

Abstract
Sepsis is a severe medical condition characterized by a systemic inflammatory response, often culminating in multiple organ dysfunction and high mortality rates. In recent years, there has been a growing recognition of the pivotal role played by mitochondrial damage in driving the progression of sepsis. Various factors contribute to mitochondrial impairment during sepsis, encompassing mechanisms such as reactive nitrogen/oxygen species generation, mitophagy inhibition, mitochondrial dynamics change, and mitochondrial membrane permeabilization. Damaged mitochondria actively participate in shaping the inflammatory milieu by triggering key signaling pathways, including those mediated by Toll-like receptors, NOD-like receptors, and cyclic GMP-AMP synthase. Consequently, there has been a surge of interest in developing therapeutic strategies targeting mitochondria to mitigate septic pathogenesis. This review aims to delve into the intricate mechanisms underpinning mitochondrial dysfunction during sepsis and its significant impact on immune dysregulation. Moreover, we spotlight promising mitochondria-targeted interventions that have demonstrated therapeutic efficacy in preclinical sepsis models.
Keywords: Sepsis, Mitochondrial dysfunction, Immune response, Mitochondria-targeted therapy
Introduction
To advance understanding of sepsis and septic shock, the task force convened by the European Society of Intensive Care Medicine and the Society of Critical Care Medicine introduced the "Sepsis-3" definition between 2014 and 2015 [1]. This update redefined sepsis as life-threatening organ dysfunction caused by a dysregulated inflammatory response to infection [1]. Sepsis contributes to 11 million deaths out of the 48.9 million reported cases, accounting for 19.7% of all global deaths in 2017 [2, 3]. The request for novel therapeutic targets in sepsis management assumes paramount importance.
Mitochondria are primary energy generators and participate in cellular processes, such as maintaining redox balance, buffering Ca2+, and initiating apoptosis [4]. A mitochondrion contains two membranes, the outer mitochondrial membrane (OMM) and the inner mitochondrial membrane (IMM), forming the matrix and intermembrane space (IMS) [5]. The mitochondrial respiratory chain comprises five complexes localized within the IMM: complex I, II, III, IV, and V. Complexes I–IV receive electrons, creating an electrochemical gradient of proton across the IMM referred to as mitochondrial membrane potential (ΔΨm). This ΔΨm is utilized by the complex V (F1F0 ATP synthase) to drive the production of ATP [6].
Accumulative evidence suggests that mitochondria play crucial roles in the pathogenesis of sepsis [7, 8]. Both laboratory studies and clinical investigations have documented the increase in mitochondrial damage during sepsis [9, 10]. Mitochondrial dysfunction occurs when mitochondria, fail to function properly upon damage. Mitochondria are not only involved in energy production, but also play critical roles in regulating cell metabolism and several cellular processes. The effects of mitochondrial dysfunction are enormous, including energy deficiency and activation of multiple pathways that alter cell function and fate. In the context of sepsis, the damaged mitochondria actively partake in shaping the inflammatory response via various signaling pathways [11]. Advances in mitochondria-targeted therapeutics have demonstrated that improving mitochondrial quality can reduce sepsis-induced inflammation, organ failure, and consequent mortality [12–14]. Thus, regulating mitochondrial quality is emerging as a promising strategy for sepsis treatment.
This review seeks to explore the complex mechanisms underlying mitochondrial dysfunction in sepsis and its profound influence on immune dysregulation. Additionally, we highlight promising interventions targeted at mitochondria that have shown therapeutic effectiveness in preclinical sepsis models. By comprehensively understanding the interplay between mitochondrial integrity and immune responses, we strive to pave the way for the development of novel and effective therapeutic approaches in combating sepsis-associated morbidity and mortality.
Mitochondrial stress and damage during sepsis
Sepsis triggers significant stress to mitochondria within immune cells and various tissues, leading to structural distortions, potential loss, and a marked decrease in respiratory activity. This section explores the specifics of the mitochondrial damage and the underlying mechanisms.
Mitochondrial damage
Despite the limited amount, clinical data clearly indicate mitochondrial dysfunction during sepsis. For instance, Japiassu and colleagues demonstrated that peripheral blood mononuclear cells (PBMCs) from septic shock patients have impaired mitochondrial ATP synthase activity [15]. Garrabou et al. observed inhibition of respiratory complexes in the PBMCs from septic patients without shock or multi-organ failure (MOF), suggesting that mitochondrial impairment precede these symptoms [16]. The platelets from septic patients showed lower activity of mitochondrial nicotinamide adenine dinucleotide dehydrogenase (NADH), complex I, I/III, and IV [17]. In addition to blood cells, muscle tissue of septic patients has also been reported with mitochondrial dysfunction and ATP depletion [18, 19]. Severe cardiac impairment has been observed in the non-surviving septic patients, with histologic analysis of heart sections showing mitochondrial cristae derangement (Table 1) [20].
Table 1.
Mitochondrial damage during sepsis
| Species | Inducer | Tissue/cell | Time/stage | Mitochondrial assessments | Refs. |
|---|---|---|---|---|---|
| Human | N.A | PBMCs | With shock | Oxygen consumption reduced; ATP synthase activity declined | [15] |
| Human | N.A | PBMCs | Early stages without shock or MOF | CI, CIII, and CIV activity decreased; ΔΨm reduced; Oxygen consumption reduced; Circulating free mtDNA in plasma increased | [16] |
| Human | N.A | Platelet | Severe sepsis or septic shock | Mitochondrial NADH, CI, CI & III, and CIV, succinate dehydrogenase (SDH) activity decreased | [17] |
| Human | N.A | Leg and serratus anterior muscle | With MOF and requiring mechanical ventilation |
Leg muscle: CI activity unchanged; CIV activity and ATP reduced; morphology unchanged Serratus anterior muscle: CI activity decreased; CIV activity, ATP, and morphology unchanged The observed changes probably because of reduced mitochondrial mass |
[28] |
| Human | N.A | Vastus lateralis muscle | With MOF | Structure swollen or damaged; Respiratory protein subunits and transcripts reduced | [19] |
| Human | N.A | Heart | Post-mortem | Mitochondrial cristae impaired | [20] |
| Mouse | LPS | Heart | 24 h | Mitochondrial number and volume reduced; Mitochondrial displayed internal vesicles, disrupted structure, and loss of cristae; expression of OXPHOS genes decreased | [29] |
| Mouse | LPS | Kidney | 18 h | Mitochondrial swollen and cristae reduced; Cytochrome c oxidase protein level and activity decreased; Mitochondrial gene expression was suppressed | [30] |
| Mouse | LPS | Liver | 0–48 h | mtDNA levels/integrity, complex I activity, and ATP level reduced; mtROS increased | [31] |
| Mouse | LPS | Leg muscle | 28 days | Mitochondrial respiration reduced; CV activity was reduced; Mitochondrial fission increased; | [32] |
| Mouse | CLP | Brain | 24 h | Oxidative phosphorylation uncoupled; ΔΨm dissipated; CIV activity reduced; CI-III activity unchanged | [33] |
| Mouse | CLP | Heart | 12 h | mtDNA copy number reduced; mitochondrial respiration reduced; ATP level declined; mitochondrial biogenesis marker PGC1β reduced; mitochondrial Ca2+ uptake capacity declined | [34] |
| Mouse | CLP | Kidney | 6–36 h | ROS increased; CI, CII & CIII activity and ATP decreased; CIV activity unchanged | [35] |
| Rat | LPS | Heart | 0–24 h | Mitochondria displayed structural abnormalities; CI, II, and IV activities reduced; ATP synthesis decreased | [36] |
| Rat | LPS | Liver | 2 h | Mitochondrial oxygen consumption reduced despite the total oxygen consumption unchanged; mitochondria became swollen and pale, with indistinct cristae and disrupted membranes; ATP/ADP ratio unchanged | [37] |
| Rat | S. pneumoniae | Heart | 24 h | ROS increased; mtDNA damaged; mitochondrial membrane disrupted; CI, II-III, IV and V activity reduced | [38] |
| Cat | LPS | Liver | 4 h | Respiratory activity was impaired; mitochondrial ultrastructural injured (swollen and membrane disruption) | [39] |
| Baboon | E. coli | Heart | 72 h or after death | CI, and II activities reduced; CIII was less affected | [25] |
MOF multi-organ failure. CI, II, III, IV, V: mitochondrial respiratory complex I, II, III, IV, V. ΔΨm: mitochondrial membrane potential
In animal models, cecal ligation and puncture (CLP), endotoxins/bacteria administration, and colon ascendens stent peritonitis (CASP) are commonly utilized to trigger sepsis [21]. Studies using these models support the clinical findings, highlighting the substantial impact of sepsis on mitochondrial homeostasis (Table 1). However, it is essential to acknowledge the presence of conflicting data showing unchanged or even increased mitochondrial activity in some cases, as previously discussed [22, 23]. Variables such as the nature of the septic insult, the severity of sepsis, timing of assessments, and methods of measurement may contribute to the discrepancies [24–26].
In addition, the common practice of using isolated cells or mitochondria from septic tissues for complex activity assays may not accurately reflect the in vivo mitochondrial status. This issue arises from the absence of circulating substances, such as cytokines, in vitro, which play pivotal roles in regulating mitochondrial respiration [16]. For instance, Boulos et al. demonstrated that serum from septic patients significantly reduced endothelial cell mitochondrial respiration compared to serum from healthy individuals, revealing the impact of circulating substances on mitochondrial function during sepsis [27].
Furthermore, normalization methods profoundly influence the evaluation of mitochondrial function. In a previous study, Fredriksson et al. measured the complex I activities in intercostal muscle mitochondria from septic patients and observed a 60% decrease when normalized to the dry weight of the muscle. However, no difference was found when normalization was conducted against citrate synthase activity. This was because the citrate synthase activity also declined during sepsis [28], emphasizing the importance of meticulous methodological consideration in such analyses.
Mechanisms of mitochondrial damage
Mitochondrial injury during sepsis is regulated by several factors, including reactive nitrogen species (RNS)/reactive oxygen species (ROS), mitophagy, mitochondrial dynamics, and mitochondrial membrane permeabilization, etc. In this section, we will examine the regulatory roles of these factors in mitochondrial health amidst septic conditions.
RNS/ROS burst
One of the well-exploited triggers of mitochondrial damage is nitric oxide (⋅NO), an RNS that is produced by inducible nitric oxide synthase (iNOS) [40]. ⋅NO could be converted to other reactive nitrogen species such as nitrite (NO2−) and nitrogen dioxide (·NO2) (Fig. 1) [41]. Escames et al. have investigated the role of iNOS in skeletal muscle mitochondria using a CLP mouse model. They observed a reduction in mitochondrial respiratory complex activity accompanied by elevated expression of iNOS during sepsis. In contrast, the iNOS-deficient mice did not exhibit such mitochondrial impairment [40]. A similar observation has been observed in the liver mitochondria of LPS-treated mice [42]. In line with these, inhibition of mitochondrial respiration induced by septic serum could be mitigated by the nitric oxide synthase inhibitor [27]. These data collectively suggest that iNOS play a crucial role in mitochondrial damage during sepsis.
Fig. 1.

Pathways of RNS and ROS production in sepsis. Excessive nitric oxide is generated by both cytosolic and mitochondrial iNOS during sepsis. The nitric oxide can be converted to other reactive nitrogen species (RNS), such as nitrite and nitrogen dioxide. On the other hand, excessive reactive oxygen species (ROS) are produced by NOXs and mitochondrial electron transport chain (ETC). Nitric oxide and superoxide react to form additional derivatives, notably the highly toxic peroxynitrite. The accumulation of these reactive species inflicts damage to cellular components, including lipids, proteins, and nucleic acids, thereby compromising mitochondrial respiration and integrity
Sepsis also stimulates the formation of ROS. Superoxide radical (O2·−) originates from nicotinamide adenine dinucleotide phosphate oxidases (NOXs) and mitochondria ETC [43]. O2·− is considered unstable and can be converted to other types of ROS (Fig. 1) [44]. On the other hand, declines in the antioxidative system happen during sepsis. For example, sirtuin proteins, which can protect cells from oxidative stress, have been found to decrease after septic insults [45, 46]. Oxidative stress occurs when ROS level overwhelms the antioxidant defense system in cells, causing damage to lipids, proteins, and nucleic acids [47].
Mechanistically, ·NO selectively binds to complex IV, where it competes with O2 for the binuclear CuB/cytochrome a3 center, resulting in a reversible inhibition of the complex IV [48]. While ·NO is relatively unreactive, its reaction with ROS gives rise to a series of more reactive derivatives [49]. Peroxynitrite (ONOO–) is one of the highly toxic derivatives (Fig. 1). Peroxynitrite can cause impairments to mitochondria via oxidation, irreversibly inhibiting mitochondrial complex I, CII, CIV, ATP synthase, and several critical enzymes. Consequently, the RNS/ROS exert a profound inhibitory effect on mitochondrial respiration and contribute to mitochondrial damage during sepsis [50].
Mitophagy
Mitophagy is a specialized form of autophagy that selectively degrades damaged mitochondria (Fig. 2a) [51]. Impaired mitophagy prevents mitochondrial turnover, leading to the accumulation of dysfunctional mitochondria [52]. Growing evidence shows that inhibition of mitophagy occurs in immune cells during the inflammatory response. Yu et al. found that Caspase-1 in macrophages cleaves Parkin to inhibit mitophagy upon inflammasome activation [53]. Similarly, Patoli et al. demonstrated mitophagy inhibition in the LPS/IFN-γ-treated macrophage cells, CLP mouse model, and septic patients, revealing caspases 1/11 dependent PINK1 degradation in the early stage of inflammation [54]. Additionally, inflammation-induced pro-IL-1α can interact with cardiolipin, preventing it from serving as a mitophagy receptor [55]. AMP-activated protein kinase (AMPK) is a regulator of autophagy, which positively regulates mitochondrial clearance by activating unc-51 like autophagy activating kinase 1 (ULK1) [56]. TLR4 signaling has been reported to prevent the activation of AMPK in neutrophil and macrophage [57]. Thus, lack of AMPK activation could be another reason for the accumulation of damaged mitochondria in sepsis.
Fig. 2.

Pathways of mitophagy and mitochondrial dynamics. a The most extensively studied mitophagy pathway involves PTEN-induced kinase 1 (PINK1) and E3 ubiquitin ligase Parkin. Upon ΔΨm loss, PINK1 is stabilized on the OMM, where it recruits and activates Parkin. Parkin subsequently ubiquitinates mitochondrial surface proteins, signaling the autophagosome to encapsulate the dysfunctional mitochondrion. Adaptor proteins including p62, Optineurin (OPTN), calcium binding and coiled-coil domain-containing protein 2 (NDP52), tax1-binding protein 1 (TAX1BP1), and next to BRCA1 gene 1 protein (NBR1) play a role in linking the ubiquitin chains to microtubule-associated protein 1 light chain 3 (LC3) on phagophores. On the other hand, some mitochondrial receptor proteins (or lipids) such as FUN14 domain containing 1 (FUNDC1), prohibitin (PHB), BCL2 interacting protein3 (BNIP3), Nip3-like protein X (NIX), cardiolipin, and syntaxin 17 (STX17) can recruit phagophore membranes independent of ubiquitin. The autophagosome finally fuses with a lysosome for the degradation and recycling of mitochondria. b Mitochondrial dynamics is regulated by several dynamin-related GTPases, including Mfn1/2, OPA1, Drp1, Fis1, MiD49/51, and Mff. Mfn1/2 facilitate the fusion of the OMM, while OPA1 is responsible for the fusion of the IMM. The fission process is initiated by the endoplasmic reticulum (ER)-mediated pre-constriction of mitochondria. Drp1 is recruited to the pre-constricted sites by receptors (Mff, Fis1, and MiD49/51). After binding to OMM, Drp1 oligomerizes into ring-shaped filaments. Hydrolysis of GTP by Drp1 results in constriction and closure of the ring. Dnm2 is finally recruited to the constricting site to complete the fission. In addition to the ER, lysosome and Golgi-derived vesicles have also been reported to regulate mitochondrial fission
Interestingly, activation of mitophagy also occurs during immune response. Ip and colleagues showed that interleukin 10 (IL-10) promotes mitophagy by inhibiting the mammalian target of rapamycin (mTOR) in macrophages [58]. Moreover, nuclear factor κB (NF-κB) facilitates mitophagy by inducing the expression of autophagic receptor p62. This NF-κB-p62-mitophagy pathway exists as a self-limiting mechanism to prevent excessive inflammation and maintain homeostasis [59]. Sestrin 2 (SESN2) also play a role in the p62-dependent mitophagy [60]. Extended LPS stimulation upregulates the protein level of SESN2, which interacts with p62 to facilitate its aggregation on mitochondria. Besides, SESN2 activates the autophagic machinery by raising the ULK1 protein level [60]. Overall, the accumulation of damaged mitochondria is likely to result from both forward and reverse regulation of mitophagy. How these different mitophagic pathways are spatiotemporally coordinated in sepsis is still elusive. Given the critical role of damaged mitochondria in promoting immune response (which will be discussed in the later section), it is plausible that early-stage inhibition of mitophagy promotes the accumulation of dysfunctional mitochondria and benefit bacterial defense [54], while late-stage activation of mitophagy represents a cellular attempt to mitigate inflammation and restore host homeostasis [59].
Unlike in immune cells, the overall mitophagy in various organs is enhanced during sepsis. The heart samples from LPS-challenged mice have shown decreased mitochondrial number and volume, suggesting autophagic removal of mitochondria [29, 61]. Immunoblot analysis of kidney tissue from LPS or CLP-treated mice has shown a decrease in mitochondrial protein levels (TOM20 and TIM23) compared with control mice, providing substantial evidence that mitophagy is induced during the sepsis-caused acute kidney injury [62]. It was also demonstrated that the mitophagy is mainly mediated by the PINK1/Parkin/OPTN pathway [62]. Reductions of mitochondrial mass were also reported in the liver of CLP or LPS-treated septic mice [63]. These studies collectively suggest that mitophagy is enhanced in various organs during sepsis.
Mitochondrial dynamics
Mitochondria constantly undergo fusion and fission processes (Fig. 2b). Fusion forms interconnected mitochondrial networks, promoting content exchange, which is essential for the integrity of the mitochondrial genome and proteome. Conversely, fission fragments mitochondria, aiding in the elimination of dysfunctional parts [64, 65]. Dysregulation of mitochondrial dynamics is a crucial mechanism that induces mitochondrial stress [66].
During sepsis, excessive mitochondrial fragmentation happens in various tissues, revealing the change in mitochondrial dynamics [62, 67, 68]. To exploit the influence of mitochondrial fission in sepsis, Gonzalez et al. treated rats with the Drp1 inhibitor mdivi-1 before CLP administration. This treatment restored mitochondrial shape and prevented the reduction of complex activities and apoptosis in hepatocytes [68]. Similar beneficial effects of mdivi-1 have been confirmed in other studies [13, 69, 70]. Pharmacological administration of alternative fission inhibitors, such as P110 (inhibit Drp1/Fis1 interaction), also showed protection to mitochondria under septic stress [71, 72]. These findings demonstrate that excessive fission significantly contributes to the impairment of mitochondria during sepsis. Inhibition of this process, therefore, is an effective method to restore mitochondrial function.
Mitochondrial membrane permeabilization
Mitochondrial membrane pores cause mitochondrial swelling, content loss, and ΔΨm dissipation [73]. Three principal mechanisms involved in pore formation during sepsis are mitochondrial outer membrane permeabilization (MOMP), mitochondrial permeability transition (MPT), and more recently characterized gasdermins (GSDMs)-mediated mitochondrial membrane opening (Fig. 3).
Fig. 3.
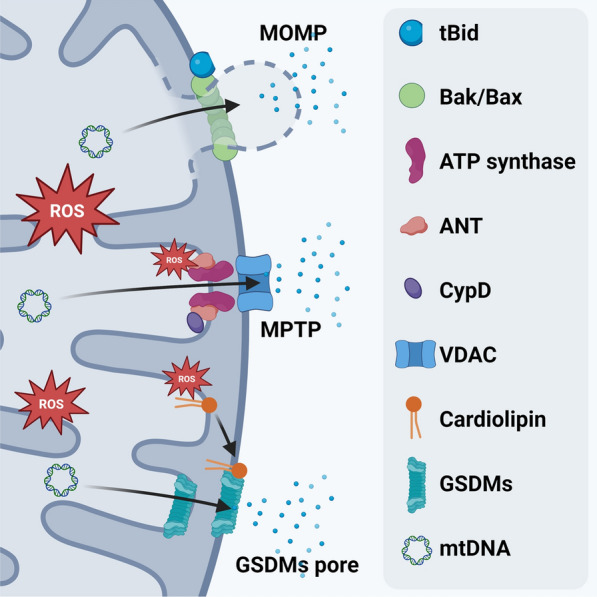
Mechanisms of mitochondrial membrane permeabilization in sepsis. Three principal mechanisms are implicated in sepsis-related mitochondrial membrane pore formation. Mitochondrial outer membrane permeabilization (MOMP) is mediated by Bcl-2 family proteins Bax/Bak and regulated tBid. Large BAK/BAX pores allow the protrusion of IMM into the cytosol, therefore, enabling the escape of mitochondrial matrix contents. Mitochondrial permeability transition pore (MPTP) represents another type of mitochondrial pore in sepsis. Despite the opening mechanism of MPTP is still elusive, mitochondrial components such as F1FO (F)-ATP synthase, ANT, and Cyp-D have been confirmed to play pivotal roles in this process. Gasdermins (GSDMs) are well-known to form pores on plasma membrane during immune response. Recent advances have demonstrated that GSDMs target both the inner and outer mitochondrial membranes via their strong binding preference to cardiolipin. ROS promote the externalization of cardiolipin from IMM to OMM, thus, positively regulate GSDMs-mediate pore formation. Overall, mitochondrial membrane permeabilization leads to mitochondrial swelling, content loss, and dissipation of membrane potential during sepsis
MOMP is mediated by Bcl-2 family proteins Bax and Bak, which open the OMM in response to apoptotic signals. MOMP leads to mitochondrial damage and release of intermembrane space molecules like cytochrome C [74]. Large Bax/Bak pores also allow the IMM to protrude out of mitochondria, releasing matrix contents [75, 76]. Cytokines such as INF-γ and TNF-α could be recognized by the cell membrane death receptors, thereby activating caspase-8 during sepsis [77]. The activated caspase-8 cleaves Bid, generating a truncated form of Bid (tBid), which translocates to mitochondria and activates Bax/Bak to induce MOMP and cell death [78–80]. Chuang et al. have observed increased tBid in mitochondrial fractions of various tissues from the CLP-treated mouse, while Bid deficiency significantly reduced the downstream cell death [81]. In addition to caspase-8, caspase-1 has also been reported to cleave Bid and induce MOMP during the inflammatory response of macrophage [82].
The occurrence of MPT has been described in several septic models [83–86]. Pharmacological inhibition of MPT by cyclosporin A (CsA) can significantly attenuate mtDNA release and mitochondrial dysfunction in these models, suggesting MPT’s crucial role in sepsis-induced mitochondrial damage [83–85]. Although the exact molecular composition and mechanism of MPT remains a topic of ongoing research, key components are believed to include the F1FO (F)-ATP synthase, adenine nucleotide translocator (ANT), cyclophilin D (Cyp-D), and voltage-dependent anion channel (VDAC) [87]. These components assemble into a supramolecular pore (MPTP) at the interface of the IMM and OMM [88]. Elevated ROS levels can induce MPTP opening through oxidative modifications of MPT constituents or other mitochondrial proteins [89, 90].
GSDMs are key players in immune response, particularly in mediating pyroptosis. During inflammation, GSDMs are cleaved by caspases to release their active N-terminal fragments (GSDMs-N). GSDMs-N translocate to the plasma membrane, forming pores and disrupting the membrane [91]. Recent findings indicate that GSDMs also target mitochondrial membranes, representing a new pathway of mitochondrial permeabilization [92–97]. Time-lapse imaging analysis revealed that mitochondrial damage occurs much earlier than the opening of cell membrane, suggesting that GSDMs-N permeabilizes the mitochondria before cell membrane [92, 93]. This phenomenon has been attributed to the strong binding preference of GSDMs-N to mitochondrial cardiolipin [92]. Of note, ROS promote the externalization of cardiolipin from IMM to OMM, explaining the accumulation of GSDMD-N on OMM, as cardiolipin predominantly localizes in the IMM under normal conditions [95]. Overall, GSDM-mediated pore formation represents a novel pathway of mitochondrial permeabilization during immune response.
Damaged mitochondria are potent triggers of immune response and metabolic reprogramming
The innate immune response plays a key role in the pathophysiology of sepsis. Upon activation, pattern recognition receptors (PRRs) initiate signaling cascades, leading to the nuclear translocation of NF-κB, interferon regulatory factor (IRF), and other transcription factors. These transcription factors initiate the production of pro-inflammatory cytokines, chemokines, type I interferons (IFNs), etc. [98]. In the past few decades, damaged mitochondria have been widely demonstrated as a crucial regulator of innate immunity, particularly through the NLRP3, TLR9, and cGAS pathways. Thus, mitochondria damage is not simply a passive outcome under stress conditions, but actively contributes to the inflammatory response that is essential for the defense of pathogens. However, in the context of sepsis, excessive or persistent inflammation can cause a fatal inflammatory imbalance in the body, which ultimately leads to tissue and organ damage [99]. In this section, we will highlight the roles of mitochondria in regulating inflammatory pathways.
NLRP3 inflammasome pathway
NLRP3 inflammasome activation
The NLRP3 inflammasome activates pro-caspase-1, which cleaves pro-inflammatory cytokines such as interleukin-1β (IL-1β) and interleukin-18 (IL-18) to promote their maturation [100]. Current studies, mainly in monocytes/macrophages, have revealed several regulatory roles of mitochondria in NLRP3 inflammasome activation. Mitochondrial ROS (mtROS) have been demonstrated as essential molecules for NLRP3 inflammasome activation. In sepsis models, inhibition of mtROS reduces the NLRP3 inflammasome-mediated cytokine production [101–103]. Several studies have provided insights into the mechanisms by which ROS modulate NLRP3 activation. In 2010, Zhou et al. identified thioredoxin (TRX)-interacting protein (TXNIP) as a binding partner of NLRP3. Under basal conditions, TXNIP binds to TRX, being unavailable to NLRP3. ROS promote NLRP3 activation by triggering the dissociation of TXNIP from TRX [104]. In another study, mtROS were confirmed to facilitate NLRP3 inflammasome activation through promoting its deubiquitination [105]. Interestingly, Bauernfeind and colleagues reported that ROS increase the NLRP3 expression instead of activating it, suggesting a translational regulation of NLRP3 by ROS [106].
mtDNA also regulates NLRP3 inflammasome activation. Nakahira et al. demonstrated that preventing MPT-mediated mtDNA release significantly reduces IL-1β secretion in macrophages after LPS + ATP challenge [83]. Interestingly, NLRP3 inflammasome is preferentially activated by oxidized mtDNA (ox-mtDNA), rather than normal mtDNA [107]. Zhong et al. found that TLR4 signalling promotes de novo mtDNA synthesis to sustain the generation of ox-mtDNA, since the newly synthesized mtDNA is not packaged and is highly susceptible to oxidation [108]. Before being released into cytosol, the ox-mtDNA undergoes a cleavage process to become 500–650 bp fragments. Xian et al. recently identified the flap structure-specific endonuclease 1 (FEN1) as the key mediator of this process [109]. AIM2 inflammasome can also be activated by the mtDNA. Unlike NLPR3 inflammasome, AIM2 inflammasome is activated by normal DNA to ox-mtDNA [107, 108].
Spatial association between mitochondria and NLRP3
The spatial association of NLRP3 inflammasome with organelles is critical for its activation. Early studies posited mitochondria or mitochondria-associated membrane (MAM) as the docking sites of NLRP3, while more recent publications have implicated trans-Golgi network (TGN) and microtubule-organizing center (MTOC) [110–114].
Zhou et al. reported that inflammatory stimuli trigger NLRP3 to co-localize with mitochondria and MAM in 2010 [112]. This spatial vicinity may facilitate the sensing of mitochondria-derived inflammatory signals [112, 115]. Microtubules also play a role in the mitochondria-NLRP3 association. Mechanistically, mitochondrial dysfunction during inflammation reduces NAD+ (oxidized form of nicotinamide adenine dinucleotide) generation, which in turn inhibits NAD+-dependent α-tubulin deacetylase sirtuin 2, leading to an increase in α-tubulin acetylation. The acetylated α-tubulin subsequently promoted dynein-dependent transport of mitochondria to NLRP3 [116]. Inflammatory stimuli also trigger the transport of NLRP3 towards mitochondria, which is orchestrated by microtubule-affinity regulating kinase 4 (MARK4). Depletion of MARK4 impairs NLRP3 spatial arrangement and inflammasome activation [111].
Mitochondrial antiviral-signaling protein (MAVS) and cardiolipin mediate the anchorage of NLRP3 on mitochondria [117–121]. MAVS lacking mitochondrial targeting domain cannot recruit NLRP3 or induce NLRP3 oligomerization, suggesting its mitochondrial localization is essential for NLRP3 inflammasome activation [120]. Given the well-established role of MAVS in recognizing virus RNA, it is plausible that microbial RNA plays a role in promoting the MAVS-dependent recruitment of NLRP3 during bacterial infection [121, 122]. In line with this, virus or Escherichia coli-induced activation of NLRP3 inflammasome was confirmed to greatly depend on MAVS [121], whereas LPS + ATP or LPS + nigericin (without microbial RNA) induced NLRP3 inflammasome activation is only partially affected by MAVS [117, 120, 121]. Cardiolipin also interacts with NLRP3 and contributes to the inflammasome activation [118]. A recent study suggested that cardiolipin recruits both NLRP3 and caspase-1, serving as a platform for inflammasome assembly [119].
Several recent publications have highlighted the involvement of Golgi apparatus and MTOC in NLRP3 inflammasome activation [113, 114, 123]. It is likely that NLRP3 protein is first transported to meet mitochondria along the microtubules, after which NLRP3 redistributes to the adjacent Golgi apparatus [115, 124]. Then, the NLRP3 is transported to the MTOC and reorganized into a single speck structure. At the MTOC, NLRP3 engages with NEK7, a centrosome-localized kinase that is essential for NLRP3 inflammasome activation (Fig. 4) [111, 114, 123, 125, 126].
Fig. 4.

Mitochondria-regulated inflammatory pathways in sepsis. Pattern recognition receptors (PRRs) of immune cells recognize pathogen-associated molecular patterns (PAMPs) from microbes or damage-associated molecular patterns (DAMPs) released by damaged host cells. a Mitochondria orchestrate the activation of the NLRP3 inflammasome. mtROS and ox-mtDNA promote the NLRP3 inflammasome activation after their release from the damaged mitochondria. Additionally, mitochondrial antiviral-signaling protein (MAVS) and cardiolipin mediate the spatial association between mitochondria and NLRP3, which may facilitate a rapid and efficient sensing of the inflammatory signals from the damaged mitochondria by NLRP3. Early studies posited mitochondria or mitochondria-associated membrane (MAM) as the docking sites of NLRP3, while more recent publications have indicated the involvement of Golgi and microtubule-organizing center (MTOC). It could be plausible that NLRP3 proteins are first transported to meet mitochondria, after which NLRP3 redistributes to the adjacent Golgi apparatus as oligomeric cages. Then, the NLRP3 cages are transported to MTOC and reorganized into a single inflammasome speck. Activated NLRP3 inflammasome mediates the activation of pro-caspase-1, which proteolytically processes GSDMs and pro-inflammatory cytokines. The matured cytokines are released from the GSDMs pores to transmit inflammatory signals. b Mitochondria regulate the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway. cGAS is activated upon mtDNA binding, leading to the synthesis of cyclic GMP-AMP (cGAMP). cGAMP then induces a conformational change in STING, leading to its activation at ER. Activated STING translocates to Golgi, where it activates transcription factors interferon regulatory factor 3 (IRF3) and nuclear factor κB (NF-κB) to initiate the production of type I interferons and other pro-inflammatory cytokines that are required for effective immune response against pathogens. c Mitochondria regulate the TLR9 pathway. mtDNA shares similarities with bacterial DNA, thus can be recognized by TLR9. As with other TLRs and cGAS-STING axis, TLR9 pathway activates IRF3 and NF-κB to regulate the inflammatory response
cGAS-STING pathway activation
The cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway is another mitochondria-regulated inflammatory pathway. cGAS detects cytosolic double-stranded DNA and initiates an innate immune response [127] (Fig. 4). Both normal and oxidized mtDNA can be recognized by cGAS [128], but ox-mtDNA is more resistant to degradation by the cytosolic nuclease 3' repair exonuclease 1, increasing its likelihood of interaction with cGAS [129].
In the CLP mouse model, Huang et al. have demonstrated that mtDNA can be released via the GSDMD pore to activate cGAS-STING pathway [94]. This pathway not only enhances the inflammatory response but also suppresses lung endothelial cell proliferation, which compromises the recovery capacity of host. Notably, the deletion of cGas gene provided mice with substantial protection from lung injury 72 h post-CLP, suggesting that cGAS-STING pathway is crucial in septic lung inflammation [94].
Studies have shown that circulating cell-free mtDNA levels are elevated in septic patients and animal models, correlating with poor prognosis [130–133]. Like cytosolic mtDNA, circulating mtDNA activates the cGAS-STING pathway and promotes inflammation during sepsis [133]. In CLP mice, the mtDNA-cGAS-STING signal promotes intestinal inflammation and gut barrier dysfunction, which could be significantly attenuated by removing circulating mtDNA with DNase I [134]. Mechanistically, host nuclear DNA, mtDNA and bacterial DNA can be recognized by cGAS, while to what extent mtDNA contributes to the cGAS-STING pathway activation awaits investigation [135, 136].
TLR9 pathway activation
TLR9 is initially known for sensing unmethylated CpG DNA motifs (consisting of a central cytosine-guanine dinucleotide plus flanking regions) from bacterial and viral DNA during innate immune response [137, 138]. However, a subsequent study suggests that TLR9 also recognizes endogenous mtDNA, which shares similarities with bacterial DNA [139]. This interaction stands for another inflammatory pathway regulated by mitochondria in sepsis (Fig. 4).
Experimental models have shown that intravenous administration of mitochondrial debris can elicit an immune response comparable to that of CLP treatment. However, Tlr9 knockout (KO) or DNase pretreatment of the mitochondrial debris greatly attenuates the inflammatory response, suggesting the crucial role of the mtDNA-TLR9 axis in sepsis [140]. In line with this, TLR9 inhibitor OND-I suppresses caspase 1 activation and IL–1β production in LPS-challenged cardiomyocytes [38]. Moreover, Tlr9 KO reduced renal dysfunction and splenic apoptosis after CLP treatment, and increased survival at 96 h [141]. Interestingly, Plitas and colleagues found that Tlr9 KO mice exhibited better bacterial clearance and greater survival than wide type (WT) mice after CLP treatment. The protective effects were associated with increased recruitment of granulocytes to the peritoneum by dendritic cells (DCs) [142]. These findings suggest that TLR9 might have a major role in the immunopathogenesis of polymicrobial sepsis compared to other TLRs [142].
Metabolic reprogramming
During sepsis, a widespread alteration in the metabolic pattern occurs in virtually all cell types, leading to a shift from oxidative phosphorylation (OXPHOS) to glycolysis—a phenomenon known as “metabolic reprogramming” [143]. The metabolic reprogramming is driven by the downregulation of OXPHOS due to mitochondrial dysfunction and the upregulation of glycolysis through the enhancement of glycolytic pathways [144]. This shift in metabolism profoundly affects immune response and organ function in sepsis.
In the immune context, glycolysis, albeit less efficient than OXPHOS, allows rapid generation of ATP and synthesis of metabolic intermediates necessary for immune cell activation and proliferation [145]. Glycolytic inhibitors, such as 2-deoxyglucose (2-DG), have been shown to reduce systemic inflammation in sepsis, indicating their potential in therapeutic interventions [146, 147]. The metabolic reprogramming is also implicated in the development MOF. An early histopathologic study of patients dying of sepsis has revealed that the actual cell death in most organs is minimal, despite severe clinical signs of MOF [148]. This observation indicates that the organ failure is functional rather than structural [149]. Given that mitochondrial dysfunction results in less efficient ATP production, cells in affected organs face an energy crisis during sepsis. Consequently, metabolic reprogramming and the resultant bioenergetic failure are recognized as critical contributors to MOF in sepsis [150]. A key question regarding MOF in sepsis is whether it represents an adaptive or maladaptive event. Singer et al. have suggested that sepsis-induced MOF might be an adaptation by the host to increase the chances of cell survival against the infection. This response mirrors a state of “hibernation” in which non-essential functions are downregulated [9, 149].
Mitochondria-based therapeutic interventions
Effective treatments are crucial for aiding critically ill patients with sepsis. Previous research has shown that sepsis survivors responded early to the illness with induction of mitochondrial biogenesis and antioxidant defense, indicating the potential of maintaining mitochondrial homeostasis for sepsis therapy [19].
Antioxidants
Given the roles of ROS in triggering mitochondrial damage and inflammation, antioxidants have been extensively studied as therapeutic agents against sepsis. Mitochondria-targeted antioxidants have shown better effects on reducing inflammation compared to the untargeted equivalents [101, 151, 152]. The most frequently used mitochondria-specific antioxidants include MitoQ, MitoVitE, and MitoTempol (Fig. 5) [153].
Fig. 5.

Mitochondria-based therapeutic interventions. Mitochondria-targeted antioxidants such as MitoQ, MitoVitE, and MitoTempol are selectively targeted to mitochondria by conjugating to a lipophilic cation decyl-triphenylphosphonium (TPP+). The positive charge of cation enables the molecules to accumulate in mitochondrial matrix, therefore, specifically quench the mitochondrial ROS. Non-antioxidants Urolithin A, Mdivi-1, and Cyclosporin A improve mitochondrial quality by promoting mitophagy, inhibiting mitochondrial fission, and blocking MPTP, respectively. Metformin plays multiple roles on mitochondria, including enhancing mitophagy, reducing mtROS, preventing ox-mtDNA production, and inhibiting mitochondrial protein translation. These compounds target different aspects of mitochondria and exhibit great potential in reducing sepsis-induced mitochondrial damage, excessive inflammation, and organ dysfunction
MitoQ
MitoQ is synthesized by conjugating ubiquinone with the lipophilic cation decyl-triphenylphosphonium (TPP+). Although it has not received approval from the U.S. Food and Drug Administration (FDA), MitoQ has been the subject of several clinical trials aiming to explore its potential benefits for treating oxidative pathologies [154]. Lowes et al. demonstrated that MitoQ protects mitochondria during sepsis, and suppresses pro-inflammatory cytokine release, leading to reduced acute liver and renal dysfunction [102]. The beneficial effects have also been confirmed in other organs, including the lung, intestinal barrier, skeletal muscle, and heart, leading to the increased survival rate of septic animals [12, 155–157]. Notably, MitoQ treatment 6 h after sepsis induction yielded comparable outcomes in comparison with immediate treatment in the diaphragm muscle. This property makes MitoQ a good candidate for clinical application, considering the typical delay between sepsis onset and treatment initiation [157].
MitoVitE
MitoVitE is a modified form of vitamin E, attached to the TPP+ [158]. Zang et al. examined its effects on cardiac dysfunction in a rat pneumonia-related sepsis model, and confirmed that a single dose of MitoVitE protected cardiac mitochondria, suppressed cytokine burst, and neutrophil infiltration in the myocardium, ultimately improving the cardiac function [101]. Another study using LPS/peptidoglycan (PepG)-induced sepsis rat model revealed that MitoVitE offers similar protection as MitoQ in improving mitochondrial health, mitigating inflammation, and reducing organ dysfunction [159].
MitoTempol
MitoTempol, constructed by combining the piperidine nitroxide (Tempol) with TPP+, is another mitochondria-targeted antioxidant [160]. In a rat model of fecal peritonitis, MitoTempol administration reduced IL-1β level, renal oxidative stress, and improved renal function [161]. Interestingly, while this study showed no beneficial effect on the core body temperature and survival rate [161], another investigation revealed that even a 6 h delayed MitoTempol therapy significantly improved core body temperature and increased the survival rate from 40 to 83% in CLP mice [35]. These contradictory results may be related to MitoTempol dosage. Insufficient dosing may fail to provide benefits, whereas excessive dosing may lead to over-accumulation of this cationic agent, resulting in mitochondrial damage [35]. Thus, fine-tuning the dosage is essential to minimize side effects and maximize therapeutic benefits.
NAD+
NAD+ is known for its functions in redox metabolism, immune response, aging, and DNA repair [162]. NAD+ shortage occurs during sepsis, prompting the exploration of NAD+ supplementation as a potential therapeutic strategy [163, 164]. NAD+ precursors, such as nicotinamide mononucleotide (NMN) and nicotinamide riboside (NR), are emerging as novel candidates for sepsis treatment. These precursors, naturally present in food and widely used as health supplements, have shown potential in preclinical studies by mitigating mitochondrial dysfunction, oxidative stress, inflammatory response, and multiorgan injury associated with sepsis [164–166]. The precise regulatory mechanisms of NAD+ in sepsis remain incompletely understood. As a critical co-enzyme, NAD+ plays a role in alleviating oxidative stress, in part through the activation of sirtuins, like Sirt3, a mitochondria-localized sirtuin [164, 165].
Non-antioxidants
Several non-antioxidant agents targeting mitophagy, mitochondrial dynamics, mitochondrial permeabilization, and other pathways have been reported to mitigate sepsis (Fig. 5).
Urolithin A
Urolithin A (UA) is a natural anti-aging compound known for inducing mitophagy [167]. It has attracted significant attention for the role in regulating inflammation [168, 169]. Two clinical trials in elderly individuals have shown that UA is safe, bioavailable, and well-tolerated [170, 171].
UA has been reported to sustain mitochondrial health in cardiomyocytes challenged with LPS in vitro [172]. In mice, UA administration alleviated the LPS-induced cardiac depression [173]. Interestingly, this protection was diminished upon FUNDC1 knockout, indicating UA activates mitophagy in a FUNDC1-dependent manner [173]. UA pre-administration was also found to alleviate pulmonary injury upon LPS challenge, and improve the survival rate [174]. Overall, while research on UA for sepsis treatment is still nascent, the existing evidence suggests significant therapeutic potential. Future research using more appropriate sepsis models would be recommended, as LPS treatment does not recapitulate the complex pathophysiological consequence of human sepsis [175, 176].
Mdivi-1
Mdivi-1 was first identified as an inhibitor of mitochondrial fission via chemical screening in 2008, [177]. Thereafter, numerous articles have been published on Mdivi-1, confirming its potency in inhibiting mitochondrial fission, improving mitochondrial health, and protecting cells from stress under various circumstances [178]. The wide application of Mdivi-1 promotes the development of novel therapeutic strategies for mitochondria-related diseases, including sepsis.
Mdivi-1 was shown to protect liver and brain function in the CLP mice model [179, 180]. Moreover, Zhu et al. showed that Mdivi-1 significantly restored the function of multiple organs in CLP-treated rats, and extended the average survival time from 8.83 h to 60.3 h. In this study, Mdivi-1 was administered 12 h after sepsis, which mirrors a clinically relevant scenario [13]. Other fission inhibitors, like P100 and irisin, have also been reported to mitigate organ injury and reduce mortality after sepsis [71, 72, 181].
Cyclosporin A
CsA is an FDA approved compound that has been utilized as an anti-inflammatory drug to treat ocular surface diseases [182]. Cyclosporin A has been widely used as an immunosuppressant to prevent immune responses against transplanted organs in clinic [183]. CsA potentially inhibits immune response via two pathways. First, CsA inhibits MPTP by interacting with Cyp-D, thus, reducing DAMPs release from mitochondria [184]. Second, CsA forms a complex with cyclophilin A to inhibit calcineurin. This inhibition thereafter impairs the nuclear translocation of nuclear factor of activated T cells (NFAT) and NFκB in immune cells, leading to reduced production of iNOS, inflammatory cytokines and prostaglandins [185].
Over the past two decades, CsA has been reported to attenuate mtDNA release and mitochondrial damage in various septic models [83–85, 186]. These studies confirmed the protective effects of CsA on sepsis-induced organ dysfunction and mortality [83–85, 186]. CsA and NIM811 (a cyclophilin non-binding derivate of CsA that inhibits MPTP) rather than tacrolimus (a potent immunosuppressive compound which does not inhibit MPTP) provided protection against septic insult, suggesting that the beneficial effects of CsA were predominantly related to the inhibition of MPTP [84, 187]. However, Joshi et al. reported that tacrolimus provided similar cardiac protection as CsA upon LPS treatment [188]. This discrepancy maybe due to the severity of mitochondrial injury; CsA's MPTP inhibitory function becomes critical only when mitochondrial damage is severe.
Metformin
Metformin has been widely used to treat type 2 diabetes, whose clinical experience and trial data have raised almost no safety concerns [189, 190]. Several studies have highlighted the capacity of metformin in combating sepsis. For instance, Liu et al. demonstrated that metformin prevents excessive inflammation and the development of immunosuppression, increasing bacterial clearance in the lungs of septic mice [191]. Metformin also preserves brain, lung, liver, and colon barrier function by suppressing sepsis-induced inflammatory responses in animal models [192–194]. In human patients, several retrospective studies have been performed to evaluate the effect of metformin on sepsis. However, some of these studies revealed that metformin users (mainly diabetic patients) had lower in-hospital mortality than nonusers [195–197], while others showed that pre-admission of metformin did not change the mortality in septic patients [198, 199]. Two meta-analyses on these published cohort data were, therefore, performed to confirm the contribution of metformin in the mortality in septic adult patients. The results supported the positive effect of metformin in lowering the mortality of sepsis [200, 201]. Recently, three clinical trials (NCT05979038, NCT06181422, NCT05900284) evaluating the efficiency and safety of metformin in sepsis patients are ongoing, which will improve our understanding about the clinical feasibility of metformin in sepsis therapy.
The pharmacological mechanisms of metformin are multifaceted. Metformin regulates inflammation through both AMPK-dependent and independent pathways [202]. Mechanistically, metformin activates AMPK and suppresses the nutrient sensor mechanistic target of rapamycin (mTOR) complex 1 (mTORC1), which promotes autophagy. Autophagic elimination of damaged mitochondria subsequently limits NLRP3 inflammasome activation [203, 204]. AMPK also phosphorylate the PGC-1α to promote mitochondrial biogenesis [205]; or regulate transcription factors to reduce ROS and cytokine production [206, 207]. The activation of AMPK by metformin appears to be dose-dependent: Low-dose metformin drives AMPK activation through the lysosomal pathway, while high-dose metformin takes effect by inhibiting the complex I of respiratory chain, resulting in ATP shortage [208]. Xian et al. have revealed that the metformin-triggered ATP shortage can prevent the production of ox-mtDNA, thus, reducing NLRP3 inflammasome activation [204]. Metformin also diminishes IL-6 secretion, likely via the suppression of JNK and p38 MAPK [204]. Very recently, Marlies Cortés and colleagues uncovered that metformin’s anti-inflammatory effects rely on the expression of ZEB1, which restricts amino acid uptake, thereby downregulating the mTORC1 signaling and mitochondrial protein translation, leading to the inhibition of both acute and chronic inflammation [209].
Conclusion
Sepsis is a significant medical challenge characterized by a systemic inflammatory response. During sepsis, factors such as RNS/ROS, impaired mitophagy, altered mitochondrial dynamics, and membrane permeabilization result in the accumulation of defective mitochondria in immune cells and tissues. These damaged mitochondria promote the immune response via key pathways governed by NLRP3, TLR9, and cGAS. Preclinical studies have identified several agents that target mitochondrial quality control to reduce mitochondrial damage. These agents effectively attenuate inflammation and mortality, offering a novel approach for sepsis treatment.
Acknowledgements
Figures used in this review are made in BioRender.com.
Abbreviations
- AKI
Acute kidney injury
- ALI
Acute lung injury
- AMPK
AMP-activated protein kinase
- ANT
Adenine nucleotide translocator
- BNIP3
BCL2 interacting protein3
- CALCOCO2/NDP52
Calcium binding and coiled-coil domain-containing protein 2
- CASP
Colon ascendens stent peritonitis
- cGAS
Cyclic GMP-AMP synthase
- CLP
Cecal ligation and puncture
- CsA
Cyclosporin A
- Cyp-D
Cyclophilin D
- DAMPs
Damage-associated molecular patterns
- DCs
Dendritic cells
- Drp1
Dynamin-related protein 1
- ER
Endoplasmic reticulum
- ETC
Electron transport chain
- FDA
U.S. food and drug administration
- FEN1
Flap structure-specific endonuclease 1
- Fis1
Fission 1
- FUNDC1
FUN14 domain containing 1
- GSDMs
Gasdermins
- ICUs
Intensive care units
- IFNs
Interferons
- IL
Interleukin
- IMS
Intermembrane space
- IRF
Interferon regulatory factor
- KO
Knockout
- LC3
Microtubule-associated protein 1 light chain 3
- L-OPA1
Long OPA1 isoforms
- LPS
Lipopolysaccharides
- MAM
Mitochondria-associated membrane
- MARK4
Microtubule-affinity regulating kinase 4
- MAVS
Mitochondrial antiviral-signaling protein
- Mff
Mitochondrial fusion factor
- Mfn1/2
Mitofusin 1/2
- MOF
Multi-organ failure
- MOMP
Mitochondrial outer membrane permeabilization
- MPT
Mitochondrial permeability transition
- mtDNA
Mitochondrial DNA
- MTOC
Microtubule-organizing center
- mTOR
Mammalian target of rapamycin
- mtROS
Mitochondrial ROS
- NADH
Nicotinamide adenine dinucleotide dehydrogenase
- NBR1
Next to BRCA1 gene 1 protein
- NFAT
Nuclear factor of activated T cells
- NF-κB
Nuclear factor κB
- NIX
Nip3-like protein X
- NLRs
NOD-like receptors
- NOXs
Nicotinamide adenine dinucleotide phosphate oxidases
- OMM
Outer mitochondrial membrane
- OPA1
Optic atrophy 1
- OPTN
Optineurin
- ox-mtDNA
Oxidized mitochondrial DNA
- PAMPs
Pathogen-associated molecular patterns
- PBMCs
Peripheral blood mononuclear cells
- PepG
Peptidoglycan
- PGC-1 β
Peroxisome-proliferator-activated receptor γ coactivator 1 β
- PHB
Prohibitin
- PINK1
PTEN-induced kinase 1
- PRRs
Pattern recognition receptors
- RNS
Reactive nitrogen species
- SESN2
Sestrin 2
- S-OPA1
Short OPA1 isoforms
- STING
Stimulator of interferon genes
- STX17
Syntaxin 17
- TAX1BP1
Tax1-binding protein 1
- tBid
Truncated form of Bid
- TCA
Tricarboxylic acid
- Tempol
Piperidine nitroxide
- TFAM
Mitochondrial transcription factor A
- TGN
Trans-Golgi network
- TLRs
Toll-like receptors
- TPP+
Decyl-triphenylphosphonium
- TRX
Thioredoxin
- TXNIP
Thioredoxin-interacting protein
- UA
Urolithin A
- UCPs
Uncoupling proteins
- ULK1
Unc-51 like autophagy activating kinase 1
- VDAC
Voltage-dependent anion channel
- WT
Wide type
- ΔΨm
Mitochondrial membrane potential
Author contributions
Dongxue Hu and Harshini Sheeja Prabhakaran contributed to the data collection of the article. Dongxue Hu, Harshini Sheeja Prabhakaran, Weifeng He, and Yih-Cherng Liou contributed to the conception, preparation and organization of this article. Yuan-Yuan Zhang and Gaoxing Luo contributed to the organization and constructive discussions. Weifeng He and Yih-Cherng Liou revised the draft of the manuscript.
Funding
This work is financially supported by MOE Tier2 and Tier1 (A-8000985 and A-8000412) grants from the Ministry of Education (MOE), Singapore, awarded to Y-C. Liou.
Availability of data and materials
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Dongxue Hua and Harshini Sheeja Prabhakaran contributed equally to this work.
Contributor Information
Weifeng He, Email: whe761211@hotmail.com.
Yih-Cherng Liou, Email: dbslyc@nus.edu.sg.
References
- 1.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):801–10. 10.1001/jama.2016.0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer S, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet. 2020;395(10219):200–11. 10.1016/S0140-6736(19)32989-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Srzic I, Nesek Adam V, Tunjic Pejak D. Sepsis definition: what’s new in the treatment guidelines. Acta Clin Croat. 2022;61(Suppl 1):67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Misrani A, Tabassum S, Yang L. Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease. Front Aging Neurosci. 2021;13:617588. 10.3389/fnagi.2021.617588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schenkel LC, Bakovic M. Formation and regulation of mitochondrial membranes. Int J Cell Biol. 2014;2014:709828. 10.1155/2014/709828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vercellino I, Sazanov LA. The assembly, regulation and function of the mitochondrial respiratory chain. Nat Rev Mol Cell Biol. 2022;23(2):141–61. 10.1038/s41580-021-00415-0 [DOI] [PubMed] [Google Scholar]
- 7.Kong C, Song W, Fu T. Systemic inflammatory response syndrome is triggered by mitochondrial damage (review). Mol Med Rep. 2022;25(4):1–8. 10.3892/mmr.2022.12663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lira Chavez FM, Gartzke LP, van Beuningen FE, Wink SE, Henning RH, Krenning G, Bouma HR. Restoring the infected powerhouse: Mitochondrial quality control in sepsis. Redox Biol. 2023;68:102968. 10.1016/j.redox.2023.102968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singer M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence. 2014;5(1):66–72. 10.4161/viru.26907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang W, Jiang H, Wu G, Huang P, Wang H, An H, Liu S, Zhang W. The pathogenesis and potential therapeutic targets in sepsis. MedComm (2020). 2023;4(6):e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marchi S, Guilbaud E, Tait SWG, Yamazaki T, Galluzzi L. Mitochondrial control of inflammation. Nat Rev Immunol. 2023;23(3):159–73. 10.1038/s41577-022-00760-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li R, Ren T, Zeng J. Mitochondrial coenzyme Q protects sepsis-induced acute lung injury by activating PI3K/Akt/GSK-3beta/mTOR pathway in rats. Biomed Res Int. 2019;2019:5240898. 10.1155/2019/5240898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu Y, Kuang L, Wu Y, Deng H, She H, Zhou Y, Zhang J, Liu L, Li T. Protective effects of inhibition of mitochondrial fission on organ function after sepsis. Front Pharmacol. 2021;12:712489. 10.3389/fphar.2021.712489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tzanavari T, Varela A, Theocharis S, Ninou E, Kapelouzou A, Cokkinos DV, Kontaridis MI, Karalis KP. Metformin protects against infection-induced myocardial dysfunction. Metabolism. 2016;65(10):1447–58. 10.1016/j.metabol.2016.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Japiassu AM, Santiago AP, d’Avila JC, Garcia-Souza LF, Galina A, Castro Faria-Neto HC, Bozza FA, Oliveira MF. Bioenergetic failure of human peripheral blood monocytes in patients with septic shock is mediated by reduced F1Fo adenosine-5’-triphosphate synthase activity. Crit Care Med. 2011;39(5):1056–63. 10.1097/CCM.0b013e31820eda5c [DOI] [PubMed] [Google Scholar]
- 16.Garrabou G, Moren C, Lopez S, Tobias E, Cardellach F, Miro O, Casademont J. The effects of sepsis on mitochondria. J Infect Dis. 2012;205(3):392–400. 10.1093/infdis/jir764 [DOI] [PubMed] [Google Scholar]
- 17.Protti A, Fortunato F, Artoni A, Lecchi A, Motta G, Mistraletti G, Novembrino C, Comi GP, Gattinoni L. Platelet mitochondrial dysfunction in critically ill patients: comparison between sepsis and cardiogenic shock. Crit Care. 2015;19(1):39. 10.1186/s13054-015-0762-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fredriksson K, Rooyackers O. Mitochondrial function in sepsis: respiratory versus leg muscle. Crit Care Med. 2007;35(9 Suppl):S449-453. 10.1097/01.CCM.0000278048.00896.4B [DOI] [PubMed] [Google Scholar]
- 19.Carre JE, Orban JC, Re L, Felsmann K, Iffert W, Bauer M, Suliman HB, Piantadosi CA, Mayhew TM, Breen P, et al. Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am J Respir Crit Care Med. 2010;182(6):745–51. 10.1164/rccm.201003-0326OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soriano FG, Nogueira AC, Caldini EG, Lins MH, Teixeira AC, Cappi SB, Lotufo PA, Bernik MM, Zsengeller Z, Chen M, et al. Potential role of poly(adenosine 5’-diphosphate-ribose) polymerase activation in the pathogenesis of myocardial contractile dysfunction associated with human septic shock. Crit Care Med. 2006;34(4):1073–9. 10.1097/01.CCM.0000206470.47721.8D [DOI] [PubMed] [Google Scholar]
- 21.Kingsley SM, Bhat BV. Differential paradigms in animal models of sepsis. Curr Infect Dis Rep. 2016;18(9):26. 10.1007/s11908-016-0535-8 [DOI] [PubMed] [Google Scholar]
- 22.Singer M, Brealey D. Mitochondrial dysfunction in sepsis. Biochem Soc Symp. 1999;66:149–66. 10.1042/bss0660149 [DOI] [PubMed] [Google Scholar]
- 23.Corrêa TD, Jakob SM, Takala J. Mitochondrial function in sepsis. Crit Care Horizons. 2015;1:31–41. [Google Scholar]
- 24.Brealey D, Karyampudi S, Jacques TS, Novelli M, Stidwill R, Taylor V, Smolenski RT, Singer M. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am J Physiol Regul Integr Comp Physiol. 2004;286(3):R491-497. 10.1152/ajpregu.00432.2003 [DOI] [PubMed] [Google Scholar]
- 25.Gellerich FN, Trumbeckaite S, Hertel K, Zierz S, Müller-Werdan U, Werdan K, Redl H, Schlag G. Impaired energy metabolism in hearts of septic baboons: diminished activities of Complex I and Complex II of the mitochondrial respiratory chain. Shock (Augusta, GA). 1999;11(5):336–41. 10.1097/00024382-199905000-00006 [DOI] [PubMed] [Google Scholar]
- 26.Brealey D, Singer M. Mitochondrial dysfunction in sepsis. Curr Infect Dis Rep. 2003;5(5):365–71. 10.1007/s11908-003-0015-9 [DOI] [PubMed] [Google Scholar]
- 27.Boulos M, Astiz ME, Barua RS, Osman M. Impaired mitochondrial function induced by serum from septic shock patients is attenuated by inhibition of nitric oxide synthase and poly(ADP-ribose) synthase. Crit Care Med. 2003;31(2):353–8. 10.1097/01.CCM.0000050074.82486.B2 [DOI] [PubMed] [Google Scholar]
- 28.Fredriksson K, Hammarqvist F, Strigard K, Hultenby K, Ljungqvist O, Wernerman J, Rooyackers O. Derangements in mitochondrial metabolism in intercostal and leg muscle of critically ill patients with sepsis-induced multiple organ failure. Am J Physiol Endocrinol Metab. 2006;291(5):E1044-1050. 10.1152/ajpendo.00218.2006 [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Feng YF, Liu XT, Li YC, Zhu HM, Sun MR, Li P, Liu B, Yang H. Songorine promotes cardiac mitochondrial biogenesis via Nrf2 induction during sepsis. Redox Biol. 2021;38:101771. 10.1016/j.redox.2020.101771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A, Zsengeller ZK, Akhavan-Sharif MR, Khankin EV, Saintgeniez M, et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest. 2011;121(10):4003–14. 10.1172/JCI58662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choumar A, Tarhuni A, Letteron P, Reyl-Desmars F, Dauhoo N, Damasse J, Vadrot N, Nahon P, Moreau R, Pessayre D, et al. Lipopolysaccharide-induced mitochondrial DNA depletion. Antioxid Redox Signal. 2011;15(11):2837–54. 10.1089/ars.2010.3713 [DOI] [PubMed] [Google Scholar]
- 32.Hansen ME, Simmons KJ, Tippetts TS, Thatcher MO, Saito RR, Hubbard ST, Trumbull AM, Parker BA, Taylor OJ, Bikman BT. Lipopolysaccharide disrupts mitochondrial physiology in skeletal muscle via disparate effects on sphingolipid metabolism. Shock. 2015;44(6):585–92. 10.1097/SHK.0000000000000468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.d’Avila JC, Santiago AP, Amancio RT, Galina A, Oliveira MF, Bozza FA. Sepsis induces brain mitochondrial dysfunction. Crit Care Med. 2008;36(6):1925–32. 10.1097/CCM.0b013e3181760c4b [DOI] [PubMed] [Google Scholar]
- 34.Kokkinaki D, Hoffman M, Kalliora C, Kyriazis ID, Maning J, Lucchese AM, Shanmughapriya S, Tomar D, Park JY, Wang H, et al. Chemically synthesized Secoisolariciresinol diglucoside (LGM2605) improves mitochondrial function in cardiac myocytes and alleviates septic cardiomyopathy. J Mol Cell Cardiol. 2019;127:232–45. 10.1016/j.yjmcc.2018.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patil NK, Parajuli N, MacMillan-Crow LA, Mayeux PR. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: mitochondria-targeted antioxidant mitigates injury. Am J Physiol Renal Physiol. 2014;306(7):F734-743. 10.1152/ajprenal.00643.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vanasco V, Saez T, Magnani ND, Pereyra L, Marchini T, Corach A, Vaccaro MI, Corach D, Evelson P, Alvarez S. Cardiac mitochondrial biogenesis in endotoxemia is not accompanied by mitochondrial function recovery. Free Radic Biol Med. 2014;77:1–9. 10.1016/j.freeradbiomed.2014.08.009 [DOI] [PubMed] [Google Scholar]
- 37.Markley MA, Pierro A, Eaton S. Hepatocyte mitochondrial metabolism is inhibited in neonatal rat endotoxaemia: effects of glutamine. Clin Sci. 2002;102(3):337–44. 10.1042/cs1020337 [DOI] [PubMed] [Google Scholar]
- 38.Yao X, Carlson D, Sun Y, Ma L, Wolf SE, Minei JP, Zang QS. Mitochondrial ROS Induces Cardiac Inflammation via a Pathway through mtDNA Damage in a Pneumonia-Related Sepsis Model. PLoS ONE. 2015;10(10):e0139416. 10.1371/journal.pone.0139416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crouser ED, Julian MW, Blaho DV, Pfeiffer DR. Endotoxin-induced mitochondrial damage correlates with impaired respiratory activity. Crit Care Med. 2002;30(2):276–84. 10.1097/00003246-200202000-00002 [DOI] [PubMed] [Google Scholar]
- 40.Escames G, Lopez LC, Tapias V, Utrilla P, Reiter RJ, Hitos AB, Leon J, Rodriguez MI, Acuna-Castroviejo D. Melatonin counteracts inducible mitochondrial nitric oxide synthase-dependent mitochondrial dysfunction in skeletal muscle of septic mice. J Pineal Res. 2006;40(1):71–8. 10.1111/j.1600-079X.2005.00281.x [DOI] [PubMed] [Google Scholar]
- 41.Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci USA. 2000;97(16):8841–8. 10.1073/pnas.97.16.8841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garcia JA, Ortiz F, Miana J, Doerrier C, Fernandez-Ortiz M, Rusanova I, Escames G, Garcia JJ, Acuna-Castroviejo D. Contribution of inducible and neuronal nitric oxide synthases to mitochondrial damage and melatonin rescue in LPS-treated mice. J Physiol Biochem. 2017;73(2):235–44. 10.1007/s13105-017-0548-2 [DOI] [PubMed] [Google Scholar]
- 43.Lopes-Pires ME, Frade-Guanaes JO, Quinlan GJ. Clotting dysfunction in sepsis: a role for ROS and potential for therapeutic intervention. Antioxidants (Basel). 2021;11(1):88. 10.3390/antiox11010088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee YM, He W, Liou YC. The redox language in neurodegenerative diseases: oxidative post-translational modifications by hydrogen peroxide. Cell Death Dis. 2021;12(1):58. 10.1038/s41419-020-03355-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao WY, Zhang L, Sui MX, Zhu YH, Zeng L. Protective effects of sirtuin 3 in a murine model of sepsis-induced acute kidney injury. Sci Rep. 2016;6:33201. 10.1038/srep33201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Labiner HE, Sas KM, Baur JA, Sims CA. Sirt3 deletion increases inflammation and mortality in polymicrobial sepsis. Surg Infect (Larchmt). 2023;24(9):788–96. 10.1089/sur.2023.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharma P, Jha AB, Dubey RS, Pessarakli M. Reactive oxygen species, oxidative damage, and antioxidative defense mechanism in plants under stressful conditions. J Bot. 2012;2012:217037. [Google Scholar]
- 48.Shiva S, Brookes PS, Patel RP, Anderson PG, Darley-Usmar VM. Nitric oxide partitioning into mitochondrial membranes and the control of respiration at cytochrome c oxidase. Proc Natl Acad Sci USA. 2001;98(13):7212–7. 10.1073/pnas.131128898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown GC, Borutaite V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim Biophys Acta. 2004;1658(1–2):44–9. 10.1016/j.bbabio.2004.03.016 [DOI] [PubMed] [Google Scholar]
- 50.Brown GC. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim Biophys Acta. 2001;1504(1):46–57. 10.1016/S0005-2728(00)00238-3 [DOI] [PubMed] [Google Scholar]
- 51.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14. 10.1038/nrm3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li A, Gao M, Liu B, Qin Y, Chen L, Liu H, Wu H, Gong G. Mitochondrial autophagy: molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 2022;13(5):444. 10.1038/s41419-022-04906-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, Horng T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci USA. 2014;111(43):15514–9. 10.1073/pnas.1414859111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patoli D, Mignotte F, Deckert V, Dusuel A, Dumont A, Rieu A, Jalil A, Van Dongen K, Bourgeois T, Gautier T, et al. Inhibition of mitophagy drives macrophage activation and antibacterial defense during sepsis. J Clin Invest. 2020;130(11):5858–74. 10.1172/JCI130996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dagvadorj J, Mikulska-Ruminska K, Tumurkhuu G, Ratsimandresy RA, Carriere J, Andres AM, Marek-Iannucci S, Song Y, Chen S, Lane M, et al. Recruitment of pro-IL-1alpha to mitochondrial cardiolipin, via shared LC3 binding domain, inhibits mitophagy and drives maximal NLRP3 activation. Proc Natl Acad Sci USA. 2021;118(1):15. 10.1073/pnas.2015632118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19(2):121–35. 10.1038/nrm.2017.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tadie JM, Bae HB, Deshane JS, Bell CP, Lazarowski ER, Chaplin DD, Thannickal VJ, Abraham E, Zmijewski JW. Toll-like receptor 4 engagement inhibits adenosine 5’-monophosphate-activated protein kinase activation through a high mobility group box 1 protein-dependent mechanism. Mol Med. 2012;18(1):659–68. 10.2119/molmed.2011.00401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356(6337):513–9. 10.1126/science.aal3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164(5):896–910. 10.1016/j.cell.2015.12.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim MJ, Bae SH, Ryu JC, Kwon Y, Oh JH, Kwon J, Moon JS, Kim K, Miyawaki A, Lee MG, et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy. 2016;12(8):1272–91. 10.1080/15548627.2016.1183081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun Y, Yao X, Zhang QJ, Zhu M, Liu ZP, Ci B, Xie Y, Carlson D, Rothermel BA, Sun Y, et al. Beclin-1-dependent autophagy protects the heart during sepsis. Circulation. 2018;138(20):2247–62. 10.1161/CIRCULATIONAHA.117.032821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y, Zhu J, Liu Z, Shu S, Fu Y, Liu Y, Cai J, Tang C, Liu Y, Yin X, et al. The PINK1/PARK2/optineurin pathway of mitophagy is activated for protection in septic acute kidney injury. Redox Biol. 2021;38:101767. 10.1016/j.redox.2020.101767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crouser ED, Julian MW, Huff JE, Struck J, Cook CH. Carbamoyl phosphate synthase-1: a marker of mitochondrial damage and depletion in the liver during sepsis. Crit Care Med. 2006;34(9):2439–46. 10.1097/01.CCM.0000230240.02216.21 [DOI] [PubMed] [Google Scholar]
- 64.Sabouny R, Shutt TE. Reciprocal regulation of mitochondrial fission and fusion. Trends Biochem Sci. 2020;45(7):564–77. 10.1016/j.tibs.2020.03.009 [DOI] [PubMed] [Google Scholar]
- 65.Held NM, Houtkooper RH. Mitochondrial quality control pathways as determinants of metabolic health. BioEssays. 2015;37(8):867–76. 10.1002/bies.201500013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tilokani L, Nagashima S, Paupe V, Prudent J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. 2018;62(3):341–60. 10.1042/EBC20170104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang S, Xu Y, Zhu J, Ma J, Niu Q, Wang X. Carbon monoxide attenuates LPS-induced myocardial dysfunction in rats by regulating the mitochondrial dynamic equilibrium. Eur J Pharmacol. 2020;889:173726. 10.1016/j.ejphar.2020.173726 [DOI] [PubMed] [Google Scholar]
- 68.Gonzalez AS, Elguero ME, Finocchietto P, Holod S, Romorini L, Miriuka SG, Peralta JG, Poderoso JJ, Carreras MC. Abnormal mitochondrial fusion-fission balance contributes to the progression of experimental sepsis. Free Radic Res. 2014;48(7):769–83. 10.3109/10715762.2014.906592 [DOI] [PubMed] [Google Scholar]
- 69.Liu R, Wang SC, Li M, Ma XH, Jia XN, Bu Y, Sun L, Yu KJ. An inhibitor of DRP1 (Mdivi-1) alleviates LPS-induced septic AKI by inhibiting NLRP3 inflammasome activation. Biomed Res Int. 2020;2020:2398420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deng S, Zhang L, Mo Y, Huang Y, Li W, Peng Q, Huang L, Ai Y. Mdivi-1 attenuates lipopolysaccharide-induced acute lung injury by inhibiting MAPKs, oxidative stress and apoptosis. Pulm Pharmacol Ther. 2020;62:101918. 10.1016/j.pupt.2020.101918 [DOI] [PubMed] [Google Scholar]
- 71.Haileselassie B, Mukherjee R, Joshi AU, Napier BA, Massis LM, Ostberg NP, Queliconi BB, Monack D, Bernstein D, Mochly-Rosen D. Drp1/Fis1 interaction mediates mitochondrial dysfunction in septic cardiomyopathy. J Mol Cell Cardiol. 2019;130:160–9. 10.1016/j.yjmcc.2019.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tan Y, Ouyang H, Xiao X, Zhong J, Dong M. Irisin ameliorates septic cardiomyopathy via inhibiting DRP1-related mitochondrial fission and normalizing the JNK-LATS2 signaling pathway. Cell Stress Chaperones. 2019;24(3):595–608. 10.1007/s12192-019-00992-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Norenberg MD, Rao KV. The mitochondrial permeability transition in neurologic disease. Neurochem Int. 2007;50(7–8):983–97. 10.1016/j.neuint.2007.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Westphal D, Kluck RM, Dewson G. Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 2014;21(2):196–205. 10.1038/cdd.2013.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, Geoghegan ND, Chappaz S, Davidson S, San Chin H, et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. 2018;359(6378):eaao6047. 10.1126/science.aao6047 [DOI] [PubMed] [Google Scholar]
- 76.Riley JS, Quarato G, Cloix C, Lopez J, O’Prey J, Pearson M, Chapman J, Sesaki H, Carlin LM, Passos JF, et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018;37(17):e99238. 10.15252/embj.201899238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. 10.1080/01926230701320337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94(4):491–501. 10.1016/S0092-8674(00)81590-1 [DOI] [PubMed] [Google Scholar]
- 79.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111(3):331–42. 10.1016/S0092-8674(02)01036-X [DOI] [PubMed] [Google Scholar]
- 80.Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou JC. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144(5):891–901. 10.1083/jcb.144.5.891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chung CS, Venet F, Chen Y, Jones LN, Wilson DC, Ayala CA, Ayala A. Deficiency of Bid protein reduces sepsis-induced apoptosis and inflammation, while improving septic survival. Shock. 2010;34(2):150–61. 10.1097/SHK.0b013e3181cf70fb [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heilig R, Dilucca M, Boucher D, Chen KW, Hancz D, Demarco B, Shkarina K, Broz P. Caspase-1 cleaves Bid to release mitochondrial SMAC and drive secondary necrosis in the absence of GSDMD. Life Sci Alliance. 2020;3(6):e202000735. 10.26508/lsa.202000735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–30. 10.1038/ni.1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Larche J, Lancel S, Hassoun SM, Favory R, Decoster B, Marchetti P, Chopin C, Neviere R. Inhibition of mitochondrial permeability transition prevents sepsis-induced myocardial dysfunction and mortality. J Am Coll Cardiol. 2006;48(2):377–85. 10.1016/j.jacc.2006.02.069 [DOI] [PubMed] [Google Scholar]
- 85.Crouser ED, Julian MW, Joshi MS, Bauer JA, Wewers MD, Hart JM, Pfeiffer DR. Cyclosporin A ameliorates mitochondrial ultrastructural injury in the ileum during acute endotoxemia. Crit Care Med. 2002;30(12):2722–8. 10.1097/00003246-200212000-00017 [DOI] [PubMed] [Google Scholar]
- 86.Hu Y, Yan JB, Zheng MZ, Song XH, Wang LL, Shen YL, Chen YY. Mitochondrial aldehyde dehydrogenase activity protects against lipopolysaccharide-induced cardiac dysfunction in rats. Mol Med Rep. 2015;11(2):1509–15. 10.3892/mmr.2014.2803 [DOI] [PubMed] [Google Scholar]
- 87.Bernardi P, Gerle C, Halestrap AP, Jonas EA, Karch J, Mnatsakanyan N, Pavlov E, Sheu S-S, Soukas AA. Identity, structure, and function of the mitochondrial permeability transition pore: controversies, consensus, recent advances, and future directions. Cell Death Differ. 2023;30(8):1869–85. 10.1038/s41418-023-01187-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bonora M, Giorgi C, Pinton P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat Rev Mol Cell Biol. 2022;23(4):266–85. 10.1038/s41580-021-00433-y [DOI] [PubMed] [Google Scholar]
- 89.Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. 2007;47:143–83. 10.1146/annurev.pharmtox.47.120505.105122 [DOI] [PubMed] [Google Scholar]
- 90.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94(3):909–50. 10.1152/physrev.00026.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang S, Moreau F, Chadee K. Gasdermins in innate host defense against entamoeba histolytica and other protozoan parasites. Front Immunol. 2022;13:900553. 10.3389/fimmu.2022.900553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10(1):1689. 10.1038/s41467-019-09397-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, Parthoens E, Lamkanfi M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019;26(1):146–61. 10.1038/s41418-018-0106-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huang LS, Hong Z, Wu W, Xiong S, Zhong M, Gao X, Rehman J, Malik AB. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity. 2020;52(3):475-486 e475. 10.1016/j.immuni.2020.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Miao R, Jiang C, Chang WY, Zhang H, An J, Ho F, Chen P, Zhang H, Junqueira C, Amgalan D, et al. Gasdermin D permeabilization of mitochondrial inner and outer membranes accelerates and enhances pyroptosis. Immunity. 2023;56(11):2523-2541 e2528. 10.1016/j.immuni.2023.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lin PH, Lin HY, Kuo CC, Yang LT. N-terminal functional domain of Gasdermin A3 regulates mitochondrial homeostasis via mitochondrial targeting. J Biomed Sci. 2015;22(1):44. 10.1186/s12929-015-0152-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kondolf HC, D’Orlando DA, Dubyak GR, Abbott DW. Protein engineering reveals that gasdermin A preferentially targets mitochondrial membranes over the plasma membrane during pyroptosis. J Biol Chem. 2023;299(2):102908. 10.1016/j.jbc.2023.102908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–20. 10.1016/j.cell.2010.01.022 [DOI] [PubMed] [Google Scholar]
- 99.Nong Y, Wei X, Yu D. Inflammatory mechanisms and intervention strategies for sepsis-induced myocardial dysfunction. Immun Inflamm Dis. 2023;11(5):e860. 10.1002/iid3.860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Abais JM, Xia M, Zhang Y, Boini KM, Li PL. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal. 2015;22(13):1111–29. 10.1089/ars.2014.5994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zang QS, Sadek H, Maass DL, Martinez B, Ma L, Kilgore JA, Williams NS, Frantz DE, Wigginton JG, Nwariaku FE, et al. Specific inhibition of mitochondrial oxidative stress suppresses inflammation and improves cardiac function in a rat pneumonia-related sepsis model. Am J Physiol Heart Circ Physiol. 2012;302(9):H1847-1859. 10.1152/ajpheart.00203.2011 [DOI] [PubMed] [Google Scholar]
- 102.Lowes DA, Thottakam BM, Webster NR, Murphy MP, Galley HF. The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free Radic Biol Med. 2008;45(11):1559–65. 10.1016/j.freeradbiomed.2008.09.003 [DOI] [PubMed] [Google Scholar]
- 103.Xia Y, Cao Y, Sun Y, Hong X, Tang Y, Yu J, Hu H, Ma W, Qin K, Bao R. Calycosin alleviates sepsis-induced acute lung injury via the inhibition of mitochondrial ROS-mediated inflammasome activation. Front Pharmacol. 2021;12:690549. 10.3389/fphar.2021.690549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136–40. 10.1038/ni.1831 [DOI] [PubMed] [Google Scholar]
- 105.Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. 2012;287(43):36617–22. 10.1074/jbc.M112.407130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol. 2011;187(2):613–7. 10.4049/jimmunol.1100613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–14. 10.1016/j.immuni.2012.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, Wong J, Ding S, Seki E, Schnabl B, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560(7717):198–203. 10.1038/s41586-018-0372-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xian H, Watari K, Sanchez-Lopez E, Offenberger J, Onyuru J, Sampath H, Ying W, Hoffman HM, Shadel GS, Karin M. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity. 2022;55(8):1370-1385 e1378. 10.1016/j.immuni.2022.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nozaki K, Li L, Miao EA. Innate sensors trigger regulated cell death to combat intracellular infection. Annu Rev Immunol. 2022;40:469–98. 10.1146/annurev-immunol-101320-011235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li X, Thome S, Ma X, Amrute-Nayak M, Finigan A, Kitt L, Masters L, James JR, Shi Y, Meng G, et al. MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule-dependent mechanism. Nat Commun. 2017;8:15986. 10.1038/ncomms15986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–5. 10.1038/nature09663 [DOI] [PubMed] [Google Scholar]
- 113.Chen J, Chen ZJ. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature. 2018;564(7734):71–6. 10.1038/s41586-018-0761-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, Deng Q, Orning P, Alam HB, Maliga Z, et al. HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science. 2020;369(6510):eaas8995. 10.1126/science.aas8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pandey A, Shen C, Feng S, Man SM. Cell biology of inflammasome activation. Trends Cell Biol. 2021;31(11):924–39. 10.1016/j.tcb.2021.06.010 [DOI] [PubMed] [Google Scholar]
- 116.Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14(5):454–60. 10.1038/ni.2550 [DOI] [PubMed] [Google Scholar]
- 117.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153(2):348–61. 10.1016/j.cell.2013.02.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39(2):311–23. 10.1016/j.immuni.2013.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Elliott EI, Miller AN, Banoth B, Iyer SS, Stotland A, Weiss JP, Gottlieb RA, Sutterwala FS, Cassel SL. Cutting edge: mitochondrial assembly of the NLRP3 inflammasome complex is initiated at priming. J Immunol. 2018;200(9):3047–52. 10.4049/jimmunol.1701723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, Yu JW, Alnemri ES. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol. 2013;191(8):4358–66. 10.4049/jimmunol.1301170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Franchi L, Eigenbrod T, Munoz-Planillo R, Ozkurede U, Kim YG, Arindam C, Gale M Jr, Silverman RH, Colonna M, Akira S, et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J Immunol. 2014;193(8):4214–22. 10.4049/jimmunol.1400582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ren Z, Ding T, Zuo Z, Xu Z, Deng J, Wei Z. Regulation of MAVS expression and signaling function in the antiviral innate immune response. Front Immunol. 2020;11:1030. 10.3389/fimmu.2020.01030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Andreeva L, David L, Rawson S, Shen C, Pasricha T, Pelegrin P, Wu H. NLRP3 cages revealed by full-length mouse NLRP3 structure control pathway activation. Cell. 2021;184(26):6299-6312 e6222. 10.1016/j.cell.2021.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang Z, Meszaros G, He WT, Xu Y, de Fatima MH, Mailly L, Mihlan M, Liu Y, Puig Gamez M, Goginashvili A, et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J Exp Med. 2017;214(9):2671–93. 10.1084/jem.20162040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Xiao L, Magupalli VG, Wu H. Cryo-EM structures of the active NLRP3 inflammasome disc. Nature. 2023;613(7944):595–600. 10.1038/s41586-022-05570-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fu J, Wu H. Structural mechanisms of NLRP3 inflammasome assembly and activation. Annu Rev Immunol. 2023;41:301–16. 10.1146/annurev-immunol-081022-021207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ablasser A, Chen ZJ. cGAS in action: Expanding roles in immunity and inflammation. Science. 2019;363(6431):eaat8657. 10.1126/science.aat8657 [DOI] [PubMed] [Google Scholar]
- 128.Xian H, Karin M. Oxidized mitochondrial DNA: a protective signal gone awry. Trends Immunol. 2023;44(3):188–200. 10.1016/j.it.2023.01.006 [DOI] [PubMed] [Google Scholar]
- 129.Gehrke N, Mertens C, Zillinger T, Wenzel J, Bald T, Zahn S, Tuting T, Hartmann G, Barchet W. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity. 2013;39(3):482–95. 10.1016/j.immuni.2013.08.004 [DOI] [PubMed] [Google Scholar]
- 130.Nakahira K, Kyung SY, Rogers AJ, Gazourian L, Youn S, Massaro AF, Quintana C, Osorio JC, Wang Z, Zhao Y et al. Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med 2013;10(12):e1001577; discussion e1001577. [DOI] [PMC free article] [PubMed]
- 131.Wang L, Zhou W, Wang K, He S, Chen Y. Predictive value of circulating plasma mitochondrial DNA for Sepsis in the emergency department: observational study based on the Sepsis-3 definition. BMC Emerg Med. 2020;20(1):25. 10.1186/s12873-020-00320-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schneck E, Edinger F, Hecker M, Sommer N, Pak O, Weissmann N, Hecker A, Reichert M, Markmann M, Sander M, et al. Blood levels of free-circulating mitochondrial DNA in septic shock and postsurgical systemic inflammation and its influence on coagulation: a secondary analysis of a prospective observational study. J Clin Med. 2020;9(7):2056. 10.3390/jcm9072056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liu Q, Wu J, Zhang X, Li X, Wu X, Zhao Y, Ren J. Circulating mitochondrial DNA-triggered autophagy dysfunction via STING underlies sepsis-related acute lung injury. Cell Death Dis. 2021;12(7):673. 10.1038/s41419-021-03961-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hu Q, Ren H, Li G, Wang D, Zhou Q, Wu J, Zheng J, Huang J, Slade DA, Wu X, et al. STING-mediated intestinal barrier dysfunction contributes to lethal sepsis. EBioMedicine. 2019;41:497–508. 10.1016/j.ebiom.2019.02.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Xu L, Li M, Yang Y, Zhang C, Xie Z, Tang J, Shi Z, Chen S, Li G, Gu Y, et al. Salmonella induces the cGAS-STING-dependent type I interferon response in murine macrophages by triggering mtDNA release. MBio. 2022;13(3):e0363221. 10.1128/mbio.03632-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang RX, Kang R, Tang DL. STING1 in sepsis: mechanisms, functions, and implications. Chin J Traumatol. 2022;25(1):1–10. 10.1016/j.cjtee.2021.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408(6813):740–5. 10.1038/35047123 [DOI] [PubMed] [Google Scholar]
- 138.Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med. 2003;198(3):513–20. 10.1084/jem.20030162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–7. 10.1038/nature08780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tsuji N, Tsuji T, Ohashi N, Kato A, Fujigaki Y, Yasuda H. Role of Mitochondrial DNA in septic AKI via toll-like receptor 9. J Am Soc Nephrol. 2016;27(7):2009–20. 10.1681/ASN.2015040376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yasuda H, Leelahavanichkul A, Tsunoda S, Dear JW, Takahashi Y, Ito S, Hu X, Zhou H, Doi K, Childs R, et al. Chloroquine and inhibition of Toll-like receptor 9 protect from sepsis-induced acute kidney injury. Am J Physiol Renal Physiol. 2008;294(5):F1050–8. 10.1152/ajprenal.00461.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Plitas G, Burt BM, Nguyen HM, Bamboat ZM, DeMatteo RP. Toll-like receptor 9 inhibition reduces mortality in polymicrobial sepsis. J Exp Med. 2008;205(6):1277–83. 10.1084/jem.20080162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Liu J, Zhou G, Wang X, Liu D. Metabolic reprogramming consequences of sepsis: adaptations and contradictions. Cell Mol Life Sci. 2022;79(8):456. 10.1007/s00018-022-04490-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liu C, Wei W, Huang Y, Fu P, Zhang L, Zhao Y. Metabolic reprogramming in septic acute kidney injury: pathogenesis and therapeutic implications. Metabolism. 2024;158:155974. 10.1016/j.metabol.2024.155974 [DOI] [PubMed] [Google Scholar]
- 145.Van Wyngene L, Vandewalle J, Libert C. Reprogramming of basic metabolic pathways in microbial sepsis: therapeutic targets at last? EMBO Mol Med. 2018;10(8):e8712. 10.15252/emmm.201708712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pan T, Sun S, Chen Y, Tian R, Chen E, Tan R, Wang X, Liu Z, Liu J, Qu H. Immune effects of PI3K/Akt/HIF-1alpha-regulated glycolysis in polymorphonuclear neutrophils during sepsis. Crit Care. 2022;26(1):29. 10.1186/s13054-022-03893-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tan C, Gu J, Chen H, Li T, Deng H, Liu K, Liu M, Tan S, Xiao Z, Zhang H, et al. Inhibition of aerobic glycolysis promotes neutrophil to influx to the infectious site via CXCR2 in sepsis. Shock. 2020;53(1):114–23. 10.1097/SHK.0000000000001334 [DOI] [PubMed] [Google Scholar]
- 148.Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27(7):1230–51. 10.1097/00003246-199907000-00002 [DOI] [PubMed] [Google Scholar]
- 149.Singer M, De Santis V, Vitale D, Jeffcoate W. Multiorgan failure is an adaptive, endocrine-mediated, metabolic response to overwhelming systemic inflammation. Lancet. 2004;364(9433):545–8. 10.1016/S0140-6736(04)16815-3 [DOI] [PubMed] [Google Scholar]
- 150.Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies NA, Cooper CE, Singer M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360(9328):219–23. 10.1016/S0140-6736(02)09459-X [DOI] [PubMed] [Google Scholar]
- 151.Dhanasekaran A, Kotamraju S, Kalivendi SV, Matsunaga T, Shang T, Keszler A, Joseph J, Kalyanaraman B. Supplementation of endothelial cells with mitochondria-targeted antioxidants inhibit peroxide-induced mitochondrial iron uptake, oxidative damage, and apoptosis. J Biol Chem. 2004;279(36):37575–87. 10.1074/jbc.M404003200 [DOI] [PubMed] [Google Scholar]
- 152.McCormick B, Lowes DA, Colvin L, Torsney C, Galley HF. MitoVitE, a mitochondria-targeted antioxidant, limits paclitaxel-induced oxidative stress and mitochondrial damage in vitro, and paclitaxel-induced mechanical hypersensitivity in a rat pain model. Br J Anaesth. 2016;117(5):659–66. 10.1093/bja/aew309 [DOI] [PubMed] [Google Scholar]
- 153.Gioscia-Ryan RA, LaRocca TJ, Sindler AL, Zigler MC, Murphy MP, Seals DR. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J Physiol. 2014;592(12):2549–61. 10.1113/jphysiol.2013.268680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kalyanaraman B, Cheng G, Hardy M, You M. OXPHOS-targeting drugs in oncology: new perspectives. Expert Opin Ther Targets. 2023;27(10):939–52. 10.1080/14728222.2023.2261631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhang S, Zhou Q, Li Y, Zhang Y, Wu Y. MitoQ modulates lipopolysaccharide-induced intestinal barrier dysfunction via regulating Nrf2 signaling. Mediat Inflamm. 2020;2020:3276148. 10.1155/2020/3276148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Supinski GS, Murphy MP, Callahan LA. MitoQ administration prevents endotoxin-induced cardiac dysfunction. Am J Physiol Regul Integr Comp Physiol. 2009;297(4):R1095-1102. 10.1152/ajpregu.90902.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Supinski GS, Schroder EA, Wang L, Morris AJ, Callahan LAP. Mitoquinone mesylate (MitoQ) prevents sepsis-induced diaphragm dysfunction. J Appl Physiol (1985). 2021;131(2):778–87. 10.1152/japplphysiol.01053.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Smith RA, Porteous CM, Coulter CV, Murphy MP. Selective targeting of an antioxidant to mitochondria. Eur J Biochem. 1999;263(3):709–16. 10.1046/j.1432-1327.1999.00543.x [DOI] [PubMed] [Google Scholar]
- 159.Lowes DA, Webster NR, Murphy MP, Galley HF. Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Br J Anaesth. 2013;110(3):472–80. 10.1093/bja/aes577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Trnka J, Blaikie FH, Smith RA, Murphy MP. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radic Biol Med. 2008;44(7):1406–19. 10.1016/j.freeradbiomed.2007.12.036 [DOI] [PubMed] [Google Scholar]
- 161.Arulkumaran N, Pollen SJ, Tidswell R, Gaupp C, Peters VBM, Stanzani G, Snow TAC, Duchen MR, Singer M. Selective mitochondrial antioxidant MitoTEMPO reduces renal dysfunction and systemic inflammation in experimental sepsis in rats. Br J Anaesth. 2021;127(4):577–86. 10.1016/j.bja.2021.05.036 [DOI] [PubMed] [Google Scholar]
- 162.Navas LE, Carnero A. NAD(+) metabolism, stemness, the immune response, and cancer. Signal Transduct Target Ther. 2021;6(1):2. 10.1038/s41392-020-00354-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Liaudet L, Mabley JG, Soriano FG, Pacher P, Marton A, Hasko G, Szabo C. Inosine reduces systemic inflammation and improves survival in septic shock induced by cecal ligation and puncture. Am J Respir Crit Care Med. 2001;164(7):1213–20. 10.1164/ajrccm.164.7.2101013 [DOI] [PubMed] [Google Scholar]
- 164.Cao T, Ni R, Ding W, Ji X, Fan GC, Zhang Z, Peng T. Nicotinamide mononucleotide as a therapeutic agent to alleviate multi-organ failure in sepsis. J Transl Med. 2023;21(1):883. 10.1186/s12967-023-04767-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hong G, Zheng D, Zhang L, Ni R, Wang G, Fan GC, Lu Z, Peng T. Administration of nicotinamide riboside prevents oxidative stress and organ injury in sepsis. Free Radic Biol Med. 2018;123:125–37. 10.1016/j.freeradbiomed.2018.05.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ye M, Zhao Y, Wang Y, Xie R, Tong Y, Sauer JD, Gong S. NAD(H)-loaded nanoparticles for efficient sepsis therapy via modulating immune and vascular homeostasis. Nat Nanotechnol. 2022;17(8):880–90. 10.1038/s41565-022-01137-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Felix AA, Williams EG, Jha P, Lo Sasso G, Huzard D, et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med. 2016;22(8):879–88. 10.1038/nm.4132 [DOI] [PubMed] [Google Scholar]
- 168.Yang Y, Hu Q, Kang H, Li J, Zhao X, Zhu L, Tang W, Wan M. Urolithin A protects severe acute pancreatitis-associated acute cardiac injury by regulating mitochondrial fatty acid oxidative metabolism in cardiomyocytes. MedComm (2020). 2023;4(6):e459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Abdelazeem KNM, Kalo MZ, Beer-Hammer S, Lang F. The gut microbiota metabolite urolithin A inhibits NF-kappaB activation in LPS stimulated BMDMs. Sci Rep. 2021;11(1):7117. 10.1038/s41598-021-86514-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Liu S, D’Amico D, Shankland E, Bhayana S, Garcia JM, Aebischer P, Rinsch C, Singh A, Marcinek DJ. Effect of urolithin a supplementation on muscle endurance and mitochondrial health in older adults: a randomized clinical trial. JAMA Netw Open. 2022;5(1):e2144279. 10.1001/jamanetworkopen.2021.44279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Andreux PA, Blanco-Bose W, Ryu D, Burdet F, Ibberson M, Aebischer P, Auwerx J, Singh A, Rinsch C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat Metab. 2019;1(6):595–603. 10.1038/s42255-019-0073-4 [DOI] [PubMed] [Google Scholar]
- 172.Zhu P, Chen Y, Wang J, Lin G, Wang R, Que Y, Zhou J, Xu G, Luo J, Du Y. Receptor-interacting protein kinase 3 suppresses mitophagy activation via the yes-associated protein/transcription factor EB pathways in septic cardiomyopathy. Front Cardiovasc Med. 2022;9:856041. 10.3389/fcvm.2022.856041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wang Y, Jasper H, Toan S, Muid D, Chang X, Zhou H. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biol. 2021;45:102049. 10.1016/j.redox.2021.102049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jiao P, Wang Y, Ren G, Chu D, Li Y, Yang Y, Sang T. Urolithin A exerts a protective effect on lipopolysaccharide-induced acute lung injury by regulating HMGB1-mediated MAPK and NF-kappaB signaling pathways. Naunyn Schmiedebergs Arch Pharmacol. 2024;397(8):5765–77. 10.1007/s00210-024-02977-0 [DOI] [PubMed] [Google Scholar]
- 175.Libert C, Ayala A, Bauer M, Cavaillon JM, Deutschman C, Frostell C, Knapp S, Kozlov AV, Wang P, Osuchowski MF, et al. Part II: minimum quality threshold in preclinical sepsis studies (MQTiPSS) for types of infections and organ dysfunction endpoints. Shock. 2019;51(1):23–32. 10.1097/SHK.0000000000001242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Joffre J, Hellman J, Ince C, Ait-Oufella H. Endothelial responses in sepsis. Am J Respir Crit Care Med. 2020;202(3):361–70. 10.1164/rccm.201910-1911TR [DOI] [PubMed] [Google Scholar]
- 177.Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14(2):193–204. 10.1016/j.devcel.2007.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Manczak M, Kandimalla R, Yin X, Reddy PH. Mitochondrial division inhibitor 1 reduces dynamin-related protein 1 and mitochondrial fission activity. Hum Mol Genet. 2019;28(2):177–99. 10.1093/hmg/ddy335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zhang Q, Liu Z, Huang X, Heng X, Wu J, Chen Z, Guo X, Fan J, Huang Q. Mdivi-1 alleviates sepsis-induced liver injury by inhibiting sting signaling activation. Shock. 2024;62(1):95–102. 10.1097/SHK.0000000000002349 [DOI] [PubMed] [Google Scholar]
- 180.Fu Q, Zhang YB, Shi CX, Jiang M, Lu K, Fu ZH, Ruan JP, Wu J, Gu XP. GSDMD/Drp1 signaling pathway mediates hippocampal synaptic damage and neural oscillation abnormalities in a mouse model of sepsis-associated encephalopathy. J Neuroinflamm. 2024;21(1):96. 10.1186/s12974-024-03084-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Haileselassie B, Joshi AU, Minhas PS, Mukherjee R, Andreasson KI, Mochly-Rosen D. Mitochondrial dysfunction mediated through dynamin-related protein 1 (Drp1) propagates impairment in blood brain barrier in septic encephalopathy. J Neuroinflammation. 2020;17(1):36. 10.1186/s12974-019-1689-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.de Paiva CS, Pflugfelder SC, Ng SM, Akpek EK. Topical cyclosporine A therapy for dry eye syndrome. Cochrane Database Syst Rev. 2019;9(9):CD010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Molnar AO, Fergusson D, Tsampalieros AK, Bennett A, Fergusson N, Ramsay T, Knoll GA. Generic immunosuppression in solid organ transplantation: systematic review and meta-analysis. BMJ. 2015;350:h3163. 10.1136/bmj.h3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Luna-Sanchez M, Bianchi P, Quintana A. Mitochondria-induced immune response as a trigger for neurodegeneration: a pathogen from within. Int J Mol Sci. 2021;22(16):8523. 10.3390/ijms22168523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Liddicoat AM, Lavelle EC. Modulation of innate immunity by cyclosporine A. Biochem Pharmacol. 2019;163:472–80. 10.1016/j.bcp.2019.03.022 [DOI] [PubMed] [Google Scholar]
- 186.Xiao Z, Jia B, Zhao X, Bi S, Meng W. Attenuation of lipopolysaccharide-induced acute lung injury by cyclosporine-A via suppression of mitochondrial DNA. Med Sci Monit. 2018;24:7682–8. 10.12659/MSM.909909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fauvel H, Marchetti P, Obert G, Joulain O, Chopin C, Formstecher P, Neviere R. Protective effects of cyclosporin A from endotoxin-induced myocardial dysfunction and apoptosis in rats. Am J Respir Crit Care Med. 2002;165(4):449–55. 10.1164/ajrccm.165.4.2105084 [DOI] [PubMed] [Google Scholar]
- 188.Joshi MS, Julian MW, Huff JE, Bauer JA, Xia Y, Crouser ED. Calcineurin regulates myocardial function during acute endotoxemia. Am J Respir Crit Care Med. 2006;173(9):999–1007. 10.1164/rccm.200411-1507OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Flory J, Lipska K. Metformin in 2019. JAMA. 2019;321(19):1926–7. 10.1001/jama.2019.3805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lv Z, Guo Y. Metformin and its benefits for various diseases. Front Endocrinol (Lausanne). 2020;11:191. 10.3389/fendo.2020.00191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Liu Z, Bone N, Jiang S, Park DW, Tadie JM, Deshane J, Rodriguez CA, Pittet JF, Abraham E, Zmijewski JW. AMP-activated protein kinase and glycogen synthase kinase 3beta modulate the severity of sepsis-induced lung injury. Mol Med. 2016;21(1):937–50. 10.2119/molmed.2015.00198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Fan SY, Zhao ZC, Liu XL, Peng YG, Zhu HM, Yan SF, Liu YJ, Xie Q, Jiang Y, Zeng SZ. Metformin mitigates sepsis-induced acute lung injury and inflammation in young mice by suppressing the S100A8/A9-NLRP3-IL-1beta signaling pathway. J Inflamm Res. 2024;17:3785–99. 10.2147/JIR.S460413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Tang G, Yang H, Chen J, Shi M, Ge L, Ge X, Zhu G. Metformin ameliorates sepsis-induced brain injury by inhibiting apoptosis, oxidative stress and neuroinflammation via the PI3K/Akt signaling pathway. Oncotarget. 2017;8(58):97977–89. 10.18632/oncotarget.20105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Liang H, Song H, Zhang X, Song G, Wang Y, Ding X, Duan X, Li L, Sun T, Kan Q. Metformin attenuated sepsis-related liver injury by modulating gut microbiota. Emerg Microbes Infect. 2022;11(1):815–28. 10.1080/22221751.2022.2045876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Doenyas-Barak K, Beberashvili I, Marcus R, Efrati S. Lactic acidosis and severe septic shock in metformin users: a cohort study. Crit Care. 2016;20:10. 10.1186/s13054-015-1180-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Green JP, Berger T, Garg N, Suarez A, Hagar Y, Radeos MS, Panacek EA. Impact of metformin use on the prognostic value of lactate in sepsis. Am J Emerg Med. 2012;30(9):1667–73. 10.1016/j.ajem.2012.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Jochmans S, Alphonsine JE, Chelly J, Vong LVP, Sy O, Rolin N, Ellrodt O, Monchi M, Vinsonneau C. Does metformin exposure before ICU stay have any impact on patients’ outcome? A retrospective cohort study of diabetic patients. Ann Intensive Care. 2017;7(1):116. 10.1186/s13613-017-0336-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.van Vught LA, Scicluna BP, Hoogendijk AJ, Wiewel MA, Klein Klouwenberg PM, Cremer OL, Horn J, Nurnberg P, Bonten MM, Schultz MJ, et al. Association of diabetes and diabetes treatment with the host response in critically ill sepsis patients. Crit Care. 2016;20(1):252. 10.1186/s13054-016-1429-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Park J, Hwang SY, Jo IJ, Jeon K, Suh GY, Lee TR, Yoon H, Cha WC, Sim MS, Carriere KC, et al. Impact of metformin use on lactate kinetics in patients with severe sepsis and septic shock. Shock. 2017;47(5):582–7. 10.1097/SHK.0000000000000782 [DOI] [PubMed] [Google Scholar]
- 200.Liang H, Ding X, Li L, Wang T, Kan Q, Wang L, Sun T. Association of preadmission metformin use and mortality in patients with sepsis and diabetes mellitus: a systematic review and meta-analysis of cohort studies. Crit Care. 2019;23(1):50. 10.1186/s13054-019-2346-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Yang Q, Zheng J, Chen W, Chen X, Wen D, Chen W, Xiong X, Zhang Z. Association between preadmission metformin use and outcomes in intensive care unit patients with sepsis and type 2 diabetes: a cohort study. Front Med (Lausanne). 2021;8:640785. 10.3389/fmed.2021.640785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Foretz M, Guigas B, Viollet B. Metformin: update on mechanisms of action and repurposing potential. Nat Rev Endocrinol. 2023;19(8):460–76. 10.1038/s41574-023-00833-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Howell JJ, Hellberg K, Turner M, Talbott G, Kolar MJ, Ross DS, Hoxhaj G, Saghatelian A, Shaw RJ, Manning BD. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 2017;25(2):463–71. 10.1016/j.cmet.2016.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Xian H, Liu Y, Rundberg Nilsson A, Gatchalian R, Crother TR, Tourtellotte WG, Zhang Y, Aleman-Muench GR, Lewis G, Chen W, et al. Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity. 2021;54(7):1463-1477 e1411. 10.1016/j.immuni.2021.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA. 2007;104(29):12017–22. 10.1073/pnas.0705070104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kim J, Kwak HJ, Cha JY, Jeong YS, Rhee SD, Kim KR, Cheon HG. Metformin suppresses lipopolysaccharide (LPS)-induced inflammatory response in murine macrophages via activating transcription factor-3 (ATF-3) induction. J Biol Chem. 2014;289(33):23246–55. 10.1074/jbc.M114.577908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wu W, Wang S, Liu Q, Shan T, Wang Y. Metformin protects against lps-induced intestinal barrier dysfunction by activating AMPK pathway. Mol Pharm. 2018;15(8):3272–84. 10.1021/acs.molpharmaceut.8b00332 [DOI] [PubMed] [Google Scholar]
- 208.Ma T, Tian X, Zhang B, Li M, Wang Y, Yang C, Wu J, Wei X, Qu Q, Yu Y, et al. Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature. 2022;603(7899):159–65. 10.1038/s41586-022-04431-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Cortes M, Brischetto A, Martinez-Campanario MC, Ninfali C, Dominguez V, Fernandez S, Celis R, Esteve-Codina A, Lozano JJ, Sidorova J, et al. Inflammatory macrophages reprogram to immunosuppression by reducing mitochondrial translation. Nat Commun. 2023;14(1):7471. 10.1038/s41467-023-42277-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No datasets were generated or analysed during the current study.