

Abstract
Congenital nasal pyriform aperture stenosis is a newly defined clinical entity that causes nasal airway obstruction in neonates due to the narrowing of the pyriform aperture, which is the narrowest and most anterior portion of the nasal airway. As newborns are obligate nasal breathers except when crying, a child with bilateral nasal pyriform aperture obstruction presents as an acute airway emergency, resulting in apnea and cyanosis. This entity should be kept in the differential diagnosis of any neonate or infant presenting with signs and symptoms of upper airway obstruction.
Keywords: Choanal atresia, congenital nasal airway obstruction, nasal pyriform aperture stenosis
INTRODUCTION
The most common cause of nasal obstruction in newborns is mucosal edema; less frequent ones include congenital nasal pyriform aperture stenosis, bilateral choanal atresia, and stenosis of the nasal bones.[1] Nasal pyriform aperture stenosis is due to excessive growth of the nasal processes of the maxilla, where the anterior 1/3rd of the nasal airway is obscured. The paired openings connecting nasal cavities to the nasopharynx are the nasal choanae, which sometimes fail to recanalize due to some membranous or bony obstruction.[2,3] As newborns are obligate nasal breathers except when crying, a child with a bilateral nasal obstruction presents as an acute airway emergency.[4] In our case report of a preterm, low birth weight baby who had obstruction of the anterior nasal cavity on one side with pyriform aperture stenosis and choanal atresia on the other side with alar agenesis, there was soft tissue in the left posterior nasal cavity (Meningocoel). Our aim is to review the differential diagnosis, associated anomalies, and management of bilateral nasal obstruction in newborns that present as acute airway emergencies.
CASE REPORT
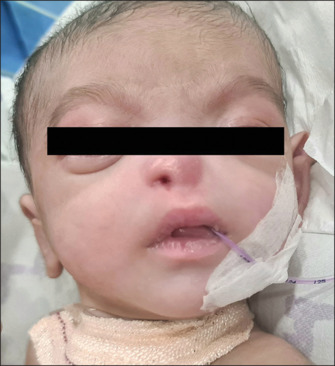
Here we present a case of a preterm (35 weeks) single baby boy born to a gravida 1 para 1 mother with a low birth weight, delivered by cesarean section, who had membranous or pyriform aperture stenosis on the right and choanal atresia with alar agenesis on the left side, hypertelorism, and accessory auricle on the right. He was shifted to our hospital with a tracheostomy in situ from outside [Figure 1]. As per the history given by the mother, the baby developed respiratory distress and apnoeic spells, for which he was initially given mechanical ventilation, then eventually shifted to head box oxygen with an endotracheal tube in situ. The baby was desaturated and developed possible cardiogenic shock (the exact nature of the shock was not mentioned in the medical records of the previous hospital), for which he was resuscitated with a normal saline bolus and inotropes. The chest X-ray showed pneumonic patches. He had multiple episodes of convulsions, and a calcium correction was done in view of hypocalcemia. A tracheostomy was done outside. Blood culture showed the growth of Pseudomonas stutzeri sensitive to Meropenam, which was given for 14 days. Again, the child developed respiratory distress; bronchoalveoalar lavage sent for testing showed growth of Acinetobacter baumanii; sensitive antibiotics were given but not relieved; BAL repeated after 2 weeks showed growth of Burkholderia cepacian; and sensitive antibiotics were upgraded. For repeated convulsions, a CSF study was done, which came out to be normal. However, anticonvulsants with mitochondrial cocktails were started after review by a neurologist, after which convulsions were controlled. When the baby was shifted to our institute in the NICU from the child specialty hospital, he was 45 days old and 2.8 kg in weight. He was receiving feeding through an orogastric tube. There was no external nasal or oral deformity except for left nasal alar agenesis. The baby was active, maintaining oxygen saturation with a no. 3.5 uncuffed tracheostomy tube in situ. There were no obvious signs of respiratory distress. The trial to pass the infant feeding tube through the right nasal cavity failed. CT nose and paranasal sinuses done outside showed posterior nasal narrowing on both sides with obstruction on the left side. Maxillary and ethmoid sinuses were also developed with poorly formed bony outlines. The CT nose and paranasal sinuses showed a narrow anterior nasal cavity with alar agenesis on the left, and in the axial plane, the distance between the nasal processes of both maxilla was <0.75 mm [Figure 2]. The coronal section showed a hypoplastic left nasal cavity with a deviated nasal septum to the left, and superior to this level, it is filled with soft tissue density with a poorly visualized bony interface between the intracranial and superior left nasal cavities. The sella and suprasellar regions appear normal [Figure 3].
Figure 1.
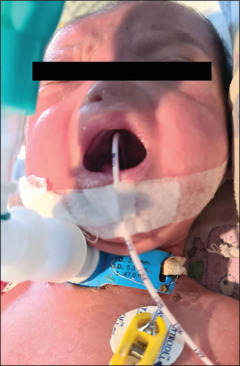
Preoperative picture of the tracheostomised baby with nasal pyriform aperture stenosis
Figure 2.
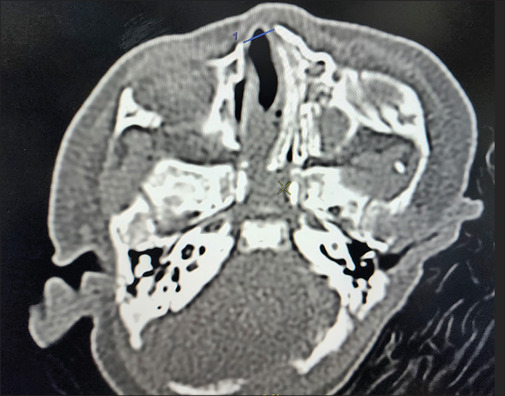
Axial section showed the distance between nasal processes of both maxilla was <0.75 mm
Figure 3.
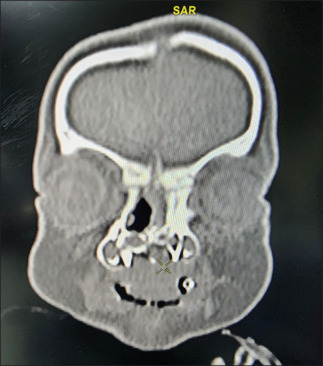
Coronal section showed hypoplastic left nasal cavity with deviated nasal septum to left
After assessing the clinical and radiological findings and after discussion with pediatric consultants, neurosurgeons, and anesthesiologists, a team decision was taken to proceed with surgery to address the right nasal obstruction for a narrowed pyriform aperture through an endonasal approach. Preoperative examination showed membranous obstruction in the anterior nasal valve area and partial bony and partial membranous obstruction on the right side. Division of pyriform area obstruction is done with the help of malleable uterine sound, and the position of its tip at choanae is confirmed by direct visualization of choanae by placing a 70° Hopkin’s rod endoscope transorally, followed by serial dilatations with a intravenous cannula, a thicker needle, and a progressive suction catheter. Then an endotracheal tube no. 2.5 was left as a stent in the right nasal cavity [Figure 4]. After 2 weeks, the stent was removed, and serial dilatations started. After 6 weeks of surgery, the tracheostomy tube was decannulated, and strapping was done [Figure 5]. Slowly, dilatations were progressed up to the no. 3.5 endotracheal tube. After 8 months of surgery, the child is better, breathes through the nose, and takes a diet orally. As per discussion with the neurosurgeon on the other side, alar agenesis and meningocele will be operated on after the child is 1 year old, or at least until he attains 10 kg of body weight.
Figure 4.
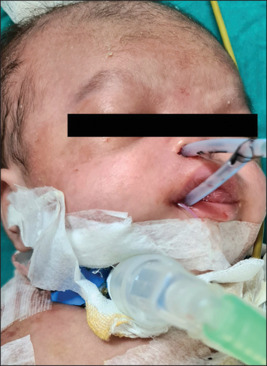
Endotracheal tube no. 2.5 was left as a stent in the right nasal cavity
Figure 5.
6 weeks of post-surgery tracheostomy tube was decannulated and strapping was done
DISCUSSION
The most common cause of nasal obstruction in newborns is mucosal edema; less frequent causes include congenital nasal pyriform aperture stenosis, bilateral choanal atresia, stenosis of the nasal bones, etc.[1] Anatomically, the pyriform aperture in the nose is formed by the nasal bones superiorly, the junction of the horizontal process of the maxilla and the anterior nasal spine inferiorly, and the lateral process of the maxilla laterally. This lateral process overgrowth leads to pyriform aperture stenosis. During fetal development, nasal choanae develop between the third and seventh weeks by rupture of the vertical epithelial fold between the olfactory groove and stomodeum. Due to incomplete resorption of the nasopharyngeal mesoderm, persistence of the nasobuccal membrane, buccopharyngeal membrane, or local misdirection of neural crest cell migration, this leads to congenital choanal atresia or nasal obstruction in newborn.[3,5] The incidence of congenital neonatal airway malformation is between 1:5000 and 1:8000 live births. Mostly unilateral (60%) than bilateral (40%). The male-female ratio is 1:2. Choanal atresia may be associated with various syndromes and anomalies, of which the most common is CHARGE syndrome (coloboma, heart disease, atresia choanae, growth and mental retardation, genital hypoplasia, and ear anomalies). Newborns are said to be obligate nasal breathers, so the symptoms of nasal obstruction become evident during feeding. Clinical presentation includes difficulty in breathing with episodes of acute respiratory distress with cyanosis, which relieves on crying and choking episodes on trial for oral feed as they are unable to breathe and swallow at the same time.[4] Differential diagnosis also includes choanal atresia, nasal trauma leading to septal hematoma, meningocele, meningoencephalocele, haemangiomas, or other skull base malformations.[5] Congenital nasal pyriform aperture stenosis was described by Douglas in 1952, and in 1989, it was mentioned as a cause of respiratory distress in children by E. Brown.[5,6] The diagnosis of the stenosis is confirmed if CT of the nose and paranasal sinuses in the axial planes at the level of the inferior meatus reveals a distance of <11 mm between the nasal processes, and clinically, a 2.2 mm fiberscope also cannot be passed through the anterior nasal cavity.[7,8] It might present as an isolated condition or as a part of a craniofacial abnormality like holoprosencephaly. It is recommended to investigate the hypothalamopituitary adrenal axis because anomalies of hypophysis can be observed in 40% of patients, leading to developmental defects.[9] Management includes conservative management followed by surgical intervention to develop the airway either through an endonasal or sublabial approach. In newborns due to poor exposure, small dimension, and risk of injury, the transnasal approach is not preferred.[10,11] The Prime aim of the management of bilateral nasal obstruction is to establish a secured airway. The management depends on the prognosis of the child, the severity of symptoms, and the level of obstruction. Conservative management with nasal humidification, steroids, aspirations, decongestant spray, etc., is given in cases with multiple anomalies and a poor prognosis[10] but in cases with severe initial presentations like respiratory distress and difficult feeding surgical management should be considered. Surgical access includes a transnasal or sublabial approach. In our case also we prepared for the sublabial approach by raising the subperiosteal flap, exposing the pyriform aperture area, and then dilating the aperture by drilling with a diamond burr and then giving stent.[12] But for multiple issues from difficult intra-venous access to various other difficulties like respiratory tract infection where general anaesthesia could not be given, we proceeded with the endonasal approach by serial dilation starting with intra venous cannula then thicker needle and progressive suction catheter increasing the size till we introduced the no. 2.5 endotracheal tube which we left as a stent.[10,13] Slowly in 2 weeks with serial dilatations we introduced the no. 3.5 endotracheal tube which we continued for the next few weeks.[14] The child comes for a follow-up every month (around 8 months now), no restenosis noted, taking oral feed, active, and playful. The complications associated with surgery include poor exposure and risk of trauma in the intranasal approach whereas trauma to periosteum, dental roots, nasolacrimal duct, nasal mucosa, and formation of granulation tissues post-surgery in the sublabial approach too. There are always risks of restenosis also. Gungor and Reierson reported a case of Pyriform stenosis dilation with a 7 mm balloon and then stenting for 12 days. There was no restenosis reported till 1-year follow-up.[13] The satisfactory opening is said to be achieved when we can push a 3.5 mm endotracheal tube through the nasal cavity which in our patient can be passed till now - 8 months of post-operative period.
CONCLUSION
Securing the airway in a child with bilateral nasal obstruction remains a matter of prime concern. The most important differential diagnosis to be kept in mind includes trauma, mucosal edema, congenital nasal pyriform aperture stenosis which is rare but lethal, bilateral choanal atresia, or mass effect due to lesions inside the nasal cavity. Surgical correction and stenting lead to successful management in these cases. A tracheostomy followed by management of nasal obstruction saved the child in our case. The child is thriving well with the patent Right nasal cavity. Our plan for the left side with alar agenesis and meningoencephalocoele is to proceed with a neurosurgeon when the child is at least 1 year of age or 10 kg by body weight.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Sesenna E, Leporati M, Brevi B, Oretti G, Ferri A. Congenital nasal pyriform aperture stenosis: Diagnosis and management. Ital J Pediatr. 2012;38:28–33. doi: 10.1186/1824-7288-38-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vishvanathan V, Wynne DM. Congenital nasal pyriform aperture stenosis: A report of 10 cases and literature review. Int J Pediatr Otorhinolaryngol. 2012;76:28–30. doi: 10.1016/j.ijporl.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 3.Zawawi F, McVey MJ, Campisi P. The pathogenesis of choanal atresia. JAMA Otolaryngol Head Neck Surg. 2018;144:758–9. doi: 10.1001/jamaoto.2018.1246. [DOI] [PubMed] [Google Scholar]
- 4.Smith MM, Island SL. Pediatric nasal obstruction. Otolaryngol Clin North Am. 2018;51:971–85. doi: 10.1016/j.otc.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Brown OE, Myer CM III, Manning SC. Congenital nasal pyriform aperture stenosis. Laryngoscope. 1989;99:86–91. doi: 10.1288/00005537-198901000-00016. [DOI] [PubMed] [Google Scholar]
- 6.Douglas B. The relief of vestibular nasal obstruction by partial resection of the nasal process of the superior maxilla. Plastic Reconstr Surg. 1952;9:42–51. [PubMed] [Google Scholar]
- 7.Belden CJ, Mancuso AA, Schmalfuss IM. CT features of congenital nasal pyriform aperture stenosis: Initial experience. Radiology. 1999;213:495–501. doi: 10.1148/radiology.213.2.r99oc38495. [DOI] [PubMed] [Google Scholar]
- 8.Mohan S, Fuller JC, Ford SF, Lindsay RW. Diagnostic and therapeutic management of nasal airway obstruction: Advances in diagnosis and treatment. JAMA Facial Plast Surg. 2018;20:409–18. doi: 10.1001/jamafacial.2018.0279. [DOI] [PubMed] [Google Scholar]
- 9.Beregszaszi M, Leger J, Garel C, Simon D, Francois M, Hassan M, et al. Nasal pyriform aperture stenosis and absence of the anterior pituitary gland: Report of two cases. J Pediatr. 1996;128:858–61. doi: 10.1016/s0022-3476(96)70343-8. [DOI] [PubMed] [Google Scholar]
- 10.Meleca JB, Anne S, Hopkins B. Reducing the need for general anesthesia in the repair of choanal atresia with steroid- eluting stents: A case series. Int J Pediatr Otorhinolaryngol. 2019;118:185–7. doi: 10.1016/j.ijporl.2019.01.004. [DOI] [PubMed] [Google Scholar]
- 11.Murray S, Luo L, Quimby A, Barrowman N, Vaccaro JP, Caulley L. Immediate versus delayed surgery in congenital choanal atresia: A systematic review. Int J Pediatr Otorhinolaryngol. 2019;119:47–53. doi: 10.1016/j.ijporl.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Chan J, Ullas G, Narapa Reddy NVS. Completely endoscopic approach using a skeeter drill to treat bilateral congenital choanal atresia in a 33 week born pre-term baby. Indian J Otolaryngol Head Neck Surg. 2018;70:608–10. doi: 10.1007/s12070-018-1406-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gungor AA, Reierson DA. Balloon dilatation for congenital nasal pyriform aperture stenosis (CNPAS): A novel conservative technique. Am J Otolaryngol. 2014;35:439–42. doi: 10.1016/j.amjoto.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 14.Wine TM, Dedham K, Chi DH. Congenital nasal pyriform aperture stenosis: Is there a role for nasal dilatation? JAMA Otolaryngol Head Neck Surg. 2014;140:352–6. doi: 10.1001/jamaoto.2014.53. [DOI] [PubMed] [Google Scholar]