

Abstract
Coupling reactions between aryl electrophiles and alkyl/perfluoroalkyl precursors have inspired elegant methodologies that leverage electrochemical, photochemical, or thermal activation modalities. This work consolidates these myriad strategies to a single set of conditions and enables previously unknown alkyl-aryl couplings. These reactions rely on the discovery of unusually persistent organonickel complexes that serve as stoichiometric platforms for C(sp2)-C(sp3) coupling. Aryl, heteroaryl, or vinyl complexes of Ni can be inexpensively prepared on multigram scale by mild electroreduction from the corresponding C(sp2) electrophile. Organonickel complexes can be isolated and stored or telescoped directly to reliably diversify drug-like molecules. Finally, the procedure was miniaturized to μL scales by integrating soluble battery chemistries as redox initiators, which enabled a high-throughput exploration of substrate diversity.
Introduction
The development of C(sp2)-C(sp3) bond-forming reactions between aryl and alkyl fragments has unveiled new retrosynthetic strategies that complement those of mature chemistries for C(sp2)-C(sp2) coupling and has opened new chemical space for molecular design.1–3 Recent efforts have focused on enabling reactions of abundant aryl electrophiles (Fig. 1a, blue boxes) with structurally-diverse alkyl reagents (red boxes) sourced from perfluoroalkanes4,5 or accessible building blocks such as halides,6,7 carboxylic acids,8–11 and amines.12–16 As illustrated, couplings of these reagent classes constitute hundreds of potential reaction methodologies (gray lines) to generate thousands of new molecules. As a result, potentially new chemical space becomes accessible with every new methodology that bridges these key substrate classes.
Fig. 1. Convergent strategy for C-C coupling.
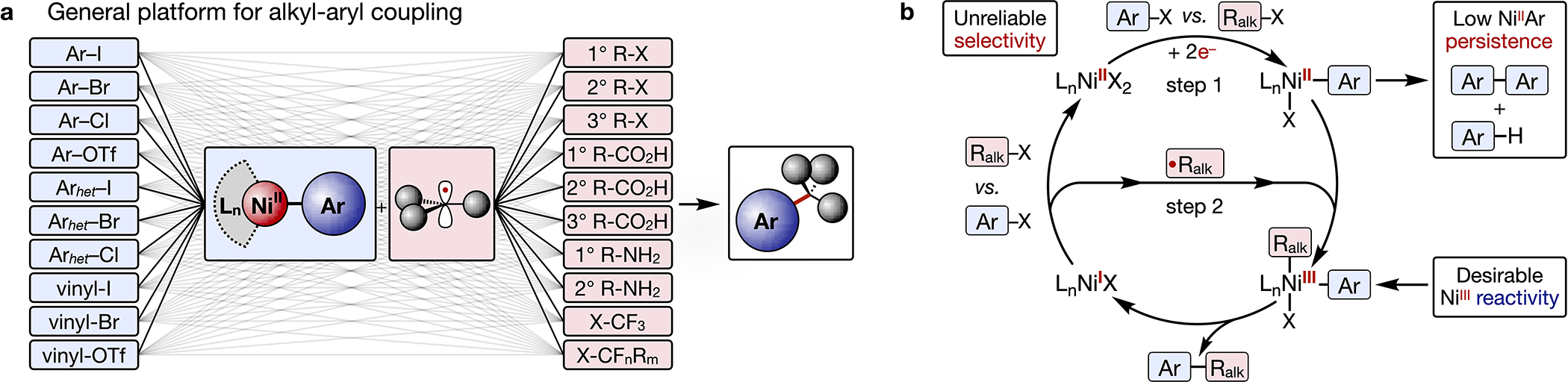
(a) General approach to aryl-alkyl coupling reactions. (b) Proposed mechanism for Ni-catalyzed XEC.
Despite this potential, reactions to perform these couplings are known for only a fraction of the illustrated combinations. Of the couplings that are known, many are catalyzed by Ni and are often performed as reductive processes with electrophilic forms of the alkyl reagent, such as pyridinium salts in place of amines 12–15 or redox-active esters (RAEs) in place of carboxylic acids.8–10 However, each protocol is highly optimized and requires its own distinct set of reagents, catalysts, and activation conditions (electrochemical, photochemical, chemical). Extending these methodologies to new electrophiles is often challenging, and applications to functionalization of drug-like compounds with significant molecular complexity can be unreliable.
A mechanism frequently invoked for cross electrophile coupling (XEC) reactions of various electrophiles (Fig. 1b) underscores why reaction systems must be so finely tuned.7 Specifically, a Ni complex must undergo a series of elementary reactions in a specific sequence with high fidelity to preferentially generate cross- over homo-coupled products. However, pairings of substrates with mismatched reactivities can derail this sequence. As examples, selective aryl over alkyl activation (step 1) can be challenging if an unreactive aryl chloride is paired with a highly electrophilic alkyl pyridinium or RAE, which is one reason these couplings and many others in Fig. 1a are currently unknown. Consequently, there is no general approach that can be easily implemented by one user to create a library of complex organic molecules with high reliability from a diverse set of alkyl precursors.
To address these limitations, this work describes an organometallic platform that can be applied to couple any combination of aryl and alkyl electrophile in Fig. 1a – many of which constitute previously-unknown methodologies – through a common set of reaction conditions. The convergent strategy relies on electrochemistry to controllably decouple the key steps of competitive substrate activation and product formation. Reactions are stoichiometric in an inexpensive Ni complex and are performed as two-step, one-pot procedures. The complex is prepared in 100 g batches from abundant starting materials and undergoes oxidative addition of aryl electrophiles upon electroreduction to form unusually persistent NiIIAr complexes of unprotected natural products, pharmaceuticals, and nucleosides. (Fig. 1b, step 1). The alkyl electrophile is then added, and further electrolysis triggers radical formation and C-C coupling (step 2). Coupled products with drug-like complexity are easily prepared in 20–60 mg quantities (0.15 mmol) from mL-scale reactions, and the complex can be easily recovered by filtration following aqueous workup of the reaction. Finally, the methodology was miniaturized to μL scales by integrating soluble battery chemistries as redox initiators. These microscale reactions enabled high throughput experimentation (HTE) and were employed in over 500 reactions that probe a vast chemical space of coupling partners.
Our work was inspired by the new chemical reactions that are now possible because of the creative implementation of stoichiometric reagents in synthesis.17–20 One such example relies on stoichiometric reactions of Pd complexes to fill synthetic gaps in coupling reactions of aryl halides and nucleophiles.20 Although our approach similarly relies on a stoichiometric metal, reactions with organonickel complexes have distinct advantages over those with Pd. Ni is orders of magnitude less expensive than Pd and requires inexpensive ligands for coupling reactions.21 Ni-mediated reactions leverage the reactivity of high-valent NiIII over the reactivity imparted to PdII by a designer ligand (Fig. 1b). Additionally, the capture and coupling of radicals (1e− fragments) at NiIIAr complexes provides a fundamental complement to couplings of nucleophiles (2e− fragments) at PdIIAr complexes. With the proper design, inexpensive and recoverable Ni complexes could serve as modern organometallic reagents for the reinvigorated field of radical chemistry.22
Separate from these advantages, stoichiometric XEC couplings with Ni pose distinct challenges compared to redox-neutral couplings with Pd. Redox-neutral reactions employ substrates with orthogonal reactivities (nucleophile and electrophile), and catalytic reactions that fail are typically successful if enough Pd is added.20 In contrast, substrates in XEC reactions are both electrophiles. When the rates of activation for the two electrophiles at Ni are mismatched – as is the case for many combinations in Fig. 1A – cross-coupled products do not form regardless of how much Ni is added because one electrophile is preferentially consumed over the other (vide infra, Fig. 5B control experiments).6
Results and Discussion
Guided by these insights, our design of a general processes for XEC required that an organonickel complex could be controllably formed from one electrophile and that the complex would persist until the second electrophile is added. Electrochemistry could provide the reductive control needed for the sequential reactions, but the poor persistence of organonickel complexes was an unsolved problem that has impeded such a strategy. In contrast to PdIIAr complexes, NiIIAr complexes often undergo a variety of uni- and bi-molecular decomposition processes to form homo-coupled or protodemetalated products (Fig. 1b).23 Known NiIIAr complexes that undergo radical coupling are limited to analogs with electron-withdrawing groups or ortho substituents on the aryl ring to provide electronic or steric stabilization.24–27 Identifying a ligand architecture for Ni that could support unbiased (hetero)aryl fragments of pharmaceutical complexity while enabling radical-based couplings was essential for developing the targeted strategy as well as for improving our fundamental understanding of organonickel chemistry.
We first investigated the reactivity and subsequent persistence of nickel-ligand complexes that are frequently employed as catalysts for XEC according to the protocol illustrated in Fig. 2.28–30 Preformed Ni complexes were electroreduced in the presence of an unbiased aryl electrophile (4-F-bromobenzene) under conditions known for electrocatalytic XEC. Following controlled reduction by 2e− equivalents vs. Ni, yields of the desired NiIIAr and undesired biaryl (Ar-Ar) or arene (Ar-H) products were quantified by 19F NMR spectroscopy. Summarized in the graphs below each ligand, reactions with complexes of tridentate trpy or PyBCamCN ligands failed to reach complete conversion (vs. Ni). These results are consistent with slow rates of oxidative addition. Most of what aryl bromide had been activated was converted to the protodehalogenated product Ar-H (black bar), and none of the organonickel complex was detected. In contrast, high conversion of the aryl bromide was observed in reactions with complexes of the bidentate ligand, tbbpy. However, none of the organonickel complex was observed and nearly all of the electrophile underwent homocoupling to form biaryl (gray bar). Competitive biaryl formation is a hallmark reactivity of bipyridyl-ligated organonickel complexes.28
Fig. 2. Evaluation of Ni Complexes.

Stoichiometric electroreduction reactions of pre-formed LnNiII complexes and 4-F-bromobenzene monitored by 19F NMR spectroscopy.
We next evaluated bis(pyridylamino)isoindoline (BPI) as a ligand for the study. We previously developed this class of ligand for electrocatalytic XEC because it incorporates an anionic donor that could promote oxidative addition, compared to neutral tridentate ligands, and offers a tridentate architecture that could offer greater stability for Ni intermediates than bidentate ligands.31 However, catalytic methodologies with these BPI complexes are limited – like most others – to couplings of aryl bromides and 1°/2° alkyl bromides. Here, near-quantitative formation of NiIIAr was observed following the electrolysis of the BPI complex and aryl bromide. A serendipitous feature of BPI is the phthalimide-like motif born in its architecture. Recent reports have shown that phthalimide additives can stabilize organonickel complexes of tbbpy, likely by replacing the halide ligand with phthalimide as the second X-type ligand of a NiIIAr intermediate.32,33 To probe this effect, we repeated the stoichiometric study with tbbpy but added an equimolar loading of phthalimide. As shown in Fig. 2 (right), the addition of phthalimide slightly reduced biaryl formation to 82%, compared to the phthalimide-free reactions, and generated low but measurable quantities of NiIIAr (17%, red bar). The yield of NiIIAr is increased further to 38% when the sodium phthalimide salt is employed in place of phthalimide, but biaryl is still formed in significant yields (63%, see the Supplementary Information). These latter two experiments highlight the stabilizing effect of an X-type moiety that is built into the ligand.
We probed the reactivity of the generated organonickel complex by adding radical precursors to the solution in the form of C(sp3) electrophiles and electrolyzing further (Fig. 2, second step). Reactions with cyclohexyl RAE or trifluoromethyl thianthrenium (TTCF3) generated C-C coupled products (blue and purple bars) in the high yields that match the yield of (BPI)NiIIAr formed in the first step. Yields from coupling reactions with the other four ligand systems were low to undetectable in most instances and similarly mirrored the formed quantities of LNiIIAr. These results, in combination with control experiments that ruled out the possibility of catalytic coupling from the BPI complex (vide infra, Fig. 5b), indicate that a persistent NiIIAr capable of capturing a radical intermediate is critical for these challenging reactions.
The promising results of our analytical studies led us to investigate whether the NiIIAr complexes could be prepared in synthetically useful scales and stored. This would allow the complexes to be implemented in synthesis like any conventional organometallic reagent but with 1e− reactivity towards radicals, rather than 2e− reactivity towards electrophiles. Illustrated in Fig. 3a, we first explored a conventional synthetic approach that relied on oxidative addition of the aryl electrophile with a Ni0 precursor, followed by ligand exchange (route A). This strategy was used to prepare the NiII(indomethacin) complex 1 in 76% yield on gram scale. However, the ligand exchange step is highly inconsistent, likely because of the poor solubility of the Na+BPI− salt. Attempted reactions with heteroaryl electrophiles were particularly susceptible to biheteroaryl formation because of slow dissolution of the BPI− salt, and the reactive NiII(HetAr) intermediates frequently decomposed in solution. Additionally, both the Ni(COD)2 precursor and base needed to deprotonate BPI-H (sodium hexamethyldisilazide) dramatically increase the cost and complexity of preparing these organometallic reagents.
Fig. 3. Synthesis and Reactivity of Organonickel Complexes.
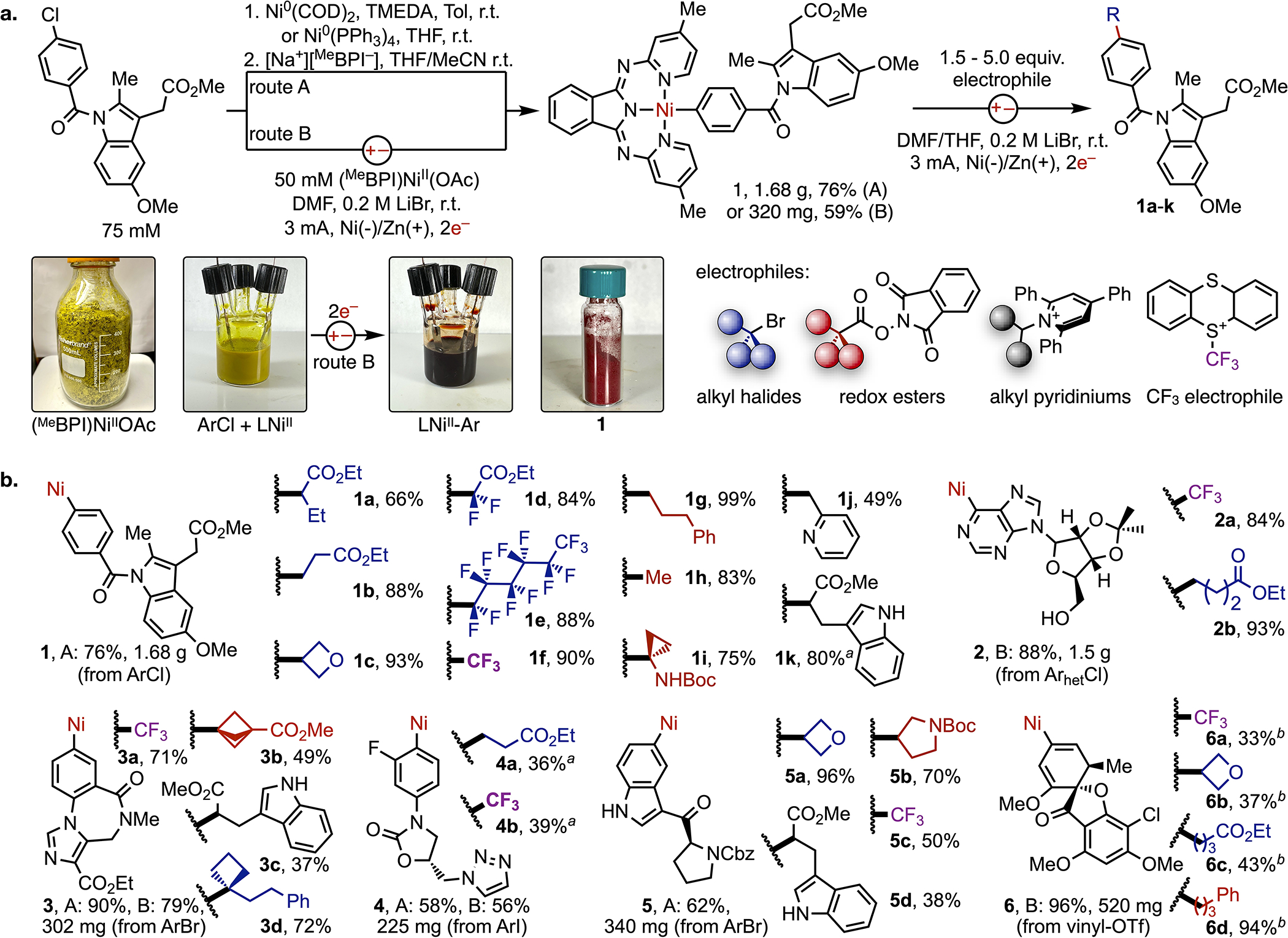
(a) Chemical and electrochemical (pictured) synthetic routes to BPI-ligated aryl complexes. Subsequent couplings of isolated complexes and alkyl electrophiles (red box) are detailed in the second step. (b) Isolated yields of complexes and organic products generated by couplings of isolated complexes on 0.15 mmol scale. Yields of the complex are reported from route A or B, where applicable. aThe product was isolated as an inseparable mixture. NMR spectroscopy was used to determine the yield of target product in the isolated mixture. bYield was determined by 1H NMR spectroscopy.
These limitations led us to evaluate the feasibility of translating the electrochemical assays performed in Fig. 2 into one-step electrosynthetic routes to access bulk quantities of organonickel complexes from the inexpensive (BPI)NiII(OAc) precursor and an aryl halide (route B). Reduction of the bright yellow mixture in DMF by 2e− generates a homogenous solution with a dark red color matching that of 1 (Fig. 3a pictures). No reaction was observed when the electrodes were submerged but without an applied potential, and zinc powder similarly failed to promote the reductive process. The electrochemical reaction can be performed on multigram scale (see the Supplementary Information, Fig. S9, S11-S13), and the complex can be easily isolated by precipitation from the DMF/LiBr electrolyte solution with degassed water. Filtration and recrystallization of the precipitate yields the complex as a red solid, which can be briefly handled in air (hours) or stored under nitrogen for at least 8 months.
In addition to a dramatic reduction in cost by over two orders of magnitude (see SI Fig. S10 for comparison), the electrosynthetic route (B) proved more reliable for generating aryl complexes with a range of electrophiles than the two-step route (A). Illustrated in Fig. 3b, we isolated organonickel complexes from reactions of aryl or heteroraryl chlorides (1, 2) and of electrophiles containing unprotected protic functionality (2, 5). Additionally, stable vinyl complexes (6) can be prepared from vinyl triflates, which expands the scope of potential C(sp2) electrophiles beyond just aryl halides to include carbonyl precursors. Collectively, these six examples constitute complexes of drugs (1), nucleosides (2), pharmaceutical mimics (3-5), and natural products (6).
We evaluated the reactivity of complexes 1-6 with TTCF3 and electrophilic forms of the most common C(sp3) sources (Fig. 3a, second step): alkyl bromides (halide), redox-active esters (carboxylic acids), and pyridiniums (amines). The organometallic complexes and electrophiles were weighed into undivided electrochemical cells on 0.15 mmol scales and electrolyzed further at room temperature in a DMF/THF solution with LiBr or KPF6 as electrolytes. An entire substrate scope can be developed for each complex. Examples that cover key coupling combinations from each class of alkyl substrate are summarized in Fig. 3b, and colors are used to indicate the alkyl source (blue = bromide, red = RAE, black = pyridinium). Collectively, these transformations represent many of the desired cross-combinations detailed in Fig. 1a and are performed under identical conditions.
Focusing on one complex, reactions of organonickel 1 form products 1a-k in high isolated yields. These reactions constitute couplings of an aryl chloride with a range of 1°/2°/3° alkyl or fluoroalkyl electrophiles. Catalytic reactions of many of these substrate combinations are unknown, even in highly optimized systems. As examples, alpha-bromo esters (1a, 1d), benzyl pyridiniums (1j), or perfluoroalkyl halides (1d, 1e) are extremely reactive and outcompete activation of aryl chlorides in catalytic processes. Usable quantities (30–60 mg) of otherwise inaccessible products can be generated for roughly $0.03 of (BPI)NiII(OAc) per stoichiometric reaction. Additionally, the complex precipitates from solution upon aqueous workup of the reaction mixtures and is collected by filtration (94% recovery, see SI Fig. S22). Inspired by the low cost and sustainable recovery of (BPI)NiII(OAc), we demonstrated a gramscale reaction of an organonickel complex (see the SM) to parallel reactions of conventional organometallic (e.g. organoboron) reagents on scales that are frequently performed in medicinal and academic chemistry.
Isolated organonickel complexes are particularly useful for diversification of specific (hetero)aryl leads or for frequent installation of privileged fragments (e.g., pyridyl) into alkyl systems. However, this methodology is likely to be more readily adopted if the organonickel complex could be formed in situ from the C(sp2) electrophile and telescoped for coupling reactions without isolation. Such a two-step, one-pot procedure would mimic how chemists prepare and use Grignard reagents but would be applicable to radical couplings. The direct electrosynthesis of NiIIAr complexes is particularly enabling for this strategy because the solvent and electrolyte are identical to that which is needed for the C-C coupling step. Described in Fig. 4 (top), we first electrolyzed a 3 mL solution (0.15 mmol scale) of (BPI)NiII(OAc) and a C(sp2) electrophile to generate the organonickel complex. An alkyl electrophile was then added to the resulting red solution, and the mixture was electrolyzed for an additional 2e− equivalents to effect C-C coupling.
Fig. 4. Telescoped Reactions.
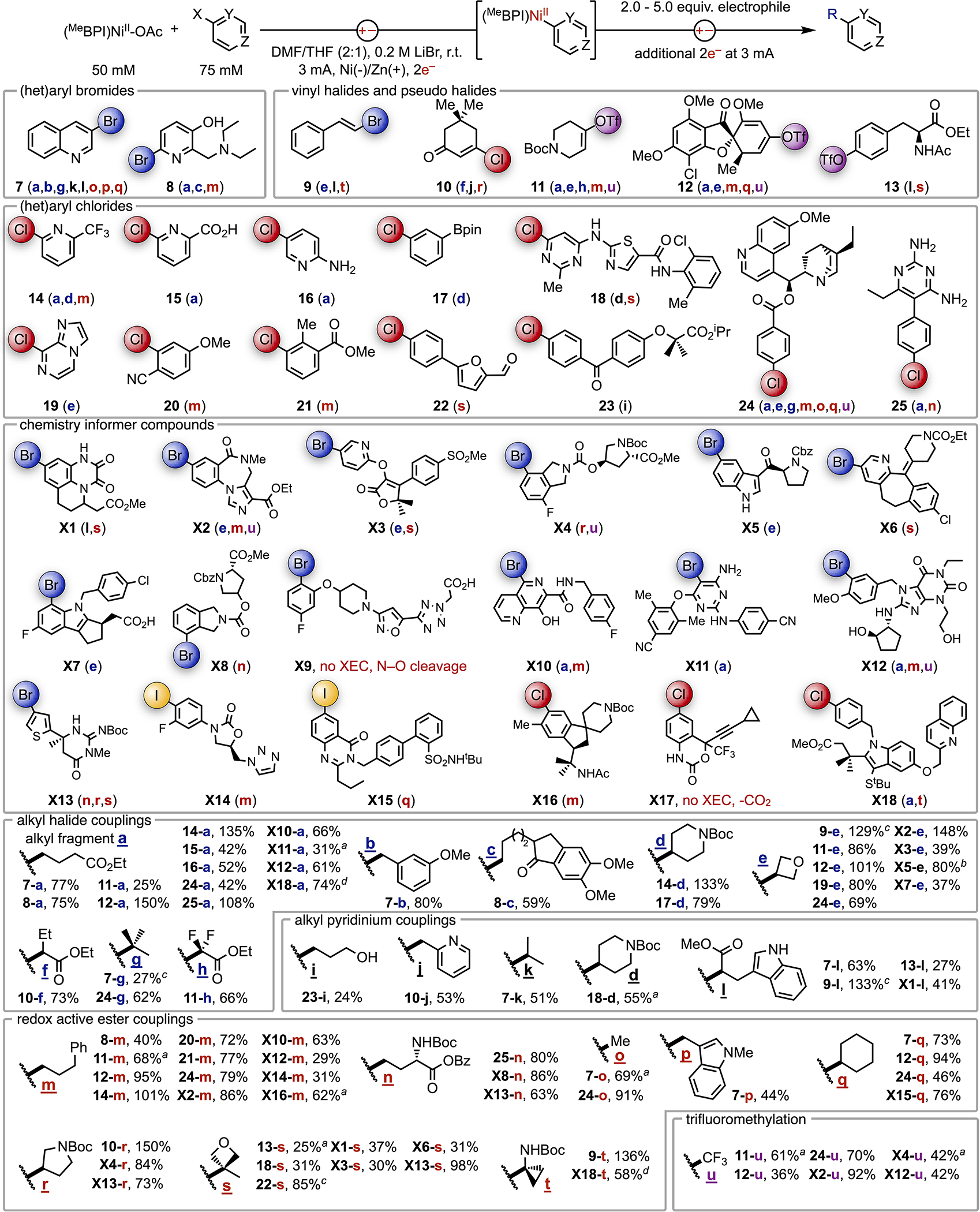
Isolated yields are reported from reactions without isolation of the NiIIAr intermediate (0.15 mmol scale). Yields can exceed 100% if a partial turnover of the metal occurs with the 1.5 equiv aryl electrophile. The letter denoting the alkyl fragment coupled to each C(sp2) electrophile is listed next to the number of the aryl fragment. The color of the letter corresponds to the class of alkyl reagent: blue = alkyl bromide, black = alkyl pyridinium, red = alkyl RAE, purple = TTCF3. aThe product was isolated as an inseparable mixture. NMR spectroscopy was used to determine the yield of target product in the isolated mixture. bYield was determined by 1H NMR spectroscopy. cIsolated as a mixture of stereoisomers. dThe complex with a methoxy-substituted BPI ligand was used in place of the standard complex.
The telescoped procedure requires just over 5 h and was applied to reactions with C(sp2) electrophiles containing drug-like complexity (heteroarenes), unprotected functional groups (free -OH, -CO2H, -NHR), or poor leaving groups (OTf, Cl). As illustrated in Fig. 4, we challenged the system by targeting reactions of aryl chlorides (14-25). The few aryl bromides selected for the scope are heteroarenes (7,8) with basic amines and unprotected alcohols (8). Many of the simpler substrates (20-22) are chloride analogs of bromoarenes identified by the Doyle group as representing diverse chemical space.34 Vinyl electrophiles were also selected (9-12), along with a triflated tyrosine (13). Finally, we included the chemistry informer library pioneered by Merck & Co., Inc., Rahway, NJ to further challenge the methodology (X1-X18).35
Reactions of these diverse C(sp2) electrophiles with the various classes of alkyl electrophile revealed an exceptionally broad range of successful couplings. Isolated yields from nearly 80 coupling reactions that represent distinct combinations of substrate classes are reported at the bottom of Fig. 4. Additionally, the letter of the coupled alkyl fragment (a-u) is listed beside the number of the corresponding C(sp2) partner and is colored according to the source of alkyl electrophile. Couplings with 1°/2°/3° alkyl bromides (a-h), including reductively sensitive fragments such as f and h, could be performed under a single set of conditions. Additionally, telescoped trifluoromethylation (u) of vinyl triflates (11, 12), aryl chlorides (24), and complex aryl bromides (X2, X4, X12) was achieved at room temperature under the same conditions as those for coupling of alkyl bromides.
Reactions of alkyl pyridinium reagents (i-l) are more challenging than those of other radical precursors, which we attribute to slow C-N fragmentation.36 Nonetheless, useful quantities of products were isolated from reactions of both 1° and 2° alkyl pyridiniums, including benzyl pyridinium j. Benzyl amines are common in pharmaceutical intermediates, but catalytic XEC reactions of benzyl pyridiniums are rare because of their rapid activation in preference to aryl activation.16
We found reactions of RAEs to be highly reliable for couplings of 1°/2°/3° alkyl fragments. Decarboxylative installation of amino acid fragments (n,r), methyl groups (o), or dimethylamino bioisosteres (t) onto C(sp2) sites are all tolerated.37 All carbon quaternary centers can also be constructed from telescoped couplings of tertiary fragments (s) and C(sp2)-chloride (18, 22) or -triflate (13) electrophiles: coupling combinations that were unsuccessful in catalytic approaches.6
Finally, we evaluated the chemistry informer library X1-X18 under the telescoped conditions. Isolated cross-products were obtained from all but two informer compounds, X9 and X17, which underwent competitive side reactions as noted in Fig. 4. Informers with ortho substituents are generally limited to couplings of 1° alkyl electrophiles (a, m, n) or 2° alkyl electrophiles of small rings. Without this steric restriction, tertiary (s) and complex secondary (l,r) alkyl electrophiles can be coupled. The general compatibility of protic and basic functionality highlights the mild conditions of the electroreductive activation. Moreover, the exceptional success rate for coupling of informer compounds (16 of 18) underscores the reliability of the methodology. Notably, this is the highest success rate achieved with the informer library to date in the context of cross-couplings.
The reactions described in Fig. 4 represent single-cell reactions that yield products on 20–60 mg scale (0.15 mmol, 3 mL volume). Although this is useful for preparing targeted products, we sought to miniaturize the coupling methodology to increase reaction throughput for medicinal chemistry applications. In doing so, we could also perform a more systematic evaluation of the reaction scope. As illustrated in Fig. 5a, solutions of isolated or pre-electrolyzed complexes could be easily distributed into wells containing distinct alkyl electrophiles (El(1) to El(n)). However, the challenge to miniaturization lies in how to insert electrodes and reliably electrolyze each individual cell in a straightforward manner.
Fig. 5. High Throughput Experimentation.
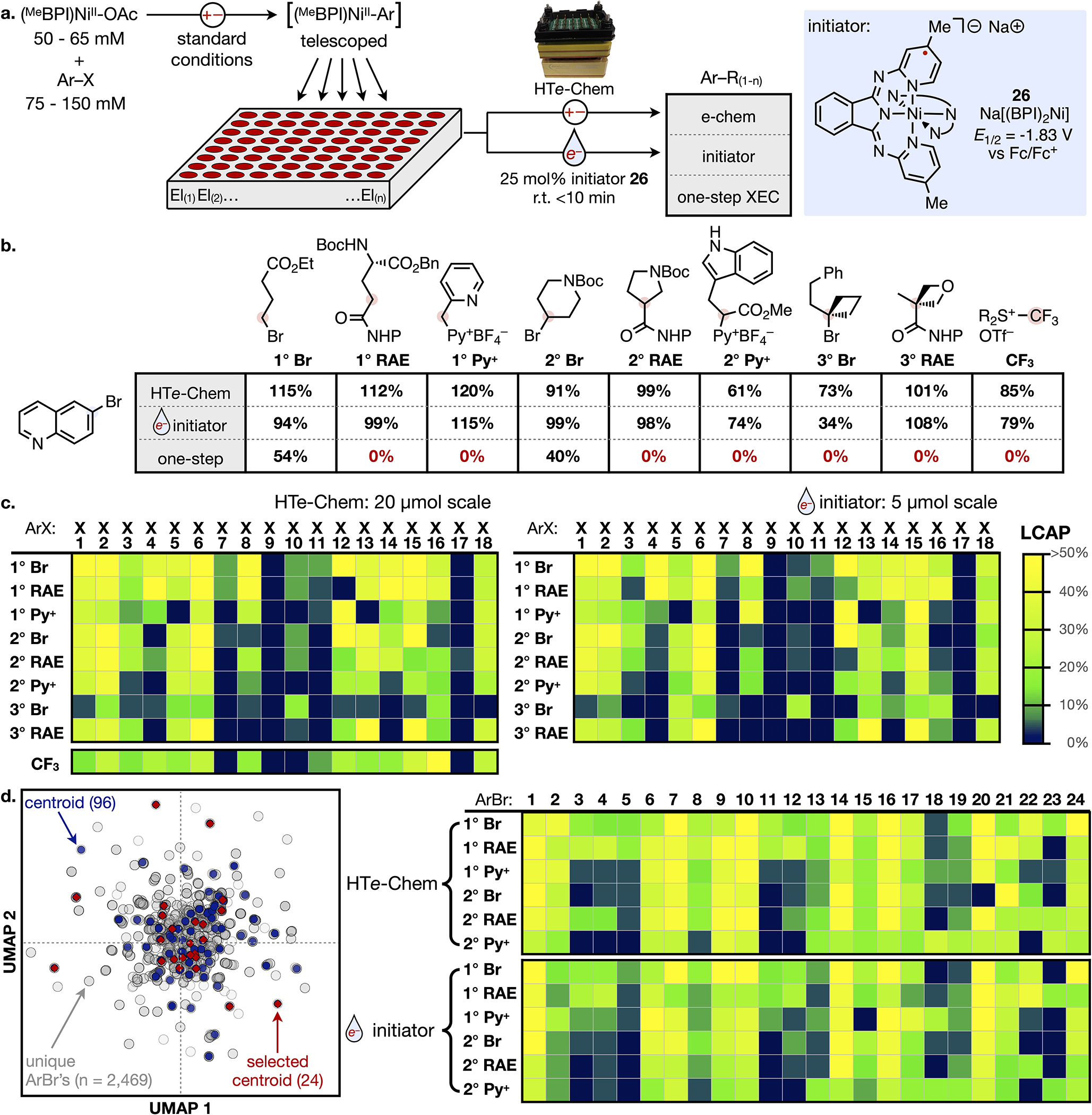
(a) Illustrated workflow for parallel reactions performed in HTe-Chem or with initiator 26. Control experiments for reactions performed in one step with both electrophiles in solution are reported in the 3rd row. (b) Results for couplings of a pre-formed Ni-quinoline complex employing the three synthetic approaches. Yields are reported vs. 1 equivalent Ni and can exceed 100% because of the excess (1.5 equivalents) aryl bromide in the telescoped procedure. (c) LCAP results from couplings of informer compounds X1 – X18. (d) (left) Chemical space map of parameterized ArBr’s (gray), cluster centroids (blue), and selected centroids (red). (right) LCAP results from couplings of telescoped ArBr’s and initiated by HTe-Chem or 26. LCAP results are color-coded according to the scale in Figure 5c.
Parallel electrolysis reactors have recently been developed to address these limitations.8,38,39 We selected the HTe-Chem reactor as a platform that simultaneously electrolyzes 24 wells and requires nearly an order of magnitude less volume (390 μL) than the standard reactions.38 We also explored a complementary approach that could further reduce the required solution volume and increase throughput by eliminating the need for electrolysis in the product forming step. This approach leverages insights from the proposed mechanism in Fig. 1b, which postulates Ni(I) as the product of C-C bond formation. The Ni(I) product could serve as the reductive activator (E1/2 = −1.65 V vs. Fc/Fc+) to generate another alkyl radical and propagate the reaction. This hypothesis of radical propagation was supported by studies of yield as a function of e− equivalents for the coupling step (see SI Fig. S7). High yields of product required just 0.05 to 0.5 e− equivalents, depending on the class of alkyl electrophile. As a result, the organonickel complexes can be viewed as being spring-loaded for coupling once radical formation is triggered by a substoichiometric quantity of reductant (initiator).
For soluble reductants that could initiate the coupling reaction, we turned to the redox flow battery literature, which specializes in the design of soluble compounds that store electrons (see SI Fig. S8).40 Mild reductants with potentials above −1.2 V (vs. Fc/Fc+) failed to induce product formation, and open-shell organic reductants inhibited coupling, likely because of competitive radical chemistry. Highest yields were observed from reactions dosed with the known anolyte and mediator Na+(BPI)2Ni− (26).31,41 This complex serves as a reliable e− donor (E1/2 = −1.83 V vs. Fc/Fc+) and does not react with transient alkyl radicals. Initiator 26 can be chemically synthesized and stored as a solid or generated by electrolysis in a separate cell and then dosed as a solution to initiate coupling. The coupling reactions were conducted with just 77 μL of the NiIIAr solution and were completed within minutes at room temperature. Finally, we performed successful couplings with as little as 15 μL of NiIIAr solution (see SI Fig. S21) as a proof-of-concept that the system can be miniaturized further with proper equipment for liquid handling.
The two coupling approaches were compared for reactions of bromo-quinoline with TTCF3 and 1°, 2°, or 3° analogs of all three classes of alkyl electrophile (Fig. 5b). The Ni(quinoline) complex was electroreductively generated and directly telescoped into wells with each alkyl partner. Coupled products were observed for all combinations, and the yields between electrolysis in the HTe-Chem reactor and homogeneous initiation mirror each other closely. By comparison, attempts to directly couple the electrophilic partners in a single step like in conventional XEC is only successful for the expected substrate combinations (Fig. 5b, bottom row). These results highlight the reliability of the coupling approach under a single set of conditions and the viability of the electrochemistry-free approach for HTE.
The simplification and miniaturization of C-C coupling allowed us to systematically evaluate the capabilities of the methodology with increased throughput. In all cases, the C-C coupling reactions were performed using both the HTe-Chem reactor and homogeneous initiator. We first evaluated reactions of the informer library compounds X1 – X18 against the same nine alkyl electrophiles of Fig. 5b that represent the main classes of alkyl fragments. These data are reported in Fig. 5c as product area percentages from UPLC chromatographic traces (LCAP) of the crude reaction mixtures. This rapid analytical method is most informative as an indicator of product formation, rather than a precise yield because LCAPs vary according to what other fragments are detected in the trace. As an example, the LCAP from couplings of 2° RAE and X4 is just 4% or 5% respectively using HTe-Chem or initiator, but the cross-product (X4-r) was isolated in 84% from the standard reaction of the same substrates (see Fig. 4 and SI Fig. S20 for a list comparing isolated yields vs. LCAPs). The heat maps from reactions with both HTe-Chem and initiator reveal high success rates for coupling of this challenging library. As expected, ortho-substituted informers X4 and X7 – X11 suffer reduced yields for couplings of 2° and 3° electrophiles compared to couplings of 1° analogs. Nonetheless, all informer compounds but X9 and X17 are coupled with at least three alkyl partners.
Finally, we applied the coupling methodology to reactions of aryl bromides that cover a diverse chemical space. Aryl or heteroaryl compounds in the Merck & Co., Inc. Rahway NJ building block collection that can only be obtained as bromide electrophiles (~2500) were used for these studies (Fig. 5d, left). Featurization of these aryl bromides using 2D molecular descriptors and RDKit fingerprints followed by dimensionality reduction and Kmeans clustering led to 96 clusters (centroids represented in blue in Fig. 5d).42 Twenty-four centroids (red diamonds in Fig. 5d, see SI Fig. S17 for structures) that span the chemical space were selected for HTE studies. Couplings of these aryl electrophiles with the 1° and 2° electrophiles are exceptionally reliable and formed products with LCAPs above 5% for 81% of HTe-Chem reactions and 77% of initiated reactions.
Conclusions
We demonstrate persistent organonickel complexes as useful organometallic reagents tailored for 1e− radical couplings that complement the classic 2e− reactivity of conventional organometallic reagents. Complexes are prepared from inexpensive precursors, are easily synthesized in multigram scales, and are reliable for challenging and unknown reactions under a single set of conditions. The mild electrochemical conditions used to form the organonickel complexes are compatible with a wide range of sensitive molecular functionality. Additionally, our implementation of battery chemistries to trigger coupling reactions further streamlines the utility of the complexes as platforms for HTE that can be used to access novel chemical space. We anticipate that these organometallic reagents will serve as a general platform for a range of other radical-based methods under different activation strategies, beyond just electrochemistry and formal XEC.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (NIH R35 GM138373) and Merck & Co. Inc. for supporting this work.
Footnotes
Competing Interests: The authors have filed patent applications directed to the technology described here.
Data Availability
All experimental data, analytical procedures, cell designs, and copies of spectra are available in the supplementary information.
References
- 1.Dombrowski AW, Gesmundo NJ, Aguirre AL, Sarris KA, Young JM, Bogdan AR, Martin MC, Gedeon S, Wang Y, Expanding the Medicinal Chemist Toolbox: Comparing Seven C(sp2)-C(sp3) Cross-Coupling Methods by Library Synthesis. ACS Med. Chem. Lett. 11, 597–604 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lovering F, Bikker J, Humblet C, Escape from flatland: Increasing saturation as an approach to improving clinical success. J. Med. Chem. 52, 6752–6756 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Wang X, Dai Y, Gong H, Nickel-Catalyzed Reductive Couplings. Top. Curr. Chem. 374, 43 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Le C, Chen TQ, Liang T, Zhang P, MacMillan DWC, A radical approach to the copper oxidative addition problem: Trifluoromethylation of bromoarenes. Science. 360, 1010–1014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho EJ, Senecal TD, Kinzel T, Zhang Y, Watson DA, Buchwald SL, The Palladium-Catalyzed Trifluoromethylation of Aryl Chlorides. Science. 328, 1679–1681 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamby TB, LaLama MJ, Sevov CS, Controlling Ni redox states by dynamic ligand exchange for electroreductive Csp3–Csp2 coupling. Science. 376, 410–416 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weix DJ, Methods and Mechanisms for Cross-Electrophile Coupling of Csp2 Halides with Alkyl Electrophiles. Acc. Chem. Res. 48, 1767–1775 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palkowitz MD, Laudadio G, Kolb S, Choi J, Oderinde MS, Ewing TEH, Bolduc PN, Chen T, Zhang H, Cheng PTW, Zhang B, Mandler MD, Blasczak VD, Richter JM, Collins MR, Schioldager RL, Bravo M, Dhar TGM, Vokits B, Zhu Y, Echeverria PG, Poss MA, Shaw SA, Clementson S, Petersen NN, Mykhailiuk PK, Baran PS, Overcoming Limitations in Decarboxylative Arylation via Ag-Ni Electrocatalysis. J. Am. Chem. Soc. 144, 17709–17720 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harwood SJ, Palkowitz MD, Gannett CN, Perez P, Yao Z, Sun L, Abruña HD, Anderson SL, Baran PS, Modular terpene synthesis enabled by mild electrochemical couplings. Science. 375, 745–752 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salgueiro DC, Chi BK, Guzei IA, GarcíaReynaga P, Weix DJ, Control of Redox-Active Ester Reactivity Enables a General Cross-Electrophile Approach to Access Arylated Strained Rings. Angew. Chemie - Int. Ed. 61 (2022), doi: 10.1002/anie.202205673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC, Merging photoredox with nickel catalysis: Coupling of α-carboxyl sp3-carbons with aryl halides. Science 345, 437–440 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Twitty JC, Hong Y, Garcia B, Tsang S, Liao J, Schultz DM, Hanisak J, Zultanski SL, Dion A, Kalyani D, Watson MP, Diversifying Amino Acids and Peptides via Deaminative Reductive Cross-Couplings Leveraging High-Throughput Experimentation. J. Am. Chem. Soc. 145, 5684–5695 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yi J, Badir SO, Kammer LM, Ribagorda M, Molander GA, Deaminative Reductive Arylation Enabled by Nickel/Photoredox Dual Catalysis. Org. Lett. 21, 3346–3351 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin-Montero R, Yatham VR, Yin H, Davies J, Martin R, Ni-catalyzed Reductive Deaminative Arylation at sp3 Carbon Centers. Org. Lett. 21, 2947–2951 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Yue H, Zhu C, Shen L, Geng Q, Hock KJ, Yuan T, Cavallo L, Rueping M, Nickel-catalyzed C-N bond activation: Activated primary amines as alkylating reagents in reductive cross-coupling. Chem. Sci. 10, 4430–4435 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Douthwaite JL, Zhao R, Shim E, Mahjour B, Zimmerman PM, Cernak T, Formal Cross-Coupling of Amines and Carboxylic Acids to Form sp3–sp2 Carbon–Carbon Bonds. J. Am. Chem. Soc. 145, 10930–10937 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kennedy SH, Dherange BD, Berger KJ, Levin MD, Skeletal editing through direct nitrogen deletion of secondary amines. Nature. 593, 223–227 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Holst DE, Wang DJ, Kim MJ, Guzei IA, Wickens ZK, Aziridine synthesis by coupling amines and alkenes via an electrogenerated dication. Nature. 596, 74–79 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruffoni A, Hampton C, Simonetti M, Leonori D, Photoexcited nitroarenes for the oxidative cleavage of alkenes. Nature. 610, 81–86 (2022). [DOI] [PubMed] [Google Scholar]
- 20.Uehling MR, King RP, Krska SW, Cernak T, Buchwald SL, Pharmaceutical diversification via palladium oxidative addition complexes. Science. 363, 405–408 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Corcoran EB, Pirnot MT, Lin S, Dreher SD, Dirocco DA, Davies IW, Buchwald SL, Macmillan DWC, Aryl amination using ligand-free Ni(II) salts and photoredox catalysis. Science. 353, 279–283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan M, Lo JC, Edwards JT, Baran PS, Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc. 138, 12692–12714 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diccianni J, Lin Q, Diao T, Mechanisms of Nickel-Catalyzed Coupling Reactions and Applications in Alkene Functionalization. Acc. Chem. Res. 53, 906–919 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ting SI, Garakyaraghi S, Taliaferro CM, Shields BJ, Scholes GD, Castellano FN, Doyle AG, 3 d-d Excited States of Ni(II) Complexes Relevant to Photoredox Catalysis: Spectroscopic Identification and Mechanistic Implications. J. Am. Chem. Soc. 142, 5800–5810 (2020). [DOI] [PubMed] [Google Scholar]
- 25.Lin Q, Diao T, Mechanism of Ni-Catalyzed Reductive 1,2-Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc. 141, 17937–17948 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piszel PE, Orzolek BJ, Olszewski AK, Rotella ME, Spiewak AM, Kozlowski MC, Weix DJ, Protodemetalation of (Bipyridyl)Ni(II)–Aryl Complexes Shows Evidence for Five-, Six-, and Seven-Membered Cyclic Pathways. J. Am. Chem. Soc. 145, 8517–8528 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terrett JA, Cuthbertson JD, Shurtleff VW, MacMillan DWC, Switching on elusive organometallic mechanisms with photoredox catalysis. Nature. 524, 330–334 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansen EC, Pedro DJ, Wotal AC, Gower NJ, Nelson JD, Caron S, Weix DJ, New ligands for nickel catalysis from diverse pharmaceutical heterocycle libraries. Nat. Chem. 8, 1126–1130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang P, Le CC, MacMillan DWC, Silyl Radical Activation of Alkyl Halides in Metallaphotoredox Catalysis: A Unique Pathway for Cross-Electrophile Coupling. J. Am. Chem. Soc. 138, 8084–8087 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perkins RJ, Pedro DJ, Hansen EC, Electrochemical Nickel Catalysis for Sp2-Sp3 Cross-Electrophile Coupling Reactions of Unactivated Alkyl Halides. Org. Lett. 19, 3755–3758 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Truesdell BL, Hamby TB, Sevov CS, General C(sp2)-C(sp3) Cross-Electrophile Coupling Reactions Enabled by Overcharge Protection of Homogeneous Electrocatalysts. J. Am. Chem. Soc. 142, 5884–5893 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Prieto Kullmer CN, Kautzky JA, Krska SW, Nowak T, Dreher SD, MacMillan DWC, Accelerating reaction generality and mechanistic insight through additive mapping. Science. 376, 532–539 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, Ehehalt LE, Huang Z, Beleh OM, Guzei IA, Weix DJ, Formation of C(sp2)–C(sp3) Bonds Instead of Amide C–N Bonds from Carboxylic Acid and Amine Substrate Pools by Decarbonylative Cross-Electrophile Coupling. J. Am. Chem. Soc. 145, 9951–9958 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kariofillis SK, Jiang S, Żurański AM, Gandhi SS, Martinez Alvarado JI, Doyle AG, Using Data Science To Guide Aryl Bromide Substrate Scope Analysis in a Ni/Photoredox-Catalyzed Cross-Coupling with Acetals as Alcohol-Derived Radical Sources. J. Am. Chem. Soc. 144, 1045–1055 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kutchukian PS, Dropinski JF, Dykstra KD, Li B, Dirocco DA, Streckfuss EC, Campeau LC, Cernak T, Vachal P, Davies IW, Krska SW, Dreher SD, Chemistry informer libraries: A chemoinformatics enabled approach to evaluate and advance synthetic methods. Chem. Sci. 7, 2604–2613 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tcyrulnikov S, Cai Q, Cameron Twitty J, Xu J, Atifi A, Bercher OP, Yap GPA, Rosenthal J, Watson MP, Kozlowski MC, Dissection of alkylpyridinium structures to understand deamination reactions. ACS Catal. 11, 8456–8466 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.West MS, Gabbey AL, Huestis MP, Rousseaux SAL, Ni-Catalyzed Reductive Cross-Coupling of Cyclopropylamines and Other Strained Ring NHP Esters with (Hetero)Aryl Halides. Org. Lett. 24, 8441–8446 (2022). [DOI] [PubMed] [Google Scholar]
- 38.Rein J, Annand JR, Wismer MK, Fu J, Siu JC, Klapars A, Strotman NA, Kalyani D, Lehnherr D, Lin S, Unlocking the Potential of High-Throughput Experimentation for Electrochemistry with a Standardized Microscale Reactor. ACS Cent. Sci. 7, 1347–1355 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gütz C, Klöckner B, Waldvogel SR, Electrochemical Screening for Electroorganic Synthesis. Org. Process Res. Dev. 20, 26–32 (2016). [Google Scholar]
- 40.Winsberg J, Hagemann T, Janoschka T, Hager MD, Schubert US, Redox-Flow Batteries: From Metals to Organic Redox-Active Materials. Angew. Chemie – Int. Ed. 56, 686–711 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sevov CS, Fisher SL, Thompson LT, Sanford MS, Mechanism-Based Development of a Low-Potential, Soluble, and Cyclable Multielectron Anolyte for Nonaqueous Redox Flow Batteries. J. Am. Chem. Soc. 138, 15378–15384 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Chen S-J, He CQ, Kong M, Wang J, Lin S, Krska SW, Stahl SS, Accessing three-dimensional molecular diversity through benzylic C–H cross-coupling. Nat. Synth. (2023), doi: 10.1038/s44160-023-00332-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All experimental data, analytical procedures, cell designs, and copies of spectra are available in the supplementary information.