

Abstract
PURPOSE
The choice of threshold and reliability of high tumor mutational burden (TMB) to predict outcomes and guide treatment choice for patients with metastatic melanoma receiving first-line immune checkpoint inhibitor (ICI) therapy in the real world is not well known.
METHODS
Using a deidentified nationwide (US-based) melanoma clinicogenomic database, we identified a real-world cohort of patients with metastatic melanoma (N = 497) who received first-line monotherapy anti–PD-1 (n = 240) or dual anti–PD-1 and anti–CTLA-4 ICI (n = 257) and had a tissue-based comprehensive genomic profiling test TMB score.
RESULTS
TMB-high (TMB-H; ≥10 mutations per megabase [muts/Mb], n = 352, 71%) was independently predictive of superior real-world progression-free survival and overall survival versus TMB-low (<10 mut/Mb, n = 145, 29%) in both mono ICI (hazard ratio [HR], 0.45 [95% CI, 0.32 to 0.63]; P < .001; HR, 0.61 [95% CI, 0.41 to 0.90]; P = .01, respectively) and dual ICI (HR, 0.67 [95% CI, 0.49 to 0.90]; P = .009; HR, 0.61 [95% CI, 0.42 to 0.88]; P = .007, respectively) patients. Dual ICI offered no significant advantage in BRAFwt patients and unexpectedly demonstrated greatest benefit in the TMB 10-19 mut/Mb group, identifying a TMB-very high (≥20 mut/Mb, n = 247, 50%) BRAFmut patient subgroup for whom mono ICI may be preferable.
CONCLUSION
TMB-H predicts superior outcomes on ICI while coassessment of BRAF status and TMB may inform first-line regimen choice.
Melanoma TMB predicts real-world ICI outcome but BRAF coassessment may better inform regimen choice.
INTRODUCTION
Immune checkpoint inhibitors (ICIs) have transformed the treatment landscape of metastatic melanoma, but short-lived response and immune-related toxicity remain clinical limitations for which predictive biomarkers are needed. Tumor mutational burden (TMB), defined as the number of somatic mutations per megabase (muts/Mb) of an interrogated genomic sequence, is both validated and approved as a predictive molecular biomarker for ICI in the treatment of metastatic solid cancers. It is hypothesized that TMB correlates with the chance of responding to ICI, on the basis of the notion that numbers of immunogenic neoantigens and TMB are proportional1 and latent immune responses to such neoantigens can be augmented or reinvigorated by ICI therapy.2,3 The predictive utility of TMB for ICI has been demonstrated across multiple cancer types separately,4-8 and in pan-cancer analyses.9-11 In the prospective multicohort KEYNOTE-158 study, DNA mismatch repair enyzme (MMR) proficient patients with a high TMB (≥10 mut/Mb, TMB-high [TMB-H]) demonstrated superior response rate and progression-free survival (PFS) versus patients with low TMB (<10 mut/Mb, TMB-low [TMB-L]), measured using the FoundationOne CDx assay, leading to tumor-agnostic approval of this TMB-directed strategy by the US Food and Drug Administration (FDA).12 Even in MMR deficient tumors for which ICI is highly effective,9 TMB provided additional predictive information.13 A threshold of 10 mut/Mb is supported by evidence of a plateau in objective response rates beyond this in patients with previously untreated non–small cell lung cancer receiving ICI in the CheckMate-568 study.14 Conversely, application of an overall median-based TMB cutoff resulted in substantially variable ICI predictive performance for individual cancer types and lacks applicability to individual patients in the real world.15
CONTEXT
Key Objective
Does tumor mutation burden (TMB) predict immune checkpoint inhibitor (ICI) therapy outcome in real-world patients with advanced melanoma?
Knowledge Generated
High TMB (>10 mutations per megabase [muts/Mb]) independently predicts superior real-world progression-free and overall survival after first-line ICI. Unexpectedly, the relationship with TMB is not continuous for dual ICI, identifying a group of patients with very high TMB (≥20 muts/Mb) who may be best treated with monotherapy ICI.
Relevance
Comprehensive genomic profiling including TMB should be performed prior to first-line systemic therapy for advanced melanoma, as TMB results may help refine management choices.
In metastatic melanoma, increasing evidence supports TMB as a predictive biomarker of ICI efficacy yet equipoise remains about its value in routine clinical practice. An early retrospective analysis demonstrated greater clinical benefit for patients with a higher mutational load when treated with anti–CTLA-4 ICI.16 In the CheckMate-067 phase III clinical trial of ipilimumab, nivolumab, or both in patients with unresectable or metastatic melanoma, high TMB (dichotomized at the median) was associated with improved survival and this effect was particularly apparent in BRAF wild-type patients.17 In this study, we evaluated the impact of TMB on real-world outcomes of patients with advanced melanoma treated with mono- or dual-therapy ICI in the first-line setting using a large clinical database linked to matched tumor-based genomic profiling.
METHODS
Patient Cohort
This study used deidentified data collected as part of the nationwide (US-based, approximately 280 US cancer clinics incorporating approximately 800 sites of care) Flatiron Health-Foundation Medicine Inc (FMI) melanoma clinicogenomic database (CGDB) after institutional review board (IRB) approval with waiver of informed consent on the basis of the observational, noninterventional nature of the study (WCG IRB, Protocol No. 420180044). Participant inclusion in the CGDB required at least two visits to a Flatiron Health clinic site and issuance of a Foundation Medicine comprehensive genomic profiling (CGP) testing report, the latter almost always occurring after a participant has met the clinic attendance requirement and thus being the more critical time point for determination of delayed study entry (see Statistical Analyses). Relevant clinicopathologic variables were extracted from electronic health records (EHRs) for multivariable analyses, as described further in the Data Supplement (Methods). The data cutoff date was March 31, 2022.
Eligibility for inclusion or reasons for exclusion from the final study cohort are outlined in the flowchart (Fig 1). Lines of therapy were derived from EHR treatment data using an oncologist-defined, rule-based approach.18 Patients must have had structured EHR activity within 90 days of their metastatic diagnosis date to ensure completeness of treatment data capture and therefore accuracy in the enumeration of lines of therapy. Patients must have received one of pembrolizumab, nivolumab, or ipilimumab plus nivolumab as the first line of therapy for metastatic melanoma, and needed to have a TMB score from a tissue-based FMI CGP test (FoundationOne or FoundationOne CDx).
FIG 1.

Flowchart of the melanoma study cohort. A total of 497 patients with melanoma were included in the study from 2,040 patients with melanoma from the deidentified Flatiron Health-Foundation Medicine melanoma CGDB after applying inclusion and exclusion criteria. Regimens of interest were nivolumab monotherapy, pembrolizumab monotherapy, and ipilimumab plus nivolumab combination therapy. CGDB, clinicogenomic database; EHR, electronic health record; TMB, tumor mutation burden.
CGP
Hybrid capture-based next-generation sequencing (NGS) was performed as a part of routine clinical care (Foundation Medicine, Cambridge, MA) using the 404-gene FoundationOne and the 324-gene FoundationOne CDx assays, which assess base substitutions, short insertions/deletions, rearrangements/fusions, and copy-number variations as well as genome-wide biomarkers such as TMB and microsatellite instability (MSI).19,20 For this study, TMB <10 muts/Mb was considered TMB-L, TMB ≥10 muts/Mb was considered TMB-H, and TMB ≥20 muts/Mb was considered very high TMB (TMB-VH).
Statistical Analyses
This study included both prespecified and exploratory analyses. R version 4.2.1 software program was used for all statistical analyses.21 A prospectively declared statistical analysis plan was developed and executed for all prespecified analyses consistent with International Society for Pharmacoeconomics and Outcomes Research guidelines,22 covering inclusion and exclusion criteria, potential sources of bias, primary outcome measures, handling of missing data, and all methods described below, unless otherwise noted. Prespecified analyses considered TMB dichotomized at a 10 mut/Mb threshold, in line with current, FDA-approved CDx indications for pembrolizumab.12
Handling of missing values and imputation were performed as described in the Data Supplement (Methods). All individual patient data were used in univariate and multivariable analyses after any missing value adjustments were applied. Overall survival (OS) was calculated from the start of treatment to death from any cause, and patients alive at last observation were censored. Real–world PFS (rwPFS) was calculated from the start of treatment to progression or death from any cause. Progression in this context refers to the real–world progression variable abstracted from EHRs as previously described.23 Inclusion in this study cohort required issuance of a successful CGP report, which may have occurred after the start of first line ICI therapy; thus, this data set was left-truncated for the purposes of OS analyses. To account for this, risk-set adjustment was performed, including only patients at each time point who met all inclusion criteria at that time point in Kaplan-Meier and Cox Model analyses (further details are provided in the Data Supplement, Methods). In the primary analysis, the composite end point of rwPFS was incompatible with risk-set adjustment as a patient can experience a nondeath progression event during their period of delayed entry and thus cannot be excluded from the risk set.
For this analysis, nivolumab monotherapy and pembrolizumab monotherapy were pooled together as monotherapy ICI. Patients receiving nivolumab and ipilimumab were counted as dual ICI therapy. As the data cutoff occurred <2 weeks after its FDA approval, no patients in this study received coformulated nivolumab and relatlimab-rmbw.
The prognostic value of TMB was assessed in both univariable and multivariable contexts using the Kaplan-Meier method and corresponding Cox proportional hazards models, as described in detail in the Data Supplement (Methods).
Sensitivity analyses and exploratory analyses were not prospectively declared, and included assessment of TMB at 10 and 20 mut/Mb cutoffs and the effect of stratification by BRAF V600E/K mutations, as described further in the Data Supplement (Methods).
RESULTS
Patient Cohort Characteristics
Of 2,040 patients with melanoma captured by the CGDB, a total of 497 patients with metastatic melanoma received one of the first-line ICI regimens of interest (257 dual ICI, 240 monotherapy ICI) and had a tissue TMB score (Fig 1; Data Supplement, Table S1). Patients receiving monotherapy were older (median age at treatment initiation 71.0 and 63.0 years in monotherapy and dual ICI, respectively; P < .001), and had less brain metastases (prevalence 26.2% mono v 35.0% dual ICI; P = .007), steroid exposure immediately before initiating therapy (12.5% mono v 19.8% dual ICI; P = .036), or BRAFmut disease (BRAFwt in 62.1% mono v 51.4% dual ICI; P = .005), but baseline ECOG performance status did not differ between treatment groups. Patients of this real-world cohort were frailer than those studied in clinical trials such as CheckMate-067, evidenced by a lower proportion of patients with ECOG PS ≤1 and inclusion of patients (11%) with ECOG PS ≥2 (Data Supplement, Fig S1).
Dual ICI use significantly increased over time (25.0%-28.6% of all treatment regimens before 2018 v >65% after 2021) and trended higher in community compared with academic practice settings (P = .095). CGP reports were issued a median of 9 days before initiating first-line ICI but only 26.8% of patients received their reports more than 14 days before first dose (Data Supplement, Table S1), with most patients receiving these data after their first dose of ICI (Data Supplement, Fig S2A). However, nearly all (94.0%) CGP tests were performed on specimens collected before first dose of ICI, with a median of 40 days from specimen collection to first dose (Data Supplement, Fig S2B).
Clinicopathologic Associations With TMB
Of the 497 patients in the final cohort, 145 (29%) had low TMB (TMB-L, <10 mut/Mb) and 352 (71%) had TMB-H by the conventional threshold (≥10 mut/Mb), including 247 (50%) with TMB-VH by the exploratory threshold (≥20 mut/Mb; Table 1). Patients with TMB-VH were more often male, BRAFwt, and given anti–PD-1 monotherapy. TMB-VH was enriched in tumor samples taken from brain or pulmonary metastatic sites. Patients with TMB-H were more likely to be BRAF V600K than other TMB subgroups and were least likely to have pulmonary metastases. TMB-L was associated with younger age and metastases limited to nonvisceral sites. Similar trends were observed when comparing characteristics strictly between the two subgroups defined by the conventional cutoff of TMB < or ≥10 mut/Mb (Data Supplement, Table S2). No significant association was observed between CDKN2A/B variant status or lactate dehydrogenase (LDH) level and TMB. Consistent with its role as the dominant carcinogen exposure relevant to cutaneous melanoma, the presence of ultraviolet (UV) mutation signatures was associated with high TMB, seen in only 17.9% of TMB-L patients, but 88.6% of TMB-H and 92.7% of TMB-VH patients (P < .001). The prevalence of BRAF non-V600E/K mutations increased with TMB, in keeping with these being largely nondriver BRAF mutations arising through the same nonspecific mutational processes leading to high TMB.
TABLE 1.
Clinicopathologic Characteristics of Patients With Melanoma Stratified by TMB Cutoffs of 10 or 20 Muts/Mb
| Feature | All Patients (N = 497) | TMB-L <10 mut/Mb (n = 145) | TMB-H 10-19 mut/Mb (n = 105) | TMB-VH ≥20 mut/Mb (n = 247) | Pa (overall) |
|---|---|---|---|---|---|
| Systemic regimen, No. (%) | <.001 | ||||
| Ipilimumab + nivolumab | 257 (51.7) | 89 (61.4) | 63 (60.0) | 105 (42.5) | |
| Nivolumab | 120 (24.1) | 25 (17.2) | 17 (16.2) | 78 (31.6) | |
| Pembrolizumab | 120 (24.1) | 31 (21.4) | 25 (23.8) | 64 (25.9) | |
| Mono or dual ICI therapy, No. (%) | <.001 | ||||
| Dual | 257 (51.7) | 89 (61.4) | 63 (60.0) | 105 (42.5) | |
| Mono | 240 (48.3) | 56 (38.6) | 42 (40.0) | 142 (57.5) | |
| Age at start of first-line systemic therapy, years, median (IQR) | 67.0 (59.0-75.0) | 63.0 (52.0-71.0) | 67.0 (59.0-73.0) | 70.0 (62.0-78.0) | <.001 |
| Race, No. (%) | .045 | ||||
| White | 384 (77.3) | 100 (69.0) | 83 (79.0) | 201 (81.4) | |
| Other race | 69 (13.9) | 27 (18.6) | 16 (15.2) | 26 (10.5) | |
| Unknown/not documented | 44 (8.9) | 18 (12.4) | 6 (5.7) | 20 (8.1) | |
| ECOG performance score, No. (%) | .194 | ||||
| 0 | 230 (46.3) | 73 (50.3) | 44 (41.9) | 113 (45.7) | |
| 1 | 157 (31.6) | 47 (32.4) | 32 (30.5) | 78 (31.6) | |
| 2 | 40 (8.1) | 10 (6.9) | 15 (14.3) | 15 (6.1) | |
| ≥3 | 18 (3.6) | 5 (3.4) | 3 (2.9) | 10 (4.1) | |
| Unknown | 52 (10.5) | 10 (6.9) | 11 (10.5) | 31 (12.6) | |
| Sex, No. (%) | .007 | ||||
| Female | 154 (31.0) | 57 (39.3) | 36 (34.3) | 61 (24.7) | |
| Male | 343 (69.0) | 88 (60.7) | 69 (65.7) | 186 (75.3) | |
| Practice type, No. (%) | .217 | ||||
| Academic | 59 (11.9) | 12 (8.3) | 12 (11.4) | 35 (14.2) | |
| Community | 438 (88.1) | 133 (91.7) | 93 (88.6) | 212 (85.8) | |
| BRAF mutation status, No. (%) | <.001 | ||||
| No BRAF mutation detected | 281 (56.5) | 72 (49.7) | 51 (48.6) | 158 (64.0) | |
| V600E | 109 (21.9) | 59 (40.7) | 28 (26.7) | 22 (8.9) | |
| V600K | 40 (8.1) | 3 (2.1) | 17 (16.2) | 20 (8.1) | |
| Other mutation | 56 (11.3) | 4 (2.8) | 7 (6.7) | 45 (18.2) | |
| Unknown | 11 (2.2) | 7 (4.8) | 2 (1.9) | 2 (0.8) | |
| CDKN2A/B mutation status, No. (%) | .190 | ||||
| Negative | 239 (48.1) | 63 (43.4) | 53 (50.5) | 123 (49.8) | |
| Positive | 229 (46.1) | 68 (46.9) | 48 (45.7) | 113 (45.7) | |
| Unknown | 29 (5.8) | 14 (9.7) | 4 (3.8) | 11 (4.5) | |
| Lactate dehydrogenase, No. (%) | .500 | ||||
| ≤ULN | 181 (36.4) | 47 (32.4) | 35 (33.3) | 99 (40.1) | |
| >ULN | 96 (19.3) | 30 (20.7) | 19 (18.1) | 47 (19.0) | |
| Unknown | 220 (44.3) | 68 (46.9) | 51 (48.6) | 101 (40.9) | |
| Sites of metastasis,b No. (%) | <.001 | ||||
| Nonvisceral | 74 (14.9) | 38 (26.2) | 11 (10.5) | 25 (10.1) | |
| Visceral pulmonary | 128 (25.8) | 35 (24.1) | 20 (19.0) | 73 (29.6) | |
| Visceral nonpulmonary | 124 (24.9) | 39 (26.9) | 30 (28.6) | 55 (22.3) | |
| Visceral NOS | 6 (1.2) | 0 (0.0) | 3 (2.9) | 3 (1.2) | |
| Brain | 153 (30.8) | 30 (20.7) | 38 (36.2) | 85 (34.4) | |
| Unknown | 12 (2.4) | 3 (2.1) | 3 (2.9) | 6 (2.4) | |
| Liver metastases before first-line ICI, n (%) | .910 | ||||
| Present | 120 (24.1) | 34 (23.4) | 27 (25.7) | 59 (23.9) | |
| Absent | 377 (75.9) | 111 (76.6) | 78 (74.3) | 188 (76.1) | |
| BMI, median (IQR) | 27.7 (24.4-31.6) | 27.6 (24.5-31.7) | 28.9 (25.1-32.9) | 27.4 (24.1-31.1) | .153 |
| Year of start of first-line ICI,c No. (%) | .288 | ||||
| 2015 or earlier | 14 (2.8) | 7 (50.0) | 0 (0.00) | 7 (50.0) | |
| 2016 | 52 (10.5) | 15 (28.8) | 13 (25.0) | 24 (46.2) | |
| 2017 | 48 (9.7) | 11 (22.9) | 8 (16.7) | 29 (60.4) | |
| 2018 | 81 (16.3) | 25 (30.9) | 15 (18.5) | 41 (50.6) | |
| 2019 | 99 (19.9) | 36 (36.4) | 16 (16.2) | 47 (47.5) | |
| 2020 | 99 (19.9) | 24 (24.2) | 26 (26.3) | 49 (49.5) | |
| 2021 | 96 (19.3) | 24 (25.0) | 24 (25.0) | 48 (50.0) | |
| 2022 | 8 (1.6) | 3 (37.5) | 3 (37.5) | 2 (25.0) | |
| Steroids immediately before therapy, No. (%) | .825 | ||||
| No steroids | 416 (83.7) | 123 (84.8) | 86 (81.9) | 207 (83.8) | |
| Received steroids | 81 (16.3) | 22 (15.2) | 19 (18.1) | 40 (16.2) | |
| Dominant tumor UV signature, No. (%) | <.001 | ||||
| Not observed | 22 (4.4) | 9 (6.2) | 9 (8.6) | 4 (1.6) | |
| Observed | 348 (70.0) | 26 (17.9) | 93 (88.6) | 229 (92.7) | |
| Unknown | 127 (25.6) | 110 (75.9) | 3 (2.9) | 14 (5.7) | |
| Site of TMB tumor specimen, No. (%) | <.001 | ||||
| Skin | 95 (19.1) | 30 (20.7) | 15 (14.3) | 50 (20.2) | |
| Nonvisceral | 170 (34.2) | 63 (43.4) | 43 (41.0) | 64 (25.9) | |
| Visceral pulmonary | 97 (19.5) | 13 (9.0) | 18 (17.1) | 66 (26.7) | |
| Visceral nonpulmonary | 63 (12.7) | 24 (16.6) | 14 (13.3) | 25 (10.1) | |
| Brain/CNS | 54 (10.9) | 10 (6.9) | 9 (8.6) | 35 (14.2) | |
| Other/unspecified | 18 (3.6) | 5 (3.5) | 6 (5.7) | 7 (2.8) | |
| Days from tumor specimen to start of first-line ICI, median (IQR) | 40.0 (22.0-78.0) | 37.0 (15.0-70.0) | 37.0 (22.0-72.0) | 43.0 (23.0-91.0) | .105 |
| Tumor specimen before or after initiation of ICI, No. (%) | .034 | ||||
| Before initiating ICI | 467 (94.0) | 130 (89.7) | 101 (96.2) | 236 (95.5) | |
| After initiating ICI | 30 (6.0) | 15 (10.3) | 4 (3.8) | 11 (4.5) | |
| Days from CGP report to start of first-line ICI, median (IQR) | –9.0 (–27.0 to 16.0) | –12.0 (–40.0 to 15.0) | –7.0 (–17.0 to 15.0) | –8.0 (–26.5 to 16.5) | .243 |
| CGP report availability relative to start of first-line ICI, No. (%) | .969 | ||||
| Report issued at least 14 days before first dose of ICI | 133 (26.8) | 38 (26.2) | 29 (27.6) | 66 (26.7) | |
| Report issued after 14 days before first dose of ICI | 364 (73.2) | 107 (73.8) | 76 (72.4) | 181 (73.3) |
Abbreviations: CGP, comprehensive genomic profiling; ECOG, Eastern Cooperative Oncology Group; ICI, immune checkpoint inhibitor; mut/Mb, mutation per megabase; NOS, not otherwise specified; TMB, tumor mutation burden; TMB-H, TMB-high; TMB-L, TMB-low; TMB-VH, very high TMB; ULN, upper limit of normal; UV, ultraviolet radiation.
P values indicate Kruskal-Wallis tests for continuous variables and chi-square/Fisher's exact tests for categorical variables.
Site indicated describes the highest M subcategory (M1a/b/c/d)–associated metastatic site present per patient; patients are counted once only.
Percentages shown in italics for this section are calculated row-wise (ie, percentage of all patients for that calendar year).
Prognostic Value of TMB and ICI Outcomes in First-Line Metastatic Melanoma
TMB-H was prognostic for improved rwPFS and OS in both monotherapy (hazard ratio [HR], 0.45 [95% CI, 0.32 to 0.63]; P < .001; HR, 0.61 [95% CI, 0.41 to 0.90]; P = .01, respectively) and dual ICI (HR, 0.67 [95% CI, 0.49 to 0.90]; P = .009; HR, 0.61 [95% CI, 0.42 to 0.88]; P = .007, respectively) patients (Fig 2). TMB-H remained prognostic for better rwPFS and OS outcomes even after adjusting for established prognostic features (Data Supplement, Fig S3). When repeated in exploratory subcohorts defined by the presence or absence of BRAF V600E/K mutations, a similar magnitude effect of TMB on rwPFS was seen regardless of BRAF status (TMB-H v -L, HR, 0.60 [95% CI, 0.41 to 0.89] in BRAFmut; HR, 0.54 [95% CI, 0.40 to 0.71], in BRAFwt; Data Supplement, Fig S4). OS results were likely confounded by the availability of effective second-line targeted therapy for BRAFmut patients, leading to better OS outcomes in BRAFmut patients than in their BRAFwt counterparts (Data Supplement, Fig S4). However, inspection of survival curves across the four strata defined by both TMB and BRAF status revealed this confounding effect to be relevant only in the TMB-L population. This may reflect a greater dependence on salvage therapy in the TMB-L subgroup who derive less benefit from first-line immunotherapy than TMB-H patients (Data Supplement, Fig S5).
FIG 2.
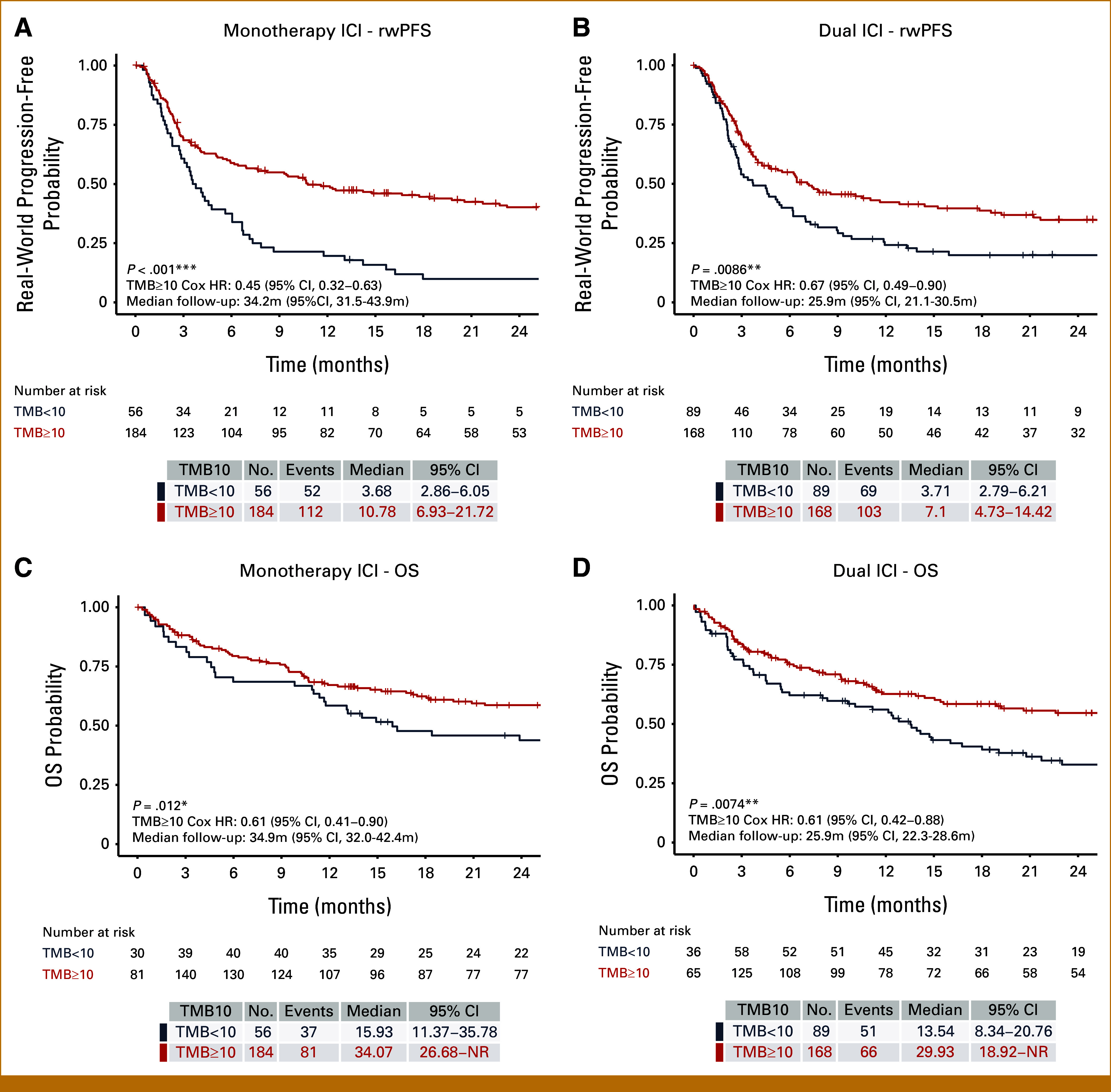
rwPFS and OS stratified by TMB using a 10 mutations per megabase cutoff (TMB10) and ICI regimen. Kaplan-Meier plots are shown stratified by TMB10 for (A) rwPFS in monotherapy ICI-treated patients, (B) rwPFS in dual ICI-treated patients, (C) OS in monotherapy ICI-treated patients, and (D) OS in dual ICI-treated patients. Significance levels: *P < .05, **P < .01, ***P < .001. HR, hazard ratio; ICI, immune checkpoint inhibitor; OS, overall survival; rwPFS, real-world progression-free survival; TMB, tumor mutation burden.
To determine whether a more stringent cutoff for TMB delivered more predictive power, we examined ICI outcomes using stepwise TMB thresholds of 10 and 20 mut/Mb, designating TMB <10 low, TMB 10-19 high, and TMB ≥20 very high. When compared with TMB <10 in patients receiving monotherapy ICI, rwPFS and OS were highest in TMB ≥20 (HR, 0.40 [95% CI, 0.28 to 0.58]; and HR, 0.58 [95% CI, 0.39 to 0.87], respectively) and intermediate for TMB10-19 (Figs 3A and 3B). In patients receiving dual ICI, both TMB 10-19 and TMB ≥20 patients demonstrated better rwPFS than TMB <10 patients but the effect was unexpectedly greatest, and statistically significant, for TMB 10-19 (HR, 0.55 [95% CI, 0.37 to 0.83]; and HR, 0.74 [95% CI, 0.53 to 1.03], for TMB 10-19 and TMB ≥20, respectively; Fig 3C). This inverted relationship between TMB and dual ICI outcome was also observed for OS (TMB 10-19: HR, 0.45 [95% CI, 0.26 to 0.76]; and TMB ≥20: HR, 0.72 [95% CI, 0.48 to 1.06], relative to TMB <10; Fig 3D). Significant clinicopathologic differences in the dual-ICI treated cohort may have contributed to this unexpected result, with more BRAFmut (V600E/K 44.4% in TMB10-19 v 23.8% in TMB ≥20; P < .001) but less brain metastases (34.9% TMB 10-19 v 44.8% TMB ≥20; P = .003) and male patients (male 65.1% TMB10-19 v 79.0% TMB ≥20; P = .007) in the TMB10-19 group (Data Supplement, Table S3). In addition, with multivariable analysis, similar effect sizes were seen comparing either TMB ≥20 or TMB 10-19 versus TMB <10, suggesting that the higher TMB groups respond similarly to dual-ICI once adjusted for clinical covariates (Data Supplement, Fig S6).
FIG 3.
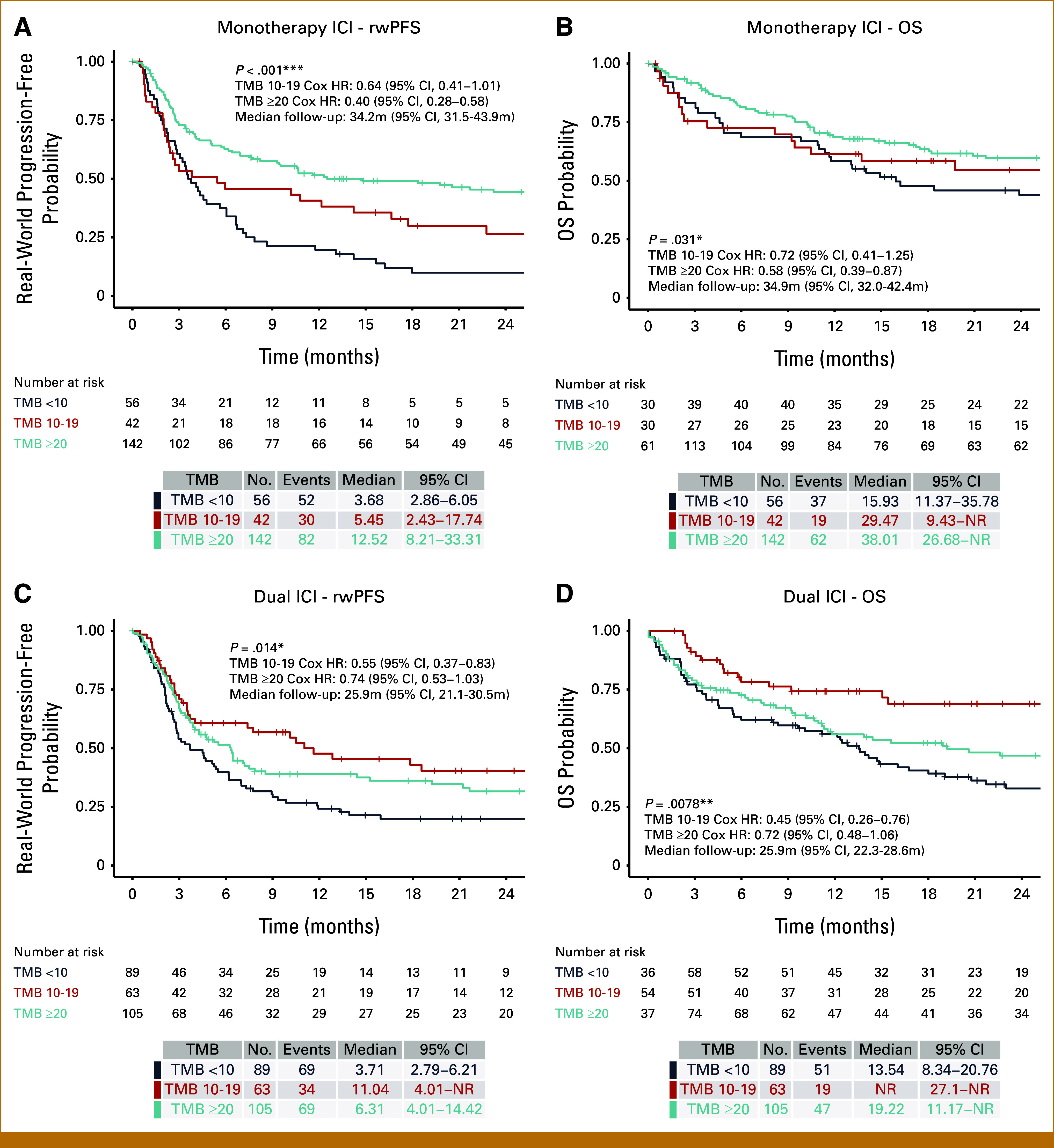
rwPFS and OS for first-line monotherapy or dual ICI stratified by TMB at 10 or 20 muts/Mb cutoffs. Kaplan-Meier plots stratified by TMB into <10, 10-19, and ≥20 muts/Mb are shown for (A) rwPFS in monotherapy ICI, (B) OS in monotherapy ICI, (C) rwPFS in dual ICI, and (D) OS in dual ICI patients. Both TMB-H and TMB-VH exhibited higher rwPFS and OS compared with TMB-L patients when treated with first-line ICI regimens. Survival outcomes were numerically greatest for TMB-VH patients after monotherapy ICI, but greatest in TMB-H patients after dual ICI. Significance levels: *P < .05, **P < .01, ***P < .001. HR, hazard ratio; ICI, immune checkpoint inhibitor; muts/Mb, mutations per megabase; OS, overall survival; rwPFS, real-world progression-free survival; TMB, tumor mutation burden; TMB-H, TMB-high; TMB-L, TMB-low; TMB-VH, very high TMB.
Characteristics of Patients With Durable Benefit
A total of 24 patients fit the definition of exceptionally durable benefit, defined as >48 months of rwPFS; 92% (22/24) had TMB-H. These patients had several characteristics suggestive of better prognosis, including fewer brain and nonpulmonary visceral metastases, more pulmonary metastases, and less elevated LDH (Data Supplement, Table S4). These patients more commonly received pembrolizumab monotherapy.
Regimen, But Not TMB, Predicts Immune-Related Adverse Events to ICI
Taking on-therapy steroid use as a proxy for immune-related adverse events, we found no difference between TMB-H and TMB-L patients (10 mut/Mb threshold; P = .28). As expected, dual-ICI patients were more likely to use steroids on therapy than mono-ICI patients (P < .001; Data Supplement, Figs S7A and S7B), findings mirrored in the time-to-steroid analysis (Data Supplement, Figs S7C-S7F).
Predictive Value of Combined TMB and BRAF Assessment
We next performed exploratory multivariable modeling to compare effectiveness of treatment regimen, adjusted by established prognostic factors, within subcohorts defined by TMB at 10 or 20 mut/Mb thresholds. In the TMB ≥20 subgroup, the HR for dual ICI versus mono ICI was 1.54 (95% CI, 1.08 to 2.19) for rwPFS and 1.52 (95% CI, 0.99 to 2.33) for OS versus TMB <20, with wider separation of HR estimates than seen when dichotomized at the 10 mut/Mb threshold, suggesting that patients with TMB ≥20 may benefit from choosing monotherapy over dual ICI (Data Supplement, Fig S8). Subdividing these cohorts further by BRAF V600 mutation status, we observed that the significant rwPFS signal in the TMB-VH cohort was driven by BRAFmut patients (HR for dual ICI, 4.92 [95% CI, 1.4 to 17.2]), supported by a similar trend in OS (Fig 4; Data Supplement, Fig S9). Notably, while the majority of BRAFwt patients were TMB-VH (203 TMB-VH v 140 not TMB-VH), the reverse was true for BRAFmut patients (44 TMB-VH v 110 not TMB-VH; Data Supplement, Fig S10).
FIG 4.

Dual versus monotherapy ICI outcomes stratified by TMB threshold and BRAF mutation status. Forest plot showing hazard ratios for dual versus monotherapy ICI in multivariable Cox proportional hazards modeling of rwPFS in subcohorts defined by TMB at various cutpoints and BRAF V600E/K mutation status as shown. Multivariable models using data for all 497 patients adjusted for age at start of therapy, race, ECOG performance status, sex, academic versus community practice type, CDKN2A/B status, LDH, sites of metastasis, and body mass index. Patients with melanoma with both a BRAF V600E/K mutation and TMB ≥20 mut/Mb may derive greater rwPFS from monotherapy than dual ICI. ECOG, Eastern Cooperative Oncology Group; ICI, immune checkpoint inhibitor; LDH, lactate dehydrogenase; mut/Mb, mutation per megabase; rwPFS, real-world progression-free survival; TMB, tumor mutation burden.
Sensitivity Analyses Limited to Patients Who Received Their CGP Results Before ICI Initiation
Sensitivity analyses repeating the main primary analyses were performed on a subcohort where all patients received their CGP results before ICI initiation (197 patients included). These findings showed directional alignment with our primary analysis results, although sample sizes and therefore statistical power were greatly reduced (Data Supplement, Figs S11-S14 and Tables S5 and S6), indicating that the statistical measures undertaken to mitigate the effects of left truncation in the data set were sufficient.
DISCUSSION
Simple biomarkers are urgently needed to guide ICI risk-benefit decision making with patients, particularly for dual ICI. In this large, multicenter, real-world study of ICI in metastatic melanoma, we confirm that high TMB is a predictor of improved survival outcomes after ICI, even after adjusting for established prognostic factors, and was a feature of patients who derived exceptionally durable benefit (>48 months of rwPFS). The continuous association between TMB and rwPFS in patients receiving monotherapy ICI was expected and consistent with other studies9,17,24-26 but not matched in patients receiving dual ICI. Patients with extreme levels of TMB (≥20 mut/Mb) benefited less from dual ICI than those with intermediate TMB (10 ≤ TMB < 20), suggesting not only that dual ICI may be unnecessary for patients with TMB-VH, but could be harmful. Furthermore, exploratory analyses identified a small subgroup of BRAFmut TMB-VH patients for whom anti–PD-1 monotherapy appears optimal but these results require further validation against the current standard-of-care recommendation for dual ICI in fit BRAFmut patients.
Our identification of inferior outcomes to dual ICI in TMB-VH patients was highly unexpected. TMB-VH patients more frequently had brain metastases at treatment initiation and were more frequently male, both likely to confer an overall worse prognosis. Conversely, TMB-VH patients less frequently harbored BRAF V600E/K mutations, consistent with findings from many cancer types, which suggest that founder driver mutations are associated with lower TMB.27 Clinicopathologic differences in our cohort may therefore partly explain these results as evidenced by the diminished differences seen after multivariable analysis.
Our data confirm and extend upon post hoc analyses of the CheckMate-067 study and a recently reported registry study including 825 patients with BRAFwt melanoma treated with first-line ICI, by showing that dual ICI confers little additional benefit to patients with metastatic melanoma without BRAF V600E/K mutations at any TMB stratum, including in patients with brain metastases at baseline.28,29 However, our real-world data incorporating TMB suggest that even in BRAF V600E/K-mutated cases, the incremental benefit of dual therapy may no longer hold true at very high levels of mutational burden. This may indicate distinct biology underlying the infrequent co-occurrence of BRAFmut and TMB-VH (9% of the total study cohort), suggesting that in these patients, BRAF V600 mutations are not classic driver events and could be expected to respond to ICI more like BRAFwt/TMB-VH than BRAFmut/TMB-L cases. Because of the small subset size, these findings should be interpreted cautiously, but further prospective evaluation of simultaneous TMB and BRAF-guided ICI regimen selection is warranted.
In our observational cohort, most patients commenced ICI before TMB results became available, indicating that treatment choice was not informed by TMB. The clinicians' reasons for requesting CGP evaluation are not known and may have been intended to inform subsequent lines of therapy. Our primary analyses therefore evaluate the relationship between TMB and ICI outcome, not whether clinician decision making on the basis of knowledge of the TMB improves survival outcomes. Nevertheless, our data suggest that coassessment of BRAF status and TMB could be informative to guide both prognostication and regimen choice in the first-line setting, thus CGP testing results should optimally be available before commencement of therapy.
It is also worth noting that our study cohort was treated before the March 2022 approval of coformulated nivolumab and relatlimab-rmbw—a LAG-3–targeting ICI—for patients with previously untreated advanced melanoma. Clinical trial data suggest that this new ICI combination has superior efficacy to nivolumab monotherapy and a safety profile favorable to ipilimumab plus nivolumab, and it has already been adopted into clinical practice guidelines as a copreferred frontline option with ipilimumab plus nivolumab.30,31 Future work may consider replicating our study to include nivolumab plus relatlimab as an alternative dual ICI arm.
Two major technical considerations are relevant to studies of TMB. First, tumor-based TMB can be assessed by a wide variety of NGS assays of whole genome, whole exome, or a large targeted gene panel,32 the last being cheaper, quicker, and more widely available.2 These assays remain methodologically and analytically unstandardized,33 making comparisons of TMB performance in different clinical settings and studies challenging,2 despite efforts to deal with assay-based diversity in TMB estimation.33 This study used the only current FDA-approved TMB companion diagnostic for an ICI. Second, although the use of a TMB biomarker threshold simplifies clinical decision making, the optimal threshold for patients with melanoma remains undefined. The ≥10 mut/Mb threshold validated using the FoundationOne CDx assay in the KEYNOTE-158 trial3 has become widely used in melanoma, but evidence supports a more proportional effect of TMB in metastatic melanoma1 and other cancer types34 treated with PD-1 inhibitors. Our data emphasize that care is required when applying strict TMB thresholds to guide treatment selection, given the impact of additional clinical covariates and potential interaction with BRAF status at high levels of TMB.
Our findings confirm the real-world utility of TMB measurement to predict ICI outcome for patients with metastatic melanoma and it may have underutilized potential to impact regimen choice when considered together with BRAF status and established prognostic clinicopathologic factors. Further research is needed to integrate TMB metrics into advanced multifactor biomarker predictive tools as this may offer patients a safe pathway to ICI regimen deintensification and resulting improved tolerability.
ACKNOWLEDGMENT
The authors thank the patients whose data made this research possible, the clinical and laboratory staff at Foundation Medicine, and the team at Flatiron Health.
SUPPORT
Supported by Foundation Medicine, a wholly owned subsidiary of Roche, which is a for-profit company and producer of FDA-regulated molecular diagnostics. Authors employed by Foundation Medicine were involved in the design and conduct of the study, analysis, interpretation of the data, preparation, review, and approval of the manuscript.
DATA SHARING STATEMENT
The data that support the findings of this study originated by Flatiron Health, Inc and Foundation Medicine, Inc. These deidentified data may be made available upon request and are subject to a license agreement with Flatiron Health and Foundation Medicine; interested researchers should contact <cgdb-fmi@flatiron.com> and <dataaccess@flatiron.com> to determine licensing terms.
AUTHOR CONTRIBUTIONS
Conception and design: Miles C. Andrews. Gerald Li, Ryon P. Graf, Jerry Mitchell, Ali Aboosaiedi, Mark Shackleton, Mahesh Iddawela, Richard S.P. Huang
Administrative support: Ali Aboosaiedi, Mark Shackleton, Richard S.P. Huang
Provision of study materials or patients: Richard S.P. Huang
Collection and assembly of data: Miles C. Andrews. Gerald Li, Richard S.P. Huang
Data analysis and interpretation: Miles C. Andrews. Gerald Li, Ryon P. Graf, Virginia A. Fisher, Harriet O’Rourke, Mark Shackleton, Mahesh Iddawela, Geoffrey R. Oxnard, Richard S.P. Huang
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Miles C. Andrews
Honoraria: MSD Australia, Pierre Fabre, MSD Australia (Inst)
Consulting or Advisory Role: BMS Australia (Inst)
Research Funding: MSD Australia (Inst)
Patents, Royalties, Other Intellectual Property: US20220016188A1 Methods and compositions for treating cancer; co-inventor, patent pending (Inst)
Gerald Li
Employment: Foundation Medicine
Stock and Other Ownership Interests: Roche
Ryon P. Graf
Employment: Foundation Medicine
Stock and Other Ownership Interests: Roche
Travel, Accommodations, Expenses: Epic Sciences, Foundation Medicine
Virginia A. Fisher
Employment: Foundation Medicine
Stock and Other Ownership Interests: Roche
Jerry Mitchell
Employment: Seagen, Foundation Medicine, Immunogen
Stock and Other Ownership Interests: Seagen, Roche, Immunogen
Ali Aboosaiedi
Employment: Foundation Medicine
Stock and Other Ownership Interests: Roche
Travel, Accommodations, Expenses: Foundation Medicine
Harriet O'Rourke
Travel, Accommodations, Expenses: Merck
Mark Shackleton
Patents, Royalties, Other Intellectual Property: Royalty payments from discoveries that led to venetoclax
Other Relationship: MSD, BMS
Mahesh Iddawela
Consulting or Advisory Role: MSD Oncology
Speakers' Bureau: BMS GmbH & Co KG
Travel, Accommodations, Expenses: Pfizer
Geoffrey R. Oxnard
This author is a member of the JCO Precision Oncology Editorial Board. Journal policy recused the author from having any role in the peer review of this manuscript.
Employment: Foundation Medicine, LOXO at Lilly
Stock and Other Ownership Interests: Roche, Lilly
Richard S.P. Huang
Employment: Roche/Foundation Medicine
Stock and Other Ownership Interests: Roche
Patents, Royalties, Other Intellectual Property: Patent on IHC, provisional patent on biomarkers and biomarker methodology
No other potential conflicts of interest were reported.
REFERENCES
- 1.Dousset L, Poizeau F, Robert C, et al. : Positive association between location of melanoma, ultraviolet signature, tumor mutational burden, and response to anti-PD-1 therapy. JCO Precis Oncol 10.1200/PO.21.00084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fumet JD, Truntzer C, Yarchoan M, et al. : Tumour mutational burden as a biomarker for immunotherapy: Current data and emerging concepts. Eur J Cancer 131:40-50, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marabelle A, Fakih M, Lopez J, et al. : Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol 21:1353-1365, 2020 [DOI] [PubMed] [Google Scholar]
- 4.Carbone DP, Reck M, Paz-Ares L, et al. : First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med 376:2415-2426, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rizvi H, Sanchez-Vega F, La K, et al. : Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol 36:633-641, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rizvi NA, Hellmann MD, Snyder A, et al. : Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348:124-128, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hellmann MD, Callahan MK, Awad MM, et al. : Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell 33:853-861.e4, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bellmunt J, de Wit R, Fradet Y, et al. : Putative biomarkers of clinical benefit with pembrolizumab in advanced urothelial cancer: Results from the KEYNOTE-045 and KEYNOTE-052 landmark trials. Clin Cancer Res 28:2050-2060, 2022 [DOI] [PubMed] [Google Scholar]
- 9.Samstein RM, Lee CH, Shoushtari AN, et al. : Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 51:202-206, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cristescu R, Aurora-Garg D, Albright A, et al. : Tumor mutational burden predicts the efficacy of pembrolizumab monotherapy: A pan-tumor retrospective analysis of participants with advanced solid tumors. J Immunother Cancer 10:e003091, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu Y, Xu J, Du C, et al. : The predictive value of tumor mutation burden on efficacy of immune checkpoint inhibitors in cancers: A systematic review and meta-analysis. Front Oncol 9:1161, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.FDA : FDA approves pembrolizumab for adults and children with TMB-H solid tumors, 2020. https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-adults-and-children-tmb-h-solid-tumors
- 13.Schrock AB, Ouyang C, Sandhu J, et al. : Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann Oncol 30:1096-1103, 2019 [DOI] [PubMed] [Google Scholar]
- 14.Ready N, Hellmann MD, Awad MM, et al. : First-line nivolumab plus ipilimumab in advanced non-small-cell lung cancer (CheckMate 568): Outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J Clin Oncol 37:992-1000, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banchereau R, Leng N, Zill O, et al. : Molecular determinants of response to PD-L1 blockade across tumor types. Nat Commun 12:3969, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Snyder A, Makarov V, Merghoub T, et al. : Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371:2189-2199, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodi FS, Wolchok JD, Schadendorf D, et al. : TMB and inflammatory gene expression associated with clinical outcomes following immunotherapy in advanced melanoma. Cancer Immunol Res 9:1202-1213, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Q, Gossai A, Monroe S, et al. : Validation analysis of a composite real-world mortality endpoint for patients with cancer in the United States. Health Serv Res 56:1281-1287, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frampton GM, Fichtenholtz A, Otto GA, et al. : Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31:1023-1031, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milbury CA, Creeden J, Yip WK, et al. : Clinical and analytical validation of FoundationOne®CDx, a comprehensive genomic profiling assay for solid tumors. PLoS One 17:e0264138, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.R Core Team : R: A Language and Environment for Statistical Computing. Vienna, Austria, R Foundation for Statistical Computing, 2022 [Google Scholar]
- 22.Berger ML, Mamdani M, Atkins D, et al. : Good research practices for comparative effectiveness research: Defining, reporting and interpreting nonrandomized studies of treatment effects using secondary data sources: The ISPOR good research practices for retrospective database analysis task force report—Part I. Value Health 12:1044-1052, 2009 [DOI] [PubMed] [Google Scholar]
- 23.Griffith SD, Tucker M, Bowser B, et al. : Generating real-world tumor burden endpoints from electronic health record data: Comparison of RECIST, radiology-anchored, and clinician-anchored approaches for abstracting real-world progression in non-small cell lung cancer. Adv Ther 36:2122-2136, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Allen EM, Miao D, Schilling B, et al. : Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350:207-211, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodman AM, Kato S, Bazhenova L, et al. : Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther 16:2598-2608, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ott PA, Bang YJ, Piha-Paul SA, et al. : T-cell-inflamed gene-expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE-028. J Clin Oncol 37:318-327, 2019 [DOI] [PubMed] [Google Scholar]
- 27.Singal G, Miller PG, Agarwala V, et al. : Association of patient characteristics and tumor genomics with clinical outcomes among patients with non-small cell lung cancer using a clinicogenomic database. JAMA 321:1391-1399, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. : Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 381:1535-1546, 2019 [DOI] [PubMed] [Google Scholar]
- 29.Franklin C, Mohr P, Bluhm L, et al. : Brain metastasis and survival outcomes after first-line therapy in metastatic melanoma: A multicenter DeCOG study on 1704 patients from the prospective skin cancer registry ADOREG. J Immunother Cancer 11:e005828, 2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tawbi HA, Schadendorf D, Lipson EJ, et al. : Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med 386:24-34, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.National Comprehensive Cancer Network : NCCN Clinical Practice Guidelines in Oncology. Melanoma: Cutaneous. Version 1.2024, 2024. https://www./NCCN.org
- 32.Buttner R, Longshore JW, Lopez-Rios F, et al. : Implementing TMB measurement in clinical practice: Considerations on assay requirements. ESMO Open 4:e000442, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vega DM, Yee LM, McShane LM, et al. : Aligning tumor mutational burden (TMB) quantification across diagnostic platforms: Phase II of the Friends of Cancer Research TMB Harmonization Project. Ann Oncol 32:1626-1636, 2021 [DOI] [PubMed] [Google Scholar]
- 34.Yarchoan M, Hopkins A, Jaffee EM: Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med 377:2500-2501, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study originated by Flatiron Health, Inc and Foundation Medicine, Inc. These deidentified data may be made available upon request and are subject to a license agreement with Flatiron Health and Foundation Medicine; interested researchers should contact <cgdb-fmi@flatiron.com> and <dataaccess@flatiron.com> to determine licensing terms.