

Abstract
PURPOSE
Fluoropyrimidine-related toxicity and mortality risk increases significantly in patients carrying certain DPYD genetic variants with standard dosing. We implemented DPYD genotyping at a multisite cancer center and evaluated its impact on dosing, toxicity, and hospitalization.
METHODS
In this prospective observational study, patients receiving (reactive) or planning to receive (pretreatment) fluoropyrimidine-based chemotherapy were genotyped for five DPYD variants as standard practice per provider discretion. The primary end point was the proportion of variant carriers receiving fluoropyrimidine modifications. Secondary end points included mean relative dose intensity, fluoropyrimidine-related grade 3+ toxicities, and hospitalizations. Fisher's exact test compared toxicity and hospitalization rates between pretreatment carriers, reactive carriers, and wild-type patients. Univariable and multivariable logistic regression identified factors associated with toxicity and hospitalization risk. Kaplan-Meier methods estimated time to event of first grade 3+ toxicity and hospitalization.
RESULTS
Of the 757 patients who received DPYD genotyping (median age 63, 54% male, 74% White, 19% Black, 88% GI malignancy), 45 (5.9%) were heterozygous carriers. Fluoropyrimidine was modified in 93% of carriers who started treatment. In 442 patients with 3-month follow-up, 64%, 31%, and 30% of reactive carriers, pretreatment carriers, and wild-type patients had grade 3+ toxicity, respectively (P = .085); 64%, 25%, and 13% were hospitalized (P < .001). Reactive carriers had 10-fold higher odds of hospitalization compared with wild-type patients (P = .001), whereas no significant difference was noted between pretreatment carriers and wild-type patients. Time-to-event of toxicity and hospitalization were significantly different between genotype groups (P < .001), with reactive carriers having the earliest onset and highest incidence.
CONCLUSION
DPYD genotyping prompted fluoropyrimidine modifications in most carriers. Pretreatment testing reduced toxicities and hospitalizations compared with reactive testing, thus normalizing the risk to that of wild-type patients, and should be considered standard practice.
Real-world evidence showed DPYD genotyping to guide fluoropyrimidine dosing was feasible across a multisite cancer center. Pretreatment testing and genotype-guided dosing reduced severe toxicities and hospitalizations. #DPYD #Pharmacogenomics #PGx
INTRODUCTION
Dose-limiting toxicities occur in about one third of patients treated with fluoropyrimidines, including 5-fluorouracil (5-FU) and capecitabine—agents widely used in GI cancers.1-3 Toxicities, including myelosuppression, diarrhea, mucositis, and hand-foot syndrome, can delay treatment, cause hospitalization, and, although rare, lead to death in approximately 0.1%-0.5% of patients.4 Fluoropyrimidines are catabolized primarily by dihydropyrimidine dehydrogenase (DPD) to inactive metabolites.5 Genetic variations in its encoding gene, DPYD, can reduce DPD activity and increase fluoropyrimidine exposure.6 Meta-analyses identified up to four- and 25-fold higher risk of treatment-related toxicity and mortality, respectively, in DPYD variant carriers receiving the standard dose fluoropyrimidine.7,8
CONTEXT
Key Objective
What is the impact of integrating in-house DPYD genotyping at a multisite cancer center on fluoropyrimidine dosing, toxicity, and hospitalizations?
Knowledge Generated
In-house DPYD genotyping resulted in fluoropyrimidine dose modifications in almost all carriers. Pretreatment genotyping and upfront dose adjustments significantly reduced severe toxicities and hospitalizations compared with patients who received reactive testing.
Relevance
These findings support widespread adoption of pretreatment DPYD genotyping and upfront dose adjustments to reduce fluoropyrimidine-related severe toxicities and hospitalizations in variant carriers.
Clinically actionable DPYD variants with moderate-to-strong evidence per the Clinical Pharmacogenetics Implementation Consortium (CPIC) include c.1905+1G>A (*2A), c.2846A>T, c.1679T>G (*13), c.1129-5923C>G, c.1236G>A (HapB3, in linkage disequilibrium with c.1129-5923C>G), and c.557A>G.9,10 The combined carrier frequency is approximately 5%-7% of the general population, as most commonly studied in Europeans, and c.557A>G is mostly found in individuals of African ancestry.9 Upfront fluoropyrimidine dose reductions in heterozygous carriers have demonstrated reduced severe toxicity rates and equivalent drug exposure compared with those in wild-type patients receiving standard dose without compromising treatment response or overall survival (OS).11-13
CPIC guidelines recommend a 50% upfront fluoropyrimidine dose reduction in patients harboring one no-function variant or one or two decreased-function variants (DPYD intermediate metabolizers) and avoiding fluoropyrimidine in patients carrying two no-function or one no-function and one decreased-function variant (DPYD poor metabolizers).9 The US Food and Drug Administration (FDA) labeling for 5-FU and capecitabine states an increased risk of severe or fatal adverse reactions in patients with impaired DPD activity,1,2 while capecitabine label was updated to consider DPYD testing.2
Nonetheless, DPYD genotyping is not routinely performed in the United States, partly due to lack of recommendations from professional societies and FDA, lack of provider awareness, limited access to comprehensive testing with rapid turnaround time, and variable reimbursement.14 On the basis of the robust literature and CPIC guidelines, we developed a clinical DPYD genotyping test available for all patients receiving or anticipated to receive a fluoropyrimidine per provider discretion.15 Herein, we evaluated the impact of an in-house DPYD genotyping test on fluoropyrimidine dosing, toxicity, and hospitalization.
METHODS
Study Design and Patient Population
We conducted a prospective observational cohort study of patients receiving DPYD testing at a multisite community-academic hybrid cancer center. Implementation of in-house DPYD genotyping test was previously described.15 DPYD testing was available, though not required, for patients receiving or planning to receive fluoropyrimidine-based chemotherapy. All procedures, treatments, and dose modifications were considered standard of care. The consent-exempt study protocol for data collection was approved by the Atrium Health Institutional Review Board.
The eligible population included patients receiving or planning to receive fluoropyrimidine-based chemotherapy from March 2020 through May 2023. The evaluable population consisted of two cohorts: (1) Implementation Cohort: eligible patients who underwent DPYD genotyping from March 2020 through May 2023 who were evaluated for demographics and implementation metrics, and (2) Outcomes Cohort: eligible patients who underwent DPYD genotyping and initiated fluoropyrimidine-based chemotherapy from March 2020 through December 2022 who were followed for 3 months and evaluated for dose intensity, toxicities, and hospitalizations (Fig 1).
FIG 1.
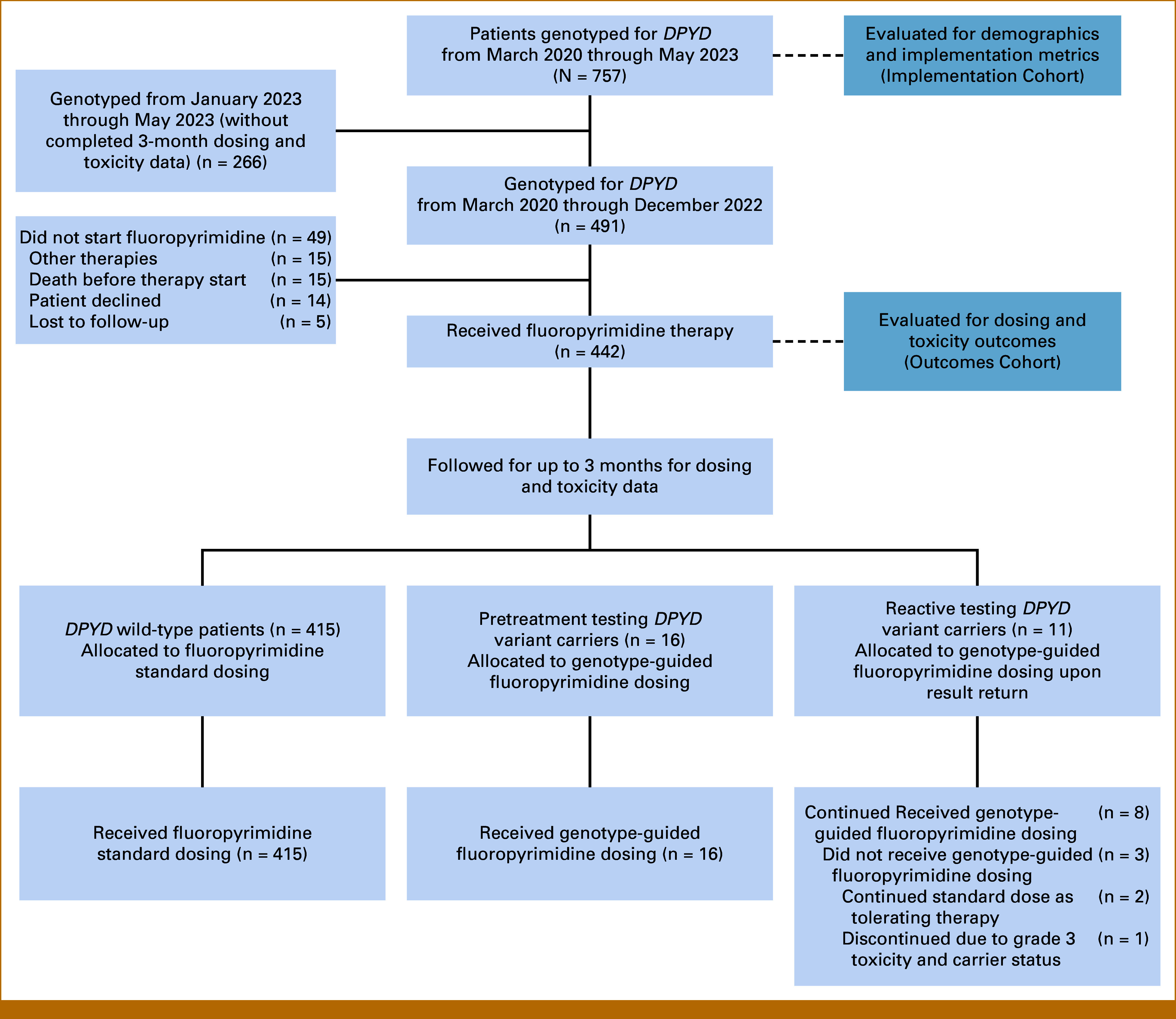
CONSORT diagram. A total of 757 patients who underwent DPYD genotyping from March 2020 through May 2023 were evaluated for demographics and implementation metrics (Implementation Cohort). Of these, 442 patients who underwent DPYD genotyping and were initiated on a fluoropyrimidine from March 2020 through December 2022 were followed for 3 months and evaluated for dosing and toxicity outcomes (Outcomes Cohort). Of these, 415 DPYD wild-type patients received standard fluoropyrimidine dosing, and 16 pretreatment testing carriers received genotype-guided dosing. Of the 11 reactive testing carriers, eight received genotype-guided dosing upon result return, two continued standard dose as tolerating therapy, and one had therapy discontinued due to carrier status and grade 3 toxicities. Reasons for nonevaluable patients are described in the diagram.
Genotyping and Return of Results
Two buccal swabs from each patient were sent to an in-house Clinical Laboratory Improvement Amendment–certified molecular biology and genomics laboratory. DNA was extracted, and TaqMan Drug Metabolism Genotyping Assays were used to detect single nucleotide polymorphisms in the DPYD gene associated with *2A (c.1905+1G>A, rs3918290), c.1679T>G (rs55886062), c.1236G>A (rs56038477, proxy for c.1129-5923C>G), c.2846A>T (rs67376798), and c.557A>G (rs115232898) alleles. Laboratory methods are described in detail in the Data Supplement. Genotyping data were analyzed with TaqMan Genotyper Software. Genotyping results were mapped to star allele nomenclature using TaqMan AlleleTyper Software and translation tables from CPIC.9
Genotype-to-phenotype translations and dosing recommendations followed the CPIC DPYD guideline.9 Discrete results were uploaded to the electronic medical record (EMR) to trigger clinical decision support, including (1) pretest alerts to prompt DPYD test ordering at fluoropyrimidine order entry for patients without DPYD results in the EMR, and (2) post-test alerts to provide dose recommendations for variant carriers. Dosing recommendations were also emailed to the oncology team for identified carriers.
Data Elements
Data were collected prospectively and manually from the EMR, including demographics; cancer diagnosis and staging; Eastern Cooperative Oncology Group performance status; fluoropyrimidine-based chemotherapy regimen at the time of testing; dates of sample collection, receipt, and result reporting; DPYD results; fluoropyrimidine dose; fluoropyrimidine-related toxicities; hospitalizations; treatment delays and discontinuations; and date(s) of events.
Relative dose intensity (RDI) was calculated for the first fluoropyrimidine cycle as the administered dose over the actual time to complete the cycle divided by the standard dose (mg/m2) over the standard time to complete the cycle for the indication and treatment regimen applicable for the patient.16
Adverse events collected from progress notes and laboratory results (eg, absolute neutrophil count) were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events version 5.0. Adverse events were categorized by the study team as hematological, GI, hand-foot syndrome, or other. Adverse events were assessed by the study team and/or oncology providers for causality as unrelated, unlikely, possible, probable, or definite as related to fluoropyrimidine. Those deemed possible, probable, or definite were included as fluoropyrimidine-related adverse events. Hospitalizations, treatment delays, and discontinuations related to fluoropyrimidine toxicities were also collected.
Statistical Analysis
Descriptive statistics were used to summarize demographics and implementation metrics, including test turnaround time, proportion of pretreatment and reactive testing, and fluoropyrimidine modifications in DPYD carriers. Analysis of variance techniques and Fisher's exact tests were used to compare baseline factors between genotype groups: wild-type patients, reactive testing carriers, and pretreatment testing carriers. Pretreatment testing was defined as specimen collection before treatment start date, whereas reactive testing was defined as specimen collection on or after treatment start date.
The primary end point was the proportion of DPYD carriers who received genotype-guided treatment modifications. Secondary end points included the mean RDI of the first cycle. Additional secondary end points included the proportion of patients with fluoropyrimidine-related grade 3+ toxicities, hospitalizations, and treatment delays and discontinuations, compared between genotype groups using Fisher's exact test or chi-square test.
Univariable and multivariable logistic regression analyses were performed to identify covariates associated with grade 3+ toxicities and hospitalizations and to assess the adjusted odds ratios (ORs) for genotype group (wild-type patients as reference). Covariates included patient demographics, diagnosis, performance status, and chemotherapy regimen. To establish a base model, factor(s) significant in the univariable models (P < .10) were included in the multivariable model, followed by backward elimination to identify independent prognostic factors. The genotype group variable was then added to the established base model to estimate adjusted ORs. Time-to-event of first grade 3+ toxicity and hospitalization were analyzed using Kaplan-Meier techniques. Graphically, cumulative incidence was estimated as 1 – Kaplan-Meier survival estimate. Comparison between genotype groups was tested using the log-rank test.
RESULTS
Patient Characteristics
From March 2020 through May 2023, 757 patients across 14 oncology clinic locations received in-house DPYD genotyping (Implementation Cohort). Patient demographics are summarized in Table 1. Most patients (88%) were treated for GI cancers and about half (55%) received 5-FU–based chemotherapy, one third (34%) received capecitabine-based chemotherapy, and the remaining (11%) did not start treatment (breakdown of chemotherapy regimens at the time of testing is summarized in the Data Supplement, Table S1). More than half (59%) received combination chemotherapy. Reasons for not starting treatment included death, initiation with alternative therapies, patient declined, and lost to follow-up (Fig 1).
TABLE 1.
Patient Demographics
| Characteristics | All Patients (N = 757) | DPYD Wild-Type (n = 712) | Pretreatment Testing Carriers (n = 32) | Reactive Testing Carriers (n = 13) | P |
|---|---|---|---|---|---|
| Age, median (range) | 63 (22-94) | 63 (22-94) | 68 (29-86) | 59 (30-82) | .501 |
| Sex, No. (%) | .022 | ||||
| Female | 347 (46) | 323 (45) | 21 (66) | 3 (23) | |
| Male | 410 (54) | 389 (55) | 11 (34) | 10 (77) | |
| Race, No. (%)a | .525 | ||||
| White | 559 (74) | 521 (73) | 25 (78) | 13 (100) | |
| Black/African American | 146 (19) | 140 (20) | 6 (19) | 0 | |
| Asian | 25 (3.3) | 24 (3.4) | 1 (3.1) | 0 | |
| Other/unknownb | 27 (3.6) | 27 (3.8) | 0 | 0 | |
| Ethnicity, No. (%)a | >.999 | ||||
| Hispanic/Latino | 32 (4.2) | 31 (4.4) | 1 (3.1) | 0 | |
| Non-Hispanic/Latino | 717 (95) | 673 (95) | 31 (97) | 13 (100) | |
| Unknown | 8 (1.1) | 8 (1.1) | 0 | 0 | |
| Cancer type, No. (%)a | .085 | ||||
| Colorectal | 348 (46) | 327 (46) | 15 (47) | 6 (46) | |
| Noncolorectal GI | 320 (42) | 304 (43) | 11 (34) | 5 (39) | |
| Breast | 35 (4.6) | 33 (4.6) | 2 (6.3) | 0 | |
| Genitourinary | 31 (4.1) | 30 (4.2) | 0 | 1 (7.7) | |
| Head and neck | 20 (2.6) | 16 (2.2) | 3 (9.4) | 1 (7.7) | |
| Unknown origin | 3 (0.4) | 2 (0.3) | 1 (3.1) | 0 | |
| Stage, No. (%)a | .732 | ||||
| 0 | 1 (0.1) | 1 (0.1) | 0 | 0 | |
| I | 31 (4.1) | 31 (4.4) | 0 | 0 | |
| II | 97 (13) | 88 (12) | 6 (19) | 3 (23) | |
| III | 225 (30) | 210 (30) | 12 (38) | 3 (23) | |
| IV | 338 (45) | 319 (45) | 12 (38) | 7 (54) | |
| Unknown | 65 (8.6) | 63 (8.8) | 2 (6.3) | 0 | |
| ECOG, No. (%)a | .927 | ||||
| 0 | 194 (26) | 181 (25) | 10 (31) | 3 (23) | |
| 1 | 327 (43) | 310 (44) | 12 (38) | 5 (39) | |
| 2 | 79 (10) | 73 (10) | 5 (16) | 1 (7.7) | |
| 3 | 18 (2.4) | 16 (2.2) | 1 (3.1) | 1 (7.7) | |
| 4 | 5 (0.7) | 5 (0.7) | 0 | 0 | |
| Unknown | 134 (18) | 127 (18) | 4 (13) | 3 (23) | |
| Treatment, No. (%)a | |||||
| Fluorouracil-based | 415 (55) | 392 (55) | 15 (47) | 8 (62) | .610 |
| Capecitabine-based | 256 (34) | 239 (34) | 12 (38) | 5 (39) | — |
| Monotherapy | 225 (30) | 210 (30) | 13 (41) | 2 (15) | .142 |
| Combination regimen | 446 (59) | 421 (59) | 14 (44) | 11 (85) | — |
| Did not start fluoropyrimidine-based treatment | 86 (11) | 81 (11) | 5 (16) | 0 | — |
| DPYD genotype, No. (%)a | |||||
| Wild type (*1/*1) | 712 (94) | 712 (100) | 0 | 0 | |
| Heterozygous carrier | 45 (5.9) | 0 | 32 (100) | 13 (100) | |
| *1/c.1236G>A (HapB3) | 23 (3.0) | 0 | 16 (50) | 7 (54) | |
| *1/c.2846A>T | 8 (1.1) | 0 | 5 (16) | 3 (23) | |
| *1/c.557A>G | 7 (0.9) | 0 | 7 (22) | 0 | |
| *1/c.1905+1G>A (*2A) | 5 (0.7) | 0 | 2 (6.3) | 3 (23) | |
| *1/c.1679T>G (*13) | 2 (0.3) | 0 | 2 (6.3) | 0 |
Abbreviation: ECOG, Eastern Cooperative Oncology Group.
Percentages may not add up to exactly 100% due to rounding.
American Indian or Alaskan Native, Native Hawaiian or Pacific Islander, or other race.
Implementation Metrics
Testing volume was low in 2020 (n = 9) and increased in 2021 (n = 83) with expansion to a regional clinic (Data Supplement, Fig S1). Testing further increased in 2022 (n = 399) after expansion to 14 oncology clinic locations and implementation of EMR interruptive alerts. In 2023, testing averaged 12 patients per week.
The median turnaround time was 6 (IQR, 3-7) days from sample collection to result and 3 (IQR, 2-6) days from sample receipt to result (Table 2). Pretreatment testing was performed in 621 (82%) patients. Of these, 60 (10%) did not have results returned by fluoropyrimidine start, with one patient identified as a heterozygous carrier. This patient initiated treatment after the December 2022 cutoff date for the Outcomes Cohort and thus was not evaluated for dose intensity and toxicities. The remaining 136 (18%) patients received reactive testing, of whom 59 (43%) had samples collected the same day as treatment start.
TABLE 2.
Implementation Metrics
| Turnaround Time | Number of Days, Median (IQR) |
|---|---|
| Overall turnaround time | 6 (3-7) |
| Time from collection to receipt | 1 (1-2) |
| Time from receipt to result | 3 (2-6) |
| Timing of Testing | Number of Patients (N = 757), No. (%)f |
| Pretreatment testing | 621 (82) |
| DPYD variant carrier rate | 32 (5.2) |
| Resulted by treatment start date | 561 (90) |
| Reactive testinga | 136 (18) |
| DPYD variant carrier rate | 13 (9.6) |
| Collected on treatment start date | 59 (43) |
| Fluoropyrimidine Modifications in DPYD Variant Carriers Upon Result Return | Number of Carriers (n = 45), No. (%)f |
| Pretreatment testing | 32 (71) |
| Dose reducedb | 27 (84) |
| Not startedc | 5 (16) |
| Reactive testing | 13 (29) |
| Dose reduced | 9 (69) |
| Discontinuedd | 1 (7.7) |
| No changee | 3 (23) |
Two patients initially had the sample collected before treatment but due to insufficient DNA required sample recollection after treatment, and results were returned after treatment start.
One pretreatment testing carrier whose results were returned after treatment start received a dose reduction starting in cycle 2. This patient was not included in the Outcomes Cohort due to the cutoff date.
Three died before treatment started, one declined chemotherapy, and one had treatment avoided due to variant and hepatic impairment.
Due to variant and grade 3 toxicities.
Tolerating therapy per provider.
fPercentages may not add up to exactly 100% due to rounding.
DPYD Variant Carriers and Treatment Modifications
Overall, 45 (5.9%) patients were identified as heterozygous DPYD variant carriers. The carrier rates were 5.2% and 9.6% in pretreatment and reactive testing groups, respectively. Variants observed were c.1236G>A (HapB3; n = 23), c.2846A>T (n = 8), c.557A>G (n = 7), c.1905+1G>A (*2A; n = 5), and c.1679T>G (*13; n = 2; Table 1). No homozygous or compound heterozygous variant carriers were identified. Most carriers (84%) were self-reported White (n = 38), 13% Black (n = 6), and 2% Asian (n = 1; Data Supplement, Table S2). Thirty-two carriers (71%) received pretreatment testing and 13 carriers (29%) received reactive testing (Table 2).
Among 32 pretreatment carriers, five did not start fluoropyrimidine (three died before treatment started, one declined chemotherapy, and one had treatment avoided in the setting of carrier status and hepatic impairment). Of the 27 pretreatment carriers who received fluoropyrimidine, all (100%) had an upfront dose reduction, including one pretreatment carrier who had results returned after treatment start and thus received a dose reduction starting in cycle 2.
Of the 13 reactive testing carriers, all of whom started standard dose, nine (69%) received dose reductions upon result return (three of whom received dose reductions before results due to toxicities or poor performance status), three (23%) had no immediate change in therapy as tolerating therapy per provider (two later required dose reduction or discontinuation due to toxicities), and one (7.7%) had treatment discontinued due to carrier status and grade 3 toxicities.
Overall, of the 40 variant carriers who initiated fluoropyrimidine chemotherapy, 37 (93%) had fluoropyrimidine dose reduced or discontinued after receiving DPYD genotype results.
Fluoropyrimidine Dosing and Toxicity Outcomes
Of the 442 patients in the Outcomes Cohort, 27 (6.1%) were heterozygous carriers. Figure 2 summarizes fluoropyrimidine treatment and outcomes for 11 reactive carriers and 16 pretreatment carriers. All pretreatment carriers in the Outcomes Cohort were identified before starting treatment and thus received upfront genotype-guided dosing. The mean RDI of the first cycle was 95% and 87% among patients receiving intravenous (IV) 5-FU and oral capecitabine, respectively. The mean RDI of the first cycle was 54% in pretreatment carriers, 95% in reactive carriers, and 93% in wild-type patients (Table 3). Among 24 carriers with dose reductions following DPYD results, five (21%) had dose escalations in subsequent cycles.
FIG 2.
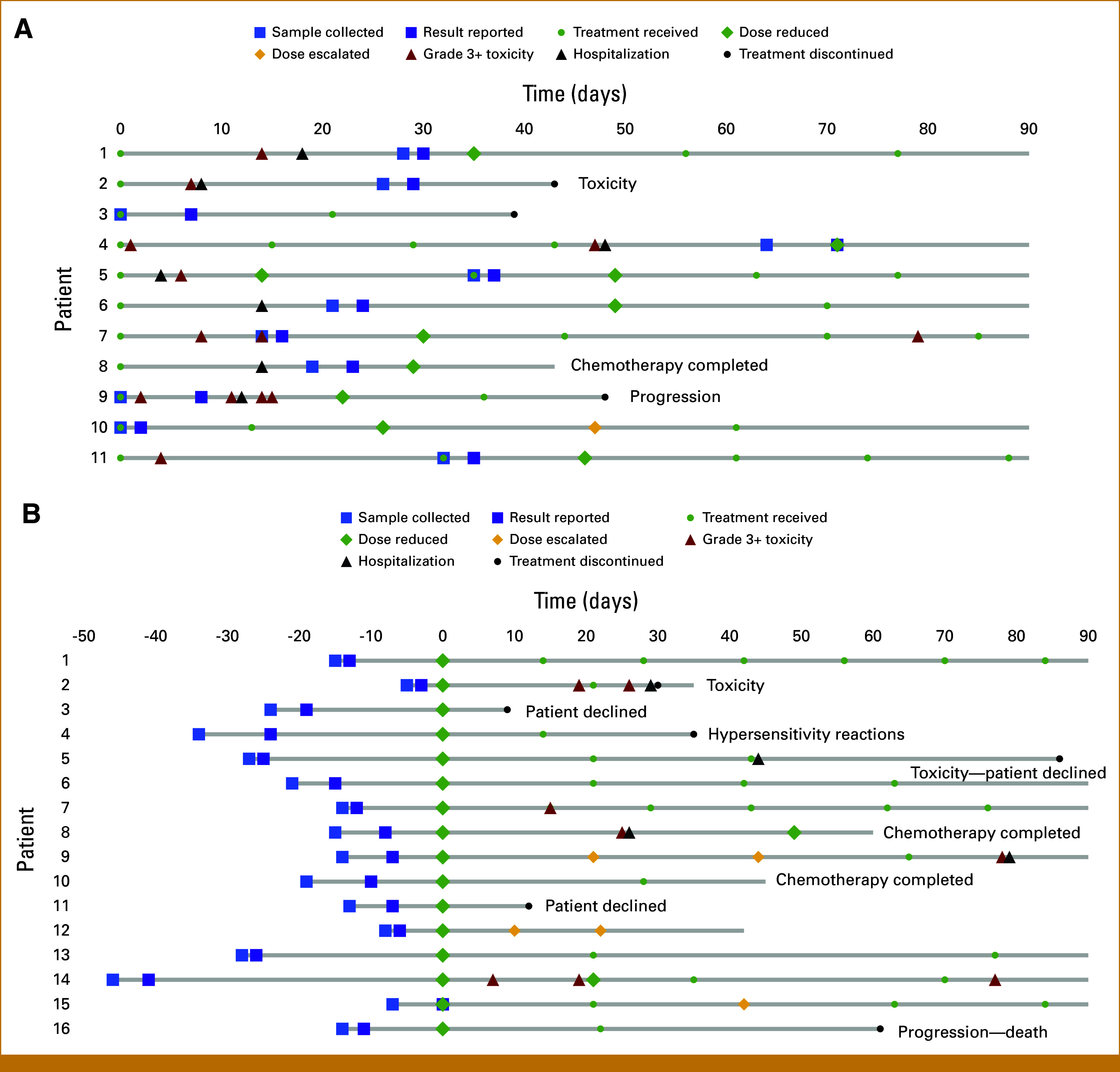
Summary of clinical course for DPYD variant carriers with 3-month follow-up data. (A) Eleven reactive carriers had samples collected for testing on or after the date of treatment initiation. (B) All 16 pretreatment carriers had samples collected and results reported by the date of treatment initiation. Timepoints of treatments, dose reductions, dose escalations, grade 3+ toxicities, hospitalizations, treatment discontinuations, and reasons for discontinuation are depicted in the figures.
TABLE 3.
Relative Dose Intensity, Fluoropyrimidine-Related Grade 3+ Toxicities, Hospitalizations, and Treatment Delays and Discontinuations
| Secondary Endpoint | All Patients (N = 442) | DPYD Wild-Type (n = 415) | Pretreatment Testing Carriers (n = 16) | Reactive Testing Carriers (n = 11) | P |
|---|---|---|---|---|---|
| RDI, first cycle, mean (range)a | 92% (12%-121%) | 93% (12%-121%) | 54% (29%-89%) | 95% (74%-102%) | — |
| Fluorouracil-based (n = 278)b | 95% (26%-107%) | 96% (26%-107%) | 52% (29%-75%) | 97% (74%-102%) | — |
| Capecitabine-based (n = 164)c | 87% (12%-121%) | 88% (12%-121%) | 56% (31%-89%) | 90% (85%-96%) | — |
| Grade 3+ toxicity, FP-related, No. (%)d | 138 (31) | 126 (30) | 5 (31) | 7 (64) | .085 |
| Hematological toxicity | 66 (15) | 62 (15) | 1 (6.3) | 3 (27) | .277 |
| GI toxicity | 77 (17) | 67 (16) | 4 (25) | 6 (55) | .006 |
| Hand-foot syndrome | 7 (1.6) | 7 (1.7) | 0 | 0 | >.999 |
| Othere | 2 (0.5) | 2 (0.5) | 0 | 0 | .937 |
| Hospitalization due to FP toxicity, No. (%)d | 64 (15) | 53 (13) | 4 (25) | 7 (64) | <.001 |
| Hematological toxicity | 11 (2.5) | 10 (2.4) | 0 | 1 (9.1) | .302 |
| GI toxicity | 56 (13) | 45 (11) | 4 (25) | 7 (64) | <.001 |
| Hand-foot syndrome | 3 (0.7) | 3 (0.7) | 0 | 0 | .906 |
| Treatment delay due to FP toxicity, No. (%)d | 116 (26) | 105 (25) | 4 (25) | 7 (64) | .017 |
| Treatment discontinuation due to FP toxicity, No. (%) | 41 (9.3) | 37 (8.9) | 3 (19) | 1 (9.1) | .281 |
Abbreviations: FOLFOX, infusional fluorouracil, leucovorin, and oxaliplatin; FP, fluoropyrimidine; RDI, relative dose intensity.
RDI was calculated as the administered dose over the actual time to complete the cycle divided by the standard dose (mg/m2) over the standard time to complete the cycle for the indication and treatment regimen applicable for the patient.
Consists of 262 wild-type patients, eight pretreatment testing carriers, and eight reactive testing carriers.
Consists of 153 wild-type patients, eight pretreatment testing carriers, and three reactive testing carriers.
dPercentages may not add up to exactly 100% due to rounding.
One DPYD wild-type patient had a cardiac arrest event after two cycles of FOLFOX. This was the only grade 5 event. One had a vagal event during cycle 1 day 1 infusion and a choking event after cycle 1 day 29 infusion of FOLFOX; cardiac work-up was negative.
Of the 442 patients, 138 (31%) reported at least one fluoropyrimidine-related grade 3+ toxicity (Table 3): 5/16 (31%) pretreatment carriers, 7/11 (64%) reactive carriers, and 126/415 (30%) wild-type patients (P = .085). Grade 3+ toxicities were significantly higher in reactive carriers compared with wild-type patients (P = .029), but no difference was noted between pretreatment carriers and wild-type patients (P = .94; Data Supplement, Table S3). Fluoropyrimidine-related toxicities led to hospitalizations in 64 (15%) patients: 4/16 (25%) pretreatment carriers, 7/11 (64%) reactive carriers, and 53/415 (13%) wild-type patients (P < .001). There was a trend toward higher hospitalization rates in reactive carriers compared with pretreatment (P = .052), no difference between pretreatment carriers and wild-type patients (P = .167), and a significant difference between reactive carriers and wild-type patients (P < .001; Data Supplement, Table S3). Grade 3+ GI toxicities (nausea, vomiting, diarrhea, or mucositis) and hospitalizations due to grade 3+ GI toxicities were higher in reactive carriers compared with pretreatment carriers and wild-type patients (Table 3).
Fluoropyrimidine delays due to fluoropyrimidine-related toxicities occurred in 25% of wild-type patients, 25% of pretreatment carriers, and 64% of reactive carriers (P = .017). Other reasons for treatment delays are reported in the Data Supplement (Table S4). Fluoropyrimidine discontinuations due to fluoropyrimidine-related toxicities were observed in 8.9% of wild-type patients, 19% of pretreatment carriers, and 9.1% of reactive carriers (P = .281).
Logistic regression modeling showed treatment route (oral v IV; OR, 1.69 [95% CI, 1.09 to 2.60]; P = .018) and regimen (monotherapy v combination; OR, 1.73 [95% CI, 1.11 to 2.69]; P = .015) were univariably associated with grade 3+ toxicities; however, only regimen was retained in the multivariable model (OR, 1.69 [95% CI, 1.08 to 2.65]; P = .022; Table 4). Although genotype category was not significantly associated with grade 3+ toxicities, reactive carriers had a nearly four-fold higher odds of grade 3+ toxicities compared with wild-type patients (OR, 3.57 [CI, 1.02 to 12.49]). Genotype category was the only independent predictor of hospitalizations (P = .001), with nearly 10-fold higher odds in reactive carriers compared with wild-type patients (OR, 9.59 [95% CI, 2.70 to 34.04]).
TABLE 4.
Factors Associated With Fluoropyrimidine-Related Grade 3+ Toxicities and Hospitalizations
| Covariate | Grade 3+ Adverse Events (N = 442) | Hospitalizations (N = 442) | ||||||
|---|---|---|---|---|---|---|---|---|
| Univariable Results | Multivariable Results | Univariable Results | Multivariable Results | |||||
| OR (CI) | P | OR (CI) | P | OR (CI) | P | OR (CI) | P | |
| DPYD genotype category | .092 | .130 | <.001 | .001 | ||||
| Wild-type v pretreatment carriers | 1.04 (0.36 to 3.06) | 1.25 (0.42 to 3.74) | 2.28 (0.71 to 7.32) | 2.02 (0.62 to 6.56) | ||||
| Wild-type v reactive carriers | 4.01 (1.15 to 13.96) | 3.57 (1.02 to 12.49) | 11.95 (3.38 to 42.22) | 9.59 (2.70 to 34.04) | ||||
| Sex | ||||||||
| Male v female | 0.84 (0.56 to 1.26) | .402 | — | — | 0.98 (0.58 to 1.67) | .946 | — | — |
| Age, years | 1.00 (0.98 to 1.01) | .625 | — | 1.01 (0.99 to 1.03) | .507 | — | ||
| Race | .675 | — | .085 | .163 | ||||
| White v Asian | 0.40 (0.09 to 1.82) | — | 0.39 (0.05 to 3.08) | 0.46 (0.06 to 3.59) | ||||
| White v Black | 1.07 (0.64 to 1.80) | — | 0.31 (0.12 to 0.79) | 0.35 (0.13 to 0.90) | ||||
| White v other | 0.99 (0.34 to 2.93) | — | NE (NE to NE) | NE (NE to NE) | ||||
| Ethnicity | .409 | — | .887 | — | ||||
| Non-Hispanic v Hispanic | 1.29 (0.53 to 3.15) | — | 1.32 (0.43 to 4.04) | — | ||||
| Non-Hispanic v other | 4.52 (0.41 to 50.25) | — | NE (NE to NE) | — | ||||
| Diagnosis | .123 | — | .314 | — | ||||
| Non-GI v colorectal | 1.05 (0.51 to 2.17) | — | 2.51 (0.74 to 8.58) | — | ||||
| Non-GI v non-colorectal GI | 1.59 (0.77 to 3.29) | — | 2.58 (0.75 to 8.90) | — | ||||
| Stage | .783 | — | .636 | — | ||||
| IV v 0/I/II/III | 0.87 (0.57 to 1.32) | — | 0.77 (0.44 to 1.33) | — | ||||
| IV v unknown | 0.85 (0.38 to 1.88) | — | 0.89 (0.32 to 2.48) | — | ||||
| ECOG | .513 | — | .784 | — | ||||
| 0/1 v 2/3/4 | 1.36 (0.74 to 2.52) | — | 1.25 (0.57 to 2.73) | — | ||||
| 0/1 v unknown | 1.24 (0.71 to 2.14) | — | 0.88 (0.41 to 1.89) | — | ||||
| Treatment route | ||||||||
| Oral v IV | 1.69 (1.09 to 2.60) | .018 | — | — | 1.15 (0.66 to 2.00) | .625 | — | — |
| Treatment regimen | ||||||||
| Monotherapy v combination | 1.73 (1.11 to 2.69) | .015 | 1.69 (1.08 to 2.65) | .022 | 1.45 (0.81 to 2.60) | .210 | — | — |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; IV, intravenous; NE, nonestimable; OR, odds ratio.
The log-rank tests for time-to-event stratified by genotype category were significant for grade 3+ toxicities and hospitalizations (P < .001), demonstrating a higher number and earlier onset of events among reactive carriers (Fig 3).
FIG 3.

Cumulative incidence of fluoropyrimidine-related grade 3+ toxicities and hospitalizations stratified by genotype groups in 442 patients. Cumulative incidence was estimated as 1—Kaplan-Meier survival estimate, and log-rank test was used to compare between genotype groups. All subjects with follow-up time >110 days were censored observations and were truncated at 110 days. (A) Grade 3+ toxicities occurred earlier and at a significantly higher rate in reactive carriers compared with pretreatment carriers and wild-type patients (P < .001). (B) Hospitalizations also occurred earlier and at a significantly higher rate in reactive carriers compared with pretreatment carriers and wild-type patients (P < .001).
DISCUSSION
In a real-world analysis of US-based patients receiving DPYD genotyping, we demonstrated that almost all variant carriers received fluoropyrimidine modifications, and pretreatment testing with genotype-guided dosing reduced severe toxicities and hospitalizations compared with reactive testing. Our findings underscore the critical relationship between DPYD variants and fluoropyrimidine toxicity, and the clinical impact of pretreatment testing and genotype-guided dosing to mitigate this risk. Recently, Hertz and Baker et al call on oncology practice guidelines to re-evaluate recommendations for pretreatment DPYD testing as standard practice.4,17 CPIC guidelines,9,13 prospective studies,11,13 and our real-world evidence of US-based patients receiving genotype-guided dosing support this call to action.
DPYD genotype–guided fluoropyrimidine dosing across multiple clinics proved feasible. Integration of EMR pretest alerts at the time of fluoropyrimidine prescribing increased testing volumes. This allowed sufficient time for testing as 90% of patients sent for pretreatment testing had results returned before starting treatment. Nonetheless, 43.4% of reactive testing patients had samples collected on the day of treatment start, indicating opportunities to improve strategies for earlier collections. We anticipate continued increase in testing volume as the DPYD test was made orderable in the EMR (October 2023) and is planned to be integrated within fluoropyrimidine-containing treatment plans. Our finding that most carriers received dose reductions align with a survey of US oncologists reporting that nearly all agreed that patients with DPD deficiency have increased toxicity risk and would modify fluoropyrimidine dosing in these patients.18
The overall carrier rate in our patient cohort, comprising 74% White and 19% Black, was 5.9%, similar to the 5%-7% reported in the CPIC DPYD frequency table.9 Our rate was slightly lower than that in a European study by Henricks et al13 (7.7%) and higher than that in a Canadian study by Wigle et al19 (3.4%); both did not include the c.557A>G variant. Although the race distribution in this study was representative of our patient population, most variant carriers, besides the c.557A>G variant, were self-reported White. Future studies focusing on non-White populations are crucial in discovering and validating clinically relevant DPYD variants in diverse populations.
Our results align with previous studies of genotype-guided fluoropyrimidine dosing. In a previous prospective study, grade 3+ toxicities significantly reduced from 73% in historical controls to 28% in *2A carriers receiving genotype-guided dosing.11 Henricks et al13 expanded on this work to genotype four relevant variants. Despite a 25%-50% dose reduction, grade 3+ toxicity was still higher in carriers (39%) compared with wild-type patients (23%). These findings prompted the European Medicines Agency and European Society of Medical Oncology to recommend pretreatment DPYD testing.20 Real-world analyses from the United Kingdom21 and Canada,19 and a meta-analysis across 17 studies22 confirmed reduced incidence of severe toxicities with DPYD genotype–guided dosing.
In our study, grade 3+ toxicity rate was not different in pretreatment carriers receiving fluoropyrimidine dose reductions (31%) compared with wild-type patients receiving standard dosing (30%). Grade 3+ toxicity rate in reactive carriers receiving initial full dose in our study (64%) was understandably lower than historical data (70%-75%) since nearly half had testing sent on the day of treatment start, and thus most received dose reductions in the second cycle, potentially avoiding severe toxicity. Hospitalizations were not evaluated in previous studies; herein, we demonstrated hospitalizations were reduced in carriers receiving pretreatment genotype-guided dosing (25%) compared with reactive carriers (64%).Treatment delays due to fluoropyrimidine toxicities were also highest in reactive carriers, potentially affecting treatment outcomes.
Reductions in toxicity, hospitalization, and treatment delay have clear implications on quality of life and health care costs. A systematic review of the cost-effectiveness of pharmacogenetic testing found that four studies evaluating DPYD testing demonstrated cost savings and one cost-effectiveness.23 Two European studies reported savings of approximately $50-$60 in US dollars per patient, which are expected to be higher in a US-based population without nationalized health care.11,24
Logistic regression analyses showed 5-FU–containing regimens were significantly associated with severe toxicities compared with oral capecitabine-containing regimens in the univariable analysis (OR, 1.69), though not significant in the multivariable analysis. As 5-FU is usually given as an IV bolus followed by continuous infusion over multiple days depending on the protocol, patients receiving oral capecitabine would have more opportunities to adjust doses mid-treatment on the basis of tolerability. This is supported by the lower first cycle RDI in patients receiving capecitabine compared with 5-FU. Additionally, combination regimens were significantly associated with severe toxicities compared with fluoropyrimidine monotherapy (OR, 1.69). Concurrent chemotherapies may have overlapping toxicities with fluoropyrimidines, which may have also contributed to the severe toxicity rate.
There are limitations to this study, such as being conducted at a single institution, thus limiting generalizability to institutions without in-house testing capabilities. Several commercial laboratories offer DPYD testing; however, it is critical to evaluate which alleles are detected to minimize false negatives.25,26 Due to the relative infrequency of DPYD variants, the sample size of carriers was small, thus limiting overall power. Nonetheless, the severe toxicity rates across genotype groups were comparable with those seen in previous studies. Patients and providers were not blinded to results, which could influence patient reporting or provider documentation of toxicities. The study was nonrandomized; thus, there was no control population who did not receive genotyping. However, the reactive carrier group served as a surrogate for toxicity with standard upfront dosing in variant carriers, interpreting that most of these patients had dose reductions in subsequent cycles. Given the real-world observational study design, outcomes data were collected from standard-of-care clinic notes and laboratory results, which can be less accurate than if collected from a prospective trial. Although reflective of the real-world setting, the population was partially heterogeneous, composed of patients with varying tumor types, treatment regimens, and dosing. RDI for all cycles was not reported due to these reasons. Nevertheless, DPYD genotype category was the only factor associated with hospitalization. Finally, our study was limited to 3-month follow-up and did not include survival end points. A recent study found that DPYD-guided dosing does not negatively affect progression-free survival (PFS) or OS in variant carriers compared with wild-type patients, although c.1236G>A variant carriers receiving a 25% reduced dose had a shorter PFS.12,27 The c.1236G>A variant, used as a proxy for the HapB3 haplotype, was also recently discovered to not be in complete linkage disequilibrium (99.85%) with the causal variant, c.1129-5923C>G.27 CPIC updated its guideline to recommend testing for the functional variant c.1129-5923C>G, which we are adding to our genotyping panel.9 Further research is needed to determine the most appropriate dose reduction and maximum tolerated dose for each variant to optimize drug exposure and response and minimize toxicity.
The diagnostic performance of our genotyping test, compared with all genotyping tests, is limited to the variants covered on the panel and may falsely identify carriers of other rare variants as wild-type. Full-gene sequencing may uncover additional variants of known or unknown function especially in diverse populations. Nonetheless, we recently reported that our in-house DPYD genotype test showed higher negative predictive value and lower false-negative rates compared with many other commercial tests that include fewer variants.26
In conclusion, results from one of the first real-world US-based experiences of in-house DPYD genotyping demonstrated that implementation across a multisite cancer hospital was feasible, resulted in fluoropyrimidine dose modifications in almost all carriers, and, most importantly, led to fewer severe toxicities and hospitalizations in those receiving pretreatment testing and genotype-guided dosing. The lack of recommendations on pretreatment testing from the FDA and oncology clinical practice guidelines continues to hinder widespread adoption in the United States.18 Nonetheless, at a minimum, all patients should have the right to be informed of DPYD testing, offered testing, and educated on the implications of testing or not testing.17
PRIOR PRESENTATION
Presented at the 2023 ASCO Annual Meeting, Chicago, IL, June 5, 2023; the 2023 PGRN Annual Meeting, Memphis, TN, June 12, 2023.
SUPPORT
Supported by Heineman Foundation of New York (J.N.P.).
AUTHOR CONTRIBUTIONS
Conception and design: Sarah A. Morris, Nury Steuerwald, James Symanowski, Donald C. Moore, Sarah Hanson, Kristen Swift, Jai N. Patel
Administrative support: Sarah Hanson, Seungjean Chai
Provision of study materials or patients: Donald C. Moore, Sarah Hanson, Kunal C. Kadakia, Seungjean Chai, Kwabena Osei-Boateng, Sini Kalapurakal, Kristen Swift, Jimmy Hwang
Collection and assembly of data: D. Grace Nguyen, Sarah A. Morris, Alicia Hamilton, Simeon O. Kwange, Nury Steuerwald, Sarah Hanson, Kaitlyn Mroz, Karine E. Lopes, Chris Larck, Laura Musselwhite, Brinda Koya, Kwabena Osei-Boateng, Sini Kalapurakal, Kristen Swift, Jimmy Hwang, Jai N. Patel
Data analysis and interpretation: D. Grace Nguyen, Sarah A. Morris, Alicia Hamilton, Simeon O. Kwange, Nury Steuerwald, James Symanowski, Sarah Hanson, Kaitlyn Mroz, Laura Musselwhite, Kunal C. Kadakia, Seungjean Chai, Kristen Swift, Jimmy Hwang, Jai N. Patel
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Alicia Hamilton
Employment: Atrium Health
Nury Steuerwald
Employment: Atrium Health
Research Funding: Atrium Health
Patents, Royalties, Other Intellectual Property: I hold a patent for a gene signature predictive of outcome in drug induced liver injury. I have not received any royalties to date from this patent
Travel, Accommodations, Expenses: Atrium Health
James Symanowski
Consulting or Advisory Role: Lilly, Immatics, CARsgen Therapeutics, Endocyte (Inst), Camurus, Astellas Pharma
Research Funding: Merck (Inst), Janssen (Inst), GlaxoSmithKline (Inst), Amgen (Inst), Incyte (Inst), Pfizer (Inst), G1 Therapeutics (Inst)
Kaitlyn Mroz
Employment: Atrium Health
Laura Musselwhite
Consulting or Advisory Role: AstraZeneca
Kristen Swift
Travel, Accommodations, Expenses: ONCC
Jimmy Hwang
Consulting or Advisory Role: Caris Centers of Excellence, Ipsen, AstraZeneca
Research Funding: Caris Centers of Excellence (Inst), Boehringer Ingelheim (Inst)
Jai N. Patel
Honoraria: VieCure, Clarified Precision Medicine
Consulting or Advisory Role: VieCure, Clarified Precision Medicine
Research Funding: Professional Compounding Centers of America (Inst), Bristol Myers Squibb (Inst), Symberix (Inst), American Association for Cancer Research (Inst), WinSanTor (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1. Fluorouracil [package insert]. Fresenius Kabi, 2016. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/202669s009lbl.pdf.
- 2. Capecitabine [package insert]. Genentech Inc, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/020896s044s045s046s047s048s049s050s051lbl.pdf.
- 3.Kuebler JP, Wieand HS, O'Connell MJ, et al. : Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: Results from NSABP C-07. J Clin Oncol 25:2198-2204, 2007 [DOI] [PubMed] [Google Scholar]
- 4.Hertz DL: Assessment of the clinical utility of pretreatment DPYD testing for patients receiving fluoropyrimidine chemotherapy. J Clin Oncol 40:3882-3892, 2022 [DOI] [PubMed] [Google Scholar]
- 5.Thorn CF, Marsh S, Carrillo MW, et al. : PharmGKB summary: Fluoropyrimidine pathways. Pharmacogenet Genomics 21:237-242, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Innocenti F, Mills SC, Sanoff H, et al. : All you need to know about DPYD genetic testing for patients treated with fluorouracil and capecitabine: A practitioner-friendly guide. JCO Oncol Pract 16:793-798, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meulendijks D, Henricks LM, Sonke GS, et al. : Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: A systematic review and meta-analysis of individual patient data. Lancet Oncol 16:1639-1650, 2015 [DOI] [PubMed] [Google Scholar]
- 8.Sharma BB, Rai K, Blunt H, et al. : Pathogenic DPYD variants and treatment-related mortality in patients receiving fluoropyrimidine chemotherapy: A systematic review and meta-analysis. Oncologist 26:1008-1016, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amstutz U, Henricks LM, Offer SM, et al. : Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin Pharmacol Ther 103:210-216, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Offer SM, Lee AM, Mattison LK, et al. : A DPYD variant (Y186C) in individuals of African ancestry is associated with reduced DPD enzyme activity. Clin Pharmacol Ther 94:158-166, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deenen MJ, Meulendijks D, Cats A, et al. : Upfront genotyping of DPYD*2A to individualize fluoropyrimidine therapy: A safety and cost analysis. J Clin Oncol 34:227-234, 2016 [DOI] [PubMed] [Google Scholar]
- 12.Knikman JE, Wilting TA, Lopez-Yurda M, et al. : Survival of patients with cancer with DPYD variant alleles and dose-individualized fluoropyrimidine therapy—A matched-pair analysis. J Clin Oncol 41:5411-5421, 2023 [DOI] [PubMed] [Google Scholar]
- 13.Henricks LM, Lunenburg C, de Man FM, et al. : DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: A prospective safety analysis. Lancet Oncol 19:1459-1467, 2018 [DOI] [PubMed] [Google Scholar]
- 14.Lau-Min KS, Varughese LA, Nelson MN, et al. : Preemptive pharmacogenetic testing to guide chemotherapy dosing in patients with gastrointestinal malignancies: A qualitative study of barriers to implementation. BMC Cancer 22:47, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morris SA, Moore DC, Musselwhite LW, et al. : Addressing barriers to increased adoption of DPYD genotyping at a large multisite cancer center. Am J Health Syst Pharm 80:1342-1349, 2023 [DOI] [PubMed] [Google Scholar]
- 16.Havrilesky LJ, Reiner M, Morrow PK, et al. : A review of relative dose intensity and survival in patients with metastatic solid tumors. Crit Rev Oncol Hematol 93:203-210, 2015 [DOI] [PubMed] [Google Scholar]
- 17.Baker SD, Bates SE, Brooks GA, et al. : DPYD testing: Time to put patient safety first. J Clin Oncol 41:2701-2705, 2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koo K, Pasternak AL, Henry NL, et al. : Survey of US medical oncologists' practices and beliefs regarding DPYD testing before fluoropyrimidine chemotherapy. JCO Oncol Pract 18:e958-e965, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wigle TJ, Povitz BL, Medwid S, et al. : Impact of pretreatment dihydropyrimidine dehydrogenase genotype-guided fluoropyrimidine dosing on chemotherapy associated adverse events. Clin Transl Sci 14:1338-1348, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de With M, Sadlon A, Cecchin E, et al. : Implementation of dihydropyrimidine dehydrogenase deficiency testing in Europe. ESMO Open 8:101197, 2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lau DK, Fong C, Arouri F, et al. : Impact of pharmacogenomic DPYD variant guided dosing on toxicity in patients receiving fluoropyrimidines for gastrointestinal cancers in a high-volume tertiary centre. BMC Cancer 23:380, 2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glewis S, Alexander M, Khabib MNH, et al. : A systematic review and meta-analysis of toxicity and treatment outcomes with pharmacogenetic-guided dosing compared to standard of care BSA-based fluoropyrimidine dosing. Br J Cancer 127:126-136, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris SA, Alsaidi AT, Verbyla A, et al. : Cost effectiveness of pharmacogenetic testing for drugs with Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines: A systematic review. Clin Pharmacol Ther 112:1318-1328, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henricks LM, Lunenburg C, de Man FM, et al. : A cost analysis of upfront DPYD genotype-guided dose individualisation in fluoropyrimidine-based anticancer therapy. Eur J Cancer 107:60-67, 2019 [DOI] [PubMed] [Google Scholar]
- 25.Hertz DL, Smith DM, Scott SA, et al. : Response to the FDA decision regarding DPYD testing prior to fluoropyrimidine chemotherapy. Clin Pharmacol Ther 114:768-779, 2023 [DOI] [PubMed] [Google Scholar]
- 26.Nguyen DG, Morris SA, Chen A, et al. : Unveiling discrepant and rare dihydropyrimidine dehydrogenase (DPYD) results using an in-house genotyping test: A case series. J Natl Compr Canc 22:e247022, 2024 [DOI] [PubMed] [Google Scholar]
- 27.Turner AJ, Haidar CE, Yang W, et al. : Updated DPYD HapB3 haplotype structure and implications for pharmacogenomic testing. Clin Transl Sci 17:e13699, 2024 [DOI] [PMC free article] [PubMed] [Google Scholar]