

SUMMARY
Oxidative phosphorylation (OXPHOS) occurs through and across the inner mitochondrial membrane (IMM). Mitochondrial membranes contain a distinct lipid composition, aided by lipid biosynthetic machinery localized in the IMM and class-specific lipid transporters that limit lipid traffic in and out of mitochondria. This unique lipid composition appears to be essential for functions of mitochondria, particularly OXPHOS, by its effects on direct lipid-to-protein interactions, membrane properties, and cristae ultrastructure. This review highlights the biological significance of mitochondrial lipids, with a particular spotlight on the role of lipids in mitochondrial bioenergetics. We describe pathways for the biosynthesis of mitochondrial lipids and provide evidence for their roles in physiology, their implications in human disease, and the mechanisms by which they regulate mitochondrial bioenergetics.
INTRODUCTION
Cellular bioenergetic homeostasis is a tightly regulated balancing act between energy supply and demand, maintained through the tight coupling between the hydrolysis and synthesis of ATP. In most eukaryotic organisms, mitochondria are the predominant supplier of ATP and can contribute >90% of all cellular ATP generation. First proposed by Peter Mitchell’s chemiosmotic theory in the early 1960s,1 mitochondria contribute heavily to the aerobic synthesis of ATP via oxidative phosphorylation (OXPHOS) and the electron transport chain (ETC), which consists of several oxidation-reduction reactions which “pump” hydrogen ions against their electrochemical gradient from the mitochondrial matrix into the intermembrane space (IMS; Figure 1). This high concentration of protons generates a chemiosmotic potential energy (ΔΨm) that, when these protons pass through ATP synthase back into the matrix, ultimately rotates the F0 and F1 subunits of ATP synthase to synthesize ATP from ADP and inorganic phosphate. For further details on the structures and functions of the ETC and F1F0-ATP synthase, we refer the reader to other reviews.1–3
Figure 1. Structures of PC, PE, and CL and their distribution in mitochondrial membranes.
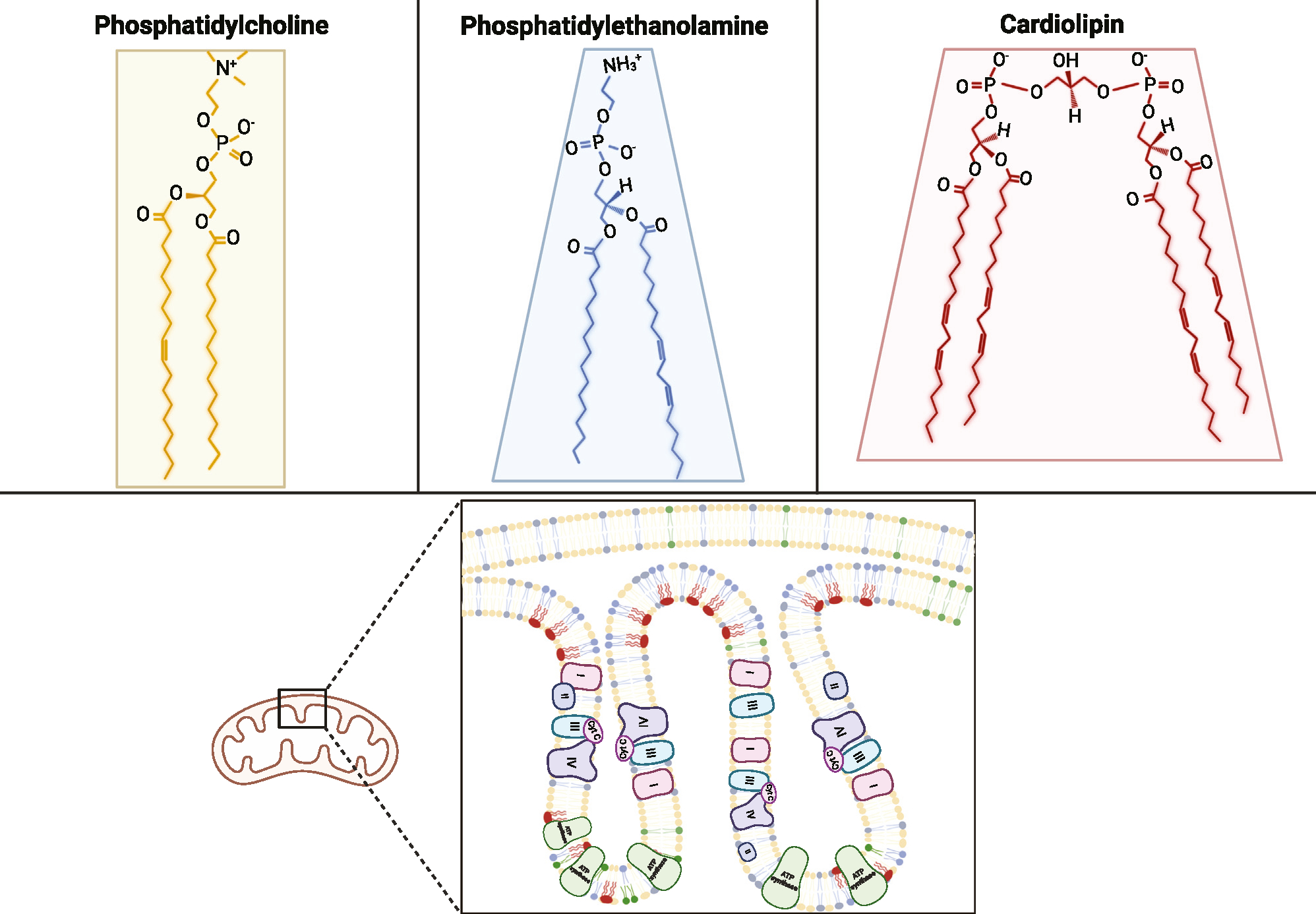
Phosphatidylcholine (PC) is a cylindrical, bilayer-forming lipid that is abundant in both outer mitochondrial membranes (OMMs) and inner mitochondrial membranes (IMMs). Phosphatidylethanolamine (PE) and cardiolipin (CL) are both conical, curvature-forming lipids that are more concentrated in cristae. CL, in particular, is almost exclusively localized in the IMM. Structures shown are 16:0/18:1-PC, 16:0/18:2-PE, and 18:2/18:2/18:2/18:2-CL (TLCL).
Mammalian mitochondria contain a unique composition of phospholipids, forming the foundational characteristics of the inner and outer mitochondrial membranes (IMM and OMM), including the formation of ΔΨm and aerobic ATP synthesis. The generation and maintenance of ΔΨm and the activity of many mitochondrial proteins—namely proteins involved in the ETC and F1F0-ATP synthase—depend heavily on the lipid composition of the IMM, as all of these proteins are embedded and operate solely in this subcellular compartment. The distinct lipid makeup of the IMM, including the presence of the mitochondria-specific lipid cardiolipin (CL), appears essential for the efficient functioning of mitochondria, including OXPHOS and many other processes that occur at the IMM or depend upon ΔΨm, such as mitochondrial Ca2+ uniporter (MCU),4 adenine nucleotide translocase (ANT),5 nicotinamide nucleotide transhydrogenase (NNT),6 and other ion transporters. Nevertheless, we will focus our discussion on the influence of mitochondrial lipids on OXPHOS, with emphases on their biochemical structures and synthesis, pathological conditions related to alterations in the mitochondrial lipidome, and the functional roles these lipid species play in regulating bioenergetics.
LIPID COMPOSITION IN THE MITOCHONDRION
Mitochondria contain two lipid membranes: an OMM and an IMM, separated by the IMS (Figure 1). Phosphatidylcholine (PC), phosphatidylethanolamine (PE), and CL comprise 75%–95% of mitochondrial membrane lipids.7–9 These phospholipids are differentially distributed in the IMM and OMM and serve unique roles in modulating mitochondrial physiology. The OMM encapsulates the outer reticular structure of the mitochondrion and selectively allows the passage of large molecules—such as adenine nucleotides, substrates for OXPHOS, and various proteins. The OMM is composed, in order of abundance, of PC, PE, phosphatidylinositol (PI), and, to a lesser extent, CL, phosphatidic acid (PA), and phosphatidylserine (PS). On the other hand, the mammalian IMM is composed mainly of PE, PC, and CL, which make up nearly 90% of its lipids, with PI and other lipids comprising <10%.10 Further, the IMM forms a dense network of cristae (lamellar invaginations into the matrix) that separate the IMS from the mitochondrial matrix and anchor proteins that drive bioenergetics—namely mitochondrial ETC complexes I–IV, F1F0-ATP synthase, the adenine nucleotide translocator, and uncoupling proteins 1–3—while preserving a tight barrier to maintain ΔΨm. The phospholipid composition of the IMM is critical for the formation of cristae folds, which increase the surface area of the IMM and support the generation of a high ΔΨm.
PC
PC is the most abundant lipid in mitochondrial membranes, forming ~50% of OMM lipids and ~40% of IMM lipids (compared with 50%–70% of the endoplasmic reticulum [ER] and plasma membranes). PC contains two fatty acids conjugated to a phosphocholine-bound glycerol backbone (Figure 1), which, due to the size of the glycerol-choline head shape and hydrocarbon tails, form a nearly cylindrical shape. This cylindrical structure of PC promotes the self-assembly of lipid bilayers in membranes so that the hydrophobic hydrocarbon chains face each other within the bilayer, whereas the polar headgroups are exposed to the exterior aqueous space surrounding the membrane. Thus, PC is integral in forming semi-permeable mitochondrial bilayers, as required for the proper assembly of β-barrel proteins,11 translocase complexes, and sorting and assembly machinery.12 Mitochondrial PC likely facilitates OXPHOS,13,14 as defects in PC biosynthesis promote irregular mitochondrial morphology and lower membrane potential.15,16
PE
PE, the second-most abundant lipid in the mitochondrial membrane, forms ~30% of the OMM and ~40% of the IMM (compared with 15%–30% of the ER and plasma membranes). PE also comprises two fatty acid chains bound to a glycerol-phosphoethanolamine backbone (Figure 1). However, the polar headgroup of PE is smaller than the choline headgroup of PC. Consequently, PE forms a conical structure and is a non-bilayer component (contributing to curvature) of the mitochondrial membranes, likely contributing to cristae formation.17 PE is known to bind to complexes I, II, III, and IV of the ETC.13,14,18–20
CL
CL, which is almost exclusively present in mitochondrial membranes, comprises ~5%–15% of the mammalian IMM, depending on the tissue and species,7 and a small percentage of the OMM. Approximately 75% of cellular CL is located within the IMM.21–23 CL is a conical diphosphatidylglycerol containing four fatty acid chains bound to a glycerol backbone (Figure 1).24 Despite the possibility for a high complexity of molecular conformations of CL, select combinations of hydrocarbon chains are more abundantly observed than others, including tetralinoleyl-CL (TLCL).25
CL plays a vital role in cristae folding and mitochondria ultrastructure26,27 due to its propensity to promote negative membrane curvature.28 CL is known to bind to complexes I, III, IV, and V.13,14,18,20,29 CL also supports the assembly of respiratory supercomplexes (see below, subsection mitochondrial electron leak)26,30–34 and other proteins embedded in the IMM, such as the ANT and uncoupling proteins (see subsection proton uncoupling).26,35–38 Augmentation or absence of CL induces dramatic phenotypic shifts in mitochondrial physiology, including impaired mitochondrial respiration26,32 and altered mitochondrial dynamics.36,39–42 Elamipretide (also known as SS-31)—a compound that has been proposed to directly interact with CL—is thought to stabilize cristae structure and mitochondrial functions.43–45
PI
PI composes a small fraction of mitochondrial membrane phospholipids (1%–7%, depending on the tissue8,9,23), most of which are located on the OMM. PI contains two fatty acid chains bound to a glycerol backbone and a phosphoinositol group. PI is ubiquitously present in mammalian cells and plays a role in lipid signaling, vesicular trafficking, and ion channel activity. The removal or masking of the PI derivative, PI(4,5)-bisphosphate, from the OMM causes mitochondrial fragmentation and mitochondrial removal via mitophagy, suggesting that PI is an important factor in the signaling pathways regulating mitochondrial morphology.46
Other lipid components of mitochondrial membranes
Mitochondrial membranes consist of several other lipids, including PS, phosphatidylglycerol (PG), PA, lysophospholipids, sterols, and sphingomyelin. These lipids comprise a small fraction of total mitochondrial membrane lipids. The functional roles of most of these lipids in mammalian mitochondria have not been well characterized, although some are critical intermediates in the biosynthesis of other phospholipids. PS (<5% of mammalian mitochondrial membrane phospholipids) is not synthesized in mitochondria but rather at the ER and is imported into mitochondria via ER-OMM membrane contact sites (see subsection lipid import).8,9,23,47,48 PS is a critical intermediate in providing PE for mammalian mitochondria because the imported PS is converted into PE in the IMM via PS decarboxylase (PSD).
BIOSYNTHESIS AND TRANSPORT OF MITOCHONDRIAL LIPIDS
The IMM is equipped with cellular machinery to generate some membrane lipids, particularly cone-shaped lipids such as PE and CL. Other lipids, such as PC, PS, and PA, are imported from the ER. Below, we describe the processes by which these lipids are synthesized or imported into mitochondria.
Lipid biosynthesis
PC, like PS, is not synthesized in mitochondria. Instead, it is generated at the ER and imported into mitochondria through the mitochondria-associated membranes (MAMs) (Figure 2). Approximately 70% of PC is synthesized by the Kennedy pathway,49 where choline undergoes sequential reactions with ATP, cytidine triphosphate (CTP), and diacylglycerol. Alternatively, PC can also be generated by methylation of PE via PE N-methyltransferase (PEMT).50,51 Some PC can also be produced by the exchange of the serine headgroup of PS with choline via PS synthase 1 (PSS1)52,53 and by acylation of lysoPC to PC via the Lands cycle and lysoPC acyltransferases (LPCATs).54
Figure 2. Mitochondrial lipid biosynthesis and trafficking.
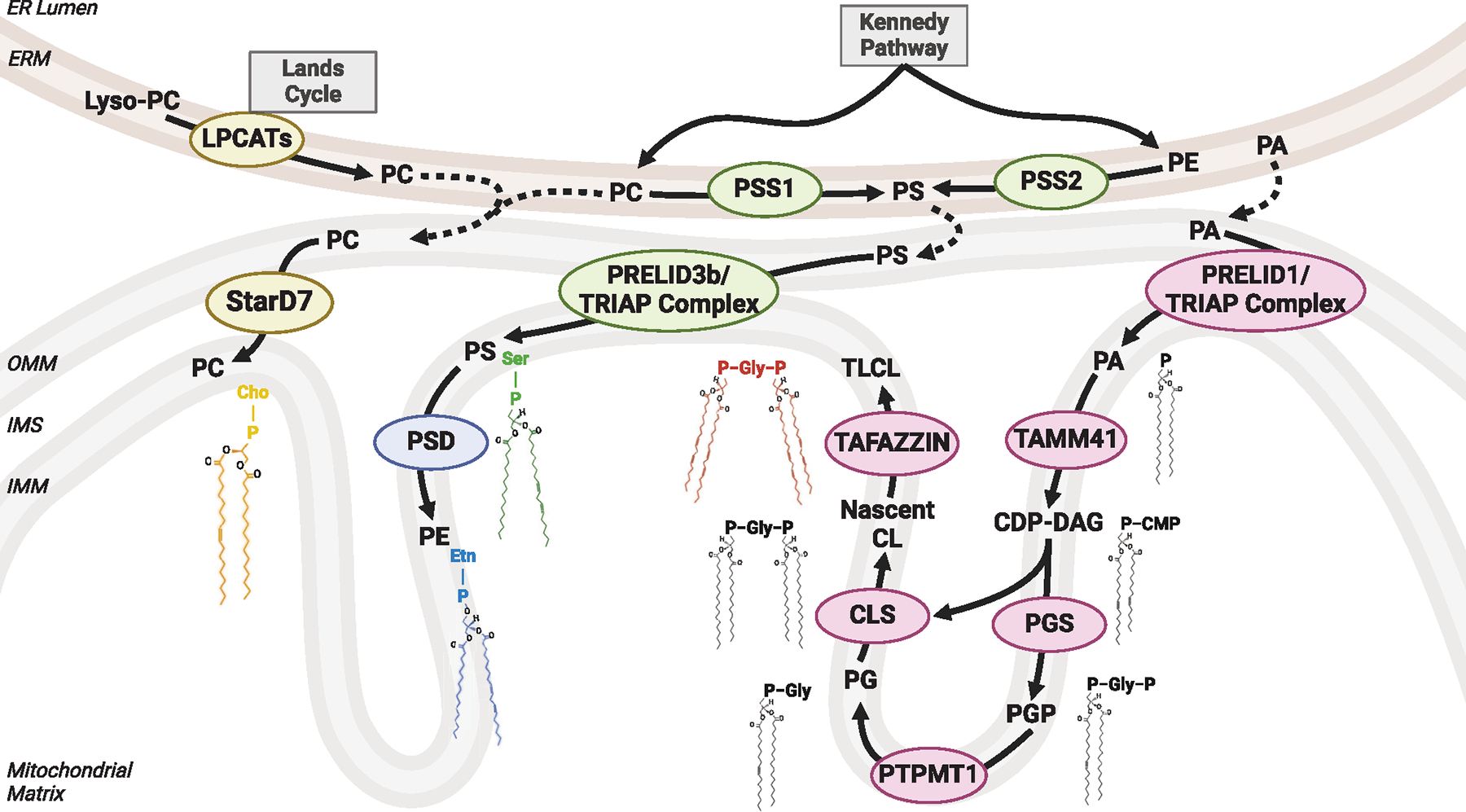
Mitochondrial PC is generated at the ER via the Kennedy pathway and other mechanisms and imported into the OMM by StarD7 (it may also play a role in translocating PC from the OMM to the IMM). PE generated at the ER does not enter mitochondria. Rather, PS generated by PSS1 or PSS2 from PC or PE is imported into mitochondria and further into the IMM by Prelid3b. Mitochondrial PS is then converted to PE via PSD. As a precursor for CL, PA is imported from OMM into IMM by Prelid1. Mitochondrial PA undergoes a series of reactions, including TAMM41, PGS, PTPMT1, and CLS. Nascent CL produced by CLS is transacylated to form TLCL by TAFAZZIN and other de/reacylases. CMP, cytidine monophosphate; Gly, glycerol; Ser, serine; Etn, ethanolamine; Cho, choline.
PE is also synthesized at the ER, but for an unclear reason, PE generated at the ER does not enter mitochondria.55 Rather, mitochondrial PE is primarily generated from PS by the PSD localized at the IMM (Figure 2).17,56 Whole-body knockout of PSD is embryonically lethal in mice, with mitochondria that are fragmented and irregularly dispersed throughout the cell.56 As an obligate substrate for PSD, the import of PS from the ER to mitochondria is required for a normal mitochondrial PE level. As described above, ER PS can be generated by PSS1 from PC or from PE by PSS2.57 Consistent with this notion, double knockout of PSS1 and PSS2 is also embryonically lethal in mice with similar mitochondrial phenotypes.57
CL biosynthesis almost exclusively occurs at IMM (Figure 2). It is generated from PA that is imported into mitochondria from the ER via the Prelid1/TRIAP complex (see below).58 PA then undergoes a series of reactions by phosphatidate cytidylyltransferase (TAMM41), PG phosphate synthase (PGS), PG phosphatase (PTPMT1), and CL synthase (CLS).59,60 Homozygous knockout of PTPMT1 in mice is embryonically lethal and drastically reduces CL and PG levels in mouse embryonic fibroblasts.59 PTPMT1 has also been implicated in several types of cancers,61,62 diabetes,63 and hypertension.64,65 Four fatty acids on CL generated by CLS often differ in lengths and degrees of desaturation, referred to as nascent CL.60 Nascent CL may undergo transacylation to form TLCL (often referred as “mature” CL) by TAFAZZIN (formerly known as TAZ) or by sequential reaction with phospholipase A2 (PLA2) and monolyso-CL acyltransferase-1 (MLCLAT1) or acyl-coenzyme A (CoA):lysocardiolipin acyltransferase-1 (ALCAT1, which resides in the ER).33,66–74 Homozygous knockout of CLS is embryonically lethal in mice.75 TAFAZZIN knockdown76,77 or knockout78,79 mice develop cardiomyopathy and malformations in mitochondria. TAFAZZIN knockdown mice also exhibit protection from obesity and hepatic steatosis.80 Mutations of CLS or TAFAZZIN in humans cause severe and often fatal conditions, most notably cardiomyopathy resulting from impaired mitochondrial respiration.81,82
Lipid import
Despite initial skepticism surrounding the discovery of mitochondria-ER contact sites in the late 1950s,48 MAMs are now recognized as an important nexus for organelle communications.83 10–30 nm in length, these suborganelle structures allow transient connectivity and inter-organelle crosstalk and exchange of molecules, including lipids.48,84–87 Emerging evidence describes mechanisms by which lipids are imported into mitochondria via MAM.88 In yeast, the vacuolar protein sorting (Vps)13 family of proteins are involved in mitochondrial lipid import.89 In humans, VPS13A and VPS13D are localized at MAM, where they similarly facilitate phospholipid import into mitochondria.89,90 In addition, newly identified mitoguardin (MIGA) and regulator of microtubule dynamics (RMDN) families of proteins appear to exhibit class-specific transport activities toward PS and PA, respectively, translocating them from the ER to the OMM.91,92
Once lipids arrive at the OMM via MAMs, they must also cross the hydrophobic barrier of the IMS (Figure 2). The mitochondrial contact site and cristae organizing system (MICOS) likely plays a role in phospholipid transfer between the OMM and IMM.93 The MICOS consists of protein complexes that form connections between the OMM and IMM to facilitate cristae junctions.94 These structures are essential for IMS lipid and protein transport. Notably, MICOS may rely on CL for proper assembly.95–98 Phospholipid transfer from the OMM and IMM presumably occurs near MICOS, mediated by class-specific lipid translocators. Steroidogenic acute regulatory protein-related lipid transfer (START)-domain-containing (StarD) family of proteins contain lipid-binding domains implicated in the intracellular lipid transport systems. StarD7-I contains a mitochondria-targeting sequence and likely facilitates the transport of PC from the OMM to the IMM.99–101 Deletion of StarD7-I decreases mitochondrial PC and impairs mitochondrial respiration.102 Homozygous knockout of StarD7 is embryonically lethal in mice,103,104 and human mutations in StarD7 have been associated with familial adult myoclonic epilepsy.105
PA and PS import is essential for CL and PE biosynthesis, respectively. In both cases, the TP53-regulated inhibitor of apoptosis gene 1 (TRIAP1) and protein of relevant evolutionary and lymphoid interest (PRELI) family of proteins form the TRIAP/PRELID complex to mediate the transfer between the OMM and the IMM. TRIAP (analogous to yeast Mdm35) and PRELI (analogous to yeast Ups) are evolutionarily conserved families of proteins residing in the IMS.58,106–108 The major isoform of TRIAP in mammals, TRIAP1, forms a protein complex together with either PRELID1, PRELID3a (or SLMO1), or PRELID3b (or SLMO2)109 and facilitates the selective shuttling of PA (in the case of PRELID1 and PRELID3a) and PS (in the case of PRELID3b) from the OMM to the IMM.58 Genetic disruption of the TRIAP/PRELI complex promotes fragmented mitochondria and reduced respiration.58 Nucleoside diphosphate kinase D (NDPK-D, also known as NME4) facilitates the transport of CL from the IMM to the OMM.110
GENETIC DISEASES ASSOCIATED WITH MITOCHONDRIAL LIPIDS
A number of human mutations in the genes of mitochondrial lipid biosynthesis have been implicated in conditions with impaired growth and development or premature death (summarized in Table 1). Examples of these conditions include Barth syndrome, which results from impaired CL synthesis82; pathogenic variants for PSD111–114; chr2-linked familial adult myoclonic epilepsy (FAME2), linked to a mutation in the STARD7 gene105; Sengers syndrome, caused by defective mutations in acylglycerol kinase (AGK) and PA synthesis115–117; and Lenz-Majewski syndrome (LMS), caused by gain-of-function mutations in the PS synthesis.53,118
Table 1.
Summary of known genetic conditions related to mitochondrial lipid biosynthesis in humans
| Condition | Affected gene | Lipid primarily affected | Clinical features |
|---|---|---|---|
|
| |||
| Barth syndrome | TAFAZZIN | cardiolipin (TLCL) | cardiomyopathy, neutropenia, blunted cardiac and skeletal muscle growth, low BMI, and adiposity |
| Pathogenic variants of PSD | PISD | phosphatidylethanolamine | congenital cataracts, skeletal dysplasia, and white matter changes |
| Mutations in STARD7 | STARD7 | unclear | myoclonic tremors and generalized tonic-clonic seizures |
| Sengers syndrome | AGK | phosphatidic acid, cardiolipin | congenital cataracts, hypertrophic cardiomyopathy, skeletal muscle weakness, and advanced exercise-induced lactic acidosis |
| Lenz-Majewski syndrome | PTDSS1 | phosphatidylserine, phosphatidylethanolamine | sclerosing bone dysplasia, intellectual disability, distinct craniofacial, dental, cutaneous, and distal limb abnormalities |
Barth syndrome
Barth syndrome is an X-linked, potentially fatal condition characterized by pediatric onset of cardiomyopathy, neutropenia, blunted cardiac and skeletal muscle growth, and exercise tolerance caused by mutations in the TAFAZZIN gene.82 Over 70% of infants with Barth syndrome develop cardiomyopathy within their first year of life, with 14% requiring heart transplantation.119 Many, but not all, patients with Barth syndrome present with low BMI and low adiposity.120 In humans, ~80% of CL in the heart is normally composed of TLCL,121,122 suggesting that cardiac function might rely on this species of CL. As observed in patients with Barth syndrome, mutations in the TAFAZZIN gene caused a 75% loss of TLCL and an increase in MLCL,123 with few alterations to other mitochondrial phospholipid species (i.e., PC, PE, PS, etc.).71 There is evidence that loss of CL remodeling disrupts the spatial organization of IMM proteins.124 A recent study using adeno-associated virus serotype 9 (AAV-9) vectors as a means of TAFAZZIN gene replacement in a mouse model of Barth syndrome showed promise as a potential therapeutic approach.78
Pathogenic variants of PSD
Mutations in PISD, a gene that encodes the PSD enzyme, have been identified to cause mitochondrial disease that clinically presents with congenital cataracts, skeletal dysplasia, and white matter changes.112,114 In one study, two female siblings were found to have compound heterozygous mutations (one missense and one splice variant) on the PISD gene. Fibroblasts derived from both sisters contained low levels of mitochondrial PE and exhibited decreased mitochondrial respiration coupled with impaired complex IV activity, lower ΔΨm, and increased mitochondrial mass compared with fibroblasts from control subjects.112,114
Other genetic conditions potentially influenced by mitochondrial lipids
Mutation in the first intron of the STARD7 gene has been implicated in FAME2.105 This condition is characterized by myoclonic tremors and generalized tonic-clonic seizures. Proton magnetic resonance spectroscopy shows increased levels of choline in the cerebellum of individuals with FAME2,125 an observation that may be predicted with dysfunctional STARD7. Nevertheless, the mutation does not appear to alter STARD7 expression in patient-derived skin fibroblasts. Thus, the relevance of mitochondrial lipids in FAME2 remains speculative.
Sengers syndrome is a rare autosomal recessive disorder characterized by congenital cataracts, hypertrophic cardiomyopathy, skeletal muscle weakness, and advanced exercise-induced lactic acidosis.117 Sengers syndrome is caused by loss-of-function mutations in the AGK gene.115,116 Homozygous mutations in the AGK gene result in a more severe form of Sengers syndrome that results in early infant mortality. Although AGK functions in a major biosynthetic pathway for PA (a necessary precursor to CL), the hypothesis that patients with Sengers syndrome exhibit alterations in mitochondrial membrane phospholipid composition has not been investigated in detail. AGK binds with the TIM22 protein import complex, which plays a role in the molecular import and assembly of various proteins, including ATP synthase.126 In cells, while global knockout of AGK resulted in mitochondrial defect, mutating the catalytic site of AGK did not recapitulate these phenotypes. Thus, Sengers syndrome may be caused by the activity of AGK independent of its effect on mitochondrial membrane lipid composition. The development of an in vivo model of Sengers syndrome is needed to confirm these findings.
LMS is clinically characterized by the compilation of sclerosing bone dysplasia, intellectual disability, and distinct craniofacial, dental, cutaneous, and distal limb abnormalities.118 Whole-genome sequencing of LMS patients suggested that the condition is caused by SNPs constellated around the gene encoding the PSS1 enzyme (PTDSS1).53 All study participants exhibited heterozygous missense, gain-of-function mutations in the PTDSS1 gene.127–129 Indeed, fibroblasts obtained from patients with LMS exhibited a higher rate of PS synthesis compared with that in control fibroblasts, indicating that overproduction of PS via PSS1 is likely the primary defect underlying LMS.53 Phenotypes associated with LMS are likely contributed by the pleiotropic role of PS, including, but not limited to, the function of PS in mitochondria.
NON-COMMUNICABLE CONDITIONS ASSOCIATED WITH MITOCHONDRIAL LIPIDS
Alterations in mitochondrial membrane lipids have also been implicated in non-communicable conditions and diseases such as exercise, metabolic-dysfunction-associated steatotic liver disease (MASLD), diabetes, and neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease.
Exercise
Exercise training is known to promote mitochondrial biogenesis in skeletal muscle.130,131 We have shown that this adaptation coincides with a remodeling of mitochondrial lipid composition.132 A greater exercise capacity, either with treadmill exercise training or in rats bred for running capacity,133 is associated with an increase in the proportion of PE in mitochondria. Conversely, physical activity induced by hindlimb unloading or reduced cage size lowers PE content in mitochondria.132,134 These changes in mitochondrial PE are likely driven by changes in PSD expression that also coincide with exercise. Loss of mitochondrial PE in skeletal muscle substantially reduces skeletal muscle contractility, supported by reduced capacities for respiration and ATP production, as well as an increase in mitochondrial electron leak that promotes oxidative stress.132
MASLD
Alterations in mitochondrial bioenergetics have been implicated in the pathogenesis of MASLD,135 but the underlying mechanisms for these changes are unclear. In mice, an increase in hepatic mitochondrial CL is observed early in obesity but decreases with the progression to MASLD.136 Mice with hepatocyte-specific deletion of CLS exhibit diet-induced steatohepatitis, though whether this effect is mediated by the functions of CL on OXPHOS is unknown.137 In contrast, mice with global deletion of TAFAZZIN are protected from MASLD, though this is confounded by their hypermetabolic phenotype.80 In rats, increased peroxidized CL has been observed with MASLD.136,138 Exogenous CL, but not peroxidized CL or other phospholipids, restored some of the OXPHOS defects associated with MASLD.137,139 How CL mediates bioenergetic alterations induced by MASLD remains to be fully elucidated.
Diabetes
In both type 1 and type 2 diabetes, the reduced insulin secretory response in pancreatic β cells contributes to hyperglycemia. Particularly for type 2 diabetes, changes in mitochondrial energetics have been implicated in the pathogenesis of defective glucose-stimulated insulin secretion.140 Mitochondrial CL may play a role in this process. In global TAFAZZIN knockdown mice, loss of TAFAZZIN promotes a ~50% reduction in insulin secretion measured ex vivo.141 Likewise, the global deletion of phospholipase A2β, one of the alternative enzymes for CL transacylation, makes β cells defective for insulin secretion.142 Tissue-specific gain- or loss-of-function studies for these enzymes are needed to confirm that these changes are due to the lack of CL in the β cells.
Neurodegenerative disease
Alterations to mitochondrial membrane phospholipids, particularly CL, may contribute to the development of Alzheimer’s disease and Parkinson’s disease. The brain appears to have a greater diversity of CL species compared with other tissues, with up to 100 different CL species reported.143,144 CL is particularly perturbed by the accumulation of tau proteins in models of Alzheimer’s disease145,146 and α-synuclein (αS) in models of Parkinson’s disease.147–153 In the early stages of Alzheimer’s disease, synaptic, but not non-synaptic, mitochondria exhibit a decreased abundance of CL (specifically TLCL) and an increased abundance of PC and lyso-PC.146 These alterations are accompanied by a decrease in the activity of complex I and lower ATP concentrations in neurons. Tau proteins appear to bind with CL directly, causing mitochondria to swell and release cytochrome c.145 Nonyl acridine orange, a molecule known to bind to CL,154 prevented tau-induced mitochondrial damage, suggesting a causal tau-CL mechanism in the pathology of Alzheimer’s disease. Similarly, αS perturbs mitochondrial lipid membranes by interacting with membranes containing greater curvature148 and acidic lipids155 (such as CL), with a higher affinity for unsaturated acyl chains. The high affinity for αS-CL binding appears to reduce complex I activity in synaptic mitochondria.156
Influence of cell types on mitochondrial membrane lipid composition
The composition of mitochondrial membranes differs between tissues in the same organism. In particular, the diversity of CL species is dramatically different between tissues in the same organism, potentially reflecting mitochondria adapted for biological processes that are specific to the cell type.157 TLCL content in mouse heart, liver, brain, and gastrocnemius muscle can range from 7.1% (brain) to 80.6% (liver) of total CL.158,159 It is not entirely clear why TLCL content is comparatively high in liver mitochondria, where its respiratory capacity is not particularly high.160 It is possible that they represent a potential requirement for TLCL in gluconeogenesis, lipogenesis, and/or transamination, which experience high flux in hepatocytes. CL is also more highly represented in the mitochondrial lipidome of cardiomyocytes, which maintain constant, high energetic demands. Alternatively, tissues with lower ATP demand, such as adipose, contain relatively low levels of CL.
INFLUENCE OF MITOCHONDRIAL LIPIDS ON BIOENERGETICS
As evidenced by the examples of human mutations in the genes of mitochondrial lipid biosynthesis, loss of these lipids likely induces defects in mitochondrial bioenergetics that contribute to their pathology. Membrane lipids likely influence OXPHOS through complex mechanisms. These mechanisms include lipid-to-protein binding that affects enzyme activity, membrane properties that affect lateral diffusion of electron carriers in IMM as well as the conductance of ions across IMM, and cristae architecture influencing suborganellar compartmentalization (Figure 3). Advances in structural biology and access to mitochondrial bioenergetic phenotyping platforms, such as high-resolution respirometry, fluorometry, electrophysiology, and microscopy, have made it possible to more deeply study the influence of mitochondrial membrane lipids on energy transduction through OXPHOS.161–166
Figure 3. Diverse roles of mitochondrial phospholipids regulating OXPHOS.
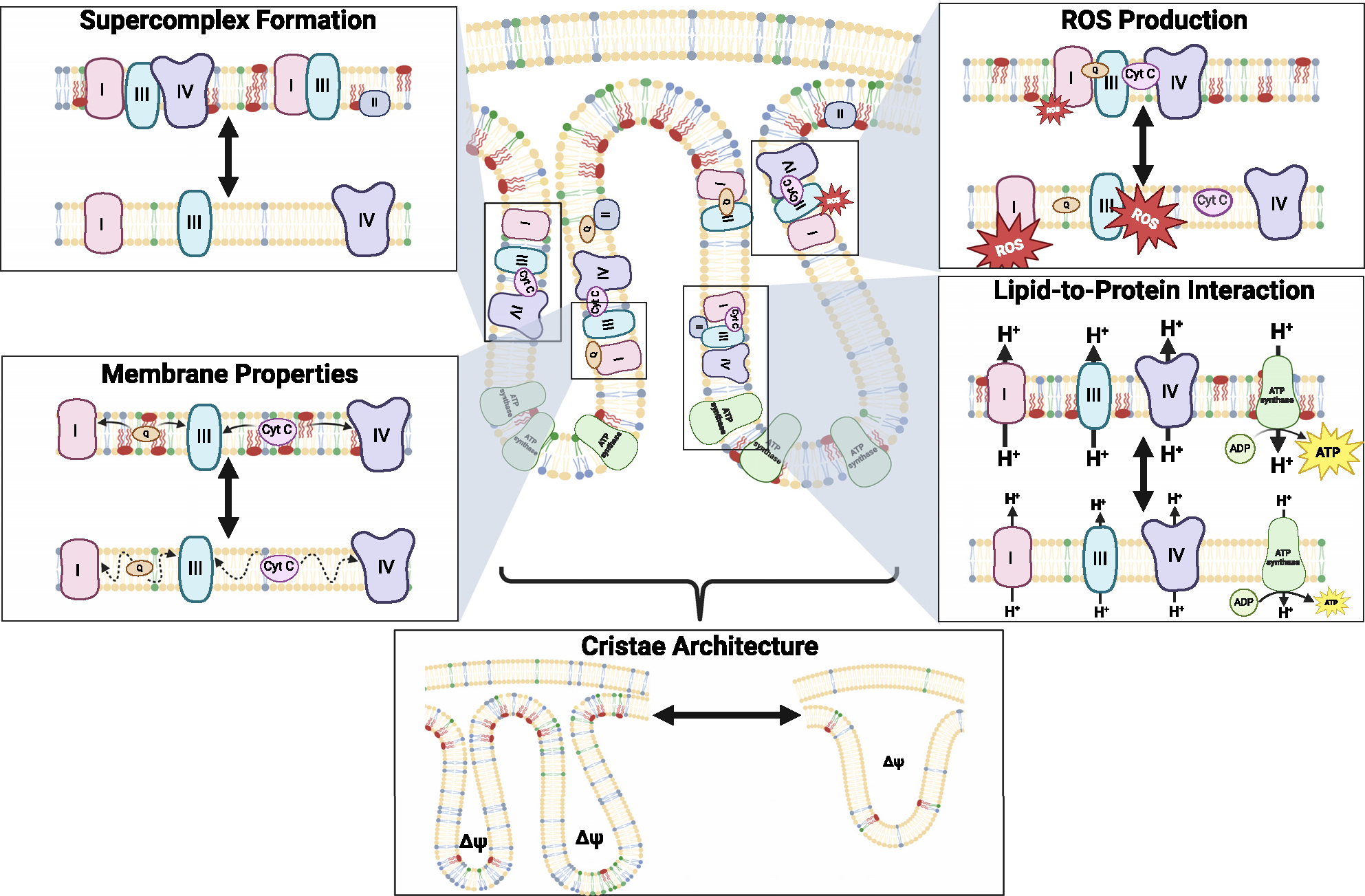
The effects of IMM lipids on bioenergetics are likely mediated by lipid-to-protein interactions that affect OXPHOS enzyme activity, membrane properties that influence lateral diffusability of electron carriers through IMM and proton conductance across the IMM, by influencing the interaction of respiratory complexes to facilitate efficient electron transfer and regulate electron leak, and cristae architecture that regulates compartmentalization of mitochondrial membrane potential.
ATP production
Respiration (oxygen consumption rate [OCR] or O2 flux [JO2]) is often measured to assess energy flux through OXPHOS. Although useful to study, the OCR is just one of the metrics for OXPHOS activity. The ETC harnesses the reduction potential between NADH (or succinate) and molecular O2 to drive the movement of electrons from complex I/II to III and to IV, while coupling the electron transfer to proton pumping from the matrix to the IMS to generate membrane potential. Oxygen can be consumed either at the terminal complex IV (4H+ + 4e− + O2 → 2H2O) or in a small fraction as a result of premature electron leak (e− + O2 → O2−). Thus, oxygen consumption is approximately, but not exactly, equimolar to energy influx to OXPHOS. In many cases, respiration is a reasonable estimation of the mitochondria’s ability to take part in ATP synthesis. Nonetheless, ATP production may be decoupled by a number of mechanisms (electron leak, proton leak, etc.), such that the direct measurement of the rate of ATP synthesis is desirable when assessing the energy output of OXPHOS.
Evidence suggests that mitochondrial CL, PE, and PC are all essential for OXPHOS. These lipids are known to directly interact with complex I–V to affect their activities.13,14,18–20,36 Mutations in TAFAZZIN resulting in Barth syndrome, which reduces TLCL, diminishes respiratory capacity in skeletal muscle mitochondria in these patients.167 Genetic ablations that negatively influence the enzymes of CL biosynthesis almost universally reduce oxygen consumption or ATP production across tissues.32,168–170 However, in the liver and in brown adipose tissue (BAT), loss of CL has no effect on ATP production.80,171,172 Similarly, loss of mitochondrial PE reduces oxygen consumption and ATP production in skeletal muscle,132,173 but not in BAT.171 It is highly intriguing that the depletion of mitochondrial lipids in different cell types promotes differential bioenergetic phenotypes. These differences are likely partly driven by the differential proteome and lipidome in these cells, as well as their differential metabolic demands. For example, locomotor activity commands a robust component of bioenergetic demands in skeletal muscle, necessitating exceptionally high ATP flux. In contrast, proton uncoupling (see below) induced by thermogenic demands primarily drives bioenergetic demands in BAT, requiring lower allocation of membrane potential energy for ATP synthesis (also discussed in the subsection influence of cell types on mitochondrial membrane lipid composition above).
Proton uncoupling
Uncoupling protein 1 (UCP1), which resides in the IMM, is largely responsible for thermogenesis in brown and beige adipocytes. UCP1 uncouples the mitochondrial membrane potential (ΔΨm) to ATP synthesis by translocating protons in IMS back into the matrix independent of complex V.174,175 This process effectively uncouples the ETC from ATP production, and the energy from the ΔΨm is dissipated as heat.
Brown and beige adipocytes are highly responsive to ambient temperature and regulate thermogenesis by modulating UCP1 transcription and activity. Membrane lipids appear to play an important role in this regulation, as CL tightly binds to UCP1.176 Cold exposure in mice induces a robust, time-dependent increase in the expression of enzymes involved in CL synthesis and transacylation.177 Loss- or gain-of-function studies show that CL positively regulates thermogenesis.171,177 However, bioenergetic phenotyping of brown adipose mitochondria isolated from CLS-deleted mice show normal UCP1-dependent respiration despite substantially compromised thermogenesis.171 Thus, while it is clear that CL is essential for thermogenesis, the exact mechanism by which it regulates UCP1 needs further clarification. In contrast, mitochondrial PE appears essential for UCP1-dependent respiration and proton conductance in brown adipocytes.171 Mitochondrial PE also robustly responds to ambient temperature. It is unknown whether PE directly binds to UCP1, but there are some reports that PE increases the protonophoric activity of UCP1.178
Mitochondrial electron leak
Electrons donated from NADH or succinate can prematurely leak to molecular O2 or other acceptors prior to doing so in a highly controlled manner in complex IV. For example, electron stalling in the Q-pool (reduced and oxidized mixture of coenzyme Q) is thought to lead to reverse electron transfer in complex I, enabling electrons to reduce O2 into O2− (oxygen radical) that in turn reacts with water to become H2O2. Excessive production of these oxidants may induce oxidative stress and they may also have important signaling roles. Similar to proton uncoupling, mitochondrial electron leak (or reduced electron transfer efficiency) can increase respiration without channeling its energy for ATP synthesis.
An increase in mitochondrial electron leak has been implicated in the pathogenesis of Barth syndrome. Some studies report an increase in mitochondrial electron leak with TAFAZZIN deficiency.168,179 On the other hand, in a study where mitochondrial electron leak was quantified from 11 potential sites (including those in the ETC and substrate catabolism), TAFAZZIN deficiency did not alter electron transfer efficiency in any of them.180 Indeed, neutralizing mitochondrial H2O2 by overexpression of mitochondria-targeted catalase did not ameliorate cardioskeletal myopathy in TAFAZZIN knockdown mice.168 Homozygous, but not heterozygous, deletion of PSD promoted mitochondrial electron leak in skeletal muscle,132,173 suggesting that a robust decrease in mitochondrial PE is required to reduce electron transfer efficiency. In BAT, neither deleting CLS nor PSD had an effect in increasing mitochondrial electron leak.171 We speculate that brown adipocytes are particularly resistant to an increase in mitochondrial electron leak due to the strong bioenergetic “pull” (energetic demand) induced by UCP1.
In mammals, up to 80%–85% of individual mitochondrial OXPHOS enzymes may be found in clusters, known as “supercomplexes” (or respirasomes).181,182 There remains some controversy regarding whether these supercomplexes represent the cause or consequence of efficient electron transfer. A recent study suggests that respirasomes are not required for maintaining normal bioenergetics.183 Other studies suggest that supercomplex assembly contributes directly to IMM curvature.184 Nevertheless, respiratory complexes appear to either permanently or transiently become close in proximity, such that the probability of electron leaking from the ETC is reduced. Lymphoblasts from patients with Barth syndrome have decreased amounts of the CI1CIII2 supercomplex.185 Likewise, mice with cardiomyocyte-specific TAFAZZIN knockout186 and inducible TAFAZZIN knockdown33,187 demonstrate lower levels of supercomplexes, suggesting that mature CL is necessary for higher-order respirasome formation. Similarly, loss of mitochondrial PE also reduces supercomplex formation in mouse muscle and in Chinese hamster ovary (CHO) cells.17,132
Cristae architecture
Mitochondrial cristae folds are a hallmark of mitochondrial morphology and are critical to OXPHOS. ETC, UCP1, and ATP synthase are all localized in the cristae.188–191 It is known that individual cristae can become biochemically separated from the rest of IMS and other cristae units by closing the cristae junction, forming cristae vesicles (Figure 4). As the two major cone-shaped lipids abundant in IMM, PE and CL are presumably highly concentrated in cristae. The concentration of these membrane lipids would be predicted to directly influence membrane curvature and, thus, the shape and volume of the cristae vesicles.
Figure 4. How phospholipids might influence membrane potential by altering cristae volume.
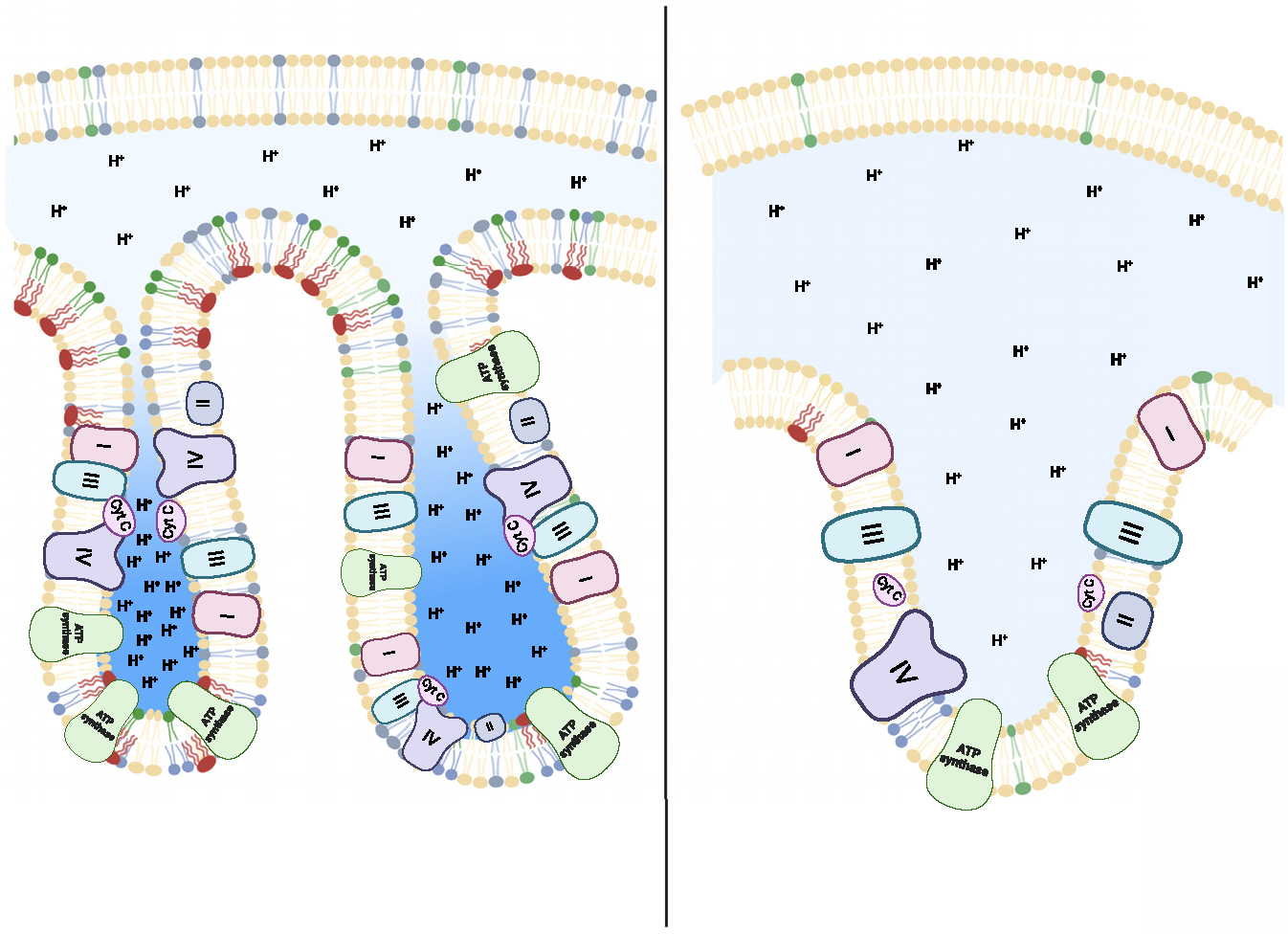
Cone-shaped phospholipids such as PE and CL create the negative curvature found in cristae. Increased abundance of these lipids (left) makes it possible for cristae to make more acute turns, resulting in smaller cristae volume. The electrochemical concentration gradient of IMM would be directly influenced by the volume of cristae vesicles, as fewer protons would be predicted to be needed to achieve the same concentration.
Why is the shape and volume of cristae vesicles important for bioenergetics? This is because membrane potential is not merely a mathematical sum of protons pumped by ETC; rather, it is that sum divided by the volume of IMS. In other words, ΔΨm is an electrochemical concentration gradient across IMM. Thus, changes in the IMS volume would be predicted to directly influence ΔΨm. We subscribe to the idea that each cristae vesicle can become segregated from other cristae vesicles and the rest of IMS (inner boundary membrane [IBM]) through the closing of the cristae junction (Figure 4), such that each cristae vesicle possesses distinct ΔΨm (heteropotential mitochondria).192–195 In this model, ΔΨm in any given mitochondrion is not uniform, giving rise to electrochemically distinct ATP-synthesizing units. If this were the case, the lipid composition of the cristae membrane would tremendously impact ΔΨm through its effect on cristae vesicle volume. Cone-shaped lipids such as PE and CL should induce membrane curvature and reduce the volume of cristae vesicles, in turn amplifying the unit of ΔΨm gained per unit of ETC flux. Consistent with this notion, mitochondria deficient in PTPMT1 (one of the enzymes of CL biosynthesis) produce cristae vesicles that are bigger in size.165 Mitochondria from patients with Barth syndrome, animal models of TAFAZZIN, and CLS deficiencies have fewer cristae folds.76,78,171,186 Similar morphological changes are observed with impairment in PSD in humans and in mouse models.17,56,132,171 Another important consideration is how the bioenergetic consequence of cristae segregation is relevant to mitochondrial dynamics, as phospholipids are also known to influence these processes.40
How morphologically dynamic are individual cristae, and how does membrane lipid composition potentially influence ΔΨm? Considering the heteropotentiality and highly dynamic nature of mitochondrial cristae architecture, it is, perhaps, logical to postulate that there exists a synergistic system by which ΔΨm and lipid composition of individual cristae co-regulate each other to dynamically remodel cristae size and shape, similar to the plasma membrane.196,197 In this model, which we term the “bagpipe hypothesis” (Figure 5), it is possible that the architecture of individual mitochondrial cristae is dynamically responsive to the magnitude of the difference in electric charge across the IMM, having a potential to temporarily diffuse its pressure by expanding cristae volume (“ballooning”). In more elastic membranes, such an effect would be immediately counteracted by the compression forces from the IMM that promote efflux back into the matrix. Thus, the framework of this bagpipe hypothesis would stipulate that the size and volume of the individual cristae folds are tightly regulated by a ΔΨm and/or lipid composition feedback system, which dictates cristae architecture, ATP production, and proton leak. Interestingly, compared with cylindrical lipids such as PC, PE is likely to increase the elasticity of biological membranes due to the higher bending rigidity of PE.198 Because this may be a property of conical lipids, it is also likely that CL increases the elasticity of biological membranes. Therefore, loss of PE and CL could increase the flaccid properties of the IMM, allowing greater expansion capacity. Further, this could be the primary reason that TLCL is deemed the main “functional” form of CL, as the saturation, number of acyl chains,199 or Ca2+ ion bonding122 in CL could considerably influence the structural, and thus the elastic, properties of the IMM.
Figure 5. The bagpipe hypothesis.
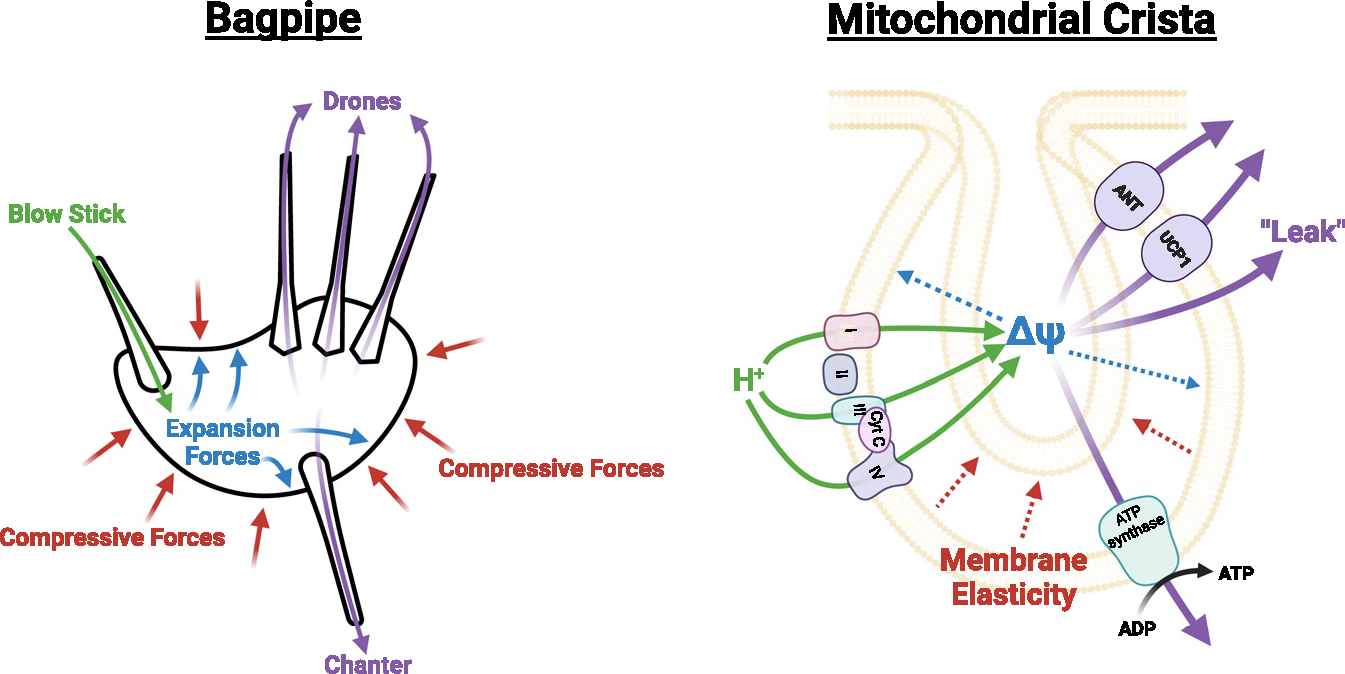
Bagpipes are played when air travels through the chanter that produces the melody, as well as through the drones that produce continuous sound. The continuous sound from the chanter and drones are enabled by air blown into the blowstick and, importantly, by compression forces to the bag. Our hypothesis proposes that mitochondrial cristae function similarly. Protons are pumped into the IMS via the ETC. As protons accumulate in the IMS, this produces an expansion force (ΔΨ), which is opposed by the IMM. The IMM membrane composition influences the elastic properties of the membrane. Highly rigid membranes retain their shape better and thus are able to generate higher membrane potentials with fewer protons in the IMS. Membranes with low rigidity are more likely to expand with a greater osmotic pressure in the IMS, stabilizing the thermodynamic gradient between the matrix and the IMS. This expansion of the IMM then requires a higher proton concentration in the IMS to drive ATP synthesis, or protons escape via various mechanisms.
How might this dynamic role of mitochondrial membrane lipids serve as a direct influence on mitochondrial bioenergetics? In membranes with greater compression forces, perhaps such as those with high CL and PE, membrane expansion is limited by the more rigid membranes. Because of this, cristae volume is also limited. Therefore, a lower proton influx into the IMS facilitated by the ETC is required to meet the ΔΨm needed for ATP production (~60 mV). On the other hand, as the proton motive force driving ATP synthesis is dictated by ΔΨm, which is influenced by cristae volume, a more compliant IMM would increase overall cristae volume, thus maintaining the thermodynamic state of the IMS. However, we might also factor in the effect that membrane expansion has on the ETC. It could be the case that, as the IMM expands, so does the distance between proteins embedded in the IMM. The expansion would then serve a dual purpose in slowing the rate of electron transfer between ETC complexes, thus generating an immediate self-regulating feedback mechanism where cristae expansion slows down H+ accumulation in the cristae, thus maintaining a relatively constant ΔΨm. The increased diffusion distance would also have implications for increased reactive oxygen species (ROS) production, as the impaired rate of electron transfer would block electron transfer between ETC proteins and carriers, increasing the likelihood that some electrons would be released from their bound chaperones. Although this thought experiment is intriguing, it would need to be thoroughly tested. Recent advancements in ultra-resolution real-time in situ imaging techniques (such as stimulated emission depletion microscopy) could provide avenues to understand the dynamic changes that occur in mitochondrial cristae in response to bioenergetic flux.
CONCLUSIONS AND PERSPECTIVES
Human mutations in genes that encode the enzymes of mitochondrial membrane biosynthesis are associated with OXPHOS dysfunction, which is detrimental to health. OXPHOS occurs through and across these IMM lipids, making them an integral component to the energy-transducing processes that yield ATP synthesis. The effects of these lipids are likely mediated by lipid-to-protein interaction that affects enzyme activity, membrane properties that influence lateral diffusability of electron carriers and proton conductance, and cristae architecture that regulates compartmentalization of mitochondrial membrane potential. Furthermore, lipids influence bioenergetics in cell-type-specific manner. In cardiac and skeletal myocytes where ATP production is the primary demand for OXPHOS, mitochondrial lipids exquisitely regulate ATP synthesis. In UCP1-positive adipocytes where there are other purposes for OXPHOS, mitochondrial lipids do not appear to strongly influence ATP synthesis. We speculate that a mitochondrial microenvironment (proteomic, metabolomic, and bioenergetic milieu) that is specific to the cell likely interacts with membrane lipids to ultimately give rise to its OXPHOS phenotype.
Recent emergence in lipid mass spectrometry, combined with tools in cell biology and mitochondrial phenotyping, enabled a greater understanding of the diverse and fundamental roles that the mitochondrial membrane phospholipids play in regulating bioenergetics. We believe these mechanisms to play an important role in how mitochondria adapt to altering cellular energetic demands and that defects in these processes may contribute to diseases associated with chronic energy inbalance. We identify two areas of research that will have the greatest impact in advancing this field. One is in structural biology to characterize the biophysical nature of the interaction of these lipids with respiratory complexes. Results from bioenergetic phenotyping could be used to guide targeted questions on interactions that are most likely to be relevant. Advances in techniques to obtain high-resolution localization of lipids is another area. Current technology does not allow suborganellar visualization of lipids, as these molecules largely lack reliable probes and spatial resolution of mass spectrometry is currently insufficient to resolve organelles. How are the membrane lipids localized in the cristae and are they clustered to form microdomain-like rafts to facilitate spatial distribution of respirasomes? The molecular resolution of these processes will likely provide the greatest insights into the functional significance of these lipids in regulating mitochondrial bioenergetics.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01 DK107397, R01 GM144613, R01 AG074535, and R01 DK127979 to K.F. and T32 DK091317 to S.T.D. The authors used BioRender to generate the figures.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Vercellino I, and Sazanov LA (2022). The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 23, 141–161. 10.1038/s41580-021-00415-0. [DOI] [PubMed] [Google Scholar]
- 2.Yoshida M, Muneyuki E, and Hisabori T (2001). ATP synthase—a marvellous rotary engine of the cell. Nat. Rev. Mol. Cell Biol. 2, 669–677. 10.1038/35089509. [DOI] [PubMed] [Google Scholar]
- 3.Nicholls DG, and Ferguson SJ (2013). Bioenergetics (Academic Press; ). https://www.elsevier.com/books/bioenergetics/nicholls/978-0-12-388425-1. [Google Scholar]
- 4.Wescott AP, Kao JPY, Lederer WJ, and Boyman L (2019). Voltage-energized Calcium-sensitive ATP Production by Mitochondria. Nat. Metab. 1, 975–984. 10.1038/s42255-019-0126-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC, Brookes PS, and Cornwall EJ (2005). The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem. J. 392, 353–362. 10.1042/BJ20050890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Earle SR, and Fisher RR (1980). Reconstitution of bovine heart mitochondrial transhydrogenase: a reversible proton pump. Biochemistry 19, 561–569. 10.1021/bi00544a026. [DOI] [PubMed] [Google Scholar]
- 7.Cortie CH, Hulbert AJ, Hancock SE, Mitchell TW, McAndrew D, and Else PL (2015). Of mice, pigs and humans: an analysis of mitochondrial phospholipids from mammals with very different maximal lifespans. Exp. Gerontol. 70, 135–143. 10.1016/j.exger.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 8.Stefanyk LE, Coverdale N, Roy BD, Peters SJ, and Leblanc PJ (2010). Skeletal Muscle Type Comparison of Subsarcolemmal Mitochondrial Membrane Phospholipid Fatty Acid Composition in Rat. J. Membr. Biol. 234, 207–215. 10.1007/s00232-010-9247-4. [DOI] [PubMed] [Google Scholar]
- 9.Tsalouhidou S, Argyrou C, Theofilidis G, Karaoglanidis D, Orfanidou E, Nikolaidis MG, Petridou A, and Mougios V (2006). Mitochondrial phospholipids of rat skeletal muscle are less polyunsaturated than whole tissue phospholipids: Implications for protection against oxidative stress. J. Anim. Sci. 84, 2818–2825. 10.2527/jas.2006-031. [DOI] [PubMed] [Google Scholar]
- 10.Comte J, Maïsterrena B, and Gautheron DC (1976). Lipid composition and protein profiles of outer and inner membranes from pig heart mitochondria. Comparison with microsomes. Biochim. Biophys. Acta 419, 271–284. 10.1016/0005-2736(76)90353-9. [DOI] [PubMed] [Google Scholar]
- 11.Schuler M-H, Di Bartolomeo F, Böttinger L, Horvath SE, Wenz L-S, Daum G, and Becker T (2015). Phosphatidylcholine Affects the Role of the Sorting and Assembly Machinery in the Biogenesis of Mitochondrial β-Barrel Proteins. J. Biol. Chem. 290, 26523–26532. 10.1074/jbc.M115.687921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuler M-H, Di Bartolomeo F, Mårtensson CU, Daum G, and Becker T (2016). Phosphatidylcholine Affects Inner Membrane Protein Translocases of Mitochondria. J. Biol. Chem. 291, 18718–18729. 10.1074/jbc.M116.722694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharpley MS, Shannon RJ, Draghi F, and Hirst J (2006). Interactions between phospholipids and NADH:ubiquinone oxidoreductase (complex I) from bovine mitochondria. Biochemistry 45, 241–248. 10.1021/bi051809x. [DOI] [PubMed] [Google Scholar]
- 14.Shinzawa-Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, Yamasaki A, Sugimura T, Kurono S, Tsujimoto K, et al. (2007). Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 26, 1713–1725. 10.1038/sj.emboj.7601618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu G, Aoyama C, Young SG, and Vance DE (2008). Early embryonic lethality caused by disruption of the gene for choline kinase alpha, the first enzyme in phosphatidylcholine biosynthesis. J. Biol. Chem. 283, 1456–1462. 10.1074/jbc.M708766200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu G, Sher RB, Cox GA, and Vance DE (2009). Understanding the muscular dystrophy caused by deletion of choline kinase beta in mice. Biochim. Biophys. Acta 1791, 347–356. 10.1016/j.bbalip.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 17.Tasseva G, Bai HD, Davidescu M, Haromy A, Michelakis E, and Vance JE (2013). Phosphatidylethanolamine Deficiency in Mammalian Mitochondria Impairs Oxidative Phosphorylation and Alters Mitochondrial Morphology. J. Biol. Chem. 288, 4158–4173. 10.1074/jbc.M112.434183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calzada E, Avery E, Sam PN, Modak A, Wang C, McCaffery JM, Han X, Alder NN, and Claypool SM (2019). Phosphatidylethanolamine made in the inner mitochondrial membrane is essential for yeast cytochrome bc1 complex function. Nat. Commun. 10, 1432. 10.1038/s41467-019-09425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D, Bartlam M, and Rao Z (2005). Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 121, 1043–1057. 10.1016/j.cell.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 20.Zhang M, Mileykovskaya E, and Dowhan W (2005). Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J. Biol. Chem. 280, 29403–29408. 10.1074/jbc.M504955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daum G (1985). Lipids of Mitochondria. Biochim. Biophys. Acta. 822, 1–42. 10.1016/0304-4157(85)90002-4. [DOI] [PubMed] [Google Scholar]
- 22.Daum G, and Vance JE (1997). Import of lipids into mitochondria. Prog. Lipid Res. 36, 103–130. 10.1016/S0163-7827(97)00006-4. [DOI] [PubMed] [Google Scholar]
- 23.Hovius R, Lambrechts H, Nicolay K, and de Kruijff B (1990). Improved methods to isolate and subfractionate rat liver mitochondria. Lipid composition of the inner and outer membrane. Biochim. Biophys. Acta 1021, 217–226. 10.1016/0005-2736(90)90036-n. [DOI] [PubMed] [Google Scholar]
- 24.Schlame M, Brody S, and Hostetler KY (1993). Mitochondrial cardiolipin in diverse eukaryotes. Comparison of biosynthetic reactions and molecular acyl species. Eur. J. Biochem. 212, 727–735. 10.1111/j.1432-1033.1993.tb17711.x. [DOI] [PubMed] [Google Scholar]
- 25.Schlame M, Horvàth L, and Vìgh L (1990). Relationship between lipid saturation and lipid-protein interaction in liver mitochondria modified by catalytic hydrogenation with reference to cardiolipin molecular species. Biochem. J. 265, 79–85. 10.1042/bj2650079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Claypool SM, Boontheung P, McCaffery JM, Loo JA, and Koehler CM (2008). The Cardiolipin Transacylase, Tafazzin, Associates with Two Distinct Respiratory Components Providing Insight into Barth Syndrome. Mol. Biol. Cell 19, 5143–5155. 10.1091/mbc.e08-09-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khalifat N, Puff N, Bonneau S, Fournier J-B, and Angelova MI (2008). Membrane Deformation under Local pH Gradient: Mimicking Mitochondrial Cristae Dynamics. Biophys. J. 95, 4924–4933. 10.1529/biophysj.108.136077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlame M, and Ren M (2009). The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim. Biophys. Acta 1788, 2080–2083. 10.1016/j.bbamem.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mileykovskaya E, Zhang M, and Dowhan W (2005). Cardiolipin in energy transducing membranes. Biochemistry (Mosc) 70, 154–158. 10.1007/s10541-005-0095-2. [DOI] [PubMed] [Google Scholar]
- 30.Fry M, and Green DE (1981). Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J. Biol. Chem. 256, 1874–1880. [PubMed] [Google Scholar]
- 31.Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, and Schägger H (2003). Cardiolipin Stabilizes Respiratory Chain Supercomplexes. J. Biol. Chem. 278, 52873–52880. 10.1074/jbc.m308366200. [DOI] [PubMed] [Google Scholar]
- 32.Prola A, Blondelle J, Vandestienne A, Piquereau J, Denis RGP, Guyot S, Chauvin H, Mourier A, Maurer M, Henry C, et al. (2021). Cardiolipin content controls mitochondrial coupling and energetic efficiency in muscle. Sci. Adv. 7, eabd6322. 10.1126/sciadv.abd6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russo S, De Rasmo D, Signorile A, Corcelli A, and Lobasso S (2022). Beneficial effects of SS-31 peptide on cardiac mitochondrial dysfunction in tafazzin knockdown mice. Sci. Rep. 12, 19847. 10.1038/s41598-022-24231-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang M, Mileykovskaya E, and Dowhan W (2002). Gluing the Respiratory Chain Together: CARDIOLIPIN IS REQUIRED FOR SUPERCOMPLEX FORMATION IN THE INNER MITOCHONDRIAL MEMBRANE. J. Biol. Chem. 277, 43553–43556. 10.1074/jbc.c200551200. [DOI] [PubMed] [Google Scholar]
- 35.Claypool SM, Oktay Y, Boontheung P, Loo JA, and Koehler CM (2008). Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J. Cell Biol. 182, 937–950. 10.1083/jcb.200801152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mehdipour AR, and Hummer G (2016). Cardiolipin puts the seal on ATP synthase. Proc. Natl. Acad. Sci. USA 113, 8568–8570. 10.1073/pnas.1609806113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Planas-Iglesias J, Dwarakanath H, Mohammadyani D, Yanamala N, Kagan VE, and Klein-Seetharaman J (2015). Cardiolipin Interactions with Proteins. Biophys. J. 109, 1282–1294. 10.1016/j.bpj.2015.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Senoo N, Chinthapalli DK, Baile MG, Golla VK, Saha B, Oluwole AO, Ogunbona OB, Saba JA, Munteanu T, Valdez Y, et al. (2024). Functional diversity among cardiolipin binding sites on the mitochondrial ADP/ATP carrier. EMBO J. 43, 2979–3008. 10.1038/s44318-024-00132-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ban T, Ishihara T, Kohno H, Saita S, Ichimura A, Maenaka K, Oka T, Mihara K, and Ishihara N (2017). Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 19, 856–863. 10.1038/ncb3560. [DOI] [PubMed] [Google Scholar]
- 40.Kameoka S, Adachi Y, Okamoto K, Iijima M, and Sesaki H (2018). Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics. Trends Cell Biol. 28, 67–76. 10.1016/j.tcb.2017.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, and Orrenius S (2002). Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 99, 1259–1263. 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schug ZT, and Gottlieb E (2009). Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim. Biophys. Acta 1788, 2022–2031. 10.1016/j.bbamem.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Mitchell W, Ng EA, Tamucci JD, Boyd KJ, Sathappa M, Coscia A, Pan MX, Han XL, Eddy NA, May ER, et al. (2020). The mitochondria-targeted peptide SS-31 binds lipid bilayers and modulates surface electrostatics as a key component of its mechanism of action. J. Biol. Chem. 295, 7452–7469. 10.1074/jbc.RA119.012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Allen ME, Pennington ER, Perry JB, Dadoo S, Makrecka-Kuka M, Dambrova M, Moukdar F, Patel HD, Han X, Kidd GK, et al. (2020). The cardiolipin-binding peptide elamipretide mitigates fragmentation of cristae networks following cardiac ischemia reperfusion in rats. Commun. Biol. 3, 389. 10.1038/s42003-020-1101-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chavez JD, Tang XT, Campbell MD, Reyes G, Kramer PA, Stuppard R, Keller A, Zhang HL, Rabinovitch PS, Marcinek DJ, et al. (2020). Mitochondrial protein interaction landscape of SS-31. Proc. Natl. Acad. Sci. USA 117, 15363–15373. 10.1073/pnas.2002250117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosivatz E, and Woscholski R (2011). Removal or masking of phosphatidylinositol(4,5)bisphosphate from the outer mitochondrial membrane causes mitochondrial fragmentation. Cell. Signal. 23, 478–486. 10.1016/j.cellsig.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stone SJ, Cui Z, and Vance JE (1998). Cloning and expression of mouse liver phosphatidylserine synthase-1 cDNA. Overexpression in rat hepatoma cells inhibits the CDP-ethanolamine pathway for phosphatidylethanolamine biosynthesis. J. Biol. Chem. 273, 7293–7302. 10.1074/jbc.273.13.7293. [DOI] [PubMed] [Google Scholar]
- 48.Vance JE (1990). Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265, 7248–7256. 10.1016/S0021-9258(19)39106-9. [DOI] [PubMed] [Google Scholar]
- 49.Kennedy EP, and Weiss SB (1956). THE FUNCTION OF CYTIDINE COENZYMES IN THE BIOSYNTHESIS OF PHOSPHOLIPIDES. J. Biol. Chem. 222, 193–214. 10.1016/S0021-9258(19)50785-2. [DOI] [PubMed] [Google Scholar]
- 50.Verkerke ARP, Ferrara PJ, Lin C-T, Johnson JM, Ryan TE, Maschek JA, Eshima H, Paran CW, Laing BT, Siripoksup P, et al. (2019). Phospholipid methylation regulates muscle metabolic rate through Ca2+ transport efficiency. Nat. Metab. 1, 876–885. 10.1038/s42255-019-0111-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vance DE, Walkey CJ, and Cui Z (1997). Phosphatidylethanolamine N-methyltransferase from liver. Biochim. Biophys. Acta 1348, 142–150. 10.1016/s0005-2760(97)00108-2. [DOI] [PubMed] [Google Scholar]
- 52.Arikketh D, Nelson R, and Vance JE (2008). Defining the Importance of Phosphatidylserine Synthase-1 (PSS1): Unexpected Viability of PSS1-Deficient Mice. J. Biol. Chem. 283, 12888–12897. 10.1074/jbc.M800714200. [DOI] [PubMed] [Google Scholar]
- 53.Sousa SB, Jenkins D, Chanudet E, Tasseva G, Ishida M, Anderson G, Docker J, Ryten M, Sa J, Saraiva JM, et al. (2014). Gain-of-function mutations in the phosphatidylserine synthase 1 (PTDSS1) gene cause Lenz-Majewski syndrome. Nat. Genet. 46, 70–76. 10.1038/ng.2829. [DOI] [PubMed] [Google Scholar]
- 54.Hishikawa D, Shindou H, Kobayashi S, Nakanishi H, Taguchi R, and Shimizu T (2008). Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity. Proc. Natl. Acad. Sci. USA 105, 2830–2835. 10.1073/pnas.0712245105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shiao YJ, Lupo G, and Vance JE (1995). Evidence That Phosphatidylserine Is Imported into Mitochondria via a Mitochondria-associated Membrane and That the Majority of Mitochondrial Phosphatidylethanolamine Is Derived from Decarboxylation of Phosphatidylserine. J. Biol. Chem. 270, 11190–11198. 10.1074/jbc.270.19.11190. [DOI] [PubMed] [Google Scholar]
- 56.Steenbergen R, Nanowski TS, Beigneux A, Kulinski A, Young SG, and Vance JE (2005). Disruption of the Phosphatidylserine Decarboxylase Gene in Mice Causes Embryonic Lethality and Mitochondrial Defects. J. Biol. Chem. 280, 40032–40040. 10.1074/jbc.m506510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bergo MO, Gavino BJ, Steenbergen R, Sturbois B, Parlow AF, Sanan DA, Skarnes WC, Vance JE, and Young SG (2002). Defining the Importance of Phosphatidylserine Synthase 2 in Mice. J. Biol. Chem. 277, 47701–47708. 10.1074/jbc.m207734200. [DOI] [PubMed] [Google Scholar]
- 58.Potting C, Tatsuta T, König T, Haag M, Wai T, Aaltonen MJ, and Langer T (2013). TRIAP1/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid. Cell Metab. 18, 287–295. 10.1016/j.cmet.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 59.Zhang J, Guan ZQ, Murphy AN, Wiley SE, Perkins GA, Worby CA, Engel JL, Heacock P, Nguyen OK, Wang JH, et al. (2011). Mitochondrial Phosphatase PTPMT1 Is Essential for Cardiolipin Biosynthesis. Cell Metab. 13, 690–700. 10.1016/j.cmet.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hostetler KY, Van den Bosch H, and Van Deenen LL (1971). Biosynthesis of cardiolipin in liver mitochondria. Biochim. Biophys. Acta 239, 113–119. 10.1016/0005-2760(71)90201-3. [DOI] [PubMed] [Google Scholar]
- 61.Bao MHR, Yang CX, Tse APW, Wei L, Lee D, Zhang MS, Goh CC, Chiu DKC, Yuen VWH, Law CT, et al. (2021). Genome-wide CRISPR-Cas9 knockout library screening identified PTPMT1 in cardiolipin synthesis is crucial to survival in hypoxia in liver cancer. Cell Rep. 34, 108676. 10.1016/j.celrep.2020.108676. [DOI] [PubMed] [Google Scholar]
- 62.Huang XD, Xiao FJ, Guo YT, Sun Y, Zhang YK, and Shi XJ (2022). Protein tyrosine phosphatase 1 protects human pancreatic cancer from erastin-induced ferroptosis. Asian J. Surg. 45, 2214–2223. 10.1016/j.asjsur.2021.11.048. [DOI] [PubMed] [Google Scholar]
- 63.Pagliarini DJ, Wiley SE, Kimple ME, Dixon JR, Kelly P, Worby CA, Casey PJ, and Dixon JE (2005). Involvement of a mitochondrial phosphatase in the regulation of ATP production and insulin secretion in pancreatic beta cells. Mol. Cell 19, 197–207. 10.1016/j.molcel.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 64.Liu C, Kraja AT, Smith JA, Brody JA, Franceschini N, Bis JC, Rice K, Morrison AC, Lu Y, Weiss S, et al. (2016). Meta-analysis identifies common and rare variants influencing blood pressure and overlapping with metabolic trait loci. Nat. Genet. 48, 1162–1170. 10.1038/ng.3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Russomanno G, Jo KB, Abdul-Salam VB, Morgan C, Endruschat J, Schaeper U, Osman AH, Alzaydi MM, Wilkins MR, and Wojciak-Stothard B (2021). miR-150-PTPMT1-cardiolipin signaling in pulmonary arterial hypertension. Mol. Ther. Nucleic Acids 23, 142–153. 10.1016/j.omtn.2020.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schlame M, Ren M, Xu Y, Greenberg ML, and Haller I (2005). Molecular symmetry in mitochondrial cardiolipins. Chem. Phys. Lipids 138, 38–49. 10.1016/j.chemphyslip.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 67.Ma BJ, Taylor WA, Dolinsky VW, and Hatch GM (1999). Acylation of monolysocardiolipin in rat heart. J. Lipid Res. 40, 1837–1845. 10.1016/S0022-2275(20)34900-2. [DOI] [PubMed] [Google Scholar]
- 68.Cao J, Liu Y, Lockwood J, Burn P, and Shi Y (2004). A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA:lysocardiolipin acyltransferase (ALCAT1) in mouse. J. Biol. Chem. 279, 31727–31734. 10.1074/jbc.m402930200. [DOI] [PubMed] [Google Scholar]
- 69.Taylor WA, and Hatch GM (2003). Purification and characterization of monolysocardiolipin acyltransferase from pig liver mitochondria. J. Biol. Chem. 278, 12716–12721. 10.1074/jbc.m210329200. [DOI] [PubMed] [Google Scholar]
- 70.Bione S, D’Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, and Toniolo D (1996). A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 12, 385–389. 10.1038/ng0496-385. [DOI] [PubMed] [Google Scholar]
- 71.Schlame M, Kelley RI, Feigenbaum A, Towbin JA, Heerdt PM, Schieble T, Wanders RJA, DiMauro S, and Blanck TJJ (2003). Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 42, 1994–1999. 10.1016/j.jacc.2003.06.015. [DOI] [PubMed] [Google Scholar]
- 72.Xu Y, Malhotra A, Ren M, and Schlame M (2006). The enzymatic function of tafazzin. J. Biol. Chem. 281, 39217–39224. 10.1074/jbc.M606100200. [DOI] [PubMed] [Google Scholar]
- 73.Schlame M, Xu Y, and Ren M (2017). The Basis for Acyl Specificity in the Tafazzin Reaction. J. Biol. Chem. 292, 5499–5506. 10.1074/jbc.M116.769182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schlame M, Acehan D, Berno B, Xu Y, Valvo S, Ren M, Stokes DL, and Epand RM (2012). The physical state of lipid substrates provides transacylation specificity for tafazzin. Nat. Chem. Biol. 8, 862–869. 10.1038/nchembio.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kasahara T, Kubota-Sakashita M, Nagatsuka Y, Hirabayashi Y, Hanasaka T, Tohyama K, and Kato T (2020). Cardiolipin is essential for early embryonic viability and mitochondrial integrity of neurons in mammals. FASEB J. 34, 1465–1480. 10.1096/fj.201901598R. [DOI] [PubMed] [Google Scholar]
- 76.Acehan D, Vaz F, Houtkooper RH, James J, Moore V, Tokunaga C, Kulik W, Wansapura J, Toth MJ, Strauss A, et al. (2011). Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J. Biol. Chem. 286, 899–908. 10.1074/jbc.M110.171439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Soustek MS, Falk DJ, Mah CS, Toth MJ, Schlame M, Lewin AS, and Byrne BJ (2011). Characterization of a transgenic short hairpin RNA-induced murine model of Tafazzin deficiency. Hum. Gene Ther. 22, 865–871. 10.1089/hum.2010.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang S, Li Y, Xu Y, Ma Q, Lin Z, Schlame M, Bezzerides VJ, Strathdee D, and Pu WT (2020). AAV Gene Therapy Prevents and Reverses Heart Failure in a Murine Knockout Model of Barth Syndrome. Circ. Res. 126, 1024–1039. 10.1161/CIRCRESAHA.119.315956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tomczewski MV, Chan JZ, Campbell ZE, Strathdee D, and Duncan RE (2023). Phenotypic Characterization of Male Tafazzin-Knockout Mice at 3, 6, and 12 Months of Age. Biomedicines 11, 638. 10.3390/biomedicines11020638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cole LK, Mejia EM, Vandel M, Sparagna GC, Claypool SM, Dyck-Chan L, Klein J, and Hatch GM (2016). Impaired Cardiolipin Biosynthesis Prevents Hepatic Steatosis and Diet-Induced Obesity. Diabetes 65, 3289–3300. 10.2337/db16-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee RG, Balasubramaniam S, Stentenbach M, Kralj T, McCubbin T, Padman B, Smith J, Riley LG, Priyadarshi A, Peng L, et al. (2022). Deleterious variants in CRLS1 lead to cardiolipin deficiency and cause an autosomal recessive multi-system mitochondrial disease. Hum. Mol. Genet. 31, 3597–3612. 10.1093/hmg/ddac040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Barth PG, Scholte HR, Berden JA, Van Der Klei-Van Moorsel JM, Luyt-Houwen IEM, Van ‘t Veer-Korthof ET, Van Der Harten JJ, and Sobotka-Plojhar MA (1983). An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 62, 327–355. 10.1016/0022-510X(83)90209-5. [DOI] [PubMed] [Google Scholar]
- 83.Dolgin E (2019). How secret conversations inside cells are transforming biology. Nature 567, 162–164. 10.1038/d41586-019-00792-9. [DOI] [PubMed] [Google Scholar]
- 84.Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, and Pozzan T (1998). Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766. 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- 85.Csordás G, Renken C, Várnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, and Hajnóczky G (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921. 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Riekhof WR, Wu J, Jones JL, and Voelker DR (2007). Identification and characterization of the major lysophosphatidylethanolamine acyltransferase in Saccharomyces cerevisiae. J. Biol. Chem. 282, 28344–28352. 10.1074/jbc.M705256200. [DOI] [PubMed] [Google Scholar]
- 87.Riekhof WR, and Voelker DR (2006). Uptake and utilization of lysophosphatidylethanolamine by Saccharomyces cerevisiae. J. Biol. Chem. 281, 36588–36596. 10.1074/jbc.M608851200. [DOI] [PubMed] [Google Scholar]
- 88.Suomalainen A, and Nunnari J (2024). Mitochondria at the crossroads of health and disease. Cell 187, 2601–2627. 10.1016/j.cell.2024.04.037. [DOI] [PubMed] [Google Scholar]
- 89.Guillén-Samander A, Leonzino M, Hanna MG, Tang N, Shen H, and De Camilli P (2021). VPS13D bridges the ER to mitochondria and peroxisomes via Miro. J. Cell Biol. 220, e202010004. 10.1083/jcb.202010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Leonzino M, Reinisch KM, and De Camilli P (2021). Insights into VPS13 properties and function reveal a new mechanism of eukaryotic lipid transport. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1866, 159003. 10.1016/j.bbalip.2021.159003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim H, Lee S, Jun Y, and Lee C (2022). Structural basis for mitoguardin-2 mediated lipid transport at ER-mitochondrial membrane contact sites. Nat. Commun. 13, 3702. 10.1038/s41467-022-31462-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hansen FM, Kremer LS, Karayel O, Bludau I, Larsson NG, Kühl I, and Mann M (2024). Mitochondrial phosphoproteomes are functionally specialized across tissues. Life Sci. Alliance 7, 7. 10.26508/lsa.202302147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mukherjee I, Ghosh M, and Meinecke M (2021). MICOS and the mitochondrial inner membrane morphology - when things get out of shape. FEBS Lett. 595, 1159–1183. 10.1002/1873-3468.14089. [DOI] [PubMed] [Google Scholar]
- 94.Eramo MJ, Lisnyak V, Formosa LE, and Ryan MT (2020). The ‘mitochondrial contact site and cristae organising system’ (MICOS) in health and human disease. J. Biochem. 167, 243–255. 10.1093/jb/mvz111. [DOI] [PubMed] [Google Scholar]
- 95.Chatzispyrou IA, Guerrero-Castillo S, Held NM, Ruiter JPN, Denis SW, IJlst L, Wanders RJ, van Weeghel M, Ferdinandusse S, Vaz FM, et al. (2018). Barth syndrome cells display widespread remodeling of mitochondrial complexes without affecting metabolic flux distribution. Biochim. Biophys. Acta Mol. Basis Dis 1864, 3650–3658. 10.1016/j.bbadis.2018.08.041. [DOI] [PubMed] [Google Scholar]
- 96.Van Strien J, Guerrero-Castillo S, Chatzispyrou IA, Houtkooper RH, Brandt U, and Huynen MA (2019). COmplexome Profiling ALignment (COPAL) reveals remodeling of mitochondrial protein complexes in Barth syndrome. Bioinformatics 35, 3083–3091. 10.1093/bioinformatics/btz025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Koob S, Barrera M, Anand R, and Reichert AS (2015). The non-glycosylated isoform of MIC26 is a constituent of the mammalian MICOS complex and promotes formation of crista junctions. Biochim. Biophys. Acta 1853, 1551–1563. 10.1016/j.bbamcr.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 98.Weber TA, Koob S, Heide H, Wittig I, Head B, van der Bliek A, Brandt U, Mittelbronn M, and Reichert AS (2013). APOOL is a cardiolipin-binding constituent of the Mitofilin/MINOS protein complex determining cristae morphology in mammalian mitochondria. PLoS One 8, e63683. 10.1371/journal.pone.0063683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Flores-Martin J, Rena V, Angeletti S, Panzetta-Dutari GM, and Genti-Raimondi S (2013). The Lipid Transfer Protein StarD7: Structure, Function, and Regulation. Int. J. Mol. Sci. 14, 6170–6186. 10.3390/ijms14036170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Horibata Y, and Sugimoto H (2010). StarD7 Mediates the Intracellular Trafficking of Phosphatidylcholine to Mitochondria. J. Biol. Chem. 285, 7358–7365. 10.1074/jbc.M109.056960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Murari A, Rhooms SK, Vimal D, Hossain KFB, Saini S, Villanueva M, Schlame M, and Owusu-Ansah E (2023). Phospholipids can regulate complex I assembly independent of their role in maintaining mitochondrial membrane integrity. Cell Rep. 42, 112846. 10.1016/j.celrep.2023.112846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Horibata Y, Ando H, Zhang P, Vergnes L, Aoyama C, Itoh M, Reue K, and Sugimoto H (2016). StarD7 Protein Deficiency Adversely Affects the Phosphatidylcholine Composition, Respiratory Activity, and Cristae Structure of Mitochondria. J. Biol. Chem. 291, 24880–24891. 10.1074/jbc.M116.736793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yang L, Lewkowich I, Apsley K, Fritz JM, Wills-Karp M, and Weaver TE (2015). Haploinsufficiency for Stard7 Is Associated with Enhanced Allergic Responses in Lung and Skin. J. Immunol. 194, 5635–5643. 10.4049/jimmunol.1500231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang L, Na CL, Luo S, Wu D, Hogan S, Huang T, and Weaver TE (2017). The Phosphatidylcholine Transfer Protein Stard7 is Required for Mitochondrial and Epithelial Cell Homeostasis. Sci. Rep. 7, 46416. 10.1038/srep46416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Corbett MA, Kroes T, Veneziano L, Bennett MF, Florian R, Schneider AL, Coppola A, Licchetta L, Franceschetti S, Suppa A, et al. (2019). Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat. Commun. 10, 4920. 10.1038/s41467-019-12671-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Osman C, Haag M, Potting C, Rodenfels J, Dip PV, Wieland FT, Brügger B, Westermann B, and Langer T (2009). The genetic interactome of prohibitins: coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J. Cell Biol. 184, 583–596. 10.1083/jcb.200810189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Connerth M, Tatsuta T, Haag M, Klecker T, Westermann B, and Langer T (2012). Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science 338, 815–818. 10.1126/science.1225625. [DOI] [PubMed] [Google Scholar]
- 108.Dee CT, and Moffat KG (2005). A novel family of mitochondrial proteins is represented by the Drosophila genes slmo, preli-like and real-time. Dev. Genes Evol. 215, 248–254. 10.1007/s00427-005-0470-4. [DOI] [PubMed] [Google Scholar]
- 109.Miliara X, Garnett JA, Tatsuta T, Abid Ali F, Baldie H, Pérez-Dorado I, Simpson P, Yague E, Langer T, and Matthews S (2015). Structural insight into the TRIAP1/PRELI-like domain family of mitochondrial phospholipid transfer complexes. EMBO Rep. 16, 824–835. 10.15252/embr.201540229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schlattner U, Tokarska-Schlattner M, Epand RM, Boissan M, Lacombe M-L, and Kagan VE (2018). NME4/nucleoside diphosphate kinase D in cardiolipin signaling and mitophagy. Lab. Invest. 98, 228–232. 10.1038/labinvest.2017.113. [DOI] [PubMed] [Google Scholar]
- 111.Aagaard Nolting L, Holling T, Nishimura G, Ek J, Bak M, Ljungberg M, Kutsche K, and Hove H (2024). Novel biallelic PISD missense variants cause spondyloepimetaphyseal dysplasia with disproportionate short stature and fragmented mitochondrial morphology. Published online May 27, 2024. Clin. Genet. 10.1111/cge.14549. [DOI] [PubMed] [Google Scholar]
- 112.Girisha KM, von Elsner L, Neethukrishna K, Muranjan M, Shukla A, Bhavani GS, Nishimura G, Kutsche K, and Mortier G (2019). The homozygous variant c.797G>A/p.(Cys266Tyr) in PISD is associated with a Spondyloepimetaphyseal dysplasia with large epiphyses and disturbed mitochondrial function. Hum. Mutat. 40, 299–309. 10.1002/humu.23693. [DOI] [PubMed] [Google Scholar]
- 113.Peter VG, Quinodoz M, Pinto-Basto J, Sousa SB, Di Gioia SA, Soares G, Ferraz Leal G, Silva ED, Pescini Gobert R, Miyake N, et al. (2019). The Liberfarb syndrome, a multisystem disorder affecting eye, ear, bone, and brain development, is caused by a founder pathogenic variant in thePISD gene. Genet. Med. 21, 2734–2743. 10.1038/s41436-019-0595-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhao T, Goedhart CM, Sam PN, Sabouny R, Lingrell S, Cornish AJ, Lamont RE, Bernier FP, Sinasac D, Parboosingh JS, et al. (2019). PISD is a mitochondrial disease gene causing skeletal dysplasia, cataracts, and white matter changes. Life Sci. Alliance 2, 2. 10.26508/lsa.201900353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Haghighi A, Haack TB, Atiq M, Mottaghi H, Haghighi-Kakhki H, Bashir RA, Ahting U, Feichtinger RG, Mayr JA, Rötig A, et al. (2014). Sengers syndrome: six novel AGK mutations in seven new families and review of the phenotypic and mutational spectrum of 29 patients. Orphanet J. Rare Dis. 9, 119. 10.1186/s13023-014-0119-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mayr JA, Haack TB, Graf E, Zimmermann FA, Wieland T, Haberberger B, Superti-Furga A, Kirschner J, Steinmann B, Baumgartner MR, et al. (2012). Lack of the Mitochondrial Protein Acylglycerol Kinase Causes Sengers Syndrome. Am. J. Hum. Genet. 90, 314–320. 10.1016/j.ajhg.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sengers RC, Trijbels JM, Willems JL, Daniels O, and Stadhouders AM (1975). Congenital cataract and mitochondrial myopathy of skeletal and heart muscle associated with lactic acidosis after exercise. J. Pediatr. 86, 873–880. 10.1016/s0022-3476(75)80217-4. [DOI] [PubMed] [Google Scholar]
- 118.Lenz WD, and Majewski F (1974). A generalized disorders of the connective tissues with progeria, choanal atresia, symphalangism, hypoplasia of dentine and craniodiaphyseal hypostosis. Birth Defects Orig. Artic. Ser. 10, 133–136. [PubMed] [Google Scholar]
- 119.Clarke SLN, Bowron A, Gonzalez IL, Groves SJ, Newbury-Ecob R, Clayton N, Martin RP, Tsai-Goodman B, Garratt V, Ashworth M, et al. (2013). Barth syndrome. Orphanet J. Rare Dis. 8, 23. 10.1186/1750-1172-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Spencer CT, Bryant RM, Day J, Gonzalez IL, Colan SD, Thompson WR, Berthy J, Redfearn SP, and Byrne BJ (2006). Cardiac and clinical phenotype in Barth syndrome. Pediatrics 118, e337–e346. 10.1542/peds.2005-2667. [DOI] [PubMed] [Google Scholar]
- 121.Sparagna GC, Chicco AJ, Murphy RC, Bristow MR, Johnson CA, Rees ML, Maxey ML, McCune SA, and Moore RL (2007). Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J. Lipid Res. 48, 1559–1570. 10.1194/jlr.M600551-JLR200. [DOI] [PubMed] [Google Scholar]
- 122.Fox CA, Ellison P, Ikon N, and Ryan RO (2019). Calcium-induced transformation of cardiolipin nanodisks. Biochim. Biophys. Acta Biomembr. 1861, 1030–1036. 10.1016/j.bbamem.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJA, and Barth PG (2000). Defective Remodeling of Cardiolipin and Phosphatidylglycerol in Barth Syndrome. Biochem. Biophys. Res. Commun. 279, 378–382. 10.1006/bbrc.2000.3952. [DOI] [PubMed] [Google Scholar]
- 124.Xu Y, Erdjument-Bromage H, Phoon CKL, Neubert TA, Ren M, and Schlame M (2021). Cardiolipin remodeling enables protein crowding in the inner mitochondrial membrane. EMBO J. 40, e108428. 10.15252/embj.2021108428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Striano P, Caranci F, Di Benedetto R, Tortora F, Zara F, and Striano S (2009). (1)H-MR spectroscopy indicates prominent cerebellar dysfunction in benign adult familial myoclonic epilepsy. Epilepsia 50, 1491–1497. 10.1111/j.1528-1167.2008.01900.x. [DOI] [PubMed] [Google Scholar]
- 126.Kang Y, Stroud DA, Baker MJ, De Souza DP, Frazier AE, Liem M, Tull D, Mathivanan S, McConville MJ, Thorburn DR, et al. (2017). Sengers Syndrome-Associated Mitochondrial Acylglycerol Kinase Is a Subunit of the Human TIM22 Protein Import Complex. Mol. Cell 67, 457–470.e5. 10.1016/j.molcel.2017.06.014. [DOI] [PubMed] [Google Scholar]
- 127.Nishijima M, Kuge O, and Akamatsu Y (1986). Phosphatidylserine biosynthesis in cultured Chinese hamster ovary cells. I. Inhibition of de novo phosphatidylserine biosynthesis by exogenous phosphatidylserine and its efficient incorporation. J. Biol. Chem. 261, 5784–5789. [PubMed] [Google Scholar]
- 128.Kuge O, Saito K, and Nishijima M (1999). Control of phosphatidylserine synthase II activity in Chinese hamster ovary cells. J. Biol. Chem. 274, 23844–23849. 10.1074/jbc.274.34.23844. [DOI] [PubMed] [Google Scholar]
- 129.Kuge O, Nishijima M, and Akamatsu Y (1986). Phosphatidylserine biosynthesis in cultured Chinese hamster ovary cells. III. Genetic evidence for utilization of phosphatidylcholine and phosphatidylethanolamine as precursors. J. Biol. Chem. 261, 5795–5798. [PubMed] [Google Scholar]
- 130.Memme JM, Erlich AT, Phukan G, and Hood DA (2021). Exercise and mitochondrial health. J. Physiol. 599, 803–817. 10.1113/JP278853. [DOI] [PubMed] [Google Scholar]
- 131.Pinto AJ, Bergouignan A, Dempsey PC, Roschel H, Owen N, Gualano B, and Dunstan DW (2023). Physiology of sedentary behavior. Physiol. Rev. 103, 2561–2622. 10.1152/physrev.00022.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Heden TD, Johnson JM, Ferrara PJ, Eshima H, Verkerke ARP, Wentzler EJ, Siripoksup P, Narowski TM, Coleman CB, Lin C-T, et al. (2019). Mitochondrial PE potentiates respiratory enzymes to amplify skeletal muscle aerobic capacity. Sci. Adv. 5, eaax8352. 10.1126/sciadv.aax8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wisløff U, Najjar SM, Ellingsen O, Haram PM, Swoap S, Al-Share Q, Fernström M, Rezaei K, Lee SJ, Koch LG, et al. (2005). Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science 307, 418–420. 10.1126/science.1108177. [DOI] [PubMed] [Google Scholar]
- 134.Siripoksup P, Cao G, Cluntun AA, Maschek JA, Pearce Q, Broth-well MJ, Jeong MY, Eshima H, Ferrara PJ, Opurum PC, et al. (2024). Sedentary behavior in mice induces metabolic inflexibility by suppressing skeletal muscle pyruvate metabolism. J. Clin. Invest. 134, e167371. 10.1172/JCI167371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, Herder C, Carstensen M, Krausch M, Knoefel WT, et al. (2015). Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 21, 739–746. 10.1016/j.cmet.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 136.Peng KY, Watt MJ, Rensen S, Greve JW, Huynh K, Jayawardana KS, Meikle PJ, and Meex RCR (2018). Mitochondrial dysfunction-related lipid changes occur in nonalcoholic fatty liver disease progression. J. Lipid Res. 59, 1977–1986. 10.1194/jlr.M085613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tu C, Xiong H, Hu Y, Wang W, Mei G, Wang H, Li Y, Zhou Z, Meng F, Zhang P, et al. (2020). Cardiolipin Synthase 1 Ameliorates NASH Through Activating Transcription Factor 3 Transcriptional Inactivation. Hepatology 72, 1949–1967. 10.1002/hep.31202. [DOI] [PubMed] [Google Scholar]
- 138.Paradies G, Paradies V, Ruggiero FM, and Petrosillo G (2014). Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 20, 14205–14218. 10.3748/wjg.v20.i39.14205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Petrosillo G, Portincasa P, Grattagliano I, Casanova G, Matera M, Ruggiero FM, Ferri D, and Paradies G (2007). Mitochondrial dysfunction in rat with nonalcoholic fatty liver Involvement of complex I, reactive oxygen species and cardiolipin. Biochim. Biophys. Acta 1767, 1260–1267. 10.1016/j.bbabio.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 140.Rutter GA, Sidarala V, Kaufman BA, and Soleimanpour SA (2023). Mitochondrial metabolism and dynamics in pancreatic beta cell glucose sensing. Biochem. J. 480, 773–789. 10.1042/BCJ20230167. [DOI] [PubMed] [Google Scholar]
- 141.Cole LK, Agarwal P, Doucette CA, Fonseca M, Xiang B, Sparagna GC, Seshadri N, Vandel M, Dolinsky VW, and Hatch GM (2021). Tafazzin Deficiency Reduces Basal Insulin Secretion and Mitochondrial Function in Pancreatic Islets From Male Mice. Endocrinology 162, bqab102. 10.1210/endocr/bqab102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhao Z, Zhang X, Zhao C, Choi J, Shi J, Song K, Turk J, and Ma ZA (2010). Protection of pancreatic beta-cells by group VIA phospholipase A(2)-mediated repair of mitochondrial membrane peroxidation. Endocrinology 151, 3038–3048. 10.1210/en.2010-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Oemer G, Lackner K, Muigg K, Krumschnabel G, Watschinger K, Sailer S, Lindner H, Gnaiger E, Wortmann SB, Werner ER, et al. (2018). Molecular structural diversity of mitochondrial cardiolipins. Proc. Natl. Acad. Sci. USA 115, 4158–4163. 10.1073/pnas.1719407115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Amoscato AA, Sparvero LJ, He RR, Watkins S, Bayir H, and Kagan VE (2014). Imaging mass spectrometry of diversified cardiolipin molecular species in the brain. Anal. Chem. 86, 6587–6595. 10.1021/ac5011876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Camilleri A, Ghio S, Caruana M, Weckbecker D, Schmidt F, Kamp F, Leonov A, Ryazanov S, Griesinger C, Giese A, et al. (2020). Tau-induced mitochondrial membrane perturbation is dependent upon cardiolipin. Biochim. Biophys. Acta Biomembr. 1862, 183064. 10.1016/j.bbamem.2019.183064. [DOI] [PubMed] [Google Scholar]
- 146.Monteiro-Cardoso VF, Oliveira MM, Melo T, Domingues MRM, Moreira PI, Ferreiro E, Peixoto F, and Videira RA (2015). Cardiolipin profile changes are associated to the early synaptic mitochondrial dysfunction in Alzheimer’s disease. J. Alzheimers Dis. 43, 1375–1392. 10.3233/JAD-141002. [DOI] [PubMed] [Google Scholar]
- 147.Gilmozzi V, Gentile G, Castelo Rueda MP, Hicks AA, Pramstaller PP, Zanon A, Lévesque M, and Pichler I (2020). Interaction of Alpha-Synuclein With Lipids: Mitochondrial Cardiolipin as a Critical Player in the Pathogenesis of Parkinson’s Disease. Front. Neurosci. 14, 578993. 10.3389/fnins.2020.578993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Middleton ER, and Rhoades E (2010). Effects of curvature and composition on alpha-synuclein binding to lipid vesicles. Biophys. J. 99, 2279–2288. 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nakamura K, Nemani VM, Wallender EK, Kaehlcke K, Ott M, and Edwards RH (2008). Optical reporters for the conformation of alpha-synuclein reveal a specific interaction with mitochondria. J. Neurosci. 28, 12305–12317. 10.1523/JNEUROSCI.3088-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, Castaño-Díez D, Schweighauser G, Graff-Meyer A, Goldie KN, et al. (2019). Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 22, 1099–1109. 10.1038/s41593-019-0423-2. [DOI] [PubMed] [Google Scholar]
- 151.Song C, Zhang J, Qi S, Liu Z, Zhang X, Zheng Y, Andersen JP, Zhang W, Strong R, Martinez PA, et al. (2019). Cardiolipin remodeling by ALCAT1 links mitochondrial dysfunction to Parkinson’s diseases. Aging Cell 18, e12941. 10.1111/acel.12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zigoneanu IG, Yang YJ, Krois AS, Haque E, and Pielak GJ (2012). Interaction of alpha-synuclein with vesicles that mimic mitochondrial membranes. Biochim. Biophys. Acta 1818, 512–519. 10.1016/j.bbamem.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ghio S, Camilleri A, Caruana M, Ruf VC, Schmidt F, Leonov A, Ryazanov S, Griesinger C, Cauchi RJ, Kamp F, et al. (2019). Cardiolipin Promotes Pore-Forming Activity of Alpha-Synuclein Oligomers in Mitochondrial Membranes. ACS Chem. Neurosci. 10, 3815–3829. 10.1021/acschemneuro.9b00320. [DOI] [PubMed] [Google Scholar]
- 154.Petit JM, Maftah A, Ratinaud MH, and Julien R (1992). 10N-nonyl acridine orange interacts with cardiolipin and allows the quantification of this phospholipid in isolated mitochondria. Eur. J. Biochem. 209, 267–273. 10.1111/j.1432-1033.1992.tb17285.x. [DOI] [PubMed] [Google Scholar]
- 155.Cole NB, Dieuliis D, Leo P, Mitchell DC, and Nussbaum RL (2008). Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp. Cell Res. 314, 2076–2089. 10.1016/j.yexcr.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Devi L, Raghavendran V, Prabhu BM, Avadhani NG, and Ananda-theerthavarada HK (2008). Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283, 9089–9100. 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lin Y, Li X, Dai M, Li Q, Shi Q, Zhang L, Huang R, Song C, and Jin S (2022). Sex Differences of Cardiolipin in Tissue Distribution Based on Targeted Lipidomic Analysis by UHPLC-QTOF-MS/MS. Molecules 27, 6988. 10.3390/molecules27206988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Song H, Wohltmann M, Bao S, Ladenson JH, Semenkovich CF, and Turk J (2010). Mice deficient in group VIB phospholipase A2 (iPLA2-gamma) exhibit relative resistance to obesity and metabolic abnormalities induced by a Western diet. Am. J. Physiol. Endocrinol. Metab. 298, E1097–E1114. 10.1152/ajpendo.00780.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Han X, Yang K, Yang J, Cheng H, and Gross RW (2006). Shotgun lipidomics of cardiolipin molecular species in lipid extracts of biological samples. J. Lipid Res. 47, 864–879. 10.1194/jlr.D500044-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.McLaughlin KL, Hagen JT, Coalson HS, Nelson MAM, Kew KA, Wooten AR, and Fisher-Wellman KH (2020). Novel approach to quantify mitochondrial content and intrinsic bioenergetic efficiency across organs. Sci. Rep. 10, 17599. 10.1038/s41598-020-74718-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Fisher-Wellman KH, Davidson MT, Narowski TM, Lin CT, Koves TR, and Muoio DM (2018). Mitochondrial Diagnostics: A Multiplexed Assay Platform for Comprehensive Assessment of Mitochondrial Energy Fluxes. Cell Rep. 24, 3593–3606.e10. 10.1016/j.celrep.2018.08.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Funai K, Summers SA, and Rutter J (2020). Reign in the membrane: How common lipids govern mitochondrial function. Curr. Opin. Cell Biol. 63, 162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bertholet AM, Chouchani ET, Kazak L, Angelin A, Fedorenko A, Long JZ, Vidoni S, Garrity R, Cho J, Terada N, et al. (2019). H+ transport is an integral function of the mitochondrial ADP/ATP carrier. Nature 571, 515–520. 10.1038/s41586-019-1400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Brand MD (2016). Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 100, 14–31. 10.1016/j.freeradbiomed.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 165.Segawa M, Wolf DM, Hultgren NW, Williams DS, van der Bliek AM, Shackelford DB, Liesa M, and Shirihai OS (2020). Quantification of cristae architecture reveals time-dependent characteristics of individual mitochondria. Life Sci. Alliance 3, 3. 10.26508/lsa.201900620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Balderas E, Eberhardt DR, Lee S, Pleinis JM, Sommakia S, Balynas AM, Yin X, Parker MC, Maguire CT, Cho S, et al. (2022). Mitochondrial calcium uniporter stabilization preserves energetic homeostasis during Complex I impairment. Nat. Commun. 13, 2769. 10.1038/s41467-022-30236-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Spencer CT, Byrne BJ, Bryant RM, Margossian R, Maisenbacher M, Breitenger P, Benni PB, Redfearn S, Marcus E, and Cade WT (2011). Impaired cardiac reserve and severely diminished skeletal muscle O2 utilization mediate exercise intolerance in Barth syndrome. Am. J. Physiol. Heart Circ. Physiol. 301, H2122–H2129. 10.1152/ajpheart.00479.2010. [DOI] [PubMed] [Google Scholar]
- 168.Johnson JM, Ferrara PJ, Verkerke ARP, Coleman CB, Wentzler EJ, Neufer PD, Kew KA, de Castro Brás LE, and Funai K (2018). Targeted overexpression of catalase to mitochondria does not prevent cardioskeletal myopathy in Barth syndrome. J. Mol. Cell. Cardiol. 121, 94–102. 10.1016/j.yjmcc.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A, Yuan H, Jiang D, Zhang D, Zangi L, et al. (2014). Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 20, 616–623. 10.1038/nm.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ferrara PJ, Lang MJ, Johnson JM, Watanabe S, McLaughlin KL, Maschek JA, Verkerke ARP, Siripoksup P, Chaix A, Cox JE, et al. (2023). Weight loss increases skeletal muscle mitochondrial energy efficiency in obese mice. Life Metab 2, load014. 10.1093/lifemeta/load014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Johnson JM, Peterlin AD, Balderas E, Sustarsic EG, Maschek JA, Lang MJ, Jara-Ramos A, Panic V, Morgan JT, Villanueva CJ, et al. (2023). Mitochondrial phosphatidylethanolamine modulates UCP1 to promote brown adipose thermogenesis. Sci. Adv. 9, eade7864. 10.1126/sciadv.ade7864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Peyta L, Jarnouen K, Pinault M, Guimaraes C, Pais de Barros JP, Chevalier S, Dumas JF, Maillot F, Hatch GM, Loyer P, et al. (2016). Reduced cardiolipin content decreases respiratory chain capacities and increases ATP synthesis yield in the human HepaRG cells. Biochim. Biophys. Acta 1857, 443–453. 10.1016/j.bbabio.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 173.Siripoksup P, Cao G, Cluntun AA, Maschek JA, Pearce Q, Lang MJ, Eshima H, Opurum PC, Mahmassani ZS, Taylor EB, et al. (2022). Phosphatidylethanolamine facilitates mitochondrial pyruvate entry to regulate metabolic flexibility. Preprint at bioRxiv. 10.1101/2022.11.01.514767. [DOI] [Google Scholar]
- 174.Cannon B, and Nedergaard J (2004). Brown adipose tissue: function and physiological significance. Physiol. Rev. 84, 277–359. 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 175.Oelkrug R, Polymeropoulos ET, and Jastroch M (2015). Brown adipose tissue: physiological function and evolutionary significance. J. Comp. Physiol. B 185, 587–606. 10.1007/s00360-015-0907-7. [DOI] [PubMed] [Google Scholar]
- 176.Lee Y, Willers C, Kunji ERS, and Crichton PG (2015). Uncoupling protein 1 binds one nucleotide per monomer and is stabilized by tightly bound cardiolipin. Proc. Natl. Acad. Sci. USA 112, 6973–6978. 10.1073/pnas.1503833112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sustarsic EG, Ma T, Lynes MD, Larsen M, Karavaeva I, Havelund JF, Nielsen CH, Jedrychowski MP, Moreno-Torres M, Lundh M, et al. (2018). Cardiolipin Synthesis in Brown and Beige Fat Mitochondria Is Essential for Systemic Energy Homeostasis. Cell Metab. 28, 159–174.e11. 10.1016/j.cmet.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jovanovic O, Pashkovskaya AA, Annibal A, Vazdar M, Burchardt N, Sansone A, Gille L, Fedorova M, Ferreri C, and Pohl EE (2015). The molecular mechanism behind reactive aldehyde action on transmembrane translocations of proton and potassium ions. Free Radic. Biol. Med. 89, 1067–1076. 10.1016/j.freeradbiomed.2015.10.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Szczepanek K, Allegood J, Aluri H, Hu Y, Chen Q, and Lesnefsky EJ (2016). Acquired deficiency of tafazzin in the adult heart: Impact on mitochondrial function and response to cardiac injury. Biochim. Biophys. Acta 1861, 294–300. 10.1016/j.bbalip.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 180.Goncalves RLS, Schlame M, Bartelt A, Brand MD, and Hotamış lıgil GS (2021). Cardiolipin deficiency in Barth syndrome is not associated with increased superoxide/H2O2 production in heart and skeletal muscle mitochondria. FEBS Lett. 595, 415–432. 10.1002/1873-3468.13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gu J, Wu M, Guo R, Yan K, Lei J, Gao N, and Yang M (2016). The architecture of the mammalian respirasome. Nature 537, 639–643. 10.1038/nature19359. [DOI] [PubMed] [Google Scholar]
- 182.Schägger H, and Pfeiffer K (2001). The ratio of oxidative phosphorylation complexes I–V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J. Biol. Chem. 276, 37861–37867. 10.1074/jbc.M106474200. [DOI] [PubMed] [Google Scholar]
- 183.Milenkovic D, Misic J, Hevler JF, Molinié T, Chung I, Atanassov I, Li X, Filograna R, Mesaros A, Mourier A, et al. (2023). Preserved respiratory chain capacity and physiology in mice with profoundly reduced levels of mitochondrial respirasomes. Cell Metab. 35, 1799–1813.e7. 10.1016/j.cmet.2023.07.015. [DOI] [PubMed] [Google Scholar]
- 184.Mühleip A, Flygaard RK, Baradaran R, Haapanen O, Gruhl T, Tobiasson V, Maréchal A, Sharma V, and Amunts A (2023). Structural basis of mitochondrial membrane bending by the I-II-III2-IV2 supercomplex. Nature 615, 934–938. 10.1038/s41586-023-05817-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.McKenzie M, Lazarou M, Thorburn DR, and Ryan MT (2006). Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients. J. Mol. Biol. 361, 462–469. 10.1016/j.jmb.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 186.Zhu S, Chen Z, Zhu M, Shen Y, Leon LJ, Chi L, Spinozzi S, Tan C, Gu Y, Nguyen A, et al. (2021). Cardiolipin Remodeling Defects Impair Mitochondrial Architecture and Function in a Murine Model of Barth Syndrome Cardiomyopathy. Circ. Heart Fail. 14, e008289. 10.1161/CIRCHEARTFAILURE.121.008289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cole LK, Mejia EM, Sparagna GC, Vandel M, Xiang B, Han X, Dedousis N, Kaufman BA, Dolinsky VW, and Hatch GM (2020). Cardiolipin deficiency elevates susceptibility to a lipotoxic hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 144, 24–34. 10.1016/j.yjmcc.2020.05.001. [DOI] [PubMed] [Google Scholar]
- 188.Gilkerson RW, Selker JML, and Capaldi RA (2003). The cristal membrane of mitochondria is the principal site of oxidative phosphorylation. FEBS Lett. 546, 355–358. 10.1016/s0014-5793(03)00633-1. [DOI] [PubMed] [Google Scholar]
- 189.Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, and Korsmeyer SJ (2002). A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2, 55–67. 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 190.Cogliati S, Enriquez JA, and Scorrano L (2016). Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 41, 261–273. 10.1016/j.tibs.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 191.Hahn A, Parey K, Bublitz M, Mills DJ, Zickermann V, Vonck J, Kühlbrandt W, and Meier T (2016). Structure of a Complete ATP Synthase Dimer Reveals the Molecular Basis of Inner Mitochondrial Membrane Morphology. Mol. Cell 63, 445–456. 10.1016/j.molcel.2016.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Wolf DM, Segawa M, Kondadi AK, Anand R, Bailey ST, Reichert AS, van der Bliek AM, Shackelford DB, Liesa M, and Shirihai OS (2019). Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J. 38, e101056. 10.15252/embj.2018101056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kondadi AK, Anand R, Hänsch S, Urbach J, Zobel T, Wolf DM, Segawa M, Liesa M, Shirihai OS, Weidtkamp-Peters S, et al. (2020). Cristae undergo continuous cycles of membrane remodelling in a MICOS-dependent manner. EMBO Rep. 21, e49776. 10.15252/embr.201949776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Bleck CKE, Kim Y, Willingham TB, and Glancy B (2018). Subcellular connectomic analyses of energy networks in striated muscle. Nat. Commun. 9, 5111. 10.1038/s41467-018-07676-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Han M, Bushong EA, Segawa M, Tiard A, Wong A, Brady MR, Momcilovic M, Wolf DM, Zhang R, Petcherski A, et al. (2023). Spatial mapping of mitochondrial networks and bioenergetics in lung cancer. Nature 615, 712–719. 10.1038/s41586-023-05793-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Bozelli JC Jr., and Epand RM (2020). Membrane Shape and the Regulation of Biological Processes. J. Mol. Biol. 432, 5124–5136. 10.1016/j.jmb.2020.03.028. [DOI] [PubMed] [Google Scholar]
- 197.Marguet D, Lenne PF, Rigneault H, and He HT (2006). Dynamics in the plasma membrane: how to combine fluidity and order. EMBO J. 25, 3446–3457. 10.1038/sj.emboj.7601204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Molugu TR, Thurmond RL, Alam TM, Trouard TP, and Brown MF (2022). Phospholipid headgroups govern area per lipid and emergent elastic properties of bilayers. Biophys. J. 121, 4205–4220. 10.1016/j.bpj.2022.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Pennington ER, Funai K, Brown DA, and Shaikh SR (2019). The role of cardiolipin concentration and acyl chain composition on mitochondrial inner membrane molecular organization and function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1864, 1039–1052. 10.1016/j.bbalip.2019.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]