2019 [DOI] [PubMed] [Google Scholar]
- 40.Harada G, Choudhury NJ, Schram AM, et al. : Mechanisms of acquired resistance to TRK inhibitors. J Clin Oncol 40:3104, 2022 [Google Scholar]
- 41.Cocco E, Lee JE, Kannan S, et al. : TRK xDFG mutations trigger a sensitivity switch from type I to II kinase inhibitors. Cancer Discov 11:126-141, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marchiò C, Scaltriti M, Ladanyi M, et al. : ESMO recommendations on the standard methods to detect NTRK fusions in daily practice and clinical research. Ann Oncol 30:1417-1427, 2019 [DOI] [PubMed] [Google Scholar]
- 43.Lim KHT, Kong HL, Chang KTE, et al. : Recommended testing algorithms for NTRK gene fusions in pediatric and selected adult cancers: Consensus of a Singapore Task Force. Asia Pac J Clin Oncol 18:394-403, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petaccia de Macedo M, Toledo Nascimento EC, Soares FA, et al. : Brazilian expert consensus for NTRK gene fusion testing in solid tumors. Clin Pathol 16:2632010X231197080, 2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu C, Si L, Wang W, et al. : Expert consensus on the diagnosis and treatment of NTRK gene fusion solid tumors in China. Thorac Cancer 13:3084-3097, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Louis WJ, O’Callaghan CJ, Krum H, et al. : Pharmaceutical benefits scheme (PBS). Med J Aust 160:306-307, 1994 [PubMed] [Google Scholar]
- 47.Laetsch TW, DuBois SG, Mascarenhas L, et al. : Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: Phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol 19:705-714, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]