

Abstract
Owing to their limited accuracy and narrow applicability, current antimicrobial peptide (AMP) prediction models face obstacles in industrial application. To address these limitations, we developed and improved an AMP prediction model using Comparing and Optimizing Multiple DEep Learning (COMDEL) algorithms, coupled with high-throughput AMP screening method, finally reaching an accuracy of 94.8% in test and 88% in experiment verification, surpassing other state-of-the-art models. In conjunction with COMDEL, we employed the phage-assisted evolution method to screen Sortase in vivo and developed a cell-free AMP synthesis system in vitro, ultimately increasing AMPs yields to a range of 0.5–2.1 g/L within hours. Moreover, by multi-omics analysis using COMDEL, we identified Lactobacillus plantarum as the most promising candidate for AMP generation among 35 edible probiotics. Following this, we developed a microdroplet sorting approach and successfully screened three L. plantarum mutants, each showing a twofold increase in antimicrobial ability, underscoring their substantial industrial application values.
Key words: Antimicrobial peptide, Deep learning, Cell-free synthesis, Probiotics, L. plantarum
Graphical abstract
This study establishes an AI-based AMPs prediction model named COMDEL for effectively mining edible AMPs and probiotics, which are subsequently synthesized and directly evolved for industrial application, respectively.
1. Introduction
Antimicrobial peptides (AMPs), a class of short amino acid polymers important for the innate immune response, are generated by a wide range of species, from prokaryotes to eukaryotes1,2. They exhibit rapid and broad-spectrum antimicrobial activity against various microorganisms, especially pathogenic ones, making them attractive substitutes for traditional antibiotics and preservatives. Importantly, AMPs are less prone to resistance development compared to conventional antibiotics, positioning them as potentially revolutionary in addressing the major concern about antibiotic overuse and the emergence of super-resistant bacteria3,4. Beyond their antimicrobial properties, recent studies have also discovered that many AMPs possess other properties involved in anti-inflammation, wound-healing and immunomodulatory, further spotlighting them as research hotspots for developing novel therapeutic agents for infectious diseases2,3.
Despite their intriguing medicinal and antiseptic potential, there are two primary challenges regarding AMP supplies. One challenge is that the relatively specific antibacterial preference of many available AMPs narrows their application range2. To address this, recent studies have leveraged computational technologies to predict and design broad-spectrum AMPs5,6. For instance, Antibp2 employs the Support Vector Machine (SVM) approach to predict and classify AMPs based on their amino acid composition, achieving high accuracy in discovering efficacious AMPs against antibiotic-resistant bacteria7. Similarly, iAMP-2L utilizes the pseudo amino acid composition and the fuzzy k-nearest neighbor algorithm to effectively distinguish and categorize AMPs according to their functions8.
In recent years, artificial intelligence (AI) approaches, particularly deep learning technologies, have been widely applied in biological fields, including protein tertiary structure prediction, enzyme molecular design, and gene editing efficiency forecasting9, 10, 11. Notably, these technologies excel at autonomously learning from sequence and structural features, offering ideal screening and design tools with the benefits of high accuracy and efficiency12. The establishment and ongoing development of AMP databases, such as APD3, have facilitated the creation and employment of a series of powerful machine learning tools based on big data classification in AMP discovery and design13, 14, 15, 16, 17, 18. For instance, Deep-ABPpred, a deep learning-based classifier designed for novel antibacterial peptides (ABPs) identification, has been applied to filter ABPs in the proteome of Streptococcus bacteriophages19.
However, current deep learning methods fall short when it comes to large-scale prediction and screening of AMPs, as evidenced by several limitations in existing studies. Firstly, these models often r often rely on oversimplified representations for feature extraction—a critical step affecting model performance20, resulting in a failure to fully capture the complexity inherent in peptides. Secondly, the absence of a unified benchmark dataset for both the training and test processes probably introduces biases in the evaluation and comparison of model performance16,19,21. Thirdly, the compatibility of existing models is limited. While they may perform well on certain datasets, they face difficulties with others due to overfitting, model complexity, and suboptimal feature selection16,22,23. Lastly, few models take toxicity factors into account, which increases the potential risk of pathogenicity in the process of AMP discovery24,25.
Another challenge lies in the complex and inefficient synthesis of AMPs, significantly driving up their production costs26,27. Currently, chemosynthesis and microbial fermentation are the two main ways for peptide synthesis. The chemosynthesis method, known as solid-phase peptide synthesis, extends the peptide chain through the sequential addition of amino acids in a cyclic manner, ultimately achieving the desired peptide sequence28. However, this method is severely limited by peptide length, and often yields insufficient quantities of AMPs in industrial production.
On the other hand, microbial fermentation has been utilized for the large-scale industrial production of certain AMPs, since several probiotics are capable of substantially synthesizing natural AMPs29,30. However, due to the inherent characteristics of AMPs, these natural AMPs from microorganisms often exhibit relatively specific antibacterial preference. For example, Nisin, the only bacteriocin permitted as a food additive worldwide and produced by Lactococcus lactis, shows specific activity against Gram-positive bacteria31.
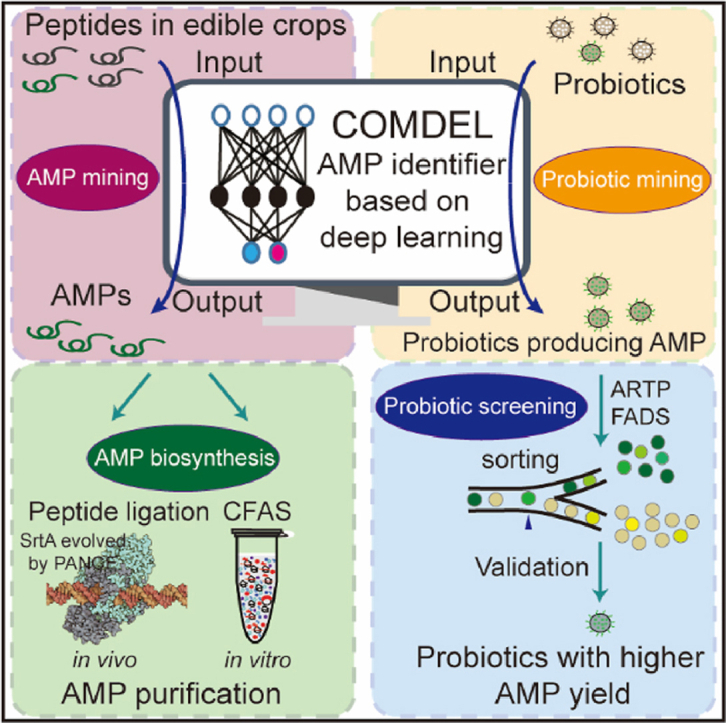
To comprehensively solve these limitations, we developed an AMP identification model, named Comparing and Optimizing Multiple DEep Learning (COMDEL), by leveraging an integrated training approach based on neural network algorithms (NNAs) and a high-throughput AMP screening method. COMDEL not only offers high accuracy but also ensures security. We then employed COMDEL in the dual tasks of screening broad-spectrum AMPs in edible crops, as well as screening probiotics for high AMP production (Fig. 1). Building upon this, we further utilized directed evolution and cell-free AMP synthesis (CFAS) technologies to significantly boost AMP yields both in vivo and in vitro.
Figure 1.

The schemes of COMDEL establishment and applications. (A) Schematic depiction of COMDEL development. The AMP and non_AMP Data were collected, followed by which the features of amino acids properties and peptide structure were extracted and filtered. Through the NNA picking and feature weighting, the model was trained and tested with feedback. This process resulted in the final version of COMDEL, characterized by high accuracy and precision. (B) Application of COMDEL in mining AMPs and probiotics. For mining AMPs and probiotics, omics data encompassing genomes, transcriptomes, and proteomes were compiled. These data were processed to deduce peptide expression levels, which were then evaluated by the COMDEL model to identify potential AMP candidates for subsequent biosynthesis and validation. In the context of natural AMP mining, COMDEL was employed to pinpoint AMP candidates with high expression in edible crops. These candidates were synthesized in vivo using an SrtA mutant evolved through the PANCE technology, and in vitro via a CFAS system. In the realm of probiotic screening, COMDEL was utilized to filter edible probiotics with elevated AMP expression. Subsequently, the probiotic mutants with increased AMP yields were obtained by using the FADS technology.
2. Materials and methods
2.1. Data collection
AMP and non-AMP data were collected as previously described16,19,21, with minor modifications. Briefly, our AMP dataset was mainly collected from three available AMP databases containing most of AMP sequences from different sources. These include 8097 sequences from ADAM17, 3414 from APD315 and 3698 from CAMPR432,33. Our focus was solely on peptides comprising typical amino acids with a length of 10–300, so the peptides containing non-standard amino acids or those less than 10 or exceeding 300 in length were eliminated. These AMP data were merged, and the duplicated and similar sequences (with an identity greater than 90%) were removed, eventually retaining 5965 sequences in the AMP dataset.
The non-AMP dataset was collected from UniProt database (https://www.uniprot.org/), excluding any entry identified as antimicrobial, antibiotic, antiviral, antifungal, effector, or excreted. We conducted a search in the UniProt database for proteins that had been manually inspected and annotated, and which ranged from 10 to 300 amino acids in length. Our search excluded proteins associated with terms such as antimicrobial, antibacterial, anti-TB, antitoxin, as well as terms like secreted, excreted, and effector. Following this refinement, we further refined our dataset by eliminating any sequences featuring non-standard amino acids and removing the duplicated and similar sequences in the dataset. After these steps, we retained a total of 5910 unique non-AMP sequences.
The AMP and non_AMP datasets were split into two sets at a ratio of 20:5, with 4772 AMPs and 4728 non-AMPs kept in the training dataset, which was utilized to build the COMDEL models. The remaining 1193 AMPs and 1182 non-AMPs were kept in the test dataset, which was used to evaluate the performance of the COMDEL models.
Probiotics representative genomes, transcriptome and proteome data were derived from DNA-seq and RNA-seq data in the NCBI GEO DataSets (https://www.ncbi.nlm.nih.gov/gds). Protein data from Glycine max and Zea mays were downloaded from the Ensembl Plants database (https://plants.ensembl.org/index.html).
2.2. Construction of the COMDEL model
To accurately extract the features, the modlAMP Python package34 was employed to calculate 56 distinct physicochemical attributes from all primary sequences, with those showing a correlation lower than 0.9 being retained. Subsequently, 13 different binary classification algorithms were tested in our model to distinguish AMPs in training dataset, including Logistic Regression (logreg)35, Decision Tree Classifier (cart), Gaussian NB (nb), Linear Discriminant Analysis (lda)36, and Quadratic Discriminant Analysis (qda)37, Support Vector Classifier Linear (svc_lr), Support Vector Classifier Radial Basis Function (svc_rbf), Support Vector Classifier Polynomial (svc_poly), Support Vector Classifier Sigmoid (svc_sig)38, Random Forest Classifier (rfc)39, Gradient Boosting Classifier (gbc)40, and Adaptive Boosting Classifier (abc)41, K-nearest neighbor (knn)42.
Considering the 56 independent physicochemical attributes were unable to robustly distinguish the AMP, we used the deep learning technology, which comprises three primary components in the model, including an embedding layer, an encoder layer, and a task layer43,44.
Initially, the embedding layer processes the input sequence and converts every amino acid in a peptide chain into a compact and dense vector that represents the particular amino acid. The primary concept of embedding is to map each type of amino acids to a distinct randomly initialized vector, which is fine-tuned based on the specific task using back propagation during model training. Each amino acid in a sequence is transformed into a dense and low-dimensional embedding vector, ensuring that every sequence in a batch is converted into a matrix composed of these vectors. Through the embedding layer, the entire sequence is uniquely represented by a matrix.
The encoder layer serves as the model's foundation, capturing the contextual information for each residue embedding vector at various positions, allowing the residue embeddings to possess distinct feature vectors based on the context, and learning the distinguishing characteristics of AMPs. At its core, the encoder layer is made up of transformers' encoders. Each encoder block consists of a multi-head attention mechanism, a feedforward network, and a pair of skip connections. The multi-head attention is a combination of several self-attention mechanisms, which is designed to learn the contextual representation of the sequence. The self-attention and multi-head attention mechanism can be mathematically described as Eqs. (1), (2), (3):
| (1) |
| (2) |
| (3) |
X is the output matrix of the embedding layer. Three different weight matrices (WQ, WK, WV) linearly transform this embedding matrix X to generate the query matrix Q, the key matrix K, and the value matrix V. The Query matrix represents the current position, the key matrix represents other positions, and the value matrix holds the information that will be weighted based on the attention scores. Only the WQ was shown above. In the context of this model, the term ‘edr’ refers to the embedding dimension of the residue, which is the dimensionality of the embedded representation. On the other hand, ‘edv’ corresponds to the embedding dimension of the value, which defines the size of the query, key, and value vectors. 'L' signifies the maximum length of the residue sequence, effectively denoting the length of an input sequence. The variable ‘i' is indicative of the count of attention heads, ranging from the first up to the ‘h'th.
Similarly, the attention weights will be computed several times with a different set of weight matrices each time. These weight matrices are WiQ, WiK and WiV, which are used to generate the query, key and value matrices for the i-th head respectively. Here, h denotes the number of heads.
Subsequently, the output of all the heads will be stitched together and mapped to the same space as the encoder input using a linear transformation using the matrix WO. This step is crucial because it ensures that the output of the attention mechanism can be directly used as the input of the next layer.
The task layer is made up of fully connected neural networks and nonlinear activation functions, transferring the representations of AMP to a probabilistic distribution of classes to make the prediction. In this layer's workflow, the input data are composed of two parts: the feature set of 56 descriptors and output of the encoder layer. These first undergo a linear transformation, followed by a ReLU activation function to introduce non-linearity. Next, the outputs will be passed through another linear transformation and another ReLU activation. Lastly, a softmax function is applied to convert the final outputs into a probability distribution, which serves as the final result.
2.3. Evaluation metrics of multiple models
To evaluate the performance of all models, we used the following metrics: accuracy (ACC), precision (Prec), matthews correlation coefficient (MCC), and area under the Receiver Operating Characteristic curve (AUC-ROC) value as in Eqs. (4), (5), (6):
| (4) |
| (5) |
| (6) |
True positives (TP) represent the number of AMPs correctly predicted as AMPs; false positives (FP) are the number of non-AMPs incorrectly predicted as AMPs; true negatives (TN) are the number of non-AMPs correctly predicted as non-AMPs; and false negatives (FN) are the number of AMPs incorrectly predicted as non-AMPs.
Accuracy (ACC) measures the overall correctness of the model across all samples, reflecting its comprehensive performance. Precision (Prec) indicates the success rate of predicting positive samples accurately. The Matthews correlation coefficient (MCC) quantifies the relationship between observed and predicted binary classifications. The AUC-ROC value represents the area under the ROC curve enclosed by the coordinate axes. An AUC value closer to 1.0 signifies a more reliable model, whereas a value of 0.5 indicates the lowest reliability and a lack of practical usefulness.
2.4. Peptide abundance calculations in probiotics
RNA-seq data were collected and analysed as previously described45. Briefly, sequencing reads were aligned to reference genomes and transcriptomes by TopHat (https://ccb.jhu.edu/software/tophat/index.shtml). ORFs were predicted by ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/), and protein abundance was calculated by their corresponding RNA level. The proteome was randomly interrupted into small fragments, and the abundance of peptides was determined based on protein abundance.
2.5. Strains, plasmids and gene synthesis
All strains and plasmids used in this study are listed in Supporting Information Table S1. The Escherichia coli strain S1030 was purchased from Addgene (Cat. No. #105063). The E. coli strains DH5α and BL21(DE3) Chemically Competent Cell were purchased from TransGen Biotech. Bacillus subtilis 168 was purchased from Biofeng. Mutagenesis plasmid MP4 (Cat. No. #69652), gIII expression vector pJC175e (Cat. No. #79219), were purchased from Addgene. pBAD18-GFP and pT7-GFP were purchased from Biofeng. M13 phage was purchased from Guangzhou Zymostar Biotech (Cat. No. ZS1004). pTET-GFP was provided from the Liu lab at the Key Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, Jiangnan University, China.
The DNA of AMP candidates, SrtA, gIII-R- pSPO-RBS-gIII-F, GFP1-10 and GFP-11 were synthesized from GUANGZHOU IGE BIOTECHNOLOGY LTD after codon optimization for E. coli in coding region.
2.6. Plasmid construction and application
The gIII-R-pSPO-RBS-gIII-F DNA was synthesized and inserted into the pJC175e plasmid to replace the gIII gene. This plasmid was used as an accessory plasmid in PANCE.
The AMP candidates listed in Supporting Information Table S2 were synthesized and inserted into the pBAD18-GFP or pT7-GFP plasmid to replace the GFP gene. These plasmids were used for AMP candidate expression in vivo and in vitro.
The GFP1-10 and GFP11 DNA were synthesized and inserted into the pTET-GFP to replace the tetO-GFP DNA. The GFP1-10 plasmid was transferred into E. coli strain BL21 (DE3), and the GFP11 plasmid was transferred into B. subtilis 168.
All these plasmids were constructed from IGE BIOTECHNOLOGY LTD. All primers used in this study are listed in Supporting Information Table S3.
2.7. Preparation of SrtA primary M13 phage for PANCE
The DNA fragment of SrtA was amplified by Hieff Canace® Gold High Fidelity DNA Polymerase (Yeasen Biotechnology Shanghai Co., Ltd., Cat. No. 10148ES60) using the synthesized gene as a template. The M13 genome skelecton without gIII was amplified by DNA Polymerase using the M13 phage as a template. The DNA fragments of SrtA was respectively cloned into the M13 genome skelecton without gIII using Hieff Clone® Plus Multi One Step Cloning Kit (Yeasen Biotechnology Shanghai Co. Ltd., Cat. No. 10912ES10). Cloning product was transformed into the E. coli strain S1030 containing the pJC175e plasmid. Transformed S1030-pJC175e was cultured at 37 °C overnight for M13 phage replication, package and release. After transient centrifugation, phage-containing supernatant was diluted and infected into fresh S1030-pJC175e strain to determinate the titer using the bilayer agarose plate method. Monoclonal phage was picked into fresh S1030-pJC175e strain for amplification. Bacterial PCR was applied to identify the correctness of fragment insertion, and the PCR product was further verified by Sanger sequencing. Primers are listed in Table S3.
2.8. Processes of PANCE for SrtA evolution
PANCE was performed according to previous report46, using the MP4 as mutagenesis plasmid. Briefly, every round of PANCE was divided into three steps. Firstly, primary M13 phage of SrtA was added into 100 μL of fresh E. coli strain S1030 containing the accessory plasmid at a final concentration of 10,000–100,000 CFU/mL. After an incubation of 20 min at 37 °C, the medium supernatant was removed by instantaneous centrifugation. Next, 10 μL of fresh Lysogeny broth (LB) medium (10 g/L tryptone, 5 g/L yeast extract, and 10 g/L sodium chloride, pH 7.0) with 100 mg/L ampicillin and 10 mmol/L l-arabinose was used to resuspend the bacterial precipitate. After incubation for 6 h at 37 °C, the medium supernatant was collected by instantaneous centrifugation. Phage titer was detected every 30–60 min until the phage titer reached 1,000,000 CFU/mL. If the phage titer remained lower than 1,000,000 CFU/mL, 100 μL of S1030-pJC175e was added to amplify the M13 phage to a titer greater than 1,000,000 CFU/mL. Finally, progeny M13 phage was obtained by collecting the supernatant of medium which was centrifuged at 10,000×g for 1 min. The progeny M13 phage was used as primary phage in the next round of PANCE.
2.9. Antimicrobial activity assay of AMP candidates
To assess whether the AMP candidates listed in Table S2 have antimicrobial activity, we constructed their codon-optimized DNA sequence onto the pBAD18-GFP vector, and transformed them into E. coli strain DH5α. A single colony strain was inoculated with 100 mg/L ampicillin and divided into six parts equally, which containing 0.01–100 mmol/L l-arabinose, respectively. After being cultured at 37 °C with a rotation speed of 200 rpm until that the optical density at 600 nm (OD600) value of one of them exceeded 1.2, the OD600 values of all the groups were detected and the value ratios of with to without l-arabinose were calculated. The survival curve was plotted by using GraphPad Prism 9 (Nonlinear regression) with a dose–response (inhibition) equation, and the minimum inhibitory concentration reached by 50% (MIC50) of l-arabinose was calculated.
For the purified peptide candidates and Nisin A (Yuanye Biotech), the indicator strains were inoculated at a concentration of approximately 500,000 CFU/mL in an appropriate medium with 0.1–1000 μmol/L peptide candidates. After being incubated at an appropriate temperature and rotate speed until that the OD600 value for the control group (without any peptide candidate) exceeded 0.8, the OD600 values of all the groups were detected and the value ratios of the groups with to without peptide candidate or Nisin A were calculated. The survival curve was plotted by using GraphPad Prism 9 (Nonlinear regression) with a dose–response (inhibition) equation, and the MIC50 value of peptide candidate was calculated.
2.10. Random 150N ORF library construction for high-throughput screening of AMPs
Two random PCRs were used to generate a random DNA library of 150 bp in the plasmid pBAD18-GFP. The first round of random PCR contains 75 random bases downstream of the start codon ATG. The second round of random PCR contained 75 random bases downstream of the first round of 75 random bases, with a spacer of 20 bps in the middle for primer binding. The random mutant plasmid library was amplified using Hieff Canace® Gold High Fidelity DNA Polymerase (Yeasen Biotechnology Shanghai Co., Ltd., Cat. No. 10148ES60) using the pBAD18-GFP as a template. The amplified PCR product was digested by DpnI and recovered using MolPure Gel Extraction Kit (Yeasen Biotechnology Shanghai Co. Ltd., Cat. No. 19101ES70), and then transformed into E. coli strain DH5α by electro-conversion to repair DNA. Total plasmid library was extracted using MolPure® Plasmid Mini Kit (Yeasen Biotechnology Shanghai Co., Ltd., Cat. No. 19001ES70) and was used as the template in the next round of random PCR. The next round PCR product underwent the same treatment and recovery processes. Finally, the total plasmids were extracted and used as the random mutant plasmid library.
2.11. Next generation sequencing for screening AMPs
The random mutant plasmid library was transformed into E. coli strain DH5α. The strain pools were cultured with or without 10 mmol/L l-arabinose at 37 °C with a rotate speed of 200 rpm until the OD600 value reached 0.6. The plasmid pool was then extracted and amplified using primers containing Illumina universal sequence and index (Yeasen Biotechnology Shanghai Co., Ltd., Cat. No. 13519ES04). The PCR product was recovered and sequenced by Illumina NovaSeq 6000 platform with the PE150 model.
After sequencing, the adaptors at both end of the sequencing reads were trimmed using Trimmomatic (http://www.usadellab.org/cms/?page=trimmomatic), followed by which Reads 1 and Reads 2 were assembled into completed sequences, and then translated from the start codon ATG to obtain the amino acid sequences. The abundance of these peptides was calculated and the ratio of peptide abundance with to without 10 mmol/L l-arabinose was calculated. Two thresholds were used to distinguish AMP candidates: a relax threshold (Foldchange value (Ara +/−) < 0.5, P value (Ara +/−) < 0.05) and a strict threshold (Foldchange value (Ara +/−) < 0.5, P value (Ara +/−) < 0.01).
2.12. Cell-free AMP synthesis (CFAS) system
The cell lysis of E. coli strain was performed according to previous reports47,48. After culturing the E. coli strain BL21(DE3) in 2 × YT medium (16 g/L tryptone, 10 g/L yeast extract, and 5 g/L sodium chloride, pH 7.0) to an OD600 value of 3, the culture was centrifuged at 5000×g and 4 °C for 15 min and washed three times with pre-chilled S30 buffer (10 mmol/L triacetate pH 8.2, 14 mmol/L magnesium acetate, 60 mmol/L potassium acetate, 2 mmol/L dithiothreitol (DTT)). Added 0.8 mL of S30 buffer per gram of wet cell mass and sonicated the cells under ice bath conditions. The sonication power is 20 W, and it stops for 20 s after working for 10 s until the suspension became clear. Added DTT at a final concentration of 3 mmol/L, and centrifuged the lysate at 12,000×g and 4 °C for 10 min.
A 50 μL CFAS reaction was assembled by mixing the following components: 50 mmol/L HEPES, pH 7.2; 1.2 mmol/L ATP, 0.85 mmol/L UTP, GTP and CTP; 34 μg/mL folinic acid; 170 μg/mL E. coli tRNA mixture; 5 μg/mL T7 RNA Polymerase; 15 μg/mL PCR product; 2 mmol/L for each of the 20 standard amino acids; 0.33 mmol/L nicotinamide adenine dinucleotide (NAD); 0.27 mmol/L coenzyme-A (CoA); 1 mmol/L putrescine; 4 mmol/L sodium oxalate; 1.5 mmol/L spermidine; 130 mmol/L potassium glutamate; 10 mmol/L ammonium glutamate; 12 mmol/L magnesium glutamate; 33 mmol/L phosphoenolpyruvate (PEP), and 30% v/v of cell extract. The CFAS reaction was incubated overnight at 37 °C, which was ready for SDS-PAGE analysis and peptide purification.
2.13. AMPs purification form CFPS reaction
The reaction system was mixed with 2 volumes of ethanol and precipitated overnight at −20 °C. After centrifuged at 12,000×g and 4 °C, the pellet was dissolved by 50 mmol/L phosphate buffer (pH 6.0), and filtered with a 0.22 μm filter membrane. Next, Sephadex G75 was equilibrated with phosphate buffer, and the dissolved pellet was added at a flow rate of 0.5 mL/min, with the effluent being collected at different stages. SP sepharose HP was equilibrated with phosphate buffer, and the protein sample was loaded. Unbound proteins were subsequently washed with 3 volumes of phosphate buffer, followed by a gradient elute with 1 mol/L NaCl-phosphate buffer. SP sepharose HP was eluted at a flow rate of 1 mL/min, and the eluate was collected. The purity and yield of AMP were determined by SDS-PAGE and the BCA Protein Assay Kit (Solabio) according to the manufacturer's instructions.
2.14. ARTP for mutant generation
The Lactobacillus plantarum strain 123 (Yunying Biotech) was cultured in MRS medium (10 g/L peptone, 10 g/L beef paste, 5 g/L yeast paste, 2 g/L diammonium hydrogen citrate, 20 g/L glucose, 0.1% (v/v) Tween-80, 5 g/L sodium acetate, 2 g/L dipotassium hydrogen phosphate (K2HPO4·3H2O), 0.58 g/L magnesium sulfate (MgSO4·7H2O), 0.25 g/L manganese sulfate (MnSO4·H2O), pH 6.2–6.6) until the OD600 value reached 0.6.
1 mL of the strain was centrifuged at 7000×g for 1 min, and then the medium supernatant was removed. The strain pellet was washed twice with 10 mL of PBS buffer (8 g/L sodium chloride, 0.2 g/L potassium chloride, 1.42 g/L disodium hydrogen phosphate, and 0.24 g/L potassium dihydrogen phosphate, pH 7.4), and subsequently suspended in 10 mL of PBS buffer. Afterwards, 10 μL of the suspension was coated on the iron piece of ARTP (Tianmu Biotechnology Co., Ltd.). A total of 3 times ARTP mutagenesis with the condition of 120 W, 15 SLM, 2 mm and 1 min was executed, followed by the piece was washed in 1 mL of MRS medium. The strain was allowed to stand for 2 h at room temperature for the preparation of microdroplets.
2.15. Preparation of microfluid chips for fluorescence-activated droplet sorting (FADS)
Microfluidic chips were built using bypoly(dimethylsiloxane) (PDMS, Dow Corning Corp.) according to standard soft-lithography methods. UV exposure was used to prepare SU8-2015 negative photoresist (MicroChem Corp.) mold a silicon wafer. A 10% (w/w) final concentration of curing agent was added to the PDMS and poured onto the mold. After degassing under vacuum condition, the mold cross-linked at 65 °C overnight. PDMS was then spalled off and punched with a 0.75 mm diameter biopsy punch, and then bound to glass microscope slide using oxygen plasma system. Finally, hydrophobic surface coating was created by injecting HFE7100 fluorinated oil (3 mol/L) with 1% (w/w) 1H,1H,2H,2H-perfluorodecyltrichlorosilane (97%; ABCR) in the 25 μm microfluidic channels.
Electrode hole of the sorting chips were filled with 51In/32.5Bi/16.5Sn low-temperature solder (Indium Corp.) and incubated at 110 °C for 30 min. The short pieces of electrical wire were inserted to the hole before the metal solidifies.
2.16. Preparation of microfluid devices for FADS
The optical setup consisted of a Compound Inverted Microscope System (Olympus) mounted on a dampening platform. A 488 nm laser was focused through the objective lens across the microfluidic chip. Emitted light from fluorescing droplets was captured and channelled back along the path of the lasers by the objective, and then separated from the laser beam and split by photomultiplier tube, which captured the light through a 510 nm bandpass filter (510/20–25; Semrock Inc.). The signal output was analyzed using a PCI-7831R Multifunction Intelligent Data Acquisition (DAQ) card (National Instruments Corporation) executing a program written in LabView 8.2 (FPGA module, National Instruments Corporation), which can identify droplets by peaks in fluorescence. A Phantom v4.2 high speed digital camera (Vision Research) was loaded on the microscope to capture light images during droplet manipulation. Liquids were injected into the chips using standard-pressure syringe pumps (Harvard Apparatus Inc.). We used aqueous droplets in HFE7500 fluorinated oil (3 mol/L) with 4% (w/w) FluoSurf surfactant (Techu Scientific).
A dropmaker chip was applied to generate 20 pL droplets at 4000 Hz by flow-focusing of the aqueous stream (5 μL/min) with two streams of HFE7500 fluorinated oil (3 mol/L) (10 μL/min) containing 4% (w/w) FluoSurf surfactant. The generated droplets flowed off-chip through PTEF tubing to a collector.
An injection chip was applied to inject biosensor (mix of GFP10-E. coli strain BL21 (DE3) and GFP11-B. subtilis strain 168) into droplets. Droplets were reloaded (1 μL/min) and spaced-out at a flow-focusing junction with HFE7500 fluorinated oil (3 mol/L) (1 μL/min). The biosensor was rejected (0.5 μL/min) and droplets at T-junction, at which a continuous voltage of 500 V was loaded. The injected droplets flowed off-chip through PTEF tubing to a collector.
The module of fluorescence-activated droplet sorting (FADS) in which droplets were reloaded (0.2 μL/min) and spaced-out at a flow-focusing junction with HFE7500 fluorinated oil (3 mol/L) (1 μL/min). The droplets were detected and analysed by the optical setup and fluorescent droplets were sorted at 500 Hz by applying an AC field pulse (30 kHz; 700–1000 V; 0.5 ms). The sorted droplets were collected in a 1.5 mL microcentrifuge tube. Under analysis mode operation, reloading module in which droplets were reloaded (0.2 μL/min) and spaced-out at a flow-focusing junction with HFE7500 fluorinated oil (3 mol/L) (1 μL/min). The droplets were detected and analysed by the optical setup at ∼1000 Hz.
2.17. Statistical analysis
Statistical analyses were performed using the two-tailed Student's t-test, one-way analysis of variance (ANOVA), or Mann–Whitney nonparametric U-test with GraphPad Prism 9.0. An asterisk (∗) indicates that a P value less than 0.01 was considered statistically significant. All experiments were performed with three replicates, and the error bars in the figure legends represent means ± SD values.
3. Results
3.1. Establishment of COMDEL model for AMP identification
As depicted in Fig. 1, we have developed an innovative AI-based approach using the methodology of Comparing and Optimizing Multiple DEep Learning (COMDEL) algorithms for the dual purposes of identifying AMPs and screening probiotics. To ensure a comprehensive AMP dataset, we amassed a collection of 10423 AMPs sourced from various species and artificial AMP databases (Fig. 2A)15,17,32,33. Subsequently, 5965 representative AMPs were retained by filtering out homologous and similar sequences, to create a robust AMP dataset (Fig. 2B). Simultaneously, to mitigate data bias, we compiled a non-AMP dataset comprising 5910 peptides that were identified for lacking antibacterial function annotations in the Uniport database. These non-AMPs were chosen based on their similarity in length distribution and amino acid composition to the AMPs (Fig. 2B). For the purpose of model training and validation, these data were randomly divided into 25 equal subsets; 20 of these were utilized for training, while the remaining 5 subsets served as a test dataset (Supporting Information Fig. S1A). Encouragingly, the peptide length distributions in both training and test datasets were similar, confirming a reasonable distribution in our methodology (Fig. S1B). The architecture of COMDEL, as detailed in Fig. 2C, comprises three main modules: the embedding layer, the encoder layer, and the task layer. Within the embedding module, each residue in an AMP sequence is represented by multiple embedding vectors. Through dimensionality reduction techniques and specifically correlation analysis, 48 of 56 independent peptide features with a correlation lower than 0.9 were narrowed down for effectively distinguishing between AMPs and non-AMPs (Supporting Information Fig. S2). Following the embedding layer, the encoder module employs a multi-head attention mechanism to adeptly capture the sequential nature of AMP data. To conclude the process, the task module utilizes various neural networks to translate the AMP representation into a probability distribution corresponding to its classification, ensuring high accuracy in AMP identification.
Figure 2.

Construction of the COMDEL model. (A) The venn diagram illustrates the AMP data collected from three AMP databases including CAMPR433, ADAM17 and APD315. (B) The pie chart displays the length distribution of the collected AMP and non-AMP data. (C) The COMDEL model consists of four main parts. The first is the ‘Embedding Module’, where each part of a protein sequence is turned into multiple data points based on its context. These data points are then standardized. Next, the ‘Encoding Module’ uses a special technique to understand complex sequence patterns, ensuring maximal utilization of every sequence portion. The third part, ‘Physicochemical Property Extraction’, pulls out 56 unique characteristics from the sequences, and these characteristics are processed using 13 different machine learning models. After these processes, their outcomes are merged with those the Encoding Module. Lastly, the ‘Task-Specific Module’ uses a group of neural networks to refine this information, turning it into probabilities for different categories. (D) AMP prediction accuracy comparison of the COMDEL model across 13 NNAs. (E) The AMP prediction performance of the COMDEL model in training and test datasets.
To account for potential biases among these 56 physicochemical features, we employed 13 NNAs as an initial screening step. This approach was designed to assess the effectiveness of these features in classifying AMPs. Concurrently, we fine-tuned the hyperparameters of natural language processing (NLP) models using independent datasets, and noted that all models converged rapidly during training. Ultimately, the Classification and Regression Tree (CART) method was selected to further improve the accuracy of our COMDEL model for its best performance in AMP prediction (Fig. 2D). In addition, hemolysis, a critical factor to consider due to its potential harm49, was comprehensively incorporated into our COMDEL model to ensure its safety in AMP mining (Supporting Information Fig. S3).
Quantitatively, we evaluated various algorithm combinations using metrics such as Precision, Recall, and the Area Under the Precision-Recall Curve (AUPRC). Remarkably, in both the training and test datasets, the precision of COMDEL rose to 95.7% and 92.4%, respectively, while its accuracy improved to 96.9% and 94.1% (Fig. 2E).
3.2. Performance comparison of COMDEL with other AMP identification models
To evaluate the performance of our COMDEL model, we conducted a comparative analysis with five other state-of-the-art AMP identification models based on machine learning approaches, including a deep learning model like the natural language processing neural network models (NLPNNM)16, and traditional machine learning models, such as AMPEP21, AMP Scanner v217,22, AMPIR24, and Deep-ABPpred19, using a unified test dataset in our collected data. The Area Under the Receiver Operating Characteristics (AUROC) curve revealed that COMDEL outperformed the others in terms of both accuracy and precision. Specifically, COMDEL boasted a higher true-positive rate and a lower false-positive rate, and consequently higher overall accuracy and precision (Fig. 3A and B). These results convincingly demonstrated that the COMDEL model is a robustly effective and reliable tool for distinguishing AMPs from sequence data.
Figure 3.

The performance comparison of COMDEL with other state-of-the-art AMP prediction methods. (A) The AUROC curve comparison of COMDEL versus other AMP prediction methods on a unified test dataset. AUC represents the Area Under the Curve. (B) The precision and accuracy comparison of COMDEL versus other AMP prediction methods on a unified test dataset. (C) The AUROC curve comparison of COMDEL versus other AMP prediction methods across peptides of varying lengths within the unified test dataset.
To delve deeper into the performance of COMDEL against competing models, we categorized the testing dataset into five categories based on peptide length. Among the three advanced AMP prediction models previously described, we observed that accuracy obviously improved with increasing peptide length, particularly in the AMPEP model (Fig. 3C). This trend might be attributed to the fact that longer peptides tend to facilitate better feature extraction, thereby enhancing model performance. Conversely, shorter peptides might pose a challenge in this aspect, as their features may not be effectively extracted. Intriguingly, this length bias seems to be greatly alleviated in our COMDEL model, indicating its superior adaptability (Fig. 3C). Further supporting this, comparison results also revealed that COMDEL consistently surpassed other methods in predicting AMPs of various lengths in the test dataset, particularly short AMPs (Fig. 3C).
3.3. Optimizing COMDEL model using high-throughput AMP screening technology
As depicted in Fig. 3C, although the COMDEL model could lessen the bias caused by peptide length, it is noteworthy that predictions for peptides shorter than 50 amino acids were still less accurate compared to those of longer peptides. This limitation was even more pronounced in the other models.
To further refine the COMDEL model, we implemented a high-throughput AMP-screening strategy aimed at effectively filtering AMPs from random peptide sequences (Fig. 4A). In this process, a plasmid pool containing 150 bp of random DNA sequences (referred to as 150N) was constructed downstream of the start codon ATG and the ribosome binding site (RBS). To mitigate the effects of unintended gene expression on host cell growth, we stringently regulated the expression of random peptide gene by using the arabinose operon. After transforming this plasmid pool into the E. coli strain DH5α and cultivating it to the logarithmic phase with or without the induction of arabinose, we harvested the plasmids and amplified the 150N sequences using universal primer pairs at its both ends (Fig. 4A).
Figure 4.

Optimization and application of the COMDEL model in AMP mining. (A) Schematic diagram of the high-throughput AMP screening method. A vector pool that contains 150N DNA sequences were controlled by the arabinose operon. The presence of bacteriostatic activity in encoded peptides results in growth inhibition of the strain upon the addition of l-arabinose. The 150N DNA is then amplified using universal primers and sequenced on the Illumina NovaSeq 6000 platform. (B) The volcano plot displays the difference in peptide expression under the conditions with and without l-arabinose by analysing sequencing data. The labels “up-relax” and “up-strict” refer to peptides with an expression increase (fold change >2) under relax (P value < 0.05) and strict (P value < 0.01) conditions, while “down-relax” and “down-strict” refer to peptides with reduced expression (fold change <0.5) under relax (P value < 0.05) and strict (P value < 0.01) conditions. (C) The effect of 10 AMP candidates screened by high-throughput method on the growth of E. coli DH5α. (D and E) The AUROC curve comparison of the COMDEL models before and after optimized in total peptides (D) and the peptides shorter than 50 amino acids (E) using the unified test dataset. (F) The effect of 50 AMP candidates predicted by COMDEL from the two edible crops on the growth of E. coli DH5α. (G) The venn diagram displays the AMP candidates predicted by the optimized COMDEL model and other methods in two edible crops. A total of 7504 and 5257 AMP candidates were identified by these four models in Glycine max and Zea mays, respectively, from datasets comprising 88,414 and 72,593 peptides. The percentages represent the proportion of AMP candidates predicted by each model to the total number of peptides. (H) The effect of the AMP candidates predicted by COMDEL and other methods on the growth of E. coli DH5α. COMDEL_u represents the AMP candidates uniquely identified by the COMDEL model, while COMDEL_e represents the AMP candidates identified by at least two models as opposed to the COMDEL model. When comparing the OD600 value of the condition with and without l-arabinose, an asterisk (∗) denotes that the data are significantly different.
Through next-generation sequencing (NGS), we discovered 415 up-regulated and 52 down-regulated peptide sequences in the relaxed model (P value < 0.05), along with 91 up-regulated and 40 down-regulated peptide sequences in the strict model (P value < 0.01) (Fig. 4B, Supporting Information Table S4). We particularly focused on the peptides that exhibited reduced expression, as these were hypothesized to potentially possess bacteriostatic properties. To confirm this hypothesis, we selected 10 down-regulated peptides that were also predicted as AMPs by the COMDEL model, for further experimental verification using antibacterial assays. The results confirmed that all the 10 down-regulated peptides displayed bacteriostatic activity (Fig. 4C, Table S2), thereby validating their potential utility as effective AMPs.
To further enhance the AMP identification accuracy of the COMDEL model, we integrated the high-throughput screening results into the training process of model construction. As a result, the overall accuracy and precision of COMDEL for total peptides impressively reached an outstanding 94.8% and 92.9%, respectively. Notably, this performance was even more remarkable for peptides shorter than 50 amino acids, where these metrics improved to 95.4% and 94.6% as a direct consequence of feature parameter optimization (Fig. 4D and E). These outcomes indicate that the performance of the COMDEL model can be substantially boosted when augmented with the NGS data. To corroborate these findings, we conducted antibacterial assays on 50 AMP candidates predicted by the optimized COMDEL model. The assays demonstrated that 44 of these peptides exhibited bacteriostatic properties, achieving a high positive verification rate of 88% (Fig. 4F, Supporting Information Fig. S4, Table S2).
In conclusion, we have successfully developed and optimized the COMDEL model, establishing it as an efficient and precise tool for AMP identification. This model exhibits considerable potential for industrial applications.
3.4. AMP mining in edible crops using COMDEL
The safety of AMPs, a critical factor for their utilization in industrial applications, remains a significant concern6,50. While hemolysis has been taken into consideration during constructing the COMDEL model, enhancing the safety of discovered AMPs remains a central focus of our ongoing research.
In an effort to maximize the safety of identified AMPs, we mined AMP candidates from soybean (Glycine max) and corn (Zea mays), two typical food crops, by using these four AMPs prediction models. Through a joint analysis of multi-omics data, a total of 7504 and 5257 peptides from Glycine max (88,424 peptides in total) and Zea mays (72,539 peptides in total), respectively, were identified as AMP candidates (Fig. 4G, Supporting Information Table S5). Among these models employed, NLPNNM, a potent model recently reported, yielded the highest number of predicted AMPs from both crops. The AMPs number identified by COMDEL was approximately half that of NLPNNM, while the other two methods could only identify a few hundred (Fig. 4G). This may be due to that COMDEL and NLPNNM are deep learning models, which possess better generalization ability than traditional machine learning methods. The NLPNNM model is the best-performing AMP prediction model currently reported, consistent to our analysis (Fig. 3B). COMDEL identified 881 (25.0%) and 205 (15.5%) unique putative AMPs in Glycine max and Zea mays, respectively, compared to NLPNNM, which identified 3636 (59.1%) and 3329 (72.1%) unique putative AMPs, as well as AMPIR (17.7% average) and AMPEP (43.2% average). Intriguingly, nearly 80% of the AMP candidates predicted by COMDEL were also recognized by NLPNNM, suggesting a high degree of concordance between the two deep learning models. Conversely, only 30% of the peptides predicted by NLPNNM were present among the AMP candidates identified by COMDEL. The majority of peptide candidates uniquely predicted by NLPNNM were not corroborated by other methods, suggesting a relatively high over-prediction rate in NLPNNM (Fig. 4G). These findings collectively indicate that the COMDEL model possesses a relatively stringent and accurate performance compared to other state-of-the-art AMP prediction models.
In Glycine max and Zea mays, a total of 16 and 21 peptides, respectively, were identified as AMP candidates across all these models, highlighting a high possibility for developing into functional AMPs (Fig. 4G, Table S5). It is noteworthy that there still existed around 1000 unique peptides candidates in Glycine max and Zea mays being exclusively identified as AMP candidates by our COMDEL model (referred to as COMDEL_u) (Fig. 4H). To empirically validate the true-positive rate of these unique candidates, we expressed 10 COMDEL_u AMP candidates in E. coli strain DH5α using the pBAD18 vector under the control of the arabinose operon. Our experiments showed that 7 of the 10 COMDEL_u peptides exhibited significant bacteriostatic activity against E. coli DH5α, underscoring that the COMDEL model has a relatively high true-positive rate in identifying AMPs that are overlooked by other models (Fig. 4H, Table S2).
Additionally, a total of 7908 AMP candidates in Glycine max and Zea mays were identified by other models but not by our COMDEL model (referred to as COMDEL_e) (Table S5). Of these, 570 AMP candidates were concurrently predicted by at least two other models (Fig. 4H). To assess the false-negative rate, we expressed 10 COMDEL_e AMP candidates in E. coli strain DH5α. Our results revealed that only one of these 10 COMDEL_e peptides significantly inhibited the growth of E. coli DH5α, implying that the COMDEL model exhibits a lower false-negative rate compared to other advanced models (Fig. 4H, Table S2).
In summary, our findings demonstrate that the COMDEL model is capable of identifying AMPs with both high accuracy and extensive coverage.
3.5. Biosynthesis of broad-spectrum AMPs derived from edible crops identified by COMDEL
As stated before, most current AMP products are derived from chemical synthesis or natural extraction methods that are both low-yield and expensive, limiting the development of large-scale AMP production28. To explore more efficient avenues for AMP synthesis, we initially used E. coli BL21 (DE3) and Pichia yeast GS115, two typical host strains for protein synthesis, to express COMDEL-identified AMPs derived from Glycine max and Zea mays with codon optimization. Unfortunately, this approach led to a significant reduction in the proliferation of these host strains upon the induction of AMP expression (Supporting Information Fig. S5A and S5B). Additionally, SDS-PAGE results corroborated this challenge, indicating the difficulty for these host strains to efficiently synthesize AMPs in vivo, probably due to their inherent antimicrobial properties (Fig. S5C and S5D).
To mitigate the adverse effects of AMPs on bacterial growth, we split AMPs into N-terminus (AMP-N) and C-terminus (AMP-C) segments, and then ligated them using the peptide ligase Sortase A (SrtA) that is widely used in peptide and protein synthesis in vitro51. Following this approach, we expressed the AMP-C, AMP-N and SrtA in the E. coli strain BL21 (DE3), and observed that the growth of E. coli harbouring all three components was only slightly inhibited compared to strains harbouring each component independently. This observation suggests that complete AMP may be successfully ligated by SrtA to generate antibacterial activity (Supporting Information Fig. S6). Nevertheless, it was evident that the E. coli growth inhibition through this process was about threefold weaker than that by expressing intact AMPs (Fig. S6). This comparatively reduced inhibition might be attributed to low ligation efficiency of SrtA, stemming from its sequence preference.
To enhance the ligation activity of SrtA for AMP synthesis, we employed the phage-assisted non-continuous evolution (PANCE)46, a cutting-edge method for directed evolution, to screen for potent SrtA mutants. This was achieved by strategically coupling SrtA's ligation activity with the abundance of pIII, a protein essential for the infectivity of M13 bacteriophages (Fig. 5A). Drawing on a recent publication52, the accessory plasmid (AP) was designed to feature a split pIII protein with both N- and C-termini, which need to be ligated into a full-length pIII by SrtA. We then replaced the native gIII gene in the M13 bacteriophage genome (named as SP) with the SrtA gene, rendering it incapable of independently infecting the host E. coli strain S1030. In parallel, a mutagenesis plasmid (MP) expressing mutagenic genes was employed to enable continuous mutation of SP containing the SrtA gene. Theoretically, an increase in the activity of the evolved SrtA mutants was expected to lead to the production of more intact pIII proteins and, consequently, a higher yield of infectious bacteriophages (Supporting Infromation Fig. S7).
Figure 5.

Establishment of AMP biosynthesis approaches. (A) A biosensor of SrtA activity linked to peptide ligation couples with the abundance of M13 essential protein pIII. In the selection phage (SP), the gIII gene is replaced with the SrtA gene. In the accessory plasmid (AP), the gIII gene is segmented into two portions according to a recent publication52, requiring ligation by SrtA to form the full-length pIII protein. (B) Detection of the M13 phage titer change during the progression of evolutionary rounds in SrtA PANCE. (C) Sanger sequencing to detect the SrtA mutations in PANCE. (D) The structures comparison of SrtA and its mutants (SrtA∗) predicted by AlphaFold2. Alterations in the side chains of mutated amino acids are delineated and presented using ChimeraX. (E) The peptide ligation efficacy of SrtA to AMP is validated by the effect on strain growth. When comparing to the OD600 value of the GFP vector, an asterisk (∗) denotes that the data under corresponding conditions are significantly different. (F) SDS-PAGE to exhibit three AMPs identified by COMDEL from the two edible crops generated by the cell-free synthesis system, and then purified by gel and ion exchange chromatography.
As the evolutionary rounds progressed, the titer of bacteriophages experienced a notable increase, indicating an enhanced activity of SrtA mutants (Fig. 5B). Through Sanger sequencing, we successfully isolated an optimized SrtA mutant (SrtA∗), harbouring the S49G and M102I mutations (Fig. 5C). Utilizing AlphaFold29, we compared the tertiary structure of SrtA∗ with that of the wild type (WT), revealing that these two mutations specifically altered the size of the active centre pocket. This structural modification is hypothesized to influence both the activity and substrate preference of SrtA (Fig. 5D). A subsequent evaluation of split AMPs ligated by SrtA∗ and WT in vivo showed that the growth rate of E. coli harbouring both SrtA∗ and split AMPs was about half that of E. coli with the wildtype SrtA and split AMPs, while little difference in growth was observed when either SrtA or SrtA∗ was expressed individually (Fig. 5E). This strongly suggests that SrtA∗ is significantly more effective than the wild type in ligating intact AMPs. These findings open up a promising approach for more efficient AMP synthesis both in vivo and in vitro. However, it's crucial to acknowledge that the requirement to split AMPs into two components imposes intrinsic limitations, leading to lower yields of the synthesized AMPs (Supporting Information Fig. S8).
Recently, cell-free protein synthesis systems have gained widespread popularity for protein production, largely due to their high efficiency and minimal cytotoxicity issues47. Given that the synthesis of broad-spectrum AMPs in vivo brings severe stresses on cell growth, we utilized a cell-free AMPs synthesis (CFAS) system for the single-step synthesis of COMDEL-identified AMPs. Results from SDS-PAGE confirmed that AMPs could be effectively synthesized in the CFAS system (Fig. 5F). Following purification through gel filtration and ion-exchange chromatography, their yields were found to range from approximately 0.5 to 2.1 g/L (Fig. 5F), presenting a promising avenue for large-scale AMP production within hours.
To evaluate the broad-spectrum efficacy of these COMDEL-identified AMPs purified from the CFAS system, we determined their effects against a panel of seven common bacteria and fungi. Our results revealed that, in comparison to nisin, the AMPs identified by our COMDEL model obviously exhibited more expansive antibacterial activities against these microorganisms expected for A. oryzae and C. glutamicum (Supporting Information Fig. S9). This underscores the great potential of these COMDEL-identified AMPs in industrial application.
3.6. Screening edible probiotics for high production of AMPs by COMDEL and FADS
Edible probiotics, notably celebrated for their safety and health benefits, have diverse applications in both food and medicine, particularly for the high production of AMPs53, 54, 55. Take this trait into account, we employed COMDEL to screen edible probiotics with exceptional potential for AMP production (Fig. 1B). We compiled the genomes, transcriptomes, and proteomes of 35 edible probiotics allowed to be used in food processing. Using a weighting algorithm, we forecasted the proteome expression level of each probiotic (Supporting Information Fig. S10). COMDEL was then applied to assess the possibility of peptides derived from these proteomes as potential AMPs. Based on the predicted expression levels, we scored the overall potential AMP intensity for each probiotic (Fig. S10). This led us to generate a comprehensive ranking according to total AMP intensity (Fig. 6A). To validate our findings, we extracted fermentation products from the top five ranked probiotics. The results corroborated that the fermentation products of these probiotics exhibited bacteriostatic properties, confirming the COMDEL prediction results. Notably, Lactobacillus plantarum emerged as the candidate with the highest AMP intensity among the top five ranked probiotics, establishing it as an excellent candidate for AMP production (Fig. 6B).
Figure 6.

Screening edible probiotic for high AMP production. (A) The rank of the edible probiotics for potential AMP intensity predicted by COMDEL. The possibility of edible probiotics to generated AMP was calculated and ranked by COMDEL according to the multi-omics data including genome, transcriptome and proteome. (B) The antibacterial activity verification of probiotic fermentation product against E. coli BL21 (DE3) and B. subtilis 168. (C) Schematic diagram of screening the L. plantarum mutants for high AMP production by FADS. A biosensor of the antibacterial activity coupled with the split GFP assembly is designed. (D) Antibacterial activity verification of the three screened L. plantarum mutants against seven microorganisms. (E) The genome map of the three screened L. plantarum mutants was drew by using the whole genome sequencing data. An asterisk (∗) indicates that the data are significantly different when comparing the OD600 value of the condition with or without the addition of the fermentation supernatant.
Although L. plantarum exhibited the most potent bacteriostatic efficacy among these probiotics, its natural AMP abundance is insufficient for extensive applications across various fields. Recognizing this limitation, we turned to fluorescence-activated droplet sorting (FADS)56—a high-throughput screening method for dominant strain selection by coupling with corresponding sensors—as a viable strategy for screening L. plantarum mutants with elevated AMP yields. To establish a correlation between bacteriostatic activity and fluorescence intensity, two strains constitutively expressing split GFP were designed to be a fluorescence sensor for AMP activity (Fig. 6C)57. The first strain is E. coli BL21 (DE3), engineered to constitutively express the GFP1-10 protein, and the second is B. subtilis 168, modified to constitutively express the GFP11 protein. The underlying principle of this design is straightforward: In case that the L. plantarum mutants produce AMPs at higher levels, the E. coli and B. subtilis cells can be lysed to release more GFP1-10 and GFP11, which subsequently assemble into active GFP.
To generate a complex pool with mutant strains, we employed atmospheric and room temperature plasma (ARTP)58, a safe and powerful mutagenesis technology, followed by employing the FADS technology to screen for L. plantarum mutants with high AMP production (Fig. 6C). We isolated 50 colonies and proceeded to extract their fermentation byproducts, which were then utilized to assess antibacterial activity. We found that numerous screened colonies displayed stronger antibacterial effects against E. coli or B. subtilis compared to the wild type. Notably, only three mutants exhibited enhanced growth inhibition against both the two bacterial strains (Supporting Information Fig. S11). To confirm these results, we analysed the bacteriostatic activity of the fermentation supernatants from these three screened L. plantarum mutants against the seven microorganisms. The data showed that, although the bacteriostatic activity against A. oryzae was not significant, these three mutants exhibited stronger and broader antibacterial activities against the other six microorganisms than wildtype L. plantarum. Moreover, the bacteriostatic activity of the three screened L. plantarum mutants (M1, M2, and M3) was more than twofold higher than that of the wild type in these six microorganisms, except for Saccharomyces cerevisiae (Fig. 6D). These findings highlight that FADS, combined with the split GFP sensors, is an effective method for screening probiotics with high antibacterial efficacy.
To decipher the gene mutations responsible for the enhanced bacteriostatic activity, we carried out whole genome sequencing (WGS) and drew a complete genome map of these three screened L. plantarum mutants (Fig. 6E). Through mutation analysis, we found that the mutations occurred in these three strains exhibited obvious specificity. There were six mutations in strain M1, seven in strain M2, and five in strain M3. Interestingly, certain mutations were shared between two of these strains, indicating their potential roles in enhancing bacteriostatic activity.
3.7. Data availability
All data generated or analyzed during this study are included in this published article.
4. Discussion
The mining and synthesis of natural broad-spectrum AMPs have long been a focal point in the research field of antibiotic alternatives. Built upon the accumulated database of AMPs, recent studies have leveraged machine learning and NNAs to create more sophisticated AMP prediction models, offering promising approaches for AMP mining14, 15, 16. Yet, they often fall short in efficiency, plagued by issues like inappropriate feature extraction, inconsistent benchmark datasets, and a limited scope in algorithms, especially when it comes to predicting short AMPs.
To overcome these limitations, we developed COMDEL, a technology that integrates deep learning to achieve efficient and accurate AMP identification. Three primary factors may contribute to the superior performance of COMDEL in identifying AMPs compared to other advanced models. Firstly, our model benefits from access to an expansive and diverse dataset available for training and test, thereby ensuring a robust and comprehensive data foundation (Fig. 2A and B). Secondly, the feature extraction and filtering processes employed in constructing the COMDEL model are more sophisticated and extensive, allowing for better generalization and predictive accuracy (Fig. S2). Thirdly, the NNA integrated into our COMDEL model is particularly well-suited for AMP prediction (Fig. 2C and D). Taken together, these superiorities strongly suggest that our COMDEL model holds excellent potential for practical application in AMP discovery.
While COMDEL outperforms other state-of-the-art machine learning-based methods in terms of its less pronounced bias towards peptide length, it still struggles with lower accuracy for peptides shorter than 50 amino acids. Recognizing this, we considered high-throughput screening techniques as a potential solution for batch screening of antimicrobial peptides. However, effective methods in this area are still underdeveloped59,60. To enhance our AMP database for optimizing COMDEL, we compiled a peptide pool with sequences shorter than 50 amino acids and screened for those with high bacteriostatic activity. Finally, the optimized COMDEL model achieved an unprecedented accuracy rate of 94.8% for total peptides and 95.4% for peptides shorter than 50 amino acids. These record-breaking performances underline COMDEL's immense potential in industrial-scale AMP prediction.
To ensure safety and feasibility, our application of COMDEL was specifically focused on two edible crops: Glycine max and Zea mays. Although the amount of data we tested was limited, in combination with proteomic data, COMDEL boasts definitely superior accuracy in AMP identification compared to other existing methods. This insight led us to consider edible biomass as a promising source for discovering safe AMPs. Moreover, we applied COMDEL to screen edible probiotics known for their high AMP expression levels. Of particular note was L. plantarum, which stood out as the most prolific natural AMP producer among 35 edible probiotics. While previous studies have highlighted L. plantarum's antibacterial traits and its AMP production during fermentation, the AMP quality and quantity are insufficient for broad application in effectively suppressing other microorganisms53. This might necessitate employing advanced directed evolution technologies like high-throughput screening methods based on flow cytometry and microdroplet, to screen for broad-spectrum AMPs, probiotics, and their mutants60,61. In pursuit of this goal, we engineered a split GFP sensor responsive to bacteriostatic activity. This innovative approach facilitated the screening of high-performing L. plantarum mutants. Our efforts culminated in identifying three L. plantarum mutants with significantly enhanced antimicrobial capabilities, highlighting their considerable potential in food processing applications.
Although COMDEL has exhibited robust performance on AMP and probiotic mining, there are still some limitations in our study. One significant limitation is that our model cannot predict the effects of unnatural amino acid and protein modifications on AMP activity, due to the fact that peptides containing these features are more difficult in synthesizing. However, these are prevalent in many natural AMPs and are frequently utilized in AMP design6,61. Incorporating these features during model development is possible in the future improvement of COMDEL. Second, our high-throughput screening method produces a limited array of AMPs, failing to encompass broad-spectrum consideration. Adequate broad-spectrum AMP screening methods and data are still urgently needed to train and optimize the AMP prediction and design models. Our model has been specifically designed to augment specific AMP data through high-throughput methods, allowing for an automatic iterative process. Within the codebase, it is capable of autonomously aggregating data from current mainstream AMP databases, facilitating the self-iteration of the model. Additionally, although we have endeavored to ensure the safety of the screened AMPs in the construction and application of COMDEL, implementing a more comprehensive and detailed AMP safety evaluation method remains essential49. Lastly, in terms of AMP synthesis, we have employed a cell-free system to achieve large-scale synthesis in hours; however, the associated costs remain prohibitively high. Therefore, developing a more efficient and inexpensive synthesis system is urgently needed for the industrial production and application of AMPs.
5. Conclusions
In summary, through utilizing deep learning and high-throughput methodologies, we developed COMDEL, an advanced AMP identifier outstanding for its exceptional accuracy, precision, and minimal bias. Furthermore, we introduced two novel and efficient methods for the synthesis of edible AMPs predicted by COMDEL: enzymatic ligation and cell-free synthesis. These innovations pave the way for the industrial-scale production of AMPs. Employing COMDEL, we have successfully screened edible probiotics for high AMP production potential and further enhanced their antibacterial ability through directed evolution. Ultimately, in an effort to broaden the accessibility of our research, we have created a web interface to showcase our COMDEL model, available at https://ai.tidetronbio.com:7782/ampPredict.html (Fig. 7). We hope that our COMDEL model will serve as a valuable tool for researchers aiming to classify and design AMPs with powerful application values.
Figure 7.

Web page preview of COMDEL. The COMDEL model is available at https://ai.tidetronbio.com:7782/ampPredict.html. It needs users to provide the peptide sequence. The possibility value ranges from 0 (non_AMP) to 1 (AMP).
Acknowledgments
The authors are grateful to Dr Jinming Cui from Guangzhou Institute of Advanced Technology for guiding the PANCE. This work was supported by a grant from the Hubei University of Science and Technology Program (No. BK202417, China), Doctoral Special Research Fund Launch Project of Jiamusi University (JMSUBZ2021-12, China), and Youth Innovative Talent Cultivation Support Plan of Jiamusi University (JMSUQP2022016, China).
Author contributions
Yu Zhang: Investigation, Methodology, Software, Validation, Visualization. Li-Hua Liu: Investigation, Methodology, Validation, Visualization. Bo Xu: Conceptualization, Data curation, Project administration, Writing – original draft. Zhiqian Zhang: Conceptualization, Funding acquisition, Project administration, Supervision. Min Yang: Investigation, Methodology, Validation, Visualization. Yiyang He: Formal analysis, Methodology, Project administration, Software, Validation, Visualization. Jingjing Chen: Visualization, Validation. Yang Zhang: Supervision, Validation. Yucheng Hu: Validation, Visualization. Xipeng Chen: Validation. Zitong Sun: Validation, Visualization. Qijun Ge: Methodology. Song Wu: Validation. Wei Lei: Software. Kaizheng Li: Validation. Hua Cui: Validation. Gangzhu Yang: Visualization. Xuemei Zhao: Methodology. Man Wang: Validation. Jiaqi Xia: Data curation, Formal analysis, Methodology, Software. Zhen Cao: Investigation, Methodology, Supervision. Ao Jiang: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing. Yi-Rui Wu: Conceptualization, Formal analysis, Project administration, Writing – original draft.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2024.05.003.
Contributor Information
Jiaqi Xia, Email: jiaqixia@whu.edu.cn.
Zhen Cao, Email: caoz@yeasen.com.
Ao Jiang, Email: jiangao@tidetronbio.com.
Yi-Rui Wu, Email: wuyirui@tidetronbio.com.
Appendix A. Supporting information
The following are the Supporting information to this article:
References
- 1.Brogden K.A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 2.Lazzaro B.P., Zasloff M., Rolff J. Antimicrobial peptides: application informed by evolution. Science. 2020;368 doi: 10.1126/science.aau5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo Y., Song Y. Mechanism of antimicrobial peptides: antimicrobial, anti-inflammatory and antibiofilm activities. Int J Mol Sci. 2021;22:11401–11420. doi: 10.3390/ijms222111401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magana M., Pushpanathan M., Santos A.L., Leanse L., Fernandez M., Ioannidis A., et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect Dis. 2020;20:e216–e230. doi: 10.1016/S1473-3099(20)30327-3. [DOI] [PubMed] [Google Scholar]
- 5.Lai Z., Yuan X., Chen H., Zhu Y., Dong N., Shan A. Strategies employed in the design of antimicrobial peptides with enhanced proteolytic stability. Biotechnol Adv. 2022;59 doi: 10.1016/j.biotechadv.2022.107962. [DOI] [PubMed] [Google Scholar]
- 6.Torres M.D.T., Sothiselvam S., Lu T.K., de la Fuente-Nunez C. Peptide design principles for antimicrobial applications. J Mol Biol. 2019;431:3547–3567. doi: 10.1016/j.jmb.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 7.Lata S., Mishra N.K., Raghava G.P. AntiBP2: improved version of antibacterial peptide prediction. BMC Bioinf. 2010;11(Suppl 1):S19. doi: 10.1186/1471-2105-11-S1-S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao X., Wang P., Lin W.Z., Jia J.H., Chou K.C. iAMP-2L: a two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal Biochem. 2013;436:168–177. doi: 10.1016/j.ab.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 9.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Madani A., Krause B., Greene E.R., Subramanian S., Mohr B.P., Holton J.M., et al. Large language models generate functional protein sequences across diverse families. Nat Biotechnol. 2023;41:1099–1106. doi: 10.1038/s41587-022-01618-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathis N., Allam A., Kissling L., Marquart K.F., Schmidheini L., Solari C., et al. Predicting prime editing efficiency and product purity by deep learning. Nat Biotechnol. 2023;41:1151–1159. doi: 10.1038/s41587-022-01613-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greener J.G., Kandathil S.M., Moffat L., Jones D.T. A guide to machine learning for biologists. Nat Rev Mol Cell Biol. 2022;23:40–55. doi: 10.1038/s41580-021-00407-0. [DOI] [PubMed] [Google Scholar]
- 13.Wang G., Vaisman I.I., van Hoek M.L. Machine learning prediction of antimicrobial peptides. Methods Mol Biol. 2022;2405:1–37. doi: 10.1007/978-1-0716-1855-4_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang G., Zietz C.M., Mudgapalli A., Wang S., Wang Z. The evolution of the antimicrobial peptide database over 18 years: milestones and new features. Protein Sci. 2022;31:92–106. doi: 10.1002/pro.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang G., Li X., Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016;44:D1087–D1093. doi: 10.1093/nar/gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma Y., Guo Z., Xia B., Zhang Y., Liu X., Yu Y., et al. Identification of antimicrobial peptides from the human gut microbiome using deep learning. Nat Biotechnol. 2022;40:921–931. doi: 10.1038/s41587-022-01226-0. [DOI] [PubMed] [Google Scholar]
- 17.Lee H.T., Lee C.C., Yang J.R., Lai J.Z., Chang K.Y. A large-scale structural classification of antimicrobial peptides. BioMed Res Int. 2015;2015 doi: 10.1155/2015/475062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maasch J., Torres M.D.T., Melo M.C.R., de la Fuente-Nunez C. Molecular de-extinction of ancient antimicrobial peptides enabled by machine learning. Cell Host Microbe. 2023;31:1260–12674 e6. doi: 10.1016/j.chom.2023.07.001. [DOI] [PubMed] [Google Scholar]
- 19.Sharma R., Shrivastava S., Kumar Singh S., Kumar A., Saxena S., Kumar Singh R. Deep-ABPpred: identifying antibacterial peptides in protein sequences using bidirectional LSTM with word2vec. Briefings Bioinf. 2021;22 doi: 10.1093/bib/bbab065. [DOI] [PubMed] [Google Scholar]
- 20.Fu H., Cao Z., Li M., Wang S. ACEP: improving antimicrobial peptides recognition through automatic feature fusion and amino acid embedding. BMC Genom. 2020;21:597. doi: 10.1186/s12864-020-06978-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhadra P., Yan J., Li J., Fong S., Siu S.W.I. AmPEP: sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci Rep. 2018;8:1697. doi: 10.1038/s41598-018-19752-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veltri D., Kamath U., Shehu A. Deep learning improves antimicrobial peptide recognition. Bioinformatics. 2018;34:2740–2747. doi: 10.1093/bioinformatics/bty179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao X., Shao Y.T., Cheng X., Stamatovic B. iAMP-CA2L: a new CNN-BiLSTM-SVM classifier based on cellular automata image for identifying antimicrobial peptides and their functional types. Briefings Bioinf. 2021;22 doi: 10.1093/bib/bbab209. [DOI] [PubMed] [Google Scholar]
- 24.Fingerhut L., Miller D.J., Strugnell J.M., Daly N.L., Cooke I.R. ampir: an R package for fast genome-wide prediction of antimicrobial peptides. Bioinformatics. 2021;36:5262–5263. doi: 10.1093/bioinformatics/btaa653. [DOI] [PubMed] [Google Scholar]
- 25.Fernandes F.C., Cardoso M.H., Gil-Ley A., Luchi L.V., da Silva M.G.L., Macedo M.L.R., et al. Geometric deep learning as a potential tool for antimicrobial peptide prediction. Front Bioinform. 2023;3 doi: 10.3389/fbinf.2023.1216362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deo S., Turton K.L., Kainth T., Kumar A., Wieden H.J. Strategies for improving antimicrobial peptide production. Biotechnol Adv. 2022;59 doi: 10.1016/j.biotechadv.2022.107968. [DOI] [PubMed] [Google Scholar]
- 27.Wen Q., Zhang L., Zhao F., Chen Y., Su Y., Zhang X., et al. Production technology and functionality of bioactive peptides. Curr Pharmaceut Des. 2023;29:652–674. doi: 10.2174/1381612829666230201121353. [DOI] [PubMed] [Google Scholar]
- 28.Mojsoska B. Solid-phase synthesis of novel antimicrobial peptoids with alpha- and beta-chiral side chains. Methods Enzymol. 2022;663:327–340. doi: 10.1016/bs.mie.2021.10.010. [DOI] [PubMed] [Google Scholar]
- 29.Wang X.J., Wang X.M., Teng D., Zhang Y., Mao R.Y., Wang J.H. Recombinant production of the antimicrobial peptide NZ17074 in Pichia pastoris using SUMO3 as a fusion partner. Lett Appl Microbiol. 2014;59:71–78. doi: 10.1111/lam.12246. [DOI] [PubMed] [Google Scholar]
- 30.Cao J., de la Fuente-Nunez C., Ou R.W., Torres M.T., Pande S.G., Sinskey A.J., et al. Yeast-based synthetic biology platform for antimicrobial peptide production. ACS Synth Biol. 2018;7:896–902. doi: 10.1021/acssynbio.7b00396. [DOI] [PubMed] [Google Scholar]
- 31.Zheng Y., Du Y., Qiu Z., Liu Z., Qiao J., Li Y., et al. Nisin variants generated by protein engineering and their properties. Bioengineering. 2022;9:251. doi: 10.3390/bioengineering9060251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waghu F.H., Barai R.S., Gurung P., Idicula-Thomas S. CAMPR3: a database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016;44:D1094–D1097. doi: 10.1093/nar/gkv1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gawde U., Chakraborty S., Waghu F.H., Barai R.S., Khanderkar A., Indraguru R., et al. CAMPR4: a database of natural and synthetic antimicrobial peptides. Nucleic Acids Res. 2023;51:D377–D383. doi: 10.1093/nar/gkac933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muller A.T., Gabernet G., Hiss J.A., Schneider G. modlAMP: python for antimicrobial peptides. Bioinformatics. 2017;33:2753–2755. doi: 10.1093/bioinformatics/btx285. [DOI] [PubMed] [Google Scholar]
- 35.Meurer W.J., Tolles J. Logistic regression diagnostics: understanding how well a model predicts outcomes. JAMA. 2017;317:1068–1069. doi: 10.1001/jama.2016.20441. [DOI] [PubMed] [Google Scholar]
- 36.Hu X., Sun Y., Gao J., Hu Y., Ju F., Yin B. Probabilistic linear discriminant analysis based on L(1)-norm and its bayesian variational inference. IEEE Trans Cybern. 2022;52:1616–1627. doi: 10.1109/TCYB.2020.2985997. [DOI] [PubMed] [Google Scholar]
- 37.Wang J., Wang L., Nie F., Li X. A novel formulation of trace ratio linear discriminant analysis. IEEE Transact Neural Networks Learn Syst. 2022;33:5568–5578. doi: 10.1109/TNNLS.2021.3071030. [DOI] [PubMed] [Google Scholar]
- 38.Zhang C., Pham M., Fu S., Liu Y. Robust multicategory support vector machines using difference convex algorithm. Math Program. 2018;169:277–305. [PMC free article] [PubMed] [Google Scholar]
- 39.Paul A., Mukherjee D.P., Das P., Gangopadhyay A., Chintha A.R., Kundu S. Improved random forest for classification. IEEE Trans Image Process. 2018;27:4012–4024. doi: 10.1109/TIP.2018.2834830. [DOI] [PubMed] [Google Scholar]
- 40.Li Y.L., Wang S. BooDet: gradient boosting object detection with additive learning-based prediction aggregation. IEEE Trans Image Process. 2022;31:2620–2632. doi: 10.1109/TIP.2022.3157453. [DOI] [PubMed] [Google Scholar]
- 41.Wang C., Xu S., Yang J. Adaboost algorithm in artificial intelligence for optimizing the IRI prediction accuracy of asphalt concrete pavement. Sensors. 2021;21:5682. doi: 10.3390/s21175682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Samet H. K-nearest neighbor finding using MaxNearestDist. IEEE Trans Pattern Anal Mach Intell. 2008;30:243–252. doi: 10.1109/TPAMI.2007.1182. [DOI] [PubMed] [Google Scholar]
- 43.Wu Z., Jiang D., Wang J., Zhang X., Du H., Pan L., et al. Knowledge-based BERT: a method to extract molecular features like computational chemists. Briefings Bioinf. 2022;23 doi: 10.1093/bib/bbac131. [DOI] [PubMed] [Google Scholar]
- 44.Prabhakar S.K., Won D.O. Medical text classification using hybrid deep learning models with multihead attention. Comput Intell Neurosci. 2021;2021 doi: 10.1155/2021/9425655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Conesa A., Madrigal P., Tarazona S., Gomez-Cabrero D., Cervera A., McPherson A., et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016;17:13. doi: 10.1186/s13059-016-0881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller S.M., Wang T., Liu D.R. Phage-assisted continuous and non-continuous evolution. Nat Protoc. 2020;15:4101–4127. doi: 10.1038/s41596-020-00410-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Des Soye B.J., Gerbasi V.R., Thomas P.M., Kelleher N.L., Jewett M.C. A highly productive, one-pot cell-free protein synthesis platform based on genomically recoded Escherichia coli. Cell Chem Biol. 2019;26 doi: 10.1016/j.chembiol.2019.10.008. 1743-54.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jewett M.C., Calhoun K.A., Voloshin A., Wuu J.J., Swartz J.R. An integrated cell-free metabolic platform for protein production and synthetic biology. Mol Syst Biol. 2008;4:220. doi: 10.1038/msb.2008.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jadhav K., Singh R., Ray E., Singh A.K., Verma R.K. Taming the devil: antimicrobial peptides for safer TB therapeutics. Curr Protein Pept Sci. 2022;23:643–656. doi: 10.2174/1389203723666220526161109. [DOI] [PubMed] [Google Scholar]
- 50.Wang C., Hong T., Cui P., Wang J., Xia J. Antimicrobial peptides towards clinical application: delivery and formulation. Adv Drug Deliv Rev. 2021;175 doi: 10.1016/j.addr.2021.05.028. [DOI] [PubMed] [Google Scholar]
- 51.Haridas V., Sadanandan S., Dheepthi N.U. Sortase-based bio-organic strategies for macromolecular synthesis. Chembiochem. 2014;15:1857–1867. doi: 10.1002/cbic.201402013. [DOI] [PubMed] [Google Scholar]
- 52.Wang T., Badran A.H., Huang T.P., Liu D.R. Continuous directed evolution of proteins with improved soluble expression. Nat Chem Biol. 2018;14:972–980. doi: 10.1038/s41589-018-0121-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seddik H.A., Bendali F., Gancel F., Fliss I., Spano G., Drider D. Lactobacillus plantarum and its probiotic and food potentialities. Probiotics Antimicrob Proteins. 2017;9:111–122. doi: 10.1007/s12602-017-9264-z. [DOI] [PubMed] [Google Scholar]
- 54.Cuevas-Gonzalez P.F., Liceaga A.M., Aguilar-Toala J.E. Postbiotics and paraprobiotics: from concepts to applications. Food Res Int. 2020;136 doi: 10.1016/j.foodres.2020.109502. [DOI] [PubMed] [Google Scholar]
- 55.Liu Q., Liu Q., Meng H., Lv H., Liu Y., Liu J., et al. Staphylococcus epidermidis contributes to healthy maturation of the nasal microbiome by stimulating antimicrobial peptide production. Cell Host Microbe. 2020;27:68–78 e5. doi: 10.1016/j.chom.2019.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baret J.C., Miller O.J., Taly V., Ryckelynck M., El-Harrak A., Frenz L., et al. Fluorescence-activated droplet sorting (FADS): efficient microfluidic cell sorting based on enzymatic activity. Lab Chip. 2009;9:1850–1858. doi: 10.1039/b902504a. [DOI] [PubMed] [Google Scholar]
- 57.Pedelacq J.D., Cabantous S. Development and applications of superfolder and split fluorescent protein detection systems in biology. Int J Mol Sci. 2019;20:3479. doi: 10.3390/ijms20143479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang X., Zhang X.F., Li H.P., Wang L.Y., Zhang C., Xing X.H., et al. Atmospheric and room temperature plasma (ARTP) as a new powerful mutagenesis tool. Appl Microbiol Biotechnol. 2014;98:5387–5396. doi: 10.1007/s00253-014-5755-y. [DOI] [PubMed] [Google Scholar]
- 59.Rathinakumar R., Wimley W.C. High-throughput discovery of broad-spectrum peptide antibiotics. FASEB J. 2010;24:3232–3238. doi: 10.1096/fj.10-157040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rathinakumar R., Walkenhorst W.F., Wimley W.C. Broad-spectrum antimicrobial peptides by rational combinatorial design and high-throughput screening: the importance of interfacial activity. J Am Chem Soc. 2009;131:7609–7617. doi: 10.1021/ja8093247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zou J., Jiang H., Cheng H., Fang J., Huang G. Strategies for screening, purification and characterization of bacteriocins. Int J Biol Macromol. 2018;117:781–789. doi: 10.1016/j.ijbiomac.2018.05.233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article.