

Abstract
The therapeutic potential of suppressing polypyrimidine tract-binding protein 1 (Ptbp1) messenger RNA by viral transduction in a post-stroke dementia mouse model has not yet been examined. In this study, 3 days after cerebral ischemia, we injected a viral vector cocktail containing adeno-associated virus (AAV)-pGFAP-mCherry and AAV-pGFAP-CasRx (control vector) or a cocktail of AAV-pGFAP-mCherry and AAV-pGFAP-CasRx-SgRNA-(Ptbp1) (1:5, 1.0 × 1011 viral genomes) into post-stroke mice via the tail vein. We observed new mCherry/NeuN double-positive neuron-like cells in the hippocampus 56 days after cerebral ischemia. A portion of mCherry/GFAP double-positive astrocyte-like glia could have been converted into new mCherry/NeuN double-positive neuron-like cells with morphological changes. The new neuronal cells integrated into the dentate gyrus and recognition memory was significantly ameliorated. These results demonstrated that the in vivo conversion of hippocampal astrocyte-like glia into functional new neurons by the suppression of Ptbp1 might be a therapeutic strategy for post-stroke dementia.
Keywords: CasRx, Hippocampal neurogenesis, In vivo direct reprogramming, Ischemic stroke, PHP.eB, Ptbp1, Recognition memory
Subject terms: Biological techniques, Neuroscience, Molecular medicine, Neurology
Introduction
The rates of mortality and disability from stroke have increased dramatically because of the increased numbers and age of the global population over the past several decades1. Acute cognitive dysfunction after stroke is well known, and the recovery of cognitive function is slower than the recovery of motor symptoms2,3. Stroke is estimated to hasten the onset of dementia by approximately 10 years4. Long-term follow-up studies of 6–12 years have reported that persistent and more rapid cognitive decline, even when corrected for its pre-stroke cognitive decline rate, also resulted in increased mortality rates5. Post-stroke dementia can be divided into two categories: one caused by damage to areas directly involved in cognitive function related to stroke, and the other by neuronal death in the hippocampus caused by reactive oxygen species (ROS) and other factors generated by the resumption of blood flow associated with treatments6,7. It has been reported that while ischemic events can cause neurogenesis in the subgranular zone of the hippocampus and the subventricular zone of the lateral ventricles transiently, the majority of newborn neurons die within 2 weeks after stroke and only 2% of mature neurons are replaced poststroke8,9. Although the antioxidant edaravone is already used in clinical practice to alleviate the neuronal death by ROS, effective treatment for post-stroke dementia remains to be established.
During the first few weeks after a stroke, neuroplasticity is enhanced by changes in gene and protein expression. Therefore, in addition to rehabilitation training, the upregulation of intrinsic neurogenesis is considered a very important phenomenon in stroke treatment10,11. The RNA-binding protein polypyrimidine tract-binding protein 1 (Ptbp1) was reported to repress neuron-specific splicing and be expressed at high levels in non-neuronal cells but downregulated in neurons12. The suppression of Ptbp1 expression derepressed many neuronal genes in non-neuronal cells and induced cellular reprogramming of cultured mouse fibroblasts and N2a cells toward the neuronal lineage13. A recent study reported that the transient suppression of Ptbp1 using an antisense oligonucleotide administered intracerebroventricularly (i.c.v.) successfully activated hippocampal neurogenesis and generated new neurons in the dentate gyrus (DG) of adult mice14. However, i.c.v. injection is highly invasive, and its effects on the diseased brain remain unknown because it is normally used for healthy young and aged wild-type mice. In the present study, therefore, we examined the therapeutic potential of suppressing Ptbp1 messenger RNA (mRNA) by viral transduction in a post-stroke dementia mouse model15.
Results
To confirm the distribution of the viral vectors, we injected AAV(PHP.eB)-pGFAP-mCherry into mice via the tail vein (Fig. S1a). mCherry positive cells were detectable throughout the whole brain and were mainly present in cells positive for GFAP, an astrocytic marker (57.5% = 163 cells/283 mCherry positive cells from nine brain sections of the cortex from three mice) and S100β (astrocytic marker; 57.0% = 106 cells/186 mCherry positive cells). Fewer mCherry positive cells were detected in cells positively stained for Tuj1 (neuronal marker; 8.5% = 17 cells/200 mCherry positive cells), MAP2 (13.7% = 32 cells/232 mCherry positive cells), NeuN (13.9% = 48 cells/345 mCherry positive cells), and Iba1 (a microglial marker; 0.4% = 1 cells/274 mCherry positive cells). However, the mCherry positive cells did not coexpress the oligodendrocyte marker Olig2 (0% = 0 cells/201 mCherry positive cells) (Fig. S1b). A quantitative analysis of the number of positive cells for mCherry and each marker was conducted. No significant differences were observed in the hippocampus (GFAP: 61.7 ± 14.7 cells/0.54 mm2, S100β: 18.3 ± 5.2 cells/0.54 mm2, Iba1: 30.7 ± 4.9 cells/0.54 mm2, Tuj1: 60.7 ± 14.1 cells/0.54 mm2, MAP2: 68.0 ± 12.8 cells/0.54 mm2, NeuN: 67.0 ± 15.3 cells/0.54 mm2). However, significant differences were identified between GFAP and Iba1 (*p < 0.05), as well as between GFAP and Olig2 (*p < 0.05), in the cortex (GFAP: 54.3 ± 15.7 cells/0.54 mm2, S100β: 35.3 ± 6.9 cells/0.54 mm2, Iba1: 0.3 ± 0.5 cells/0.54 mm2, Tuj1: 5.7 ± 1.9 cells/0.54 mm2, MAP2: 10.7 ± 3.1 cells/0.54 mm2, NeuN: 12.3 ± 3.4 cells/0.54 mm2) (Fig. S1b).
Next, we injected a cocktail of AAV(PHP.eB)-pGFAP-mCherry and AAV(PHP.eB)-pGFAP-CasRx-Ptbp1 at 3, 7, or 14 days after 30 min tMCAO to evaluate the appropriate time point for viral injection (Fig. S2a). The pixel intensity of mCherry showed a significant time-dependent decrease on day 7 (0.74 ± 0.06 pixels fold change to day 3) or day 14 (0.31 ± 0.09 pixels fold change to day 3) (Fig. S2b). On the basis of these results, day 3 was selected as the appropriate time point for viral injection in the following experiments.
Next, we injected HBSS, a cocktail of AAV(PHP.eB)-pGFAP-mCherry and AAV(PHP.eB)-pGFAP-CasRx as a control vector, or a cocktail of AAV(PHP.eB)-pGFAP-mCherry and AAV(PHP.eB)-pGFAP-CasRx-SgRNA-Ptbp1 via the tail vein 3 days after 30 min tMCAO (Fig. 1a). We checked Bederson’s score, the rotarod test, and EBST to confirm improvements in motor function; however, there were no significant differences between the three groups (Fig. 1b–e). The NORT, which is associated with hippocampal recognition memory, was performed 56 days after tMCAO. There was no significant difference in the time to approach a familiar object between the three groups (HBSS: 43.9 ± 54.1 s, CasRx: 16.4 ± 22.4 s, CasRx-Ptbp1: 29.1 ± 21.1 s). Of note, only the CasRx-Ptbp1 group showed a significant increase in the novel object time compared with the familiar object time (HBSS: 55.8 ± 74.4 s, CasRx: 8.0 ± 6.6 s, CasRx-Ptbp1: 112.2 ± 95.5 s, *p < 0.05) (Fig. 1e). Next, we checked the cerebral ischemic volume, but there was no significant difference in ischemic volume between the three groups 56 days after tMCAO (HBSS: 98.9 ± 15.9 mm3, CasRx: 96.0 ± 15.9 mm3, CasRx-Ptbp1: 94.5 ± 14.8 mm3) (Fig. 2a,b). We also checked for alterations in hippocampal neurons (CA1, CA2, and CA3), but there were no significant differences in the ischemic side and non-ischemic side between the three groups (Figs. S3, S4). In addition, there was no significant increase in the number of mCherry/NeuN double-positive cells in the cortex between the CasRx and CasRx-Ptbp1 groups (CasRx: 9.0 ± 3.0 cells/0.54 mm2, CasRx-Ptbp1: 10.0 ± 3.5 cells/0.54 mm2). However, there were significantly more mCherry/NeuN double-positive cells in the DG of the CasRx-Ptbp1 group compared with the CasRx group (Total; CasRx: 47.1 ± 30.1 cells/1.08 mm2, CasRx-Ptbp1: 117.6 ± 70.9 cells/1.08 mm2, *p < 0.05, Ipsilateral; CasRx: 23.2 ± 17.9 cells/0.54 mm2, CasRx-Ptbp1: 53.9 ± 44.6 cells/0.54 mm2, Contralatral; CasRx: 23.8 ± 14.0 cells/0.54 mm2, CasRx-Ptbp1: 63.7 ± 29.5 cells/0.54 mm2, *p < 0.05) (Fig. 3a, b). We next confirmed Ptbp1 expression in the hippocampal granule cell layer (GCL) indicated by A in Fig. 4a and in the DG excluding the GCL indicated by B in Fig. 4a. The Ptbp1 is also expressed in the GCL, and no significant difference was observed between CasRx and CasRx-ptbp1 in GCL (Ipsilateral: 1.42 ± 0.26 fold change to CasRx, Contralateral: 1.46 ± 0.41 fold change to CasRx). However, in DG excluding the GCL, the Ptbp1 expression was significantly lower in CasRx-Ptbp1 (Ipsilateral: 0.60 ± 0.27 fold change to CasRx, *p < 0.05, Contralateral: 0.63 ± 0.22 fold change to CasRx, *p < 0.05) (Fig. 4a,b).
Fig. 1.

(a) Schematic diagram of the experimental procedure. The arrowheads indicate the intravenous injection of HBSS, a cocktail of AAV(PHP.eB)-pGFAP-mCherry and AAV(PHP.eB)-pGFAP-CasRx as a control vector, or a cocktail of AAV(PHP.eB)-pGFAP-mCherry and AAV(PHP.eB)-pGFAP-CasRx-SgRNA-Ptbp1. (b) Bederson’s score, (c) the rotarod test, and (d) elevated body swing test (EBST) were compared between the three groups. (e) The examination time between a novel object and a familiar object in the novel object recognition test (NORT) was compared between groups. Note that the CasRx-Ptbp1 group showed a significant increase in the novel object time compared with the familiar object time (*p < 0.05). P-values < 0.05 were considered statistically significant.
Fig. 2.
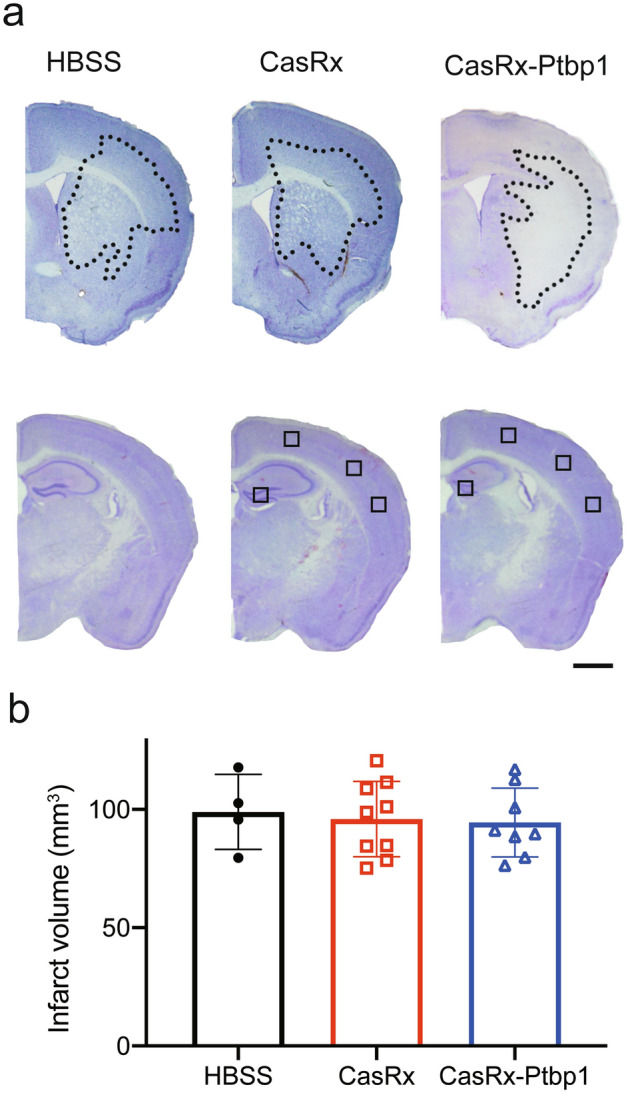
(a) Nissl staining and infarct volume 56 days after tMCAO in the three experimental groups. The use of small squares indicates the approximate area where imaging was performed. Scale bars: 1 mm. (b) Quantitative analysis of infarct volume in in the three experimental groups.
Fig. 3.

(a) Immunofluorescent staining of mCherry/NeuN/Hoechst in the cortex and hippocampus 56 days after tMCAO. Arrowheads indicate mCherry/NeuN double-positive cells. Scale bars: 50 µm and 10 µm. (b) Quantitative analysis of mCherry and NeuN double-positive cells in the cortex and hippocampus. CasRx-Ptbp1 significantly increased the number of mCherry/NeuN double-positive cells in the DG compared with the CasRx group (*p < 0.05). P-values < 0.05 were considered statistically significant.
Fig. 4.

(a) Representative images of Ptbp1 (white) in the hippocampus of the two experimental groups. Scale bars: 50 µm. (b) Pixel intensity of Ptbp1 in GCL indicated by A and the DG excluding the GCL indicated by B. The Ptbp1 expression was significantly lower in CasRx-Ptbp1 in DG excluding GCL. (*p < 0.05) P-values < 0.05 were considered statistically significant.
Discussion
In the present study, we demonstrated that AAV-pGFAP-CasRx-Ptbp1 crossed the BBB and generated new mCherry/NeuN double-positive neuron-like cells, its origin could be mCherry/GFAP double-positive astrocyte-like glia, in the hippocampus of post-stroke dementia mouse model (Figs. 3a and b, S1, S2). Moreover, the CasRx-Ptbp1 group showed a significant amelioration in recognition memory on day 56 after tMCAO (Fig. 1e).
During adult hippocampal neurogenesis, neural stem cells are activated and differentiate into proliferating intermediate neural progenitors and neuroblasts, which become post-mitotic immature dentate granule cells that eventually mature and become incorporated into the neural circuitry16. In ischemic stroke, adult neurogenesis of the DG is markedly increased in the ipsilateral and contralateral hemispheres11,17. However, the division of progenitor cells starts to decrease in the second week and returns to the control level within a few weeks18,19. Additionally, it has been demonstrated that the number of hippocampal neurogenesis and neural progenitor cells declines with age20–22. Nevertheless, even in aged mice, the reduction of Ptbp1 has been observed to result in the conversion of radial glia-like cells to granule cells, a phenomenon that has been documented in young mice and during development14. In this study, we elucidated that hippocampal neurogenesis was robustly stimulated and significantly recovered hippocampal recognition memory in the CasRx-Ptbp1 group 56 days after tMCAO (Figs. 1e, 3a and b). miR-124, a neuron-specific microRNA, suppresses Ptbp1 protein expression by inhibiting the 3′ UTR of Ptbp1 mRNA. This suppression allows the shift to neuron-specific splicing12, which might promote neuronal differentiation from astrocytes, as well as change the fate of radial glia that normally convert to astrocytes, forcing neuronal differentiation14.
The in vivo conversion of resident glial cells to functional new neurons by the ectopic expression of a single or combination of transcription factors or by the knockdown of a single gene has been attempted by many researchers14,23–33. However, some of these results have been questioned by fate-mapping experiments, which are the gold standard for definitively confirming the origin of newly generated cells34–36. The primary cause is regarded as the leaky expression of the construct into neurons under a GFAP promoter37. In the present study, we observed a phenomenon considered leakage expression and could not detect a significant increase in ectopic new neurons in the cortex (Figs. 3a and b, S1). However, we could explain why hippocampal neurogenesis was increased, and hippocampal recognition memory was recovered in the CasRx-Ptbp1 group only (Figs. 1e, 3a and b). This suggests that CRISPR-CasRx-mediated Ptbp1 knockdown may impact hippocampal astrocytes and radial glia, dramatically enhancing endogenous neurogenesis.
Currently, recombinant AAVs (rAAV) have become a significant platform for the in vivo delivery of gene therapy. A combinatorial design that coordinates the genomic characteristics and properties of the capsid and the vector is needed to create an rAAV platform for treating human diseases38. The efficacy and safety of voretigene neparvovec, which is a new drug for RPE65-mediated inherited retinal dystrophy using AAV2 discovered in human cells, and onasemnogene abeparvovec-xioi, which is a prescription for spinal muscular atrophy packaged by AAV9, have been confirmed and used for treatment39,40. However, there is still room for improvement because issues remain with these gene delivery platforms, including immunological barriers to the delivery of rAAV. In the near future, this platform might be a target for treating cerebral infarction after the further improvement of the Rep/Cap packaging plasmid pAAV-PHP.eB with a GFAP promoter.
The present study has some limitations: as in previous studies14, we observed that hippocampal recognition memory was recovered in the CasRx-Ptbp1 group only (Figs. 1e, 3a and b). However, only one type of cognitive function test was performed, so it would be beneficial to validate this result in other cognitive tests in a future study. Secondly, we have not conducted any experiments to assess the loss of function. It would have been preferable to perform such experiments using AAV vectors carrying Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) which manipulate a series of human muscarinic receptors that respond only to the synthetic ligand clozapine N-oxide (CNO)41. Thirdly, it has been reported that the knockdown of Ptbp1 increases the number of DCX-positive cells in the granule cell layer14. However, we have not directly examined whether there is an increase or decrease in neural progenitor cells. In our next study, we intend to investigate the mechanism of neurogenesis using BrdU and markers of each neural progenitor cell. Fourthly, in light of the ongoing discussions surrounding ectopic direct reprogramming, it is crucial to trace the lineage back to the origin of the cell. Our present study does not provide a definitive answer as to the precise origin of the new neurons.
In conclusion, our findings indicate that the transient suppression of Ptbp1 mRNA by viral transduction following the administration of AAV via the tail vein has the potential to facilitate the generation of new neurons from astrocyte-like glia in the hippocampus of a post-stroke dementia mouse model by the CasRx-Ptbp1 vector.
Materials and methods
Animals and pretreatment
Adult male C57BL/6 J mice (20–25 g, 7 weeks old) were purchased from Japan SLC Inc. (Shizuoka Japan). In the first pilot study, mice were injected with AAV(PHP.eB)-pGFAP-mCherry (4.6 × 1010 viral genomes (vg)) through the tail vein to evaluate whether the viral vector could cross the blood–brain barrier (BBB) and infect astrocytes in the brain. Mice were sacrificed 21 days after injection (Fig. S1). In the second pilot study, mice were injected with a cocktail of AAV(PHP.eB)-pGFAP-mCherry and AAV(PHP.eB)-pGFAP-CasRx-SgRNA-Ptbp1 (1:5, total 1.2 × 1010 vg) through the tail vein at 3, 7, or 14 days after 30 min transient middle cerebral artery occlusion (tMCAO) to determine the optimal time point of the viral vector injection to the post-stroke brain. Mice were sacrificed 21 days after injection (Fig. S2). In the first full experiment, mice were randomly divided into three groups depending on the treatment: HBSS group (150 µL, n = 4); cocktail of AAV(PHP.eB)-pGFAP-mCherry and AAV(PHP.eB)-pGFAP-CasRx (control vector) group (1:5, total 1.0 × 1011 vg, n = 9); and cocktail of AAV(PHP.eB)-pGFAP-mCherry and AAV(PHP.eB)-pGFAP-CasRx-SgRNA-Ptbp1 group (1:5, total 1.0 × 1011 vg, n = 8). Mice were injected through the tail vein at 3 days after 30 min tMCAO and sacrificed 56 days after the injection (Fig. 1a). Animal procedures were approved by the Animal Care and Committee of the Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences (approval #OKU-2021732), and all animal experiments were conducted and reported in accordance with the Okayama University guidelines on the Care and Use of the Laboratory Animals and ARRIVE guidelines.
Focal cerebral ischemia
Focal cerebral ischemia was induced in mice by tMCAO following our previous reports42,43. Briefly, mice were anesthetized with isoflurane through an inhalation mask. The left common carotid artery was exposed and a 7–0 nylon thread with a silicone-coated tip was inserted into the left middle cerebral artery (MCA). Following a 30 min period of occlusion, the silicone-coated thread was extracted in order to restore blood flow. Then, the incision was closed, and the animals recovered and were allowed free access to water and food at ambient temperature. Four sham control mice were prepared with a sham cervical operation but without inserting the thread.
AAV vectors and virus production
AAV-GFAP-NLS-CasRx-NLS-FLAG and AAV-GFAP-NLS-CasRx-NLS-FLAG-U6-DR-SgRNA_Ptbp1-DR plasmids were obtained from Addgene (#154000 and #154001, MA, USA). Each plasmid was packed by a pHelper plasmid (#240071, Agilent Technologies, CA, USA) and the Rep/Cap packaging plasmid pAAV-PHP.eB, provided by Dr. Sehara at Jichi Medical University, in HEK293TF cells. All plasmids were verified by restriction enzyme digestion before use. Packaging plasmids and a vector plasmid were transfected into HEK293TF cells using Fugene HD (E2311, Promega, WI, USA). Culture medium containing the transfection reagent was changed to serum-free medium 1 day after transfection. The AAV-containing supernatants were collected 6 days after transfection, and AAV were purified using the minimal purification method44. Viral titers were confirmed using an AAVpro® Titration Kit for Real-Time PCR Ver. 2 (#6233, Takara Bio, Shiga, Japan) with primers recognizing the inverted terminal repeat (ITR) regions.
Behavioral analysis after tMCAO
Mice were evaluated for changes in body weight and behavior at 3, 7, 14, 21, 28, 35, 42, 49, and 56 days after tMCAO. Bederson’s scale scores with minor modifications were applied in this study23,45. A rotarod test was also conducted to detect the impairment of motor function, which correlates with ischemic volume and neurological scores as described previously23. The elevated body swing test (EBST) was performed as described previously46,47. Briefly, the percentage of the number of left side swings, in which the mouse moved its head more than 90° from the vertical axis to either side to the total number of swings, was calculated in 60 s. The novel object recognition test (NORT) was performed to test recognition memory, which is thought to be linked to hippocampal damage14. The mice were habituated in a 35 × 35 × 16 cm3 open field for 2 h. After habituation, the mice were placed in the box with the same two objects (yellow ball with a diameter of 7 cm) for 10 min as training. After 1 h, the time taken for the mouse to approach the novel object (shuttlecock of badminton) and the familiar object was recorded, and then behavior was scored for the object contact time (Fig. 1b).
Tissue preparation
Each mouse was anesthetized by the intraperitoneal injection of pentobarbital (20 mg/kg), and then perfused with chilled PBS, followed by 4% paraformaldehyde (PFA) in 0.1 mol/L phosphate buffer. After post-fixation in the same fixative for 12 h at 4 °C, whole brains were cut into 50-μm-thick sections with a vibrating blade microtome (VT1000S; Leica, Wetzlar, Germany).
Infarct volume and immunofluorescent staining
Coronal brain sections were stained with cresyl violet as Nissl staining to measure the infarct and hippocampal areas using image processing software (ImageJ; National Institutes of Health, MD, USA). The infarct volume of each brain was calculated by the summation of the infarct areas of five serial brain slices, at a 0.6 mm interval, between 1.0 mm anterior and 1.5 mm posterior to the bregma48.
Mice exhibiting neuronal loss in Nissl-stained coronal sections, including the hippocampus, were excluded from the analysis to prevent the direct effects of cerebral ischemia on the hippocampus.
The small squares in Fig. 2 indicated the approximate analyzed area. In the cortex, three locations were randomly selected without distinction between the ipsilateral and contralateral sides. The hippocampus was analyzed separately for the ipsilateral and contralateral sides.
For immunofluorescent staining, free-floating sections were blocked in 5% bovine serum albumin for 2 h. Then, they were incubated at 4 °C overnight with primary antibodies. All antibodies used in this study were from commercial sources: rabbit anti-mCherry antibody (1:200, GTX128505, GeneTex, CA, USA), mouse anti-mCherry antibody (1:200, GTX630189, GeneTex), rabbit anti-glial fibrillary acidic protein (GFAP) antibody (1:500, Z0334, Dako, Glostrup, Denmark), rabbit anti-S100β antibody (1:200, GTX129573, GeneTex, CA, USA), rabbit anti-PTBP1 antibody (1:200, 12582-1-AP, Proteintech, IL, USA), rabbit anti-ionized calcium binding adapter protein 1 (Iba1) antibody (1:500, NCNP24, Wako, Osaka, Japan), rabbit anti-Olig-2 antibody (1:100, AB9610, Merck Millipore, MA, USA), mouse anti-beta III tubulin (Tuj1) antibody (1:100, sc-58888, Santa Cruz Biotechnology, TX, USA); rabbit anti-MAP2 (1:100, sc-58888, Santa Cruz Biotechnology), mouse anti-NeuN antibody (1:200, MAB377, Merck Millipore), rabbit anti-NeuN antibody (1:1000, ab104225, Abcam, Cambridge, UK), and mouse anti-NeuroD1 antibody (1:200, ab60704, Abcam). The sections were washed in PBS and then incubated with secondary antibodies Alexa Fluor 488 or 555 (1:500; Molecular Probes, MA, USA) for 2 h at room temperature.
Statistical analysis
All data are presented as the mean ± standard deviation. Statistical analyses were performed using GraphPad Prism version 8.4.3 (GraphPad Software LCC, CA, USA). After having checked for normality, we performed the Mann–Whitney U-test to compare mCherry and NeuN positive cells in the cortex and hippocampus between the CasRx and CasRx-Ptbp1 groups, and the examination time of NORT between the novel object and familiar object. We also performed a two-way repeated measure ANOVA to compare Bederson’s score, rotarod test, and EBST between the HBSS, CasRx, and CasRx-Ptbp1 groups. Other statistical analyses were performed with the Kruskal–Wallis test to compare ischemic volume, pixel intensity of mCherry and Nissl staining between the HBSS, CasRx, and CasRx-Ptbp1 groups. P-values < 0.05 were considered statistically significant.
Supplementary Information
Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP21K19572, JP22K17799, JP21K15190, and Grants-in-Aid from the Research Committees (Toba K, and Tsuji S) from the Japan Agency for Medical Research and Development. We thank J. Ludovic Croxford, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Abbreviations
- AAV
Adeno-associated virus
- BBB
Blood–brain barrier
- EBST
Elevated body swing test
- GFAP
Glial fibrillary acidic protein
- Iba-1
Ionized calcium binding adapter protein 1
- i.c.v.
Intracerebroventricular
- MCA
Middle cerebral artery
- mRNA
Messenger RNA
- NORT
Novel object recognition test
- PBS
Phosphate-buffered saline
- PFA
Paraformaldehyde
- Ptbp1
Polypyrimidine tract-binding protein 1
- rAAV
Recombinant AAVs
- tMCAO
Transient middle cerebral artery occlusion
- Tuj1
Beta III tubulin
- vg
Viral genome
Author contributions
Y.F. and T.Y. designed and performed the experiments and wrote the manuscript. R.M. and X.H. performed behavioral test and analysis. Y.N., T.Y., M.T., and K.A. provided supervision and discussion. All authors read and approved the manuscript.
Data availability
The datasets generated or analyzed during this study are included in this paper and its Supplementary Information file or are available from the corresponding author upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-71212-w.
References
- 1.Katan, M. & Luft, A. Global burden of stroke. Semin. Neurol.38, 208–211. 10.1055/s-0038-1649503 (2018). 10.1055/s-0038-1649503 [DOI] [PubMed] [Google Scholar]
- 2.Alawieh, A. M., Langley, E. F., Feng, W., Spiotta, A. M. & Tomlinson, S. Complement-dependent synaptic uptake and cognitive decline after stroke and reperfusion therapy. J. Neurosci.40, 4042–4058. 10.1523/JNEUROSCI.2462-19.2020 (2020). 10.1523/JNEUROSCI.2462-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.del Ser, T. et al. Evolution of cognitive impairment after stroke and risk factors for delayed progression. Stroke36, 2670–2675. 10.1161/01.STR.0000189626.71033.35 (2005). 10.1161/01.STR.0000189626.71033.35 [DOI] [PubMed] [Google Scholar]
- 4.De Ronchi, D. et al. The combined effect of age, education, and stroke on dementia and cognitive impairment no dementia in the elderly. Dement Geriatr. Cogn. Disord.24, 266–273. 10.1159/000107102 (2007). 10.1159/000107102 [DOI] [PubMed] [Google Scholar]
- 5.Zheng, F., Yan, L., Zhong, B., Yang, Z. & Xie, W. Progression of cognitive decline before and after incident stroke. Neurology93, e20–e28. 10.1212/WNL.0000000000007716 (2019). 10.1212/WNL.0000000000007716 [DOI] [PubMed] [Google Scholar]
- 6.Pluta, R., Januszewski, S., Jablonski, M. & Ulamek, M. Factors in creepy delayed neuronal death in hippocampus following brain ischemia-reperfusion injury with long-term survival. Acta Neurochir. Suppl.106, 37–41. 10.1007/978-3-211-98811-4_5 (2010). 10.1007/978-3-211-98811-4_5 [DOI] [PubMed] [Google Scholar]
- 7.Pluta, R., Ulamek, M. & Jablonski, M. Alzheimer’s mechanisms in ischemic brain degeneration. Anat. Rec. (Hoboken)292, 1863–1881. 10.1002/ar.21018 (2009). 10.1002/ar.21018 [DOI] [PubMed] [Google Scholar]
- 8.Arvidsson, A., Collin, T., Kirik, D., Kokaia, Z. & Lindvall, O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med.8, 963–970. 10.1038/nm747 (2002). 10.1038/nm747 [DOI] [PubMed] [Google Scholar]
- 9.Marques, B. L. et al. The role of neurogenesis in neurorepair after ischemic stroke. Semin. Cell Dev. Biol.95, 98–110. 10.1016/j.semcdb.2018.12.003 (2019). 10.1016/j.semcdb.2018.12.003 [DOI] [PubMed] [Google Scholar]
- 10.Carmichael, S. T. et al. Growth-associated gene expression after stroke: Evidence for a growth-promoting region in peri-infarct cortex. Exp. Neurol.193, 291–311. 10.1016/j.expneurol.2005.01.004 (2005). 10.1016/j.expneurol.2005.01.004 [DOI] [PubMed] [Google Scholar]
- 11.Ceanga, M., Dahab, M., Witte, O. W. & Keiner, S. Adult neurogenesis and stroke: A tale of two neurogenic niches. Front. Neurosci.15, 700297. 10.3389/fnins.2021.700297 (2021). 10.3389/fnins.2021.700297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makeyev, E. V., Zhang, J., Carrasco, M. A. & Maniatis, T. The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell27, 435–448. 10.1016/j.molcel.2007.07.015 (2007). 10.1016/j.molcel.2007.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xue, Y. et al. Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell152, 82–96. 10.1016/j.cell.2012.11.045 (2013). 10.1016/j.cell.2012.11.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maimon, R. et al. Therapeutically viable generation of neurons with antisense oligonucleotide suppression of PTB. Nat. Neurosci.24, 1089–1099. 10.1038/s41593-021-00864-y (2021). 10.1038/s41593-021-00864-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang, Z. et al. Post-ischemia common carotid artery occlusion worsens memory loss, but not sensorimotor deficits, in long-term survived stroke mice. Brain Res. Bull.183, 153–161. 10.1016/j.brainresbull.2022.03.008 (2022). 10.1016/j.brainresbull.2022.03.008 [DOI] [PubMed] [Google Scholar]
- 16.Mira, H. & Morante, J. Neurogenesis from embryo to adult —Lessons from flies and mice. Front. Cell Dev. Biol.8, 533. 10.3389/fcell.2020.00533 (2020). 10.3389/fcell.2020.00533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niv, F. et al. Aberrant neurogenesis after stroke: a retroviral cell labeling study. Stroke43, 2468–2475. 10.1161/STROKEAHA.112.660977 (2012). 10.1161/STROKEAHA.112.660977 [DOI] [PubMed] [Google Scholar]
- 18.Yagita, Y. et al. Neurogenesis by progenitor cells in the ischemic adult rat hippocampus. Stroke32, 1890–1896. 10.1161/01.str.32.8.1890 (2001). 10.1161/01.str.32.8.1890 [DOI] [PubMed] [Google Scholar]
- 19.Cuartero, M. I. et al. Abolition of aberrant neurogenesis ameliorates cognitive impairment after stroke in mice. J. Clin. Invest.129, 1536–1550. 10.1172/JCI120412 (2019). 10.1172/JCI120412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morgenstern, N. A., Lombardi, G. & Schinder, A. F. Newborn granule cells in the ageing dentate gyrus. J. Physiol.586, 3751–3757. 10.1113/jphysiol.2008.154807 (2008). 10.1113/jphysiol.2008.154807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sorrells, S. F. et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature555, 377–381. 10.1038/nature25975 (2018). 10.1038/nature25975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heine, V. M., Maslam, S., Joels, M. & Lucassen, P. J. Prominent decline of newborn cell proliferation, differentiation, and apoptosis in the aging dentate gyrus, in absence of an age-related hypothalamus-pituitary-adrenal axis activation. Neurobiol. Aging25, 361–375. 10.1016/S0197-4580(03)00090-3 (2004). 10.1016/S0197-4580(03)00090-3 [DOI] [PubMed] [Google Scholar]
- 23.Yamashita, T. et al. In vivo direct reprogramming of glial linage to mature neurons after cerebral ischemia. Sci. Rep.9, 10956. 10.1038/s41598-019-47482-0 (2019). 10.1038/s41598-019-47482-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niu, W. et al. In vivo reprogramming of astrocytes to neuroblasts in the adult brain. Nat. Cell Biol.15, 1164–1175. 10.1038/ncb2843 (2013). 10.1038/ncb2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grande, A. et al. Environmental impact on direct neuronal reprogramming in vivo in the adult brain. Nat. Commun.4, 2373. 10.1038/ncomms3373 (2013). 10.1038/ncomms3373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo, Z. et al. In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer’s disease model. Cell Stem Cell14, 188–202. 10.1016/j.stem.2013.12.001 (2014). 10.1016/j.stem.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu, Y. et al. Ascl1 converts dorsal midbrain astrocytes into functional neurons in vivo. J. Neurosci.35, 9336–9355. 10.1523/JNEUROSCI.3975-14.2015 (2015). 10.1523/JNEUROSCI.3975-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsuda, T. et al. Pioneer factor NeuroD1 rearranges transcriptional and epigenetic profiles to execute microglia-neuron conversion. Neuron101, 472–485. 10.1016/j.neuron.2018.12.010 (2019). 10.1016/j.neuron.2018.12.010 [DOI] [PubMed] [Google Scholar]
- 29.Mattugini, N. et al. Inducing different neuronal subtypes from astrocytes in the injured mouse cerebral cortex. Neuron103, 1086–1095. 10.1016/j.neuron.2019.08.009 (2019). 10.1016/j.neuron.2019.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qian, H. et al. Author correction: Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature584, E17. 10.1038/s41586-020-2583-3 (2020). 10.1038/s41586-020-2583-3 [DOI] [PubMed] [Google Scholar]
- 31.Tai, W. et al. In vivo reprogramming of NG2 glia enables adult neurogenesis and functional recovery following spinal cord injury. Cell Stem Cell28, 923–937. 10.1016/j.stem.2021.02.009 (2021). 10.1016/j.stem.2021.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Torper, O. et al. In vivo reprogramming of striatal NG2 glia into functional neurons that integrate into local host circuitry. Cell Rep12, 474–481. 10.1016/j.celrep.2015.06.040 (2015). 10.1016/j.celrep.2015.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou, H. et al. Glia-to-neuron conversion by CRISPR-CasRx alleviates symptoms of neurological disease in mice. Cell181, 590–603. 10.1016/j.cell.2020.03.024 (2020). 10.1016/j.cell.2020.03.024 [DOI] [PubMed] [Google Scholar]
- 34.Chen, W., Zheng, Q., Huang, Q., Ma, S. & Li, M. Repressing PTBP1 fails to convert reactive astrocytes to dopaminergic neurons in a 6-hydroxydopamine mouse model of Parkinson’s disease. Elife10.7554/eLife.75636 (2022). 10.7554/eLife.75636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoang, T. et al. Genetic loss of function of Ptbp1 does not induce glia-to-neuron conversion in retina. Cell Rep.39, 110849. 10.1016/j.celrep.2022.110849 (2022). 10.1016/j.celrep.2022.110849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie, Y., Zhou, J. & Chen, B. Critical examination of Ptbp1-mediated glia-to-neuron conversion in the mouse retina. Cell Rep.39, 110960. 10.1016/j.celrep.2022.110960 (2022). 10.1016/j.celrep.2022.110960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang, L. L. et al. Revisiting astrocyte to neuron conversion with lineage tracing in vivo. Cell184, 5465–5481. 10.1016/j.cell.2021.09.005 (2021). 10.1016/j.cell.2021.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang, D., Tai, P. W. L. & Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug. Discov.18, 358–378. 10.1038/s41573-019-0012-9 (2019). 10.1038/s41573-019-0012-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mendell, J. R. et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med.377, 1713–1722. 10.1056/NEJMoa1706198 (2017). 10.1056/NEJMoa1706198 [DOI] [PubMed] [Google Scholar]
- 40.Russell, S. et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet390, 849–860. 10.1016/S0140-6736(17)31868-8 (2017). 10.1016/S0140-6736(17)31868-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Urban, D. J. & Roth, B. L. DREADDs (designer receptors exclusively activated by designer drugs): Chemogenetic tools with therapeutic utility. Annu. Rev. Pharmacol. Toxicol.55, 399–417. 10.1146/annurev-pharmtox-010814-124803 (2015). 10.1146/annurev-pharmtox-010814-124803 [DOI] [PubMed] [Google Scholar]
- 42.Abe, K., Kawagoe, J., Araki, T., Aoki, M. & Kogure, K. Differential expression of heat shock protein 70 gene between the cortex and caudate after transient focal cerebral ischaemia in rats. Neurol. Res.14, 381–385. 10.1080/01616412.1992.11740089 (1992). 10.1080/01616412.1992.11740089 [DOI] [PubMed] [Google Scholar]
- 43.Yamashita, T. et al. Novel therapeutic transplantation of induced neural stem cells for stroke. Cell Transpl.26, 461–467. 10.3727/096368916X692988 (2017). 10.3727/096368916X692988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Konno, A. & Hirai, H. Efficient whole brain transduction by systemic infusion of minimally purified AAV-PHP.eB. J. Neurosci. Methods346, 108914. 10.1016/j.jneumeth.2020.108914 (2020). 10.1016/j.jneumeth.2020.108914 [DOI] [PubMed] [Google Scholar]
- 45.Bederson, J. B. et al. Rat middle cerebral artery occlusion: Evaluation of the model and development of a neurologic examination. Stroke17, 472–476. 10.1161/01.str.17.3.472 (1986). 10.1161/01.str.17.3.472 [DOI] [PubMed] [Google Scholar]
- 46.Borlongan, C. V. & Sanberg, P. R. Elevated body swing test: A new behavioral parameter for rats with 6-hydroxydopamine-induced hemiparkinsonism. J. Neurosci.15, 5372–5378 (1995). 10.1523/JNEUROSCI.15-07-05372.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaneko, N. et al. New neurons use Slit-Robo signaling to migrate through the glial meshwork and approach a lesion for functional regeneration. Sci. Adv.4, eaav0618. 10.1126/sciadv.aav0618 (2018). 10.1126/sciadv.aav0618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paxinos, G., Franklin, K. B. J. & Franklin, K. B. J. The Mouse Brain in Stereotaxic Coordinates 2nd edn. (Academic Press, 2001). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated or analyzed during this study are included in this paper and its Supplementary Information file or are available from the corresponding author upon reasonable request.