

Abstract
We present a case of presumed xeroderma pigmentosum (XP) with concomitant foveal hypoplasia. A 50-year-old male patient with extensive bilateral symblepharon-like pseudopterygia was referred for visual rehabilitation. After dermatology consultation and ophthalmologic examination, presumed XP was diagnosed. Optical coherence tomography revealed grade 2 foveal hypoplasia. The patient was referred for genetic testing because concomitant XP and foveal hypoplasia are rare. The genetic test results revealed mutations in some genes, including the hemochromatosis genes HFE, COL1A2, Lysosome Trafficking Regulator (LYST), NF1, and HMBS. The LYST gene is known to be associated with foveal hypoplasia. Since the association of foveal hypoplasia and XP has been reported in another case in the literature, we present our case to share this rare association.
Keywords: Foveal hypoplasia, LYST gene, Xeroderma pigmentosum
Introduction
Hebra and Kaposi were the first to report xeroderma pigmentosum (XP), a rare autosomal recessive condition (1). Ophthalmic involvement typically affects the ocular surface and sun-exposed periocular skin, which has been reported in 40–100% of XP patients. The manifestations of ophthalmic XP include ectropion, lagophthalmos, conjunctival injection, conjunctival melanosis, corneal scarring, keratopathy, pterygium, ocular surface, and eyelid malignancies. Photophobia and dry eyes are frequent ocular complaints (2). The best-corrected visual acuity of the patients could be influenced by any of the ocular surface complications of XP. However, visual acuity should improve after surgical correction unless there is any other concomitant posterior segment pathology or amblyopia.
XP is known to result from an anomaly in the nucleotide excision repair system and affects both sexes equally. This condition has a 100% penetrance rate, with an estimated incidence of 1/1.000.000 live births in the United States and Western Europe (3). Eight subgroups, XPA through XPG and XP-V, can result from mutations in any of the eight genes involved in identifying and repairing ultraviolet (UV)-induced DNA damage. Genetic testing identifies and verifies the XP subgroup the patient belongs to (4).
Here, we present a rare case of foveal hypoplasia in an XP patient and discuss the possible etiologies based on genetic tests.
Case Report
A 50-year-old male presented with progressive eye symptoms, including symblepharon formation and cicatricial ectropion in both eyes. Both corneas lost transparency because of the overlying pathological conjunctiva. The patient also displayed facial skin findings, including freckling, xerosis, progressive atrophy, and hyper/hypopigmentation. He had a history of basal cell carcinoma diagnosis following a skin biopsy. Based on his clinical findings, the dermatology consultation yielded a “clinically presumed XP” (Fig. 1). Photophobia, xerophthalmia, and conjunctival injection were also coexisting findings. His visual acuity was hand motion in the right eye and 0.15 in Snellen lines in the left eye. Optical coherence tomography (OCT) revealed grade 2 foveal hypoplasia (Fig. 2). We consulted him for genetic testing because foveal hypoplasia and XP are unusual to be found together.
Figure 1.
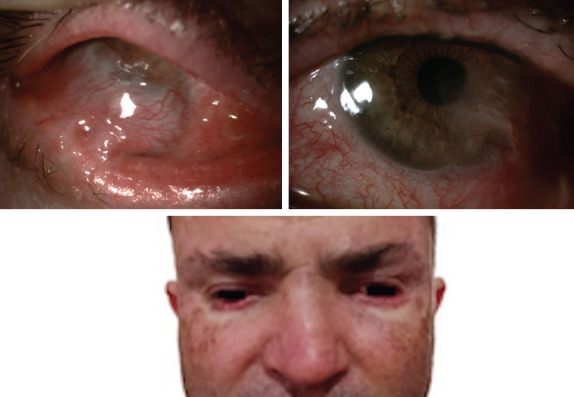
This picture depicts anterior segment photography of both eyes. Symblepharon formation was observed extending from the tarsal conjunctiva to the bulbar conjunctiva and spreading over the cornea, preventing the selection of the anterior chamber in the right eye (upper left). The left eye image shows more apparent anterior chamber detail than the right eye. Symblepharon formation extended from the inferior eyelid and inferior bulbar conjunctiva to the temporal cornea (upper right). Pigmented lesions were observed in the maxillofacial–temporal areas of the patient’s face. Upon anamnesis, it was learned that basal cell carcinoma-nodular type was diagnosed due to the biopsy taken from the pigmented lesions on the skin (bottom).
Figure 2.
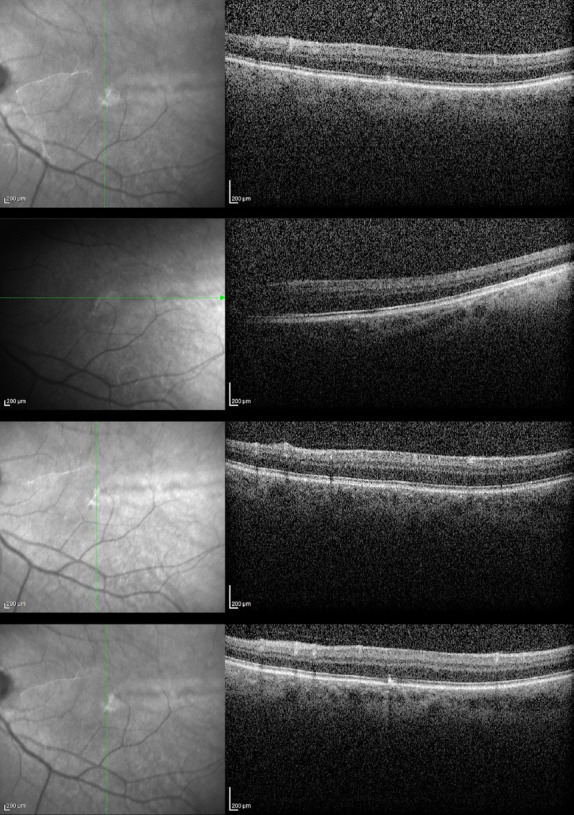
Left eye OCT images showing grade 2 foveal hypoplasia. The foveal pit and extrusion of plexiform layers were absent, but outer segment lengthening was present.
For genetic testing, peripheral blood was used to obtain genomic DNA. A skin disorders kit, Celemics Inc. (Celemics, Seoul, S. Korea), and Nextseq sequencing system (Illumina) analysis software were used. Genetic pathology was classified according to the American College of Medical Genetics and Genomics guidelines. The test yielded some positive results. A pathogenic heterozygous c.187C>G (chr6:26091179) (p. His63Asp, rs1799945) change was detected in the 2nd exon of the HFE gene. A heterozygous c.3279_3287dup (chr7:94056941, p.Pro1100_Gly1102dup) mutation of unknown clinical significance was detected in exon 49 of COL1A2. A heterozygous c.181A>G (chr1:235993537) (p.Ile61Val,rs776670065) change of unknown clinical significance was seen in the 3rd exon of Lysosome Trafficking Regulator (LYST). A heterozygous c.2585C>G (chr17:29556218) (p.Thr862Ser,rs200302954) variant with unknown clinical significance was detected in the 3rd exon of NF1. A heterozygous c.613-11C>G (chr11:118962824) change of unknown clinical significance was detected in the 9th intron of HBMS. Among these mutated genes, it was noteworthy that the LYST gene, which has been reported to be associated with foveal hypoplasia, was positive.
An ocular conjunctival excisional biopsy was performed to rehabilitate the ocular surface and expose the corneal surface, including the visual axis. Ocular surface reconstruction included amniotic membrane transplantation, and symblepharon ring fitting to prevent pseudopterygia recurrence. At the last postoperative visit, eight months after the surgery, there was a severe recurrence of the disease in the operated eye-topical treatment with %0.02 mitomycin C q.i.d. before further surgical intervention was planned. The patient was operated on after using topical mitomycin c for about 6 months. The final visual acuity was 0.05, according to the Snellen chart (Fig. 3). Written informed consent was obtained from the patient on publication of his clinical findings.
Figure 3.
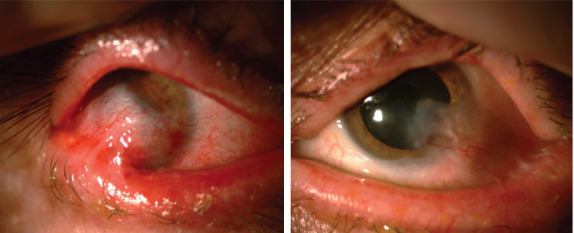
Appearance after ocular surface rehabilitation surgery. The aggressive symblepharon formation in the right eye has regressed, and the anterior chamber details have become more visible (left image). In the left eye, ocular surface findings remained the same (right image).
Discussion
Visual function depends on the intricate process of foveal maturation in humans. Several genes are known to be involved in retinal development. Foveal hypoplasia, which denotes the underdevelopment of the fovea, is frequently the result of pathogenic variations in these genes, disrupting the foveal developmental process (5). The Leicester Grading System is used to grade foveal hypoplasia. It is divided into 1 grade of atypical FH, which is linked to photoreceptor degeneration, and four grades of typical FH (grades 1-4) (Fig. 4) (5,6).
Figure 4.
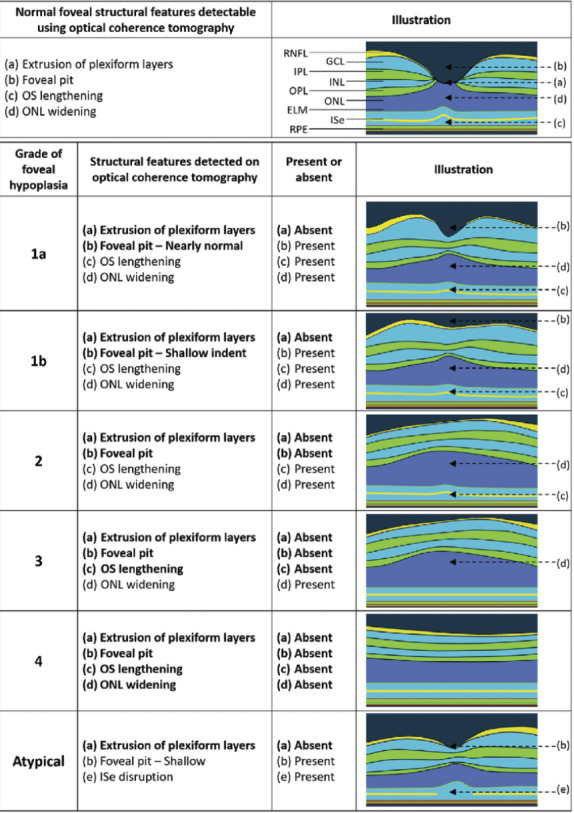
Illustration of the Leicester Grading System for Foveal Hypoplasia, highlighting OCT-detectable characteristics of a normal fovea before highlighting the characteristics of typical and atypical grades of foveal hypoplasia. The plexiform layers are extruded or displaced outward in a typical fovea. Continuation of plexiform layers is a hallmark of all degrees of foveal hypoplasia.
One possible gene that may cause foveal hypoplasia is a mutation in the LYST gene responsible for regulating lysosome and lysosome-related organelle sizes. The storage and secretory functions of lysosomal granules of leukocytes, fibroblasts, dense bodies of platelets, azurophilic granules of neutrophils, and melanosomes of melanocytes are all affected by mutations in LYST. These abnormalities cause the formation of dysfunctional lysosomes and swollen vesicles. Foveal hypoplasia is a known ophthalmological finding in Chediak-Higashi syndrome (CHS), which can be caused by LYST mutations (7). In a cell culture study, CHS cells exhibit similar characteristics to XP and XP variants. CHS fibroblasts show altered responses in depletion of NAD and elevation of diadenosine-5’,5”’-tetraphosphate (Ap4A) following DNA damage. Lowering NAD following UV irradiation is similar to control and XP variant cells. Cultured cells from these patients show increased sensitivity to irradiation with UV, characteristics also associated with cells derived from patients with XP. It is of interest, therefore, to compare alterations of cellular metabolism that occur in XP and XP-V cell lines with those of cells derived from patients with CHS to investigate possible relationships between the metabolic bases of these photosensitive, cancer-prone diseases and to identify molecular defects associated with CHS. The study concluded that CHS cells resemble XP variant cells (8). CHS was not considered clinically in our patient, but the positivity of the LYST gene and the demonstration that CHS cells give similar metabolic responses to UV exposure as XP variant cells and the presence of foveal hypoplasia suggest that a possible XP variant should be investigated in our case.
In XP, ocular disease is evident in at least 40% of the cases. Blepharospasm and photophobia are common symptoms (9). Most XP visual symptoms are related to exposure. Patients with XP rarely experience fundus abnormalities because the cornea and lens usually shield the posterior segment from UV rays. However, our patient exhibited bilateral grade 2 foveal hypoplasia. It frequently manifests as nystagmus and fluctuating visual acuity loss, as observed in our patient. Foveal hypoplasia is typically bilateral and commonly observed in conditions such as aniridia, achromatopsia, albinism, or even disorders linked to the PAX6 mutation (10,11). As a result of gene mutations reported in XP disease, there are disruptions in nucleotide excision repair and impairments in apoptosis response (12-14). The LYST gene mutation found in our case has not been reported in XP but has been described in foveal hypoplasia.
The literature review yielded only one patient with concomitant foveal hypoplasia, posterior embryotoxon, and XP (10). In this case report, a 28-year-old man has had XP since childhood. The dermatology department recommended a routine eye checkup, which revealed bilateral foveal hypoplasia. Gonioscopy showed an open angle with an anteriorly displaced Schwalbe’s line that was noticeable. There were no iridocorneal adhesions or other indications of dysgenesis of the angular structure. Grade 2 foveal hypoplasia was discovered on OCT and was indicated by the absence of a foveal pit. Artificial tears were used to treat the patient, and she was advised to wear specially designed ophthalmic UV. The provided research indicated no direct evidence of a linkage between foveal hypoplasia and XP.
HFE gene mutation in an XP patient has been reported in the literature. This patient, who was also diagnosed with hemochromatosis, was then offered a genetic test to detect the second mutation due to the known proximity of the HFE (6p22.2) and POLH (6p21.1) genes which is accountable for the XP-V variant and was found to carry a rare, but potentially harmful, homozygous variant of the POLH gene c.571A>C (p.(Thr191Pro)) (15). Our patient was also HFE positive but had no liver-related disease.
Recent advancements in high-resolution OCT imaging have unveiled characteristics of foveal hypoplasia that were not detected by conventional imaging methods. Clinical characteristics are foveal hypoplasia,-that is, a missing foveal pit, abnormal foveal or macular reflexes, and a poorly defined foveal avascular zone-and, in some cases, anterior segment abnormalities such as Axenfeld’s anomaly or posterior embryotoxon. There is a rare inherited syndrome called foveal hypoplasia, optic nerve decussation defects, and anterior segment dysgenesis that is distinct from albinism and is caused by a recessive mutation in the putative glutamine transporter gene SLC38A8 (16). On the other hand, XP is a rare genetic disorder characterized by defective DNA repair, leading to clinical and cellular hypersensitivity to UV radiation and carcinogenic agents. The association of foveal hypoplasia and XP has been reported in one case in the literature, and our case is the second case to our knowledge.
Our patient had been previously diagnosed with mucous membrane pemphigoid (MMP) elsewhere, with inconclusive treatment efforts. At this point, the distinction between these two diseases is worth mentioning. First, pigmented lesions in the maxilla frontal area of the face were compatible with XP in our patient. Another distinguishing point was the type of malposition of the eyelid. While entropion is frequently seen in MMP, bilateral cicatricial ectropion was observed in our case. In addition, shortening of the fornix, which is commonly seen in ocular MMP diseases, was not present in our case, and the fibro-adhesive changes detected were symblepharon formations from the tarsal conjunctiva to the bulbar conjunctiva and the cornea. Another distinctive finding was that the patient was much younger than the age range in which MMP is frequently seen.
Conclusion
Our case study indicates a potential connection between XP and foveal hypoplasia. This is the first study based on genetic research on a patient with both pathologies. Given the rarity of this correlation, further study of genotype-phenotype relationships may be necessary better to understand the pathophysiology of this disease and its symptoms and to provide patients with more specialized care.
Footnotes
Disclosures
Informed consent: Written informed consent was obtained from the patient for the publication of the case report and the accompanying images.
Peer-review: Externally peer-reviewed.
Conflict of Interest: None declared.
Use of AI for Writing Assistance: Not declared.
Authorship Contributions: Concept – E.K., C.A.U., M.K.; Design – E.K., C.A.U.; Supervision – E.K., C.A.U., M.K., B.L.; Resource – E.K., C.A.U., M.K.; Materials – E.K., C.A.U.; Data Collection and/or Processing – E.K., C.A.U., B.L.; Analysis and/or Interpretation – E.K., C.A.U.; Literature Search – E.K., C.A.U., B.L.; Writing – E.K., C.A.U.; Critical Reviews – E.K., C.A.U.
References
- 1.Lim R, Sethi M, Morley AM. Ophthalmic manifestations of xeroderma pigmentosum:A perspective from the United Kingdom. Ophthalmology. 2017;124:1652–61. doi: 10.1016/j.ophtha.2017.04.031. [DOI] [PubMed] [Google Scholar]
- 2.Brooks BP, Thompson AH, Bishop RJ, Clayton JA, Chan CC, Tsilou ET, et al. Ocular manifestations of xeroderma pigmentosum:Long-term follow-up highlights the role of DNA repair in protection from sun damage. Ophthalmology. 2013;120:1324–36. doi: 10.1016/j.ophtha.2012.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kleijer WJ, Laugel V, Berneburg M, Nardo T, Fawcett H, Gratchev A, et al. Incidence of DNA repair deficiency disorders in Western Europe:Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst) 2008;7:744–50. doi: 10.1016/j.dnarep.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 4.Hoesl M, Dietz K, Röcken M, Berneburg M. Vitamin D levels of XP-patients under stringent sun-protection. Eur J Dermatol. 2010;20:457–60. doi: 10.1684/ejd.2010.0968. [DOI] [PubMed] [Google Scholar]
- 5.Thomas MG, Kumar A, Mohammad S, Proudlock FA, Engle EC, Andrews C, et al. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography a predictor of visual acuity? Ophthalmology. 2011;118:1653–60. doi: 10.1016/j.ophtha.2011.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rufai SR, Thomas MG, Purohit R, Bunce C, Lee H, Proudlock FA, et al. Can structural grading of foveal hypoplasia predict future vision in infantile Nystagmus?:A longitudinal study. Ophthalmology. 2020;127:492–500. doi: 10.1016/j.ophtha.2019.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ji X, Zhao L, Umapathy A, Fitzmaurice B, Wang J, Williams DS, et al. Deficiency in Lyst function leads to accumulation of secreted proteases and reduced retinal adhesion. PLoS One. 2022;17:e0254469. doi: 10.1371/journal.pone.0254469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker JC, Ames BN. Alterations of NAD and adenylyl dinucleotide metabolism in Chediak-Higashi syndrome fibroblasts. J Inherit Metab Dis. 1988;11:221–8. doi: 10.1007/BF01800362. [DOI] [PubMed] [Google Scholar]
- 9.Ramkumar HL, Brooks BP, Cao X, Tamura D, Digiovanna JJ, Kraemer KH, et al. Ophthalmic manifestations and histopathology of xeroderma pigmentosum:Two clinicopathological cases and a review of the literature. Surv Ophthalmol. 2011;56:348–61. doi: 10.1016/j.survophthal.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El Kaissoumi L, Mrini B, Lazrek O, Boutimzine N, Cherkaoui LO. Posterior embryotoxon in xeroderma pigmentosum:A case report. J Case Rep Images Ophthalmol. 2022;5:14–7. [Google Scholar]
- 11.Karaca EE, Çubuk MÖ, Ekici F, Akçam HT, Waisbourd M, Hasanreisoğlu M. Isolated foveal hypoplasia:Clinical presentation and imaging findings. Optom Vis Sci. 2014;91:S61–5. doi: 10.1097/OPX.0000000000000191. [DOI] [PubMed] [Google Scholar]
- 12.Xue H, Ni P, Lin B, Xu H, Huang G. X-ray repair cross-complementing group 1 (XRCC1) genetic polymorphisms and gastric cancer risk:A HuGE review and meta-analysis. Am J Epidemiol. 2011;173:363–75. doi: 10.1093/aje/kwq378. [DOI] [PubMed] [Google Scholar]
- 13.Gillet LC, Schärer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev. 2006;106:253–76. doi: 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- 14.Taylor EM, Broughton BC, Botta E, Stefanini M, Sarasin A, Jaspers NG, et al. Xeroderma pigmentosum and trichothiodystrophy are associated with different mutations in the XPD (ERCC2) repair/transcription gene. Proc Natl Acad Sci. 1997;94:8658–63. doi: 10.1073/pnas.94.16.8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monte F, Garrido M, Pereira Guedes T, Reis J, Porto G, Pedroto I. Hemochromatosis and xeroderma pigmentosum:Two (Un)suspicious neighbors. GE Port J Gastroenterol. 2022;29:38–44. doi: 10.1159/000513587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahmadi K, Fracasso A, van Dijk JA, Kruijt C, van Genderen M, Dumoulin SO, et al. Altered organization of the visual cortex in FHONDA syndrome. Neuroimage. 2019;190:224–31. doi: 10.1016/j.neuroimage.2018.02.053. [DOI] [PubMed] [Google Scholar]