

ABSTRACT
Babesia and Plasmodium pathogens, the causative agents of babesiosis and malaria, are vector-borne intraerythrocytic protozoan parasites, posing significant threats to both human and animal health. The widespread resistance exhibited by these pathogens to various classes of antiparasitic drugs underscores the need for the development of novel and more effective therapeutic strategies. Antifolates have long been recognized as attractive antiparasitic drugs as they target the folate pathway, which is essential for the biosynthesis of purines and pyrimidines, and thus is vital for the survival and proliferation of protozoan parasites. More efficacious and safer analogs within this class are needed to overcome challenges due to resistance to commonly used antifolates, such as pyrimethamine, and to address liabilities associated with the dihydrotriazines, WR99210 and JPC-2067. Here, we utilized an in vitro culture condition suitable for the continuous propagation of Babesia duncani, Babesia divergens, Babesia MO1, and Plasmodium falciparum in human erythrocytes to screen a library of 50 dihydrotriazines and 29 biguanides for their efficacy in vitro and compared their potency and therapeutic indices across different species and isolates. We identified nine analogs that inhibit the growth of all species, including the P. falciparum pyrimethamine-resistant strain HB3, with IC50 values below 10 nM, and display excellent in vitro therapeutic indices. These compounds hold substantial promise as lead antifolates for further development as broad-spectrum antiparasitic drugs.
KEYWORDS: apicomplexan, nucleotide biosynthesis, antifolates, DHFR-TS, dihydrotriazine, antiparasitic drugs
INTRODUCTION
The hemoparasites Plasmodium and Babesia invade host red blood cells where they develop and multiply to cause the pathological symptoms associated with human malaria and babesiosis, respectively. While Plasmodium species are transmitted by mosquitoes, Babesia parasites are primarily transmitted by ticks. The 2023 World Health Organization report indicated that in 2022, malaria was responsible for 249 million clinical cases and 608,000 deaths worldwide, the majority of which were caused by Plasmodium falciparum (1). Human babesiosis has also a worldwide distribution with most cases reported in the United States where the disease is considered endemic and represents a significant risk to the blood supply (1–3). Several Babesia species have been shown to cause human disease (3, 4). These include Babesia microti, Babesia duncani, and Babesia MO1 in the United States and Babesia divergens and Babesia venatorum in Europe and Asia (5–8). The number of reported cases of human babesiosis has increased significantly since 2011 when babesiosis became a nationally notifiable disease (2, 5, 9). The Centers for Disease Control and Prevention (CDC) recently reported that over 14,000 cases of human babesiosis were reported between 2011 and 2018 in the United States (2). Although transmission of Babesia parasites is primarily via the tick bite, other transmission modes have also been reported, with blood transfusion being the most common (10, 11). The disease manifestations range from asymptomatic to severe, with typical clinical symptoms including chills, fever, and sweats. For people with weak immune systems (such as cancer, lymphoma, and AIDS), immunological complications, or immune therapies, the disease can be more severe and sometimes lethal (12). The current CDC-recommended treatment protocol for human babesiosis includes combination therapy of the antimalarials azithromycin with atovaquone for mild babesiosis and clindamycin with quinine for severe babesiosis (2, 13). However, these treatments are often associated with adverse events (14–16). Consequently, persistent Babesia infections and recrudescence can lead to prolonged treatment (17, 18) and the emergence of mutant parasites, which are resistant to therapies (19, 20). Resistant mutations were also identified following treatment of mice infected with B. microti or B. duncani with recommended therapies (21, 22) or were selected following drug pressure in vitro (23, 24). Altogether, these findings highlight the need for more effective therapies for the treatment of human babesiosis.
Because folate metabolism is essential for the development and survival of parasites, targeting this process by inhibiting specific enzymatic steps in the biosynthesis of folates has long been a major focus of the antiparasitic drug development efforts for both prophylaxis and treatment (25, 26). Folate cofactors are essential in several metabolic processes including the production of purine and pyrimidine for DNA replication and amino acid metabolism. First-generation antifolates have been shown to act as competitive inhibitors of bifunctional enzyme dihydrofolate reductase thymidylate synthase (DHFR-TS) enzymes from protozoan parasites and have been used as antiparasitic drugs against Plasmodium and Toxoplasma species since World War II (25, 27, 28). The use of antifolate-based treatments is often applied when parasites become resistant to other first-line antiparasitic drugs. One such antifolate is pyrimethamine (PYM) (2,4-Diamino-5-(4-chlorophenyl)−6-ethylpyrimidine), which selectively inhibits DHFR-TS enzymes of protozoan parasites (29), and is commonly used in combination with other inhibitors targeting additional steps in the folate metabolic pathway, including sulfadoxine (drug combination known as Fansidar) and dapsone (drug combination known as Maloprim) to achieve maximum efficacy (30, 31).
A major challenge to the efficacy of antifolate-based drug therapy arose with the emergence of antifolate-resistant isolates (32, 33). Parasite lines with point mutations in the DHFR-TS enzyme showed reduced susceptibility to treatment with a combination of sulfonamide and pyrimethamine (34–36). The prevalent mutations in P. falciparum DHFR-TS (PfDHFR-TS) associated with resistance to pyrimethamine include N51I, C59R, S108N, and I164L. These mutations result in decreased binding affinity of antifolates to the DHFR-TS enzyme due to spatial restrictions imposed by the drug, leading to a reduction in efficacy (37, 38). In vitro studies and analysis of field isolates have shown that S108N is one of the most common mutations in the DHFR domain of the DHFR-TS enzyme associated with pyrimethamine resistance and is strongly associated with high levels of resistance. C59R mutation is often found in combination with S108N, contributing to increased resistance. Other mutations, including N51I, I164L, and S117N, contribute to resistance, often in combination with other mutations (25, 35, 36, 39, 40).
The small molecule WR99210 is a second-generation antimalarial triazine derivative developed in the late 1960s with improved efficacy against pyrimethamine- and chloroquine-resistant parasites (41, 42). Analysis of a library of mutants of PfDHFR-TS in Escherichia coli identified a set of residues in the enzyme that are associated with either sensitivity or resistance to this drug (37, 43). Despite its excellent biological activity, WR99210 exhibited poor bioavailability and caused gastrointestinal complications in healthy subjects, leading to its withdrawal from further clinical evaluation (44, 45). Third-generation biguanide prodrugs have been reported to show improved gastrointestinal tolerability and enhanced activity against malaria parasites (42, 44, 46–48). One of these, JPC-2067 showed promise when tested against an array of pathogens, including P. falciparum, Toxoplasma gondii, Mycobacterium tuberculosis, and Nocardia species (48–50). Unlike their efficacy against Plasmodium and Toxoplasma species, pyrimethamine, sulfamethoxazole, trimethoprim, and dapsone monotherapy or the combination of Pyrimethamine-sulfadoxine have been shown to have little to no activity against B. microti infection in Mongolian jirds (51). A moderate to effective inhibition of in vitro parasite growth of Babesia gibsoni and Babesia bovis was observed with trimethoprim, methotrexate, and pyrimethamine (52, 53).
Recent advances in understanding the genomic composition of various Babesia species that infect humans have helped identify potential targets for more effective anti-babesial drugs. Anti-babesial drug discovery was further accelerated following the development of the B. duncani model of infection, which combines both a continuous in vitro culture of the parasite in human red blood cells and infection in C3H/HeJ mice (54–58). Interestingly, previous studies have shown moderate efficacy of pyrimethamine against B. duncani with an IC50 value of 940 nM, whereas WR99210 is highly potent against the parasite with an IC50 value of 0.5 nM (59).
In order to search for potent but safer analogs of WR99210, we standardized the in vitro culture systems of several intraerythrocytic parasites in human erythrocytes in order to compare drug efficacy across different species and isolates. Using this strategy, we screened a library of 79 compounds (50 dihydrotriazines and 29 biguanides), analogs of WR99210, against B. duncani, B. divergens, B. MO1 (B. divergens-like species), and P. falciparum drug-sensitive and -resistant strains. Nine analogs with IC50 values < 10 nM against all babesiosis and malaria parasites were identified and are promising early candidates for further development as broad-spectrum antiparasitic drugs.
RESULTS
Babesia DHFR-TS enzymes are suitable targets for antibabesial drug discovery
The availability of annotated proteomes from several human Babesia parasites, including B. microti, B. duncani, B. divergens, and B.sp MO1, has enabled the analysis of the primary structures of their DHFR-TS enzymes and made it possible to predict the susceptibility of these organisms to antifolates (Fig. 1A). Unlike DHFR-TS enzymes from P. falciparum (608 amino acids) and other Plasmodium species, the DHFR-TS enzymes of Babesia species are comparatively smaller, with sizes ranging between 502 and 514 amino acids. This size difference is largely due to the presence of insertions of 94 to 106 amino acids in most DHFR-TS enzymes of Plasmodium species (Fig. 1B; Fig. S1). Comparative analysis of amino acid identities and similarities within the DHFR domain revealed that the DHFR enzymes of Babesia species share the highest similarity with those of P. falciparum (59) (Fig. 1B; Fig. S1). In particular, the S108 residue associated with susceptibility to pyrimethamine in P. falciparum and other apicomplexan parasites corresponds to threonine in B. microti, B. duncani, B. divergens, B. MO1, and B. bovis (Fig. 1B). Phylogenetic analyses conducted on the complete protein sequences of DHFR-TS enzymes from 52 apicomplexan parasites revealed a consistent alignment with the evolutionary trajectory of species within the Apicomplexa phylum. The B. microti DHFR-TS appears as an out-group relative to other Piroplasmida (see Fig. 1C). Examination of the DHFR and TS moieties across various apicomplexan lineages revealed nearly identical evolutionary patterns. While DHFR moieties in most parasites (except Babesia parasites) contain insertions, no insertions or deletions were detected in the TS moieties. Furthermore, analysis of the linker region between the DHFR and TS domains (see Fig. S2) categorized them into long or short types, with the shortest linker being longer than those found in other protozoan parasites, such as kinetoplastidae (60). Taken together, these findings strongly indicate a close evolutionary relationship among DHFR-TS enzymes in Apicomplexa, encompassing species like B. duncani, B. microti, and Plasmodium spp.
Fig 1.

Conservation of the folate metabolic pathway among Babesia and Plasmodium parasites. (A) A schematic representation of folate metabolism in Babesia and Plasmodium parasites. The main enzymes (blue color) targeted by antifolates (violet color) are highlighted. Abbreviations (Abbs.): iRBC = infected red blood cell, DHP = dihydropteroate, DHPP = dihydropteroate pyrophosphate, DHPS = dihydropteroate synthase, DHF = dihydrofolate, DHFR = dihydrofolate reductase, PABA = p-aminobenzoic acid, SHMT = Serine hydroxy methyltransferase, THF = tetrahydrofolate, and TS = thymidylate synthase. (B) Alignment of a region of DHFR-TS enzymes from various Babesia species as well as pyrimethamine-sensitive (3D7) and -resistant (Dd2, HB3, and V1/S) P. falciparum strains. Residues known to be associated with susceptibility or tolerance to antifolates are marked in red. (C) Phylogenetic analysis of DHFR-TS enzymes from piroplasmids (tick-transmitted intraerythrocytic parasites) and various Plasmodium species. The tree was built using Phylogeny.fr pipeline combining Mafft, BMGE, and FastTree software applications. An advanced option was used to generate 1,000 bootstraps providing branch support. Clade supporting Piroplasmida sequences is shown in green; Plasmodium sequences are in blue, with branches of P. falciparum sequences represented in violet.
JPC-2067 inhibits parasite DHFR activity and the development of B. duncani in human erythrocytes
The in vitro activity of both JPC-2067 and its pro-drug JPC-2056 (analogs of WR99210) was assessed against B. duncani using a continuous in vitro culture system in human erythrocytes. Both compounds exhibited inhibitory activity at the nanomolar level with IC50 values of 869.4 ± 4 nM and 8.9 ± 2 nM for JPC-2056 and JPC-2067, respectively (Fig. 2A and B). We further examined the cytotoxicity profile of both compounds against four human cell lines: HeLa, HCT-116, HepG2, and HEK-293, but no discernible effect was observed up to 900 µM. The calculated therapeutic indices were determined to be >110 for JPC-2056 and > 15,000 for JPC-2067 (Table 1).
Fig 2.

Inhibition of B. duncani DHFR-TS by JPC-2067 and JPC-2056. (A and B) Chemical structures and inhibition of B. duncani intraerythrocytic development by the prodrug, JPC-2056 (A), and its active drug JPC-2067 (B). The data represent the mean of two biological replicates with three technical replicates. Values represent mean ± SD. (C-D) A dose-dependent inhibition of purified the DHFR activity of the B. duncani (C) and B. microti (D) DHFR-TS enzymes by JPC-2067. EC50 values were calculated from the inhibition curves and represent mean ± SD from three independent experiments, each performed in triplicate.
TABLE 1.
Cytotoxicity and therapeutic indices of JPC-2056 and its active metabolite JPC-2067
| Drug | IC50 B. duncani |
IC50 HeLa |
IC50 HepG2 |
IC50 HEK293 |
IC50 HCT |
Therapeutic index |
|---|---|---|---|---|---|---|
| JPC-2067 | 8.93 nM | >0.9 mM | 1.15 mM | 1 mM | 1 mM | >15,000 |
| JPC-2056 | 869 nM | > 0.9 mM | 1 mM | 1 mM | 1 mM | >110 |
Recent studies have shown that the antifolate compound WR99210 inhibits the activity of DHFR-TS enzymes from B. microti and B. duncani (59). Therefore, we examined the inhibitory activity of JPC-2056 and JPC-2067 against the Babesia enzymes. The NADPH-dependent reduction of dihydrofolate (DHF) to tetrahydrofolate (THF) catalyzed by the Babesia enzymes was measured in the absence or presence of rising concentrations of the compounds. JPC-2067 inhibited enzyme activity with EC50 values of 1.0 nM and 14.7 nM for B. duncani and B. microti, respectively (Fig. 2C and D), whereas the prodrug JPC-2056 had no discernible inhibitory activity (Fig. S3A and B).
In vivo efficacy of JPC-2056 in animal models of babesiosis
The finding that JPC-2067 is effective against B. duncani in vitro led us to investigate the activity of its prodrug JPC-2056 in the mouse model of lethal infection (C3H/HeJ mice) (57). In this model, the mice infected with B. duncani parasites exhibit 100% mortality within 6–13 days post-infection (61). To evaluate the efficacy of JPC-2056, two groups of 5 female mice each were infected with 1 × 104 B. duncani-infected erythrocytes and at day 1 post-infection (DPI-1) received either a vehicle (PEG-400) or 30 mg/kg of JPC-2056 administered orally once a day for 10 days. Mice treated with the vehicle (PEG-400) alone showed evidence of infection within 5 days and reached peak parasitemia within 10 days post-infection, with all animals succumbing to infection by DPI-10 (Fig. 3A and C). Mice treated with 30 mg/kg of JPC-2056 displayed a delay in parasite emergence and displayed reduced parasitemia over the course of the infection compared to the vehicle-treated mice. However, parasitemia in these mice continued to increase over time reaching peak levels at DPI-12 (Fig. 3A and B). These animals succumbed to lethal B. duncani infection at DPI-13 (Fig. 3A and C).
Fig 3.

In vivo efficacy of JPC-2057 in mouse models of human babesiosis. (A) Parasitemia in female C3H/HeJ mice (n = 5) infected (intravenously) with 1 × 104 B. duncani-iRBCs, which cause lethal infection in these mice, and treated with either vehicle (PEG-400) (red) or JPC-2056 (30 mg/kg, blue lines) administered via oral gavage once a day for 10 days (DPI-1 to 10). Treated mice were monitored daily and were euthanized when treatment was deemed not to be effective. Each data point depicts parasitemia measured via inspection of Giemsa-stained blood smears on the specified day. (B) Bar diagram depicting % parasitemia in the vehicle and JPC-2056 groups on the last day of treatment (DPI-10). Data are presented as mean ± SEM (n = 5 mice per group). (C) Kaplan-Meyer curve depicting survival of B. duncani-infected female C3H/HeJ mice followed by treatment with vehicle and JPC-2056. (D) Parasitemia in female CB17/SCID mice (n = 5) infected with 1 × 104 B. microti infected RBCs (LabS1 strain) and treated with either vehicle (PEG-400, red lines) or JPC-2056 (blue lines) at 30 mg/kg once a day for 10 days. (E) Bar diagram showing the % parasitemia after 40 days post-infection in the vehicle and JPC-2056-treated groups. Data are presented as mean ± SEM (n = 5 mice per group).
Building on the promising efficacy of JPC-2056 against B. duncani parasites in vivo, we extended our investigation to assess the efficacy of this compound against B. microti (ATCC LabS1) in the SCID (severely combined immunodeficient) mouse model of infection following inoculation with a 1 × 104 infected erythrocytes. Although the control group treated with the vehicle (PEG-400) alone reached high parasitemia levels approaching 60% to 70% by 20 days post-infection (DPI-20), treatment with JPC-2056 at 30 mg/kg for 10 days (DPI-1 to 10) (Fig. 3D and E) resulted in clearance of infection with no parasites detected up to DPI-45.
Dihydrotriazines and biguanides, analogs of JPC-2067, exhibit potent broad-spectrum antiparasitic activity in vitro
Given the previously reported liabilities of JPC-2067 (48), we sought to evaluate the in vitro activity of dihydrotriazine and biguanide derivatives against several intraerythrocytic parasites for which a continuous in vitro culture system in human erythrocytes has been developed. Due to the diverse culture media used over the years in various laboratories for in vitro culture of intraerythrocytic parasites, the extrapolation of in vitro efficacy and other biological data from one species to another has proven to be a major hurdle in drawing meaningful conclusions across different species. It was previously shown that B. duncani can grow continuously in human erythrocytes in DMEM-F12 supplemented with 20% FBS (DFS20). This medium contains all the nutrients present in RPMI plus additional components including, putrescine, linoleic acid, and lipoic acid, which are essential for B. duncani survival. The use of DFS20 medium could thus facilitate a more precise comparison of the susceptibility of different species under similar experimental conditions (62, 63). Because P. falciparum is traditionally maintained in RPMI-based media, we compared the growth of the P. falciparum strain 3D7 in human erythrocytes in DMEM-F12- and RPMI-based culture media. Although slight differences in parasitemia were observed on both media, similar peak parasitemia levels were reached by 96h under both conditions (Fig. S4A). Using DFS20 culture conditions, we screened a total of 79 DHT derivatives (50 dihydrotriazines and 29 biguanides) (structures represented in Tables S2 and S3) against all parasites. We assessed inhibition of parasite growth at concentrations of 5, 10, 50, and 100 nM against B. duncani (WA-1), B. divergens (Rouen87), B. MO1 (Fig. 4A through C), and P. falciparum (3D7, Dd2, and HB3 strains) (Fig. 4D through F). Among the compounds tested, biguanides exhibited greater efficacy against the P. falciparum pyrimethamine-resistant HB3 strain, but showed lesser activity against P. falciparum 3D7 and Dd2 or Babesia species. In contrast, dihydrotriazines were found to be effective against both drug-sensitive and -resistant strains of P. falciparum, with more than 76% of the compounds tested inhibiting growth by more than 90% at 100 nM (Fig. 4D through F). The efficacy of dihydrotriazines was relatively lower against Babesia species, with ~34% of the compounds found to be effective against B. duncani (WA1 strain), B. divergens (Rouen87 strain), and B. MO1 inhibiting growth by more than 90% at 100 nM. Among the 79 DHT derivatives, nine compounds (JPC-2060, JPC-3671, JPC-3681, JPC-3680, JPC-210, JPC-2748, JPC-1044, JPC-1059, and JPC-2053) were selected for their broad anti-parasitic activity and prioritized for further investigations. All nine compounds showed effective dose-dependent parasite growth inhibition with IC50 values ranging between 0.4 nM and 10 nM (Fig. S5; Table 2). The efficacy of these nine compounds against P. falciparum was similar in DFS20 and RPMI-based media (Fig. S4B; Table S3). All nine compounds were subsequently evaluated for their cytotoxicity in human cell lines (HepG2, HCT-116, HEK-293, and HeLa) and displayed low cytotoxicity. The vitro therapeutic indices for these compounds ranged between 4,000 and 100,000 (Table 2). Among these compounds, JPC-2060 emerged as the most broadly effective compound (Fig. S5), displaying high potency against P. falciparum strains (3D7: IC50 ~1.9 nM; Dd2: IC50 ~0.4 nM; HB3: IC50 ~0.6 nM), B. duncani WA1 strain (IC50 ~0.8 nM), B. divergens Rouen87 strain (IC50 ~4 nM), and B. MO1 (IC50 ~0.5 nM) with an in vitro therapeutic index of >11,000 (Table 2).
Fig 4.

Activity of DHTs and biguanides against Babesia and Plasmodium species and isolates in vitro. (A-F) In vitro efficacy of dihydrotriazines and biguanides against Babesia [B. duncani WA1 (A), B. divergens Rouen87 (B), and B.MO1 (C)] and Plasmodium species [3D7 (D), Dd2 (E), and HB3 (F)] at 5, 10, 50, and 100 nM drug concentrations. Color-coded heat maps represent mean (n = 3) percent inhibition of parasite growth, with dark blue representing 100% growth and dark red representing 100% inhibition.
TABLE 2.
In vitro efficacy and therapeutic indices of dihydrotriazine derivatives
| Compound | Chemical structure |
Babesia species | Plasmodium falciparum | Human cell lines | Therapeutic index | ||||
|---|---|---|---|---|---|---|---|---|---|
|
B. duncani WA1 (IC50 in nM) |
B. divergens Rouen87 (IC50 in nM) |
B. MO1 (IC50 in nM) |
Pf3D7 (IC50 in nM) |
PfDd2 (IC50 in nM) |
PfHB3 (IC50 in nM) |
Average IC50 (µM) |
|||
| JPC-2060 |
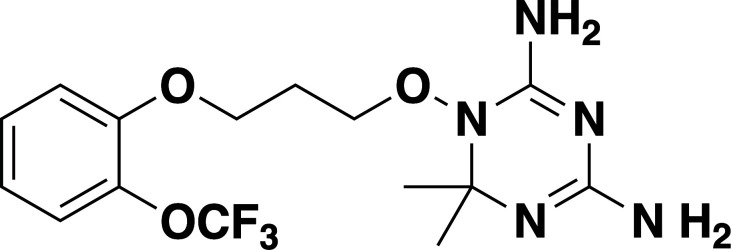
|
1 ± 0.06 | 4 ± 0.98 | 0.6 ± 0.07 | 2.3 ± 0.5 | 0.77 ± 0.4 | 0.7 ± 0.1 | 15 | >11,000 |
| JPC-3671 |
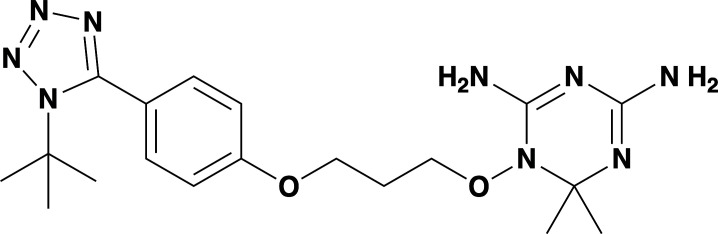
|
1.6 ± 0.5 | 24 ± 2.14 | 6.5 ± 0.70 | 1 ± 0.32 | 17 ± 1.5 | 0.4 ± 0.02 | 38 | >19,000 |
| JPC-3681 |
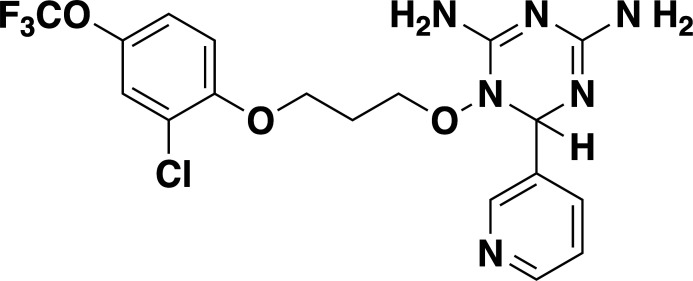
|
2 ± 0.20 | 6.4 ± 0.70 | 1 ± 0.07 | 2.3 ± 0.3 | 0.45 ± 0.03 | 0.8 ± 0.07 | 38 | >4,000 |
| JPC-3680 |
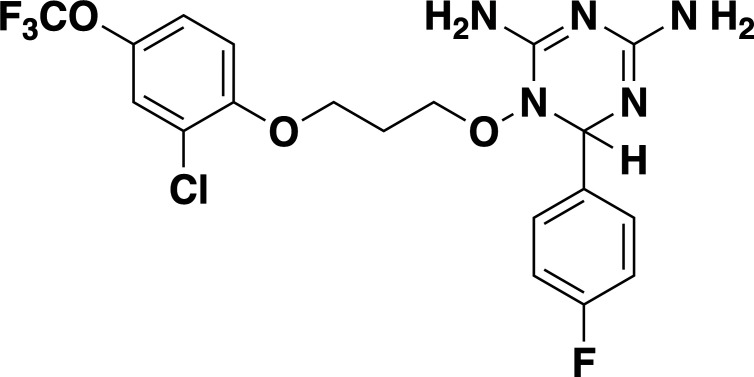
|
3.2 ± 0.21 | 40 ± 0.70 | 6 ± 0.5 | 1.7 ± 0.7 | 37 ± 1.25 | 2.1 ± 0.33 | 10 | >600 |
| JPC-210 |
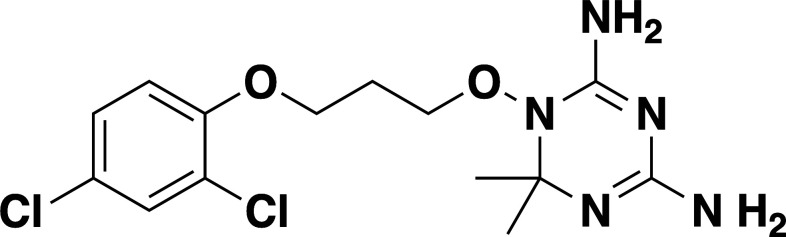
|
3.6 ± 0.42 | 3.1 ± 1.42 | 0.8 ± 0.24 | 3.1 ± 1.24 | 0.05 ± 0.01 | 0.4 ± 0.07 | 69 | >40,000 |
| JPC-2748 |
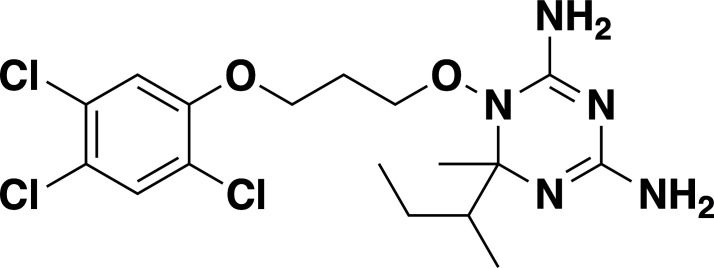
|
4.4 ± 0.01 | 3.7 ± 0.3 | 4 ± 0.74 | 3 ± 0.77 | 12 ± 1.13 | 0.41 ± 0.07 | 1.5 | >4,000 |
| JPC-1044 |
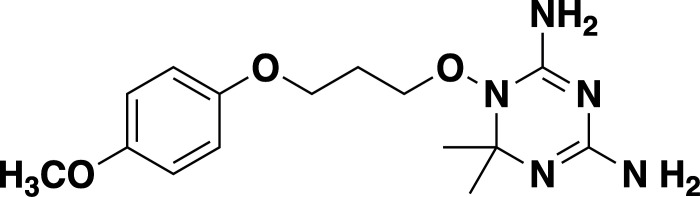
|
7.1 ± 0.32 | 12 ± 0.16 | 0.2 ± 0.15 | 3.3 ± 2 | 0.6 ± 0.14 | 0.42 ± 0.25 | 62 | >100,000 |
| JPC-1059 |
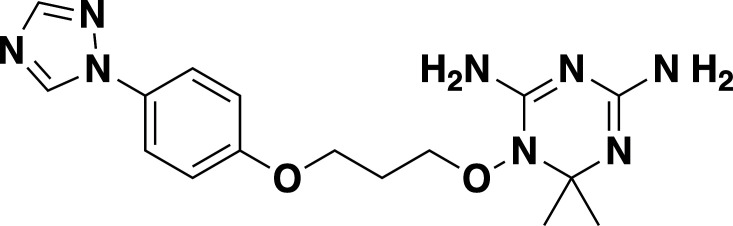
|
10.3 ± 0.70 | 3.7 ± 0.70 | 5.4 ± 1.4 | 2 ± 0.64 | 0.3 ± 0.02 | 0.3 ± 0.17 | 132 | >100,000 |
| JPC-2053 |
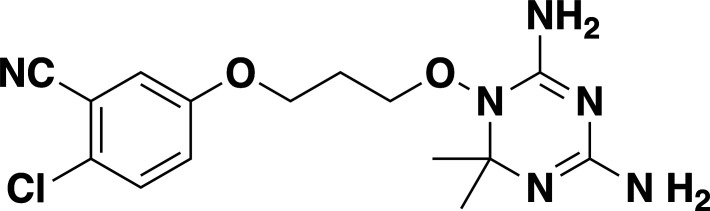
|
13.4 ± 1.41 | 6.6 ± 1.70 | 0.6 ± 0.50 | 2.6 ± 1.51 | 0.5 ± 0.045 | 0.75 ± 0.34 | 26 | >50,000 |
DISCUSSION
The DHFR-TS enzymes of Babesia species exhibit notable differences compared with those of P. falciparum, particularly in size and amino acid composition. Unlike the longer P. falciparum enzyme, Babesia DHFR-TS enzymes are comparatively smaller due to the absence of large insertions in their sequences. However, they share a high similarity with the P. falciparum enzyme, suggesting a close evolutionary relationship within the Apicomplexa phylum.
The in vitro assessment of the antifolate JPC-2067 and its pro-drug JPC-2056 revealed potent inhibitory activity against B. duncani, with nanomolar IC50 values. Importantly, these compounds exhibited high selectivity with minimal cytotoxicity against human cell lines. Furthermore, JPC-2067 effectively inhibited Babesia DHFR-TS enzymes, underscoring its mechanism of action. In vivo studies using JPC-2056 demonstrated efficacy of the compound against B. duncani in a murine model of babesiosis. Although complete parasite clearance was not achieved, treatment with JPC-2056 resulted in a significant delay in parasite burden and reduced peak parasitemia. Moreover, JPC-2056 showed remarkable efficacy against B. microti in a SCID mouse model of babesiosis, leading to the elimination of parasites without observable adverse effects.
Expanding on the efficacy of JPC-2067, we evaluated structurally related dihydrotriazines and biguanides for their antibabesial as well as antimalarial activity. However, comparing the efficacy of various compounds across different species and strains poses challenges in drug susceptibility testing. Variations in culture conditions can introduce confounding factors, making it difficult to draw accurate conclusions. Our present study addressed this challenge by employing DFS20, a culture medium that provides uniform nutrient supplementation, including essential elements such as putrescine, linoleic acid, and lipoic acid crucial for the growth of B. duncani. By utilizing DFS20 as a standardized culture medium for culturing B. duncani, B. divergens, B. MO1, and P. falciparum strains, we have created a platform conducive to more reliable cross-species and cross-strain comparisons of drug susceptibility. Using these standardized conditions, we screened a new library of 50 dihydrotriazines and 29 biguanides against all these species. Nine promising candidates were identified with IC50 values below 10 nM against all species and strains tested, and no toxicity at concentrations up to 900 µM. Both multi-drug-resistant and drug-sensitive P. falciparum strains were sensitive to the tested compounds. Among these candidates, JPC-2060 emerged as the most promising candidate for further investigation. However, treating mice infected with B. duncani with prodrugs for six of these candidates and all biguanides at 30 mg/kg for 5 days did not confer protection against this infection. Further optimization, including structural modifications and synthesis of more bioactive analogs, is thus necessary to improve bioavailability and extend efficacy studies to live animal models.
Our screening of analogs against B. duncani revealed a notable structure-activity relationship, particularly with the 2-chloro, 4-trifluoromethoxy analogs. By altering the substitution on the dihydrotriazine ring from dimethyl to aryl, we found that adding an electron-deficient aryl group at C-6 increased activity in the case of 6-(3-pyridyl) JPC-3681 and 6-(4-fluorophenyl) JPC-3680. Substituting the aryl group with an alkyl group, like 6-propyl JPC-3682, or a different pyridine, like 6-(2-pyridyl) JPC-3683, maintained some activity. Conversely, compounds with electron-rich or neutral aryl groups, such as 6-(4-methoxyphenyl) JPC-3678 and 6-phenyl JPC-3679 exhibited decreased activity. Additionally, appending nitrogen heterocycles on the terminal aryl group proved beneficial, as observed with 4-(1-(t-butyl)−1H Tetrazol-5-yl) JPC-3671 or 4-(1 H-1,2,4-triazol-1-yl) JPC-1059. These findings suggest avenues for further exploration by either combining or delving deeper into these structural regions.
In conclusion, our study underscores the importance of standardized culture conditions in facilitating accurate comparisons of drug susceptibility across different parasitic species and strains. The identification of highly potent pan-antiparasitic drugs targeting DHFR-TS enzymes represents a significant advancement in the quest for effective treatments against malaria and babesiosis. Future research should focus on further optimization of these early lead compounds to identify candidates with biological activity in animal models of babesiosis and malaria.
MATERIALS AND METHODS
Unless otherwise stated, chemicals were purchased from commercial suppliers and used as received. WR99210, JPC- derivatives, biguanides, and DHT derivatives with purity ≥95%, as measured by reversed-phase high-performance liquid chromatography (HPLC), were procured from Jacobus Pharmaceuticals.
Animal studies
Immunocompetent C3H/HeJ mice were procured from The Jackson Laboratory, and immunocompromised CB17/SCID mice were obtained from Envigo. All animal experiments strictly adhered to Yale University’s institutional guidelines for the care and use of laboratory animals and were conducted under the approval of a protocol sanctioned by the Institutional Animal Care and Use Committees (IACUC) at Yale University.
Chemical synthesis of dihydrotriazines and biguanides
The dihydrotriazines were prepared as previously described by Jensen et al. (64). The majority of the dihydrotriazines were made by acidic condensation of the related pro-drug biguanide with the corresponding ketone, with acetone (Y and Z = CH3) as the standard as shown in step (g) as described in the scheme (Fig. S6). The biguanides were prepared as described previously and in the patent literature (65, 66). Compound identity and purity were confirmed by LC/MS. The purity of all compounds at the time of testing was >95%. The structures of the compounds assayed, the observed molecular ions, and the Chemical Abstract Service numbers (CAS #) for previously reported compounds can be found in Table S1 and S2.
In vitro parasite culture of B. duncani and P. falciparum
Continuous propagation of B. duncani in human RBCs in vitro was carried out as first reported by Abraham et al. and further optimized by Singh et al., (23, 62). Briefly, parasites were cultured in a complete HL-1/DMEMF12 medium [base medium of HL-1 (Lonza, 344017)/DMEMF12 (Gibco, 11320033) supplemented with 20% heat-inactivated FBS, 2% 50X HT Media Supplement Hybrid-MaxTM (Sigma, H0137), 1% 200 mM L-Glutamine (Gibco, 25030–081), 1% 100X Penicillin/Streptomycin (Gibco, 15240-062), and 1% 10 mg/mL Gentamicin (Gibco, 15710-072)] in 5% hematocrit A+ RBCs and maintained at 37°C in a humidified chamber containing 2% O2, 5% CO2, and 93% N2. P. falciparum (3D7, Dd2, and HB3) parasites were cultured in 3%-5% human O+ RBCs in RPMI-1640 (Gibco, 110875093) complete medium containing 0.5% Albumax-II (Invitrogen), 2 mM L-glutamine, 50 mg/L hypoxanthine, 25 mM HEPES, 0.225% NaHCO3, and 10 mg/mL gentamycin. Cultures were maintained at 37°C and gassed with a sterile mixture of 4% O2, 5% CO2, and 91% N2. The culture medium was changed daily, and parasitemia was monitored by light microscope examination of Giemsa-stained thin-blood smears.
Assessment of drug cytotoxicity on human cell lines
HeLa, HepG2, HEK293, and HCT116 cell lines were obtained from the American Type Culture Collection (ATCC) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen 11995–065) containing 25 mM glucose, 1 mM sodium pyruvate, and supplemented with 5 mM HEPES, 10% FBS, and penicillin-streptomycin (50 U/mL penicillin, 50 µg/mL streptomycin). Cells were seeded in a 96-well tissue culture plate (20,000 cells per well) and allowed to adhere for 24 hours, after which they were treated with a 12-step 2-fold serial dilution of each drug starting at 1 mM as the highest final concentration. Cells maintained in the medium supplemented with either 0.1% or 10% DMSO were used as negative and positive vehicle controls, respectively. The plates were incubated at 37 ˚C for 72 h, after which each well was incubated with 0.5 mg/mL of MTT reagent (MP Chemicals #02102227) for 4 h in the dark at 37 ˚C. The formazan crystals formed by living cells were solubilized with the addition of 100 µL of DMSO to each well. We measured the OD590nm using a SpectraMax plate reader. From the obtained OD values, percent cell viability was estimated by normalizing to the mean of 10% DMSO wells (set as 100% toxicity) and mean of the vehicle control wells (set as 0% toxicity). Dose-response curves were plotted using GraphPad Prism version 9.1.2.
Enzyme inhibition assays
The inhibition of DHFR activity with selected JPC derivatives was measured as previously described (59). Briefly, the purified enzyme (0.1 µM) (supporting information) (Fig. S7) was incubated with increasing concentrations of JPC-2067 or JPC-2056 in a reaction buffer containing 300 µM DHF and 300 µM NADPH. OD340 reduction rate (OD340/min) was measured for further calculation. The % inhibition was calculated using the formula:
The IC50 was determined from a sigmoidal dose-response curve using GraphPad Prism version 9.1.2.
In vitro drug efficacy
The effect of DHT derivatives on the intraerythrocytic development of P. falciparum, B. duncani, B. divergens and B. MO1 and the determination of IC50 values were conducted as previously reported (54, 67). Briefly, the parasites cultured in vitro (0.1% parasitemia with 5% hematocrit in complete DFS20 medium or RPMI-1640) were treated with various concentrations of the compounds in a 96-well plate and incubated at 37 ˚C for either 60 h in the case of B. duncani or 72 h in the case of P. falciparum. Parasitemia was then determined using the SYBR Green-I method by adding an equal volume of parasite culture to the lysis buffer (0.008% saponin, 0.08% Triton-X-100, 20 mM Tris-HCl (pH = 7.5), and 5 mM EDTA) containing SYBR Green-I (0.01%). The mixture was then incubated at 37°C for 1 h in the dark. The fluorescence was measured at 480 nm (excitation) and 540 nm (emission) using a BioTek Synergy™ Mx Microplate Reader. Background fluorescence from uninfected erythrocytes maintained in a complete DFS20 and RPMI-1640 medium was subtracted from each sample, and the 50% inhibitory concentration (IC50) of the drug was determined by plotting drug concentrations versus parasite growth using GraphPad Prism 9.2.1. Data are shown as mean ± SD from two biological replicates with three technical replicates.
In vivo drug efficacy
Both C3H/HeJ and CB17/SCID mice aged 5–6 weeks (4 to 5 mice per group in each of the studies described herein) were injected intravenously with (1 × 104 iRBCs) of B duncani (WA-1) and B. microti (LabS1) parasites, respectively. The infected mice were treated by oral gavage over 10 days (DPI −1 to 10). During the treatment, each mouse received 100 µL of either vehicle (PEG-400) or prodrug JPC-2056 (30 mg/kg). Percent parasitemia was determined by counting at least 3,000 RBCs from Giemsa-stained thick blood smears.
Statistical analysis
Data sets were analyzed with GraphPad Prism version 9.1.2. Statistical significances were determined using unpaired t-tests.
ACKNOWLEDGMENTS
C.B.M.'s research is supported by National Institutes of Health (NIH) grants (AI123321, AI138139, AI152220, and AI136118), the Steven and Alexandra Cohen Foundation (Lyme 62 2020), and the NBIA Foundations.
Contributor Information
Choukri Ben Mamoun, Email: choukri.benmamoun@yale.edu.
Audrey Odom John, The Children's Hospital of Philadelphia, Philadelphia, Pennsylvania, USA.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/aac.00423-24.
Gene IDs, species names, proteins, and protein lengths.
Figures S1 to S7.
Supplemental methods; legends for Fig. S1 to S7.
List of dihydrotriazines and structures.
List of biguanides and structures.
In vitro efficacy (IC50 values) of DHTs against P. falciparum (3D7) in different media conditions.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. World Health O. 2023. World malaria report 2023. World Health Organization, Geneva. [Google Scholar]
- 2. Swanson M, Pickrel A, Williamson J, Montgomery S. 2023. Trends in reported babesiosis cases - United States, 2011-2019. Am J Transplant 23:582–584. doi: 10.1016/j.ajt.2023.03.013 [DOI] [PubMed] [Google Scholar]
- 3. Krause PJ. 2019. Human babesiosis. Int J Parasitol 49:165–174. doi: 10.1016/j.ijpara.2018.11.007 [DOI] [PubMed] [Google Scholar]
- 4. Vannier E, Krause PJ. 2012. Human babesiosis. N Engl J Med 366:2397–2407. doi: 10.1056/NEJMra1202018 [DOI] [PubMed] [Google Scholar]
- 5. Kumar A, O’Bryan J, Krause PJ. 2021. The global emergence of human babesiosis. Pathogens 10:1447. doi: 10.3390/pathogens10111447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiang J-F, Zheng Y-C, Jiang R-R, Li H, Huo Q-B, Jiang B-G, Sun Y, Jia N, Wang Y-W, Ma L, Liu H-B, Chu Y-L, Ni X-B, Liu K, Song Y-D, Yao N-N, Wang H, Sun T, Cao W-C. 2015. Epidemiological, clinical, and laboratory characteristics of 48 cases of “Babesia venatorum” infection in China: a descriptive study. Lancet Infect Dis 15:196–203. doi: 10.1016/S1473-3099(14)71046-1 [DOI] [PubMed] [Google Scholar]
- 7. Sun Y, Li S-G, Jiang J-F, Wang X, Zhang Y, Wang H, Cao W-C. 2014. Babesia venatorum Infection in Child, China. Emerg Infect Dis 20:896–897. doi: 10.3201/eid2005.121034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gray A, Capewell P, Loney C, Katzer F, Shiels BR, Weir W. 2019. Sheep as host species for zoonotic Babesia venatorum, United Kingdom. Emerg Infect Dis 25:2257–2260. doi: 10.3201/eid2512.190459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Renard I, Ben Mamoun C. 2021. Treatment of human babesiosis: then and now. Pathogens 10:1120. doi: 10.3390/pathogens10091120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lux JZ, Weiss D, Linden JV, Kessler D, Herwaldt BL, Wong SJ, Keithly J, Della-Latta P, Scully BE. 2003. Transfusion-associated babesiosis after heart transplant. Emerg Infect Dis 9:116–119. doi: 10.3201/eid0901.020149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leiby DA. 2006. Babesiosis and blood transfusion: flying under the radar. Vox Sang 90:157–165. doi: 10.1111/j.1423-0410.2006.00740.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vannier EG, Diuk-Wasser MA, Ben Mamoun C, Krause PJ. 2015. Babesiosis. Infect Dis Clin North Am 29:357–370. doi: 10.1016/j.idc.2015.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smith RP, Hunfeld KP, Krause PJ. 2020. Management strategies for human babesiosis. Expert Rev Anti Infect Ther 18:625–636. doi: 10.1080/14787210.2020.1752193 [DOI] [PubMed] [Google Scholar]
- 14. Krause PJ, Spielman A, Telford SR 3rd, Sikand VK, McKay K, Christianson D, Pollack RJ, Brassard P, Magera J, Ryan R, Persing DH. 1998. Persistent parasitemia after acute babesiosis. N Engl J Med 339:160–165. doi: 10.1056/NEJM199807163390304 [DOI] [PubMed] [Google Scholar]
- 15. Gonzalez LM, Rojo S, Gonzalez-Camacho F, Luque D, Lobo CA, Montero E. 2014. Severe babesiosis in immunocompetent man, Spain, 2011. Emerg Infect Dis 20:724–726. doi: 10.3201/eid2004.131409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kjemtrup AM, Conrad PA. 2000. Human babesiosis: an emerging tick-borne disease. Int J Parasitol 30:1323–1337. doi: 10.1016/s0020-7519(00)00137-5 [DOI] [PubMed] [Google Scholar]
- 17. Krause PJ, Gewurz BE, Hill D, Marty FM, Vannier E, Foppa IM, Furman RR, Neuhaus E, Skowron G, Gupta S, et al. 2008. Persistent and relapsing babesiosis in immunocompromised patients. Clin Infect Dis 46:370–376. doi: 10.1086/525852 [DOI] [PubMed] [Google Scholar]
- 18. Raffalli J, Wormser GP. 2016. Persistence of babesiosis for >2 years in a patient on rituximab for rheumatoid arthritis. Diagn Microbiol Infect Dis 85:231–232. doi: 10.1016/j.diagmicrobio.2016.02.016 [DOI] [PubMed] [Google Scholar]
- 19. Wormser GP, Prasad A, Neuhaus E, Joshi S, Nowakowski J, Nelson J, Mittleman A, Aguero-Rosenfeld M, Topal J, Krause PJ. 2010. Emergence of resistance to azithromycin-atovaquone in immunocompromised patients with Babesia microti infection. Clin Infect Dis 50:381–386. doi: 10.1086/649859 [DOI] [PubMed] [Google Scholar]
- 20. Lemieux JE, Tran AD, Freimark L, Schaffner SF, Goethert H, Andersen KG, Bazner S, Li A, McGrath G, Sloan L, Vannier E, Milner D, Pritt B, Rosenberg E, Telford S, Bailey JA, Sabeti PC. 2016. A global map of genetic diversity in Babesia microti reveals strong population structure and identifies variants associated with clinical relapse. Nat Microbiol 1:16079. doi: 10.1038/nmicrobiol.2016.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simon MS, Westblade LF, Dziedziech A, Visone JE, Furman RR, Jenkins SG, Schuetz AN, Kirkman LA. 2017. Clinical and molecular evidence of atovaquone and azithromycin resistance in relapsed Babesia microti infection associated with rituximab and chronic lymphocytic leukemia. Clin Infect Dis 65:1222–1225. doi: 10.1093/cid/cix477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lawres LA, Garg A, Kumar V, Bruzual I, Forquer IP, Renard I, Virji AZ, Boulard P, Rodriguez EX, Allen AJ, Pou S, Wegmann KW, Winter RW, Nilsen A, Mao J, Preston DA, Belperron AA, Bockenstedt LK, Hinrichs DJ, Riscoe MK, Doggett JS, Ben Mamoun C. 2016. Radical cure of experimental babesiosis in immunodeficient mice using a combination of an endochin-like quinolone and atovaquone. J Exp Med 213:1307–1318. doi: 10.1084/jem.20151519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Abraham A, Brasov I, Thekkiniath J, Kilian N, Lawres L, Gao R, DeBus K, He L, Yu X, Zhu G, Graham MM, Liu X, Molestina R, Ben Mamoun C. 2018. Establishment of a continuous in vitro culture of Babesia duncani in human erythrocytes reveals unusually high tolerance to recommended therapies. J Biol Chem 293:19974–19981. doi: 10.1074/jbc.AC118.005771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Y, Bai C, Shi W, Alvarez-Manzo H, Zhang Y. 2020. Identification of essential oils including garlic oil and black pepper oil with high activity against Babesia duncani. Pathogens 9:466. doi: 10.3390/pathogens9060466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Müller IB, Hyde JE. 2013. Folate metabolism in human malaria parasites--75 years on. Mol Biochem Parasitol 188:63–77. doi: 10.1016/j.molbiopara.2013.02.008 [DOI] [PubMed] [Google Scholar]
- 26. Nzila A. 2006. The past, present and future of antifolates in the treatment of Plasmodium falciparum infection. J Antimicrob Chemother 57:1043–1054. doi: 10.1093/jac/dkl104 [DOI] [PubMed] [Google Scholar]
- 27. Aspˆck, H. Prevention of congenital toxoplasmosis in Austria: experience of 25 years. 2000.
- 28. Montoya JG, Liesenfeld O. 2004. Toxoplasmosis. Lancet 363:1965–1976. doi: 10.1016/S0140-6736(04)16412-X [DOI] [PubMed] [Google Scholar]
- 29. Yuthavong Y. 2021. Antifolate drugs, p 1–12. In Hommel M, Kremsner PG (ed), Encyclopedia of malaria. [Google Scholar]
- 30. Metz J. 2007. Folic acid metabolism and malaria. Food Nutr Bull 28:S540–9. doi: 10.1177/15648265070284S407 [DOI] [PubMed] [Google Scholar]
- 31. Overbosch D, Schilthuis H, Bienzle U, Behrens RH, Kain KC, Clarke PD, Toovey S, Knobloch J, Nothdurft HD, Shaw D, Roskell NS, Chulay JD, Malarone International Study Team . 2001. Atovaquone-proguanil versus mefloquine for malaria prophylaxis in nonimmune travelers: results from a randomized, double-blind study. Clin Infect Dis 33:1015–1021. doi: 10.1086/322694 [DOI] [PubMed] [Google Scholar]
- 32. Costanzo MS, Hartl DL. 2011. The evolutionary landscape of antifolate resistance in Plasmodium falciparum. J Genet 90:187–190. doi: 10.1007/s12041-011-0072-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sibley CH, Price RN. 2012. Monitoring antimalarial drug resistance: applying lessons learned from the past in a fast-moving present. Int J Parasitol Drugs Drug Resist 2:126–133. doi: 10.1016/j.ijpddr.2012.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quan H, Igbasi U, Oyibo W, Omilabu S, Chen S-B, Shen H-M, Okolie C, Chen J-H, Zhou X-N. 2020. High multiple mutations of Plasmodium falciparum-resistant genotypes to sulphadoxine-pyrimethamine in Lagos, Nigeria. Infect Dis Poverty 9:91. doi: 10.1186/s40249-020-00712-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pethrak C, Posayapisit N, Pengon J, Suwanakitti N, Saeung A, Shorum M, Aupalee K, Taai K, Yuthavong Y, Kamchonwongpaisan S, Jupatanakul N. 2022. New insights into antimalarial chemopreventive activity of antifolates. Antimicrob Agents Chemother 66:e0153821. doi: 10.1128/AAC.01538-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kuesap J, Suphakhonchuwong N, Kalawong L, Khumchum N. 2022. Molecular markers for sulfadoxine/pyrimethamine and chloroquine resistance in Plasmodium falciparum in Thailand. Korean J Parasitol 60:109–116. doi: 10.3347/kjp.2022.60.2.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Japrung D, Leartsakulpanich U, Chusacultanachai S, Yuthavong Y. 2007. Conflicting requirements of Plasmodium falciparum dihydrofolate reductase mutations conferring resistance to pyrimethamine-WR99210 combination. Antimicrob Agents Chemother 51:4356–4360. doi: 10.1128/AAC.00577-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yuvaniyama J, Chitnumsub P, Kamchonwongpaisan S, Vanichtanankul J, Sirawaraporn W, Taylor P, Walkinshaw MD, Yuthavong Y. 2003. Insights into antifolate resistance from malarial DHFR-TS structures. Nat Struct Biol 10:357–365. doi: 10.1038/nsb921 [DOI] [PubMed] [Google Scholar]
- 39. Gregson A, Plowe CV. 2005. Mechanisms of resistance of malaria parasites to antifolates. Pharmacol Rev 57:117–145. doi: 10.1124/pr.57.1.4 [DOI] [PubMed] [Google Scholar]
- 40. Sridaran S, McClintock SK, Syphard LM, Herman KM, Barnwell JW, Udhayakumar V. 2010. Anti-folate drug resistance in Africa: meta-analysis of reported dihydrofolate reductase (dhfr) and dihydropteroate synthase (dhps) mutant genotype frequencies in African Plasmodium falciparum parasite populations. Malar J 9:247. doi: 10.1186/1475-2875-9-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Childs GE, Lambros C. 1986. Analogues of N-benzyloxydihydrotriazines: in vitro antimalarial activity against Plasmodium falciparum. Ann Trop Med Parasitol 80:177–181. doi: 10.1080/00034983.1986.11812002 [DOI] [PubMed] [Google Scholar]
- 42. Edstein MD, Bahr S, Kotecka B, Shanks GD, Rieckmann KH. 1997. In vitro activities of the biguanide PS-15 and its metabolite, WR99210, against cycloguanil-resistant Plasmodium falciparum isolates from Thailand. Antimicrob Agents Chemother 41:2300–2301. doi: 10.1128/AAC.41.10.2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chusacultanachai S, Thiensathit P, Tarnchompoo B, Sirawaraporn W, Yuthavong Y. 2002. Novel antifolate resistant mutations of Plasmodium falciparum dihydrofolate reductase selected in Escherichia coli. Mol Biochem Parasitol 120:61–72. doi: 10.1016/s0166-6851(01)00440-6 [DOI] [PubMed] [Google Scholar]
- 44. Shearer TW, Kozar MP, O’Neil MT, Smith PL, Schiehser GA, Jacobus DP, Diaz DS, Yang Y-S, Milhous WK, Skillman DR. 2005. In vitro metabolism of phenoxypropoxybiguanide analogues in human liver microsomes to potent antimalarial dihydrotriazines. J Med Chem 48:2805–2813. doi: 10.1021/jm049683+ [DOI] [PubMed] [Google Scholar]
- 45. Canfield CJ, Milhous WK, Ager AL, Rossan RN, Sweeney TR, Lewis NJ, Jacobus DP. 1993. PS-15: a potent, orally active antimalarial from a new class of folic acid antagonists. Am J Trop Med Hyg 49:121–126. doi: 10.4269/ajtmh.1993.49.121 [DOI] [PubMed] [Google Scholar]
- 46. Diaz DS, Kozar MP, Smith KS, Asher CO, Sousa JC, Schiehser GA, Jacobus DP, Milhous WK, Skillman DR, Shearer TW. 2008. Role of specific cytochrome P450 isoforms in the conversion of phenoxypropoxybiguanide analogs in human liver microsomes to potent antimalarial dihydrotriazines. Drug Metab Dispos 36:380–385. doi: 10.1124/dmd.106.013920 [DOI] [PubMed] [Google Scholar]
- 47. Mokmak W, Chunsrivirot S, Hannongbua S, Yuthavong Y, Tongsima S, Kamchonwongpaisan S. 2014. Molecular dynamics of interactions between rigid and flexible antifolates and dihydrofolate reductase from pyrimethamine-sensitive and pyrimethamine-resistant Plasmodium falciparum. Chem Biol Drug Des 84:450–461. doi: 10.1111/cbdd.12334 [DOI] [PubMed] [Google Scholar]
- 48. Edstein MD, Kotecka BM, Ager AL, Smith KS, DiTusa CA, Diaz DS, Kyle DE, Schiehser GA, Jacobus DP, Rieckmann KH, O’Neil MT. 2007. Antimalarial pharmacodynamics and pharmacokinetics of a third-generation antifolate--JPC2056--in cynomolgus monkeys using an in vivo in vitro model. J Antimicrob Chemother 60:811–818. doi: 10.1093/jac/dkm280 [DOI] [PubMed] [Google Scholar]
- 49. Mui EJ, Schiehser GA, Milhous WK, Hsu H, Roberts CW, Kirisits M, Muench S, Rice D, Dubey JP, Fowble JW, Rathod PK, Queener SF, Liu SR, Jacobus DP, McLeod R. 2008. Novel triazine JPC-2067-B inhibits Toxoplasma gondii in vitro and in vivo. PLoS Negl Trop Dis 2:e190. doi: 10.1371/journal.pntd.0000190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mookherjee S, Shoen C, Cynamon M. 2012. In vitro activity of JPC 2067 alone and in combination with sulfamethoxazole against nocardia species. Antimicrob Agents Chemother 56:1133–1134. doi: 10.1128/AAC.05855-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ruebush TK, Contacos PG, Steck EA. 1980. Chemotherapy of Babesia microti infections in Mongolian Jirds. Antimicrob Agents Chemother 18:289–291. doi: 10.1128/AAC.18.2.289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Aboge GO, Jia H, Terkawi MA, Goo Y-K, Nishikawa Y, Sunaga F, Namikawa K, Tsuji N, Igarashi I, Suzuki H, Fujisaki K, Xuan X. 2008. Cloning, expression, and characterization of Babesia gibsoni dihydrofolate reductase-thymidylate synthase: inhibitory effect of antifolates on its catalytic activity and parasite proliferation. Antimicrob Agents Chemother 52:4072–4080. doi: 10.1128/AAC.00384-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Begley DW, Edwards TE, Raymond AC, Smith ER, Hartley RC, Abendroth J, Sankaran B, Lorimer DD, Myler PJ, Staker BL, Stewart LJ. 2011. Inhibitor-bound complexes of dihydrofolate reductase-thymidylate synthase from Babesia bovis. Acta Crystallogr Sect F Struct Biol Cryst Commun 67:1070–1077. doi: 10.1107/S1744309111029009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chiu JE, Renard I, Pal AC, Singh P, Singh P, Vydyam P, Thekkiniath J, Kumar M, Gihaz S, Pou S, Winter RW, Dodean R, Frueh L, Nilsen AC, Riscoe MK, Doggett JS, Mamoun CB. 2021. Effective therapy targeting cytochrome bc1 prevents Babesia erythrocytic development and protects from lethal infection. Antimicrob Agents Chemother 17:e0066221. doi: 10.1128/AAC.00662-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vydyam P, Choi J-Y, Gihaz S, Chand M, Gewirtz M, Thekkiniath J, Lonardi S, Gennaro JC, Ben Mamoun C. 2023. Babesia BdFE1 esterase is required for the anti-parasitic activity of the ACE inhibitor fosinopril. J Biol Chem 299:105313. doi: 10.1016/j.jbc.2023.105313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vydyam P, Pal AC, Renard I, Chand M, Kumari V, Gennaro JC, Mamoun CB. 2024. Tafenoquine-atovaquone combination achieves radical cure and confers sterile immunity in experimental models of human babesiosis. J Infect Dis 229:161–172. doi: 10.1093/infdis/jiad315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fang T, Ben Mamoun C. 2024. Babesia duncani, a model organism for investigating intraerythrocytic parasitism and novel anti-parasitic therapeutic strategies. J Infect Dis:jiae191. doi: 10.1093/infdis/jiae191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vydyam P, Chand M, Pou S, Winter RW, Liebman KM, Nilsen A, Doggett JS, Riscoe MK, Ben Mamoun C. 2024. Effectiveness of two new endochin-like quinolones, ELQ-596 and ELQ-650, in experimental mouse models of human babesiosis. ACS Infect Dis 10:1405–1413. doi: 10.1021/acsinfecdis.4c00143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Singh P, Lonardi S, Liang Q, Vydyam P, Khabirova E, Fang T, Gihaz S, Thekkiniath J, Munshi M, Abel S, et al. 2023. Babesia duncani multi-omics identifies virulence factors and drug targets. Nat Microbiol 8:845–859. doi: 10.1038/s41564-023-01360-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. O’Neil RH, Lilien RH, Donald BR, Stroud RM, Anderson AC. 2003. Phylogenetic classification of protozoa based on the structure of the linker domain in the bifunctional enzyme, dihydrofolate reductase-thymidylate synthase. J Biol Chem 278:52980–52987. doi: 10.1074/jbc.M310328200 [DOI] [PubMed] [Google Scholar]
- 61. Pal AC, Renard I, Singh P, Vydyam P, Chiu JE, Pou S, Winter RW, Dodean R, Frueh L, Nilsen AC, Riscoe MK, Doggett JS, Ben Mamoun C. 2022. Babesia duncani as a model organism to study the development, virulence and drug susceptibility of intraerythrocytic parasites in vitro and in vivo. J Infect Dis 226:1267–1275. doi: 10.1093/infdis/jiac181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Singh P, Pal AC, Mamoun CB. 2022. An alternative culture medium for continuous in vitro propagation of the human pathogen Babesia duncani in human erythrocytes. Pathogens 11:599. doi: 10.3390/pathogens11050599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Singh P, Mamoun CB. 2023. Spermidine is the main polyamine required by intracellular parasites for survival within host erythrocytes. Microbiology. doi: 10.1101/2023.02.01.526700 [DOI]
- 64. Jensen NP, Ager AL, Bliss RA, Canfield CJ, Kotecka BM, Rieckmann KH, Terpinski J, Jacobus DP. 2001. Phenoxypropoxybiguanides, prodrugs of DHFR-inhibiting diaminotriazine antimalarials. J Med Chem 44:3925–3931. doi: 10.1021/jm010089z [DOI] [PubMed] [Google Scholar]
- 65. Jacobus D, Jensen N. 2003. Antimalarial N,N′-substituted biguanides derived from hydroxylamines.
- 66. Jacobus D. 2004. Biguanide and dihydrotriazine derivatives.
- 67. Vydyam P, Dutta D, Sutram N, Bhattacharyya S, Bhattacharyya MK. 2019. A small-molecule inhibitor of the DNA recombinase Rad51 from Plasmodium falciparum synergizes with the antimalarial drugs artemisinin and chloroquine. J Biol Chem 294:8171–8183. doi: 10.1074/jbc.RA118.005009 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene IDs, species names, proteins, and protein lengths.
Figures S1 to S7.
Supplemental methods; legends for Fig. S1 to S7.
List of dihydrotriazines and structures.
List of biguanides and structures.
In vitro efficacy (IC50 values) of DHTs against P. falciparum (3D7) in different media conditions.