

Abstract
Introduction:
Anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA) is a rare congenital anomaly. While only a few of those born with this anomaly survive into adulthood, it becomes an extremely rare diagnosis in adults.
Case presentation:
Here, the authors present a case of ALCAPA in a symptomatic adult female with angina and palpitations that was repeatedly missed on transthoracic echocardiogram.
Clinical discussion:
The adult type of ALCAPA is often missed due to non-specific changes in electrocardiogram, leading to reluctance for angiography. Therefore, identifying abnormal coronaries in echocardiogram is crucial.
Conclusion:
It is essential to consider anomalous coronary arteries as a differential diagnosis in patients with chest pain, despite their rarity. This case report highlights the role of various cardiac imaging modalities in improving the diagnostic yield of ALCAPA.
Keywords: ALCAPA, case report, coronary angiography, coronary anomalies, echocardiography
Introduction
Highlights
A 38-year-old woman presented with angina and palpitations and was eventually diagnosed with ALCAPA (anomalous origin of the left coronary artery from the pulmonary artery), a rare condition.
The diagnosis was repeatedly missed on previous transthoracic echocardiograms.
Transthoracic echocardiography, coronary angiography, and computed tomography coronary angiography findings were consistent with ALCAPA.
This case highlights the importance of considering anomalous coronaries as differential diagnoses in patients presenting with chest pain, even though it is rare.
Emphasizes the critical role of transthoracic echocardiography and other imaging modalities in diagnosing ALCAPA.
Anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA), also known as Bland-White-Garland syndrome, is a rare congenital anomaly accounting for 0.25–0.5% of all congenital cardiac anomalies1. Most cases present within the first year of life, and if untreated, only 10% of these patients survive beyond infancy2. For those who do survive, the average lifespan is limited to 35 years3.
Common presenting symptoms include angina, dyspnea, palpitations, and fatigue in 66% of cases, while ventricular arrhythmia, syncope, or sudden death occur in 17%. Fourteen percent of patients remain asymptomatic, and the remaining 12% are diagnosed only at autopsy4. On examination, 87% of patients exhibit a murmur, with 71% having a systolic murmur and the rest presenting with a continuous or ‘to and fro’ murmur, predominantly localized to the left sternal or apical region4. Various imaging modalities are employed in the diagnosis of ALCAPA including electrocardiogram (ECG), transthoracic echocardiography (TTE), computed tomography coronary angiography (CT-CAG) or magnetic resonance angiography (MRA), and invasive coronary angiography (CAG). Surgery remains the mainstay of treatment for these cases.
Here, we present a case of ALCAPA in a symptomatic adult female that was repeatedly missed on TTE, and highlight the consideration of anomalous coronaries as differential diagnoses in patients presenting with chest pain.
Case report
A 38-year-old female with no known comorbidities, a nonsmoker with no history of alcohol consumption and no significant family history, presented with complaints of increased angina and palpitations over the last few months. She had been symptomatic in recent years, leading to multiple hospital visits. She underwent echocardiography and exercise stress tests multiple times during these visits, all of which were unremarkable. She visited our outpatient department due to her persistent symptoms. On examination, she was comfortable and of average build. Her vitals were within normal limits: pulse was regular with a rate of 80 beats per minute, blood pressure of 120/80 mm Hg, respiratory rate of 18 breaths per min, and oxygen saturation of 97% in room air. Systemic examination revealed a normal position of apex beat and normal first and second heart sounds with a grade 3 of 6 pansystolic murmur at the apex radiating to the right axilla.
As the patient presented with cardiac-type chest pain, an ECG (Fig. 1) was done. It showed sinus rhythm with a rate of 60 beats per min, left axis deviation, and left ventricular hypertrophy by Cornell criteria, along with a strain pattern in the lateral leads5. Cardiac enzymes were normal. Chest X-ray was unremarkable. TTE (Fig. 2) showed normal chambers, normal wall thickness, and a dilated right coronary artery (RCA) ostium and proximal RCA with turbulent flow. However, the flow into the pulmonary arteries could not be localized. A 24-h Holter monitor showed no significant arrhythmia. Invasive CAG (Fig. 3A and B) revealed an abnormal origin of the left coronary artery (LCA) from the pulmonary artery, with the coronary supply of the left from rudimentary small vessels in the left coronary cusps and collaterals from the RCA. The RCA showed normal origin but was very large and aneurysmal, with a diameter of 15 mm. CT-CAG (Fig. 4A, B and C) revealed the RCA arising from the right coronary cusp, supplying the left coronary system via multiple collaterals. The LCA arose from the main pulmonary trunk, measuring ~13.6 mm. The left anterior descending (LAD) and left circumflex (LCX) arteries arose from the LCA. Focal aneurysmal dilation was noted on its proximal aspect, with a diameter of about 13 mm. Reflux of contrast from the left main coronary artery into the main pulmonary artery suggested a reversal of flow. Both imaging modalities showed no significant stenosis.
Figure 1.
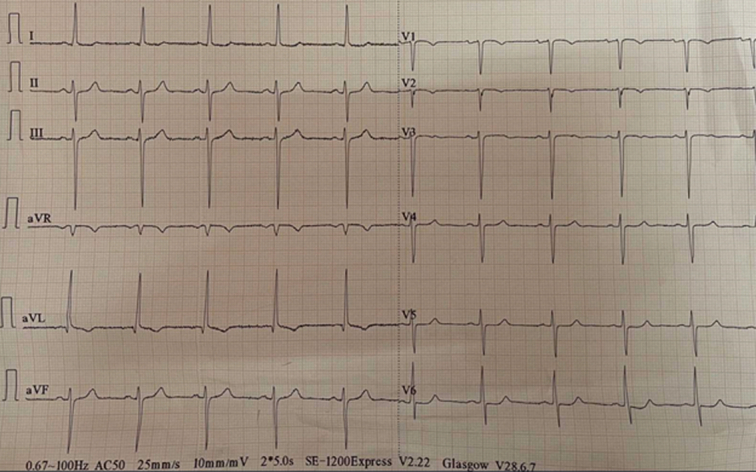
Electrocardiogram showing sinus rhythm with left axis deviation and left ventricular hypertrophy.
Figure 2.
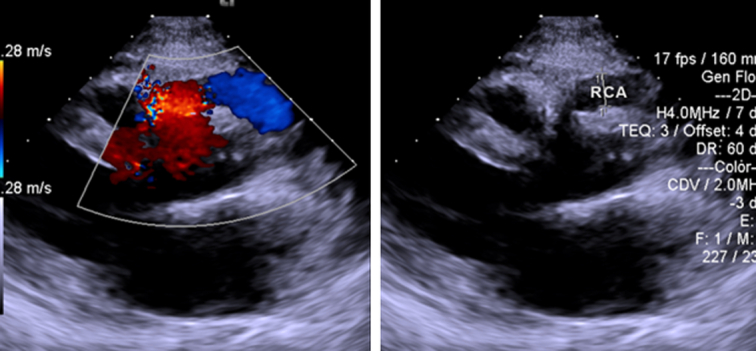
Transthoracic echocardiography showing dilated RCA ostium and proximal RCA with turbulent flow. RCA, right coronary artery.
Figure 3.
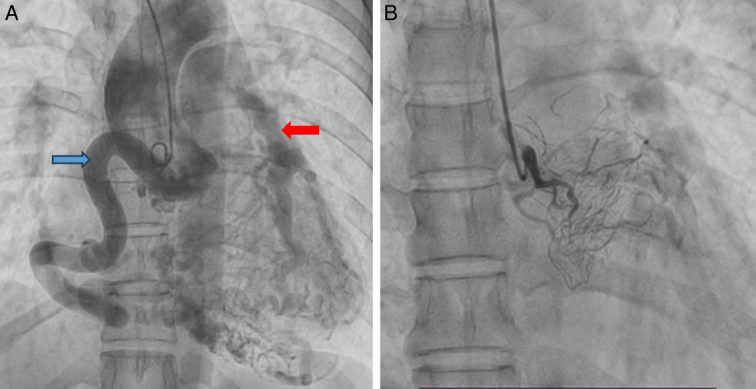
(A) Coronary angiogram showing aneurysmal right coronary artery (blue arrow) with normal origin and collaterals to the left coronary system (red arrow). (B) Coronary angiogram showing rudimentary vessels and collaterals to the left coronary system.
Figure 4.

(A, B) Reformatted computed tomography (CT) coronary angiogram images showing an anomalous left coronary artery (red arrows) arising from the pulmonary artery with a diffusely dilated and tortuous course of the main left coronary artery and its branches, along with multiple collaterals. Note the origin of the right coronary artery (blue arrows) from the right coronary cusp with a dilated and tortuous course. The right coronary artery is noted supplying the left coronary artery via multiple collaterals. (C) Oblique coronal reformatted image of the CT coronary angiogram showing the origin of the RCA and LCA with their courses. LCA, left coronary artery; RCA, right coronary artery.
The patient was counseled about her anomaly, but she refused any surgical correction. She was kept under regular follow-up. On her 6-month follow-up, her symptoms had not worsened.
Discussion
ALCAPA was established as a medical condition by Bland, White, and Garland in 1933, with autopsy confirmation in a 3-month-old infant as a syndrome with recurrent irritability, diaphoresis, dyspnea, and pallor due to cardiac ischemia6. ALCAPA can be divided into two types based on the onset of presentation: infantile and adult types7. The infantile type presents a few months after birth due to high pulmonary artery pressure during this period. Retrograde flow from the LCA occurs due to decreased pulmonary vascular resistance a few months after birth, causing ischemic symptoms. Collateral formation along with the progressive dilation of the RCA sustains life in those who survive infancy. The average lifespan is 35 years; however, there have been case reports of patients surviving for more than 50 years and even up to their 80s3,8.
Investigations in adults are prompted by cardiac signs and symptoms. ECG changes lead to further investigation. ECG findings include infarct patterns with Q waves in 50%, left ventricular hypertrophy in 28%, left axis deviation in 15%, and a normal ECG in 4% of cases4. TTE, being noninvasive and accessible, is usually the initial preferred imaging modality. TTE typically shows a dilated RCA, retrograde Doppler flow from the LCA to the pulmonary artery, and absence of the LCA ostium in the aortic root9. The diagnosis can be confirmed via CT-CAG, which offers excellent spatial resolution and is highly effective in visualizing the coronary artery anatomy. MRA may be preferred in some cases for assessing myocardial viability10. However, high cost and limited availability restrict the use of MRA. Invasive coronary angiography (CAG) is considered the gold standard imaging for obtaining detailed anatomical information5. These invasive procedures provide direct visualization of the coronary arteries and enable the assessment of hemodynamics.
The adult type is often missed due to the lack of specific ST changes in ECG and exercise stress tests, creating reluctance for angiography, especially given the financial burden in resource-limited settings like ours. In this context, the identification of abnormal coronaries in TTE is of utmost importance, and the failure by physicians to notice changes in TTE hinders diagnosis and patient care.
The conventional technique used for the management of ALCAPA in adults is coronary artery bypass graft surgery (CABG) combined with left coronary artery ligation and patch of pulmonary artery2. However, reimplantation is the preferred choice wherever suitable11. Considering the hazardous process of reimplantation, especially in conditions like a short main left coronary artery, diminished vessel elasticity for mobilization, and increased coronary frailty, CABG can still be done in these cases12. However, our patient denied any surgical repair and hence was kept on regular follow-up.
Conclusion
In conclusion, it is essential to consider anomalous coronary arteries as a differential diagnosis in patients presenting with chest pain, even though such cases are rare. Transthoracic echocardiography plays a critical role in diagnosing the abnormal origin of the LCA from the pulmonary artery, highlighting its importance in initial evaluations. Additionally, the use of other imaging modalities, such as CT-CAG and, invasive CAG is crucial for accurately establishing the diagnosis of anomalous coronaries. These advanced imaging techniques provide comprehensive details that are vital for the appropriate management and treatment of patients with this rare condition.
Patient perspective
I grew tired of visiting different hospitals and getting the same answer: nothing was wrong, despite persistent symptoms. Now, I am relieved that my disease has been diagnosed, though learning about my rare condition has left me scared, especially with the risk of sudden cardiac death. I am hesitant about surgery, but I commit to regular follow-ups and will return if my symptoms worsen.
Ethical approval
Ethical approval was taken from the Institutional Review Committee of NAIHS.
Consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Source of funding
Not applicable.
Author contribution
S.K.C.: conceptualization, manuscript writing, editing and reviewer. P.K.: contributed in conceptualization, performing literature review, mentor, and editing. A.K.: contributed in manuscript writing, literature review and as corresponding author S.P.: contributed in literature review. All authors have read and approved the manuscript.
Conflicts of interest disclosure
The authors declare no conflicts of interest.
Research registration unique identifying number (UIN)
Since it is a case report and not a research study, no clinical trials have been performed.
Guarantor
Sharada KC, Abhikanta Khatiwada.
Data availability statement
It will be open access and publicly available as per the journal guideline.
Provenance and peer review
Not applicable.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Contributor Information
Sharada KC, Email: sharadakc8888@gmail.com.
Parag Karki, Email: dr.parag.karki.md@gmail.com.
Abhikanta Khatiwada, Email: avikant12@gmail.com.
Saroj Pokhrel, Email: pokhrel.saroj35@gmail.com.
References
- 1.Pfannschmidt J, Ruskowski H, de Vivie ER. Bland-White-Garland syndrome. Clinical aspects, diagnosis, therapy. Klin Padiatr 1992;204:328–334. [DOI] [PubMed] [Google Scholar]
- 2.Wesselhoeft H, Fawcett JS, Johnson AL. Anomalous origin of the left coronary artery from the pulmonary trunk: its clinical spectrum, pathology, and pathophysiology, based on a review of 140 cases with seven further cases. Circulation 1968;38:403–425. [DOI] [PubMed] [Google Scholar]
- 3.Jurishica AJ. Anomalous left coronary artery. Am Heart J 1957;54:429–436. [DOI] [PubMed] [Google Scholar]
- 4.Yau JM, Singh R, Halpern EJ, et al. Anomalous origin of the left coronary artery from the pulmonary artery in adults: a comprehensive review of 151 adult cases and a new diagnosis in a 53-year-old woman. Clin Cardiol 2011;34:204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casale PN, Devereux RB, Alonso DR, et al. Improved sex-specific criteria of left ventricular hypertrophy for clinical and computer interpretation of electrocardiograms: validation with autopsy findings. Circulation 1987;75:565–572. [DOI] [PubMed] [Google Scholar]
- 6.Bland EF, White PD, Garland J. Congenital anomalies of the coronary arteries: report of an unusual case associated with cardiac hypertrophy. Am Heart J 1933;8:787–801. [Google Scholar]
- 7.Kanoh M, Inai K, Shinohara T, et al. Outcomes from anomalous origin of the left coronary artery from the pulmonary artery repair: long-term complications in relation to residual myocardial abnormalities. J Cardiol 2017;70:498–503. [DOI] [PubMed] [Google Scholar]
- 8.Fierens C. A 72 year old woman with ALCAPA. Heart 2000;83:2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu Y, Wang QS, Wang XF, et al. Diagnostic value of echocardiography on detecting the various types of anomalous origin of the left coronary artery from the pulmonary artery. J Thorac Dis 2020;12:319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peña E, Nguyen ET, Merchant N, et al. ALCAPA syndrome: not just a pediatric disease. Radiographics 2009;29:553–565. [DOI] [PubMed] [Google Scholar]
- 11.Dodge-Khatami A, Mavroudis C, Backer CL. Anomalous origin of the left coronary artery from the pulmonary artery: collective review of surgical therapy. Ann Thorac Surg 2002;74:946–955. [DOI] [PubMed] [Google Scholar]
- 12.Kottayil BP, Jayakumar K, Dharan BS, et al. Anomalous origin of left coronary artery from pulmonary artery in older children and adults: direct aortic implantation. Ann Thorac Surg 2011;91:549–553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
It will be open access and publicly available as per the journal guideline.