

Abstract
Die Huntington-Krankheit (HK) ist eine autosomal-dominante Erbkrankheit, die zu motorischen, kognitiven und psychiatrischen Symptomen führt. Die Diagnose kann durch den molekulargenetischen Nachweis einer verlängerten CAG-Wiederholung im Huntingtin-Gen gesichert werden. Psychische und Verhaltenssymptome sind bei der HK häufig und können Jahre vor den motorischen Symptomen auftreten. Zu den psychiatrischen Symptomen gehören Apathie, Depression, Angst, Zwangssymptome und in einigen Fällen Psychosen und Aggression. Diese können aktuell nur symptomatisch behandelt werden, da sich krankheitsmodifizierende Therapieansätze bei der HK noch in der Erprobung befinden. Die derzeitige klinische Praxis basiert auf Expertenmeinungen sowie Erfahrung mit der Behandlung ähnlicher Symptome bei anderen neurologischen und psychiatrischen Krankheiten. In diesem Artikel geben wir einen Überblick über die komplexen psychischen Manifestationen der HK, die diagnostischen Möglichkeiten und die etablierten pharmakologischen und nichtpharmakologischen Behandlungsansätze.
Schlüsselwörter: Verhaltensauffälligkeiten, Psychische Symptome, Pharmakologische Therapie, Nichtpharmakologische Therapie, Psychotherapie
Abstract
Huntington’s disease (HD) is an autosomal dominant inherited disease, which leads to motor, cognitive and psychiatric symptoms. The diagnosis can be confirmed by genetic testing for extended CAG repeats in the Huntingtin gene. Mental and behavioral symptoms are common in HD and can appear several years before the onset of motor symptoms. The psychiatric symptoms include apathy, depression, anxiety, obsessive-compulsive symptoms and, in some cases, psychoses and aggression. These are currently restricted to symptomatic treatment as disease-modifying treatment approaches are still under investigation. The current clinical practice is based on expert opinions as well as experience with the treatment of similar symptoms in other neurological and mental health diseases. This article provides an overview of the complex psychiatric manifestations of HD, the diagnostic options and the established pharmacological and nonpharmacological treatment approaches.
Keywords: Behavioral symptoms, Mental health, Pharmacological therapy, Nonpharmacological therapy, Psychotherapy
Lernziele
Nach Lektüre dieses Beitrags …
verstehen Sie die Grundlagen der Huntington-Krankheit und ihre klinischen Manifestationen,
wissen Sie, welche psychiatrischen Symptome sich bei der Huntington-Krankheit manifestieren können,
kennen Sie pharmakologische sowie nichtpharmakologische Ansätze zur Behandlung der Huntington-Krankheit,
sind Sie mit den ethischen Aspekten vertraut, die bei der Betreuung von Personen mit Huntington-Krankheit auftreten können.
Einführung
Die Entdeckung des die Huntington-Krankheit (HK) verursachenden Gens im Jahr 1993 und die damit verbundene Möglichkeit, gezielt nach innovativen Therapien zu suchen, haben in den letzten Jahren zu einer Reihe klinischer Studien geführt, die jedoch bisher keinen krankheitsmodifizierenden Therapieansatz hervorgebracht haben. Neben den motorischen und kognitiven Symptomen der HK führen vor allem psychische Symptome
psychische Symptome
und Verhaltensänderungen zu Leistungseinbußen im Alltag und schränken die Lebensqualität der Betroffenen erheblich ein. Im Folgenden sollen die psychiatrischen Symptome der HK und ihre Behandlungsmöglichkeiten
Behandlungsmöglichkeiten
dargestellt werden.
Fallbeispiel
Eine 38-jährige Patientin stellt sich bei einem niedergelassenen Arzt vor. Sie berichtet, dass sie seit einigen Monaten Konzentrationsschwierigkeiten und vermehrt Konfliktsituationen mit Kollegen am Arbeitsplatz erlebe, da sie ihre Aufgaben nicht mehr so gut erfüllen könne. Dies alles habe sich schleichend entwickelt. Sie sei reizbar und aufbrausend geworden, was zu vielen Problemen in der Familie führe. Auch mit dem Haushalt komme sie nicht mehr zurecht. Sie schlafe nachts sehr schlecht und habe 7 kg abgenommen. Ihr Partner habe sie auf leichte Zuckungen der Finger aufmerksam gemacht. Ihre Mutter sei gesund und helfe ihr im Alltag. Ihr Vater sei vor vielen Jahren bei einem Autounfall ums Leben gekommen. In den letzten Jahren habe er unter Depressionen gelitten. Der Großvater väterlicherseits sei an Demenz gestorben, habe auch ein unsicheres Gangbild gehabt, die Großmutter sei mit 75 Jahren an einem Herzinfarkt gestorben. Die Großeltern mütterlicherseits seien über 80 Jahre alt und altersentsprechend gesund. Weitere Erkrankungen in der Familie sind nicht bekannt.
Hintergrund
Die Huntington-Krankheit (HK) ist eine neurodegenerative Erkrankung, die durch eine Trias von motorischen Symptomen (vor allem Bewegungsstörungen), kognitiven Beeinträchtigungen und psychiatrischen Symptomen gekennzeichnet ist. Die Krankheit ist nach George Huntington
George Huntington
benannt, der als praktizierender Hausarzt die Krankheit und ihre Erblichkeit in mehreren Familien auf Long Island, New York, beschrieb [1].
Der HK liegt eine Mutation des Huntingtin-Gens
Huntingtin-Gens
(HTT) zugrunde, die durch übermäßige Wiederholung („repeats“
„repeats“
) dreier Basen (CAG) im Exon 1 auf Chromosom 4 (4p16.3) verursacht wird und u. a. zur Bildung des mutierten Huntingtin-Proteins (mHTT) führt, das für die Pathologie und Toxizität mitverantwortlich ist ([2]; Abb. 1). Die Länge der CAG-Wiederholungen
CAG-Wiederholungen
bestimmt, ob eine Person an HK erkrankt [3]. Die Anzahl der CAGs in der Allgemeinbevölkerung liegt im Bereich von 6 bis 35 CAG-Wiederholungen, bei ≥ 40 CAG-Wiederholungen zeigt die Mutation volle Penetranz und löst einen Krankheitsprozess aus, der unvermeidlich zu den Symptomen der HK führt [3]. Im Bereich zwischen 36 und 39 CAGs ist eine inkomplette Penetranz bekannt, d. h. die Betroffenen erkranken zu Lebzeiten nicht oder zeigen erst spät erste Symptome [3].
Abb. 1.

Pathophysiologie der Huntington-Krankheit (HK)
CAG-Wiederholungen zwischen 27 und 35 CAGs (auch als intermediäre Allele
intermediäre Allele
bezeichnet) sind in der Regel nicht mit Krankheitssymptomen assoziiert, aber die Möglichkeit der Expansion der CAG-Repeats stellt ein erhöhtes Erkrankungsrisiko für die Nachkommen dar [3, 4]. Das Vorhandensein dieser intermediären Allele ist einer der möglichen Gründe, warum Krankheitsfälle beobachtet werden, obwohl bisher niemand in der Familie erkrankt war [4]. Es besteht eine inverse Beziehung zwischen der Zahl der CAG-Wiederholungen und dem Erkrankungsalter
Erkrankungsalter
, d. h. eine höhere Wiederholungszahl bedingt ein früheres Auftreten von Symptomen und ein rascheres Fortschreiten der HK [3]. Das durchschnittliche Erkrankungsalter liegt bei ca. 45 Jahren, die HK kann aber auch schon in der Kindheit oder erst im späteren Leben auftreten.
Die HK ist in allen ethnischen Gruppen endemisch, tritt aber bei Menschen europäischer Abstammung
europäischer Abstammung
mit 17,2 Fällen pro 100.000 häufiger auf [5]. Da immer mehr Europäer ein hohes Lebensalter erreichen und die Behandlungsmöglichkeiten besser werden, ist eine Zunahme der Prävalenz wahrscheinlich [4].
Der Verlauf der HK
Verlauf der HK
kann klinisch deskriptiv in prämanifeste und manifeste Stadien eingeteilt werden, wobei auch von einer Prodromalphase (mit subklinischer Symptomatik) in den Jahren vor Ausbruch der HK gesprochen wird [6].
Darüber hinaus wurde zu Forschungszwecken eine neue Klassifikation
Klassifikation
– das Huntington’s Disease Integrated Staging System (HD-ISS) – eingeführt, die Personen von Geburt an charakterisiert, beginnend mit Stadium 0
Stadium 0
(d. h. Personen mit der genetischen Mutation für die Huntington-Krankheit ohne nachweisbare Pathologie), dem Fortschreiten der Huntington-Krankheit anhand messbarer Indikatoren (z. B. Magnetresonanztomographie, MRT) der zugrunde liegenden Pathologie (Stadium 1
Stadium 1
), einem nachweisbaren klinischen Phänotyp (Stadium 2
Stadium 2
) und schließlich dem funktionellen Abbau (Stadium 3
Stadium 3
; [7]). Es wird erwartet, dass sich diese Klassifikation in der klinischen Praxis durchsetzt.
Die Anlageträger der Mutation im HTT-Gen, umgangssprachlich auch „Genträger“
„Genträger“
genannt, unterscheiden sich klinisch und funktionell in den ersten Lebensjahren typischerweise nicht von Personen ohne Mutation im HTT-Gen [6]. Erst im Laufe des Lebens entwickeln Mutationsträger kognitive Beeinträchtigungen, psychiatrische Symptome, Verhaltensauffälligkeiten und motorische Symptome (z. B. unwillkürliche choreatische Bewegungen), die zu einer zunehmenden Beeinträchtigung im Alltag, Pflegebedürftigkeit und zum Tod führen [6, 8].
Die motorischen Symptome
motorischen Symptome
der HK lassen sich in zwei Hauptkomponenten unterteilen:
die unwillkürlichen Bewegungsstörungen (z. B. Chorea) und
die Beeinträchtigung der willkürlichen Bewegungen mit Koordinationsstörungen und Bradykinesie, die typischerweise mit Rigor, Dysarthrie, Dysphagie und Gangstörungen einhergehen, sowie Dystonien in den Spätstadien der Erkrankung [6].
Eine Ausnahme bilden die juvenilen Formen der Erkrankung, die bereits im Frühstadium mit bradykinetischen und dystonen Symptomen einhergehen [6].
Die kognitive Beeinträchtigung
kognitive Beeinträchtigung
beginnt sehr früh und schreitet wie die motorischen Symptome allmählich voran [9]. Die Merkmale der kognitiven Beeinträchtigung ähneln denen anderer striatal-subkortikaler Hirnerkrankungen
striatal-subkortikaler Hirnerkrankungen
(z. B. Parkinson-Krankheit). HK-Patienten können Probleme mit der Aufmerksamkeit, der kognitiven Flexibilität, der Planung, dem Erkennen von Emotionen und eine psychomotorische Verlangsamung haben [10]. Die Sprache bleibt bei der HK meist lange gut erhalten, aber es entwickelt sich eine zunehmende Dysarthrie, im Stadium der Demenz häufig auch Wortfindungsstörungen [6].
Neben den kognitiven Beeinträchtigungen können psychiatrische Symptome
psychiatrische Symptome
auftreten, die sich als Ängste, Depressionen, Impulskontrollstörungen, Antriebsstörungen, Apathie, Zwänge, Unruhe und Aggressivität sowie psychotische Symptome manifestieren können [10]. Obwohl motorische Symptome als die charakteristischsten Symptome der HK angesehen werden, berichten sowohl Menschen mit HK als auch ihre Familien, dass die psychischen Symptome teilweise mehr zur Beeinträchtigung der Lebensqualität
Beeinträchtigung der Lebensqualität
beitragen als die motorischen Symptome.
Die HK geht auch mit einem katabolen Zustand
katabolen Zustand
einher, und ein fortschreitender Gewichtsverlust
Gewichtsverlust
ist ein gut dokumentiertes Merkmal der Krankheit, das zusammen mit Schluckstörungen zu klinischen Komplikationen wie Mangelernährung bis hin zur Kachexie führt [11, 12].
Merke
Die HK zeichnet sich durch eine Trias von motorisch-neurologischen, kognitiven und psychiatrischen Symptomen aus.
Betroffene Areale bei der Huntington-Krankheit
Die Komplexität und Vielfalt der klinischen Symptome der HK erschließt sich nur, wenn die Gesamtheit der beteiligten Hirnareale berücksichtigt wird. Veränderungen des Gehirns sind bereits 10 bis 15 Jahre vor dem Auftreten der ersten motorischen Symptome nachweisbar und schreiten allmählich voran [9]. Die neuronale Degeneration betrifft früh das Striatum
Striatum
in den Basalganglien, insbesondere die mittelgroßen dornentragenden Projektionsneurone („medium spiny neurons“
„medium spiny neurons“
, MSN), breitet sich aber auf andere Strukturen des Gehirns aus [13, 14]. Die Basalganglien
Basalganglien
sind an einer Vielzahl von Funktionen beteiligt, darunter willkürliche und unwillkürliche Bewegungen, prozedurales Lernen, Gewohnheitsbildung, Kognition und Emotion. Ebenso spielt auch das Muster der kortikalen Degeneration eine wichtige Rolle bei der Ausprägung des klinischen Phänotyps [13].
Die Neurodegeneration vor allem der Pyramidenzellen
Pyramidenzellen
des primären motorischen Kortex trägt zu den motorischen Symptomen der HK bei. Die Schädigung der Pyramidenzellen im zingulären Kortex, wo Emotionen verarbeitet werden, ist dagegen hauptsächlich mit affektiven Symptomen assoziiert [15]. Das Symptom der Apathie ist mit einer starken Abnahme der grauen Substanz innerhalb des kortikal-subkortikalen Netzwerks verbunden, wobei die bilaterale Amygdala
bilaterale Amygdala
und der temporale Kortex am stärksten betroffen sind [16].
Im Hypothalamus wurden Veränderungen in verschiedenen Kernen dieser Region festgestellt, z. B. im Nucleus suprachiasmaticus
Nucleus suprachiasmaticus
, einer Schlüsselregion für die Regulation von Schlaf und zirkadianem Rhythmus [17]. Ebenso wurde berichtet, dass die Degeneration der wichtigsten efferenten Neuronen des Kleinhirns, der Purkinje-Zellen
Purkinje-Zellen
, auch ein Merkmal der HK mit vorherrschenden motorischen Symptomen sein kann [18]. Die psychischen Symptome der HK werden mit den Stammganglien und anderen Hirnarealen in Verbindung gebracht (Abb. 2).
Abb. 2.
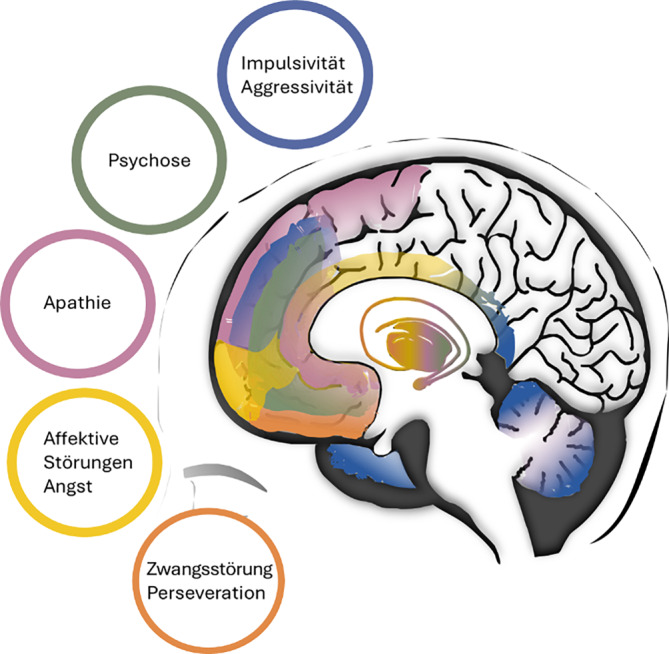
Betroffene Areale bei der Huntington-Krankheit
Merke
Die Vielfalt der klinischen Symptome der Huntington-Krankheit erschließt sich nur, wenn die Gesamtheit der beteiligten Hirnareale berücksichtigt wird.
Die HK betrifft neben den Basalganglien auch andere Hirnareale wie den Kortex, die Amygdala sowie das Kleinhirn.
Psychiatrische Symptome der Huntington-Krankheit
Die klinische Diagnose einer manifesten HK basiert aktuell auf dem Vorhandensein motorischer Symptome wie Chorea und Bradykinesie
Chorea und Bradykinesie
[19]. Psychische Symptome und Verhaltensauffälligkeiten können jedoch schon viele Jahre zuvor auftreten [9]. Die Prävalenz psychiatrischer Symptome liegt zwischen 33 und 76 % [9, 19]. Dazu gehören affektive Labilität, Depression, Ängste und zwangsähnliche Symptome, Reizbarkeit, Apathie und (seltener) Psychosen [20].
Die große Beobachtungsstudie „Registry“ [21], die auch an mehreren Zentren in Deutschland durchgeführt wurde, ergab, dass nur 27 % der Betroffenen keine psychiatrischen Symptome aufwiesen. Die restlichen 73 % litten unter mäßiger bis schwerer Apathie
Apathie
(28 %), Depression (12,7 %), Reizbarkeit und Aggressivität (13 %) sowie Zwängen und Perseverationen (13,2 %; [20]). Viele Symptome überschneiden sich und führen zu komplexen psychiatrischen Phänomenen, wie z. B. Apathie bei gleichzeitiger Impulskontrollstörung
Impulskontrollstörung
. Die psychiatrischen Symptome beeinträchtigen die Autonomie und Lebensqualität der Patienten erheblich, haben massive Auswirkungen auf das soziale Leben und können stationäre Behandlungen und/oder Unterbringung in Pflegeeinrichtungen notwendig machen [19, 22]. Sie müssen daher möglichst rechtzeitig erkannt und behandelt werden [22].
Es ist zu beachten, dass sich die HK im höheren Lebensalter (unabhängig von der Zahl der CAG-Wiederholungen) häufiger motorisch, seltener psychiatrisch manifestiert [23]. Obwohl mehr als 40 % der Patienten mindestens ein psychiatrisches oder kognitives Symptom vor den motorischen Symptomen aufwiesen, wobei Depressionen am häufigsten waren, ist bei der klinischen Diagnose von jungen Menschen
jungen Menschen
, die durch genetische Tests als HK-Mutationsträger identifiziert wurden, Vorsicht geboten [23]. Hier sollte das Gesamtbild berücksichtigt werden und eine umfassende Diagnostik in spezialisierten Ambulanzen oder Huntington-Zentren
Huntington-Zentren
durchgeführt werden.
Affektive Störungen und Suizidalität
Depressive Symptome
Depressive Symptome
zählen mit einer Prävalenz von 30–70 % zu den häufigsten Symptomen bei der HK [24, 25]. Sie treten in allen Stadien der HK auf und sind eng mit Suizidalität und anderen psychiatrischen Komorbiditäten verbunden. Die Pathophysiologie der Depression bei der HK ist noch nicht vollständig verstanden, aber neben reaktiven Anteilen sind bei der HK Störungen von Neurotransmittern
Neurotransmittern
wie Serotonin und neurophysiologischen Regelkreisen
neurophysiologischen Regelkreisen
, wie z. B. die Hypothalamus-Hypophysen-Nebennieren-Achse, nachweisbar [24]. Es werden Veränderungen von Verbindungen der Basalganglien, des präfrontalen Kortex sowie limbischer und paralimbischer Strukturen mit der Entstehung depressiver Symptome in Verbindung gebracht [24].
Insgesamt haben 20–30 % der Patienten mit HK Suizidgedanken
Suizidgedanken
und 7–10 % unternehmen einen Suizidversuch [26], im Vergleich zu 1–3 % in der Allgemeinbevölkerung [27]. Suizidgedanken bleiben oft während des gesamten Krankheitsverlaufs bestehen; Depressionen, Angstzustände oder Impulskontrollstörungen erhöhen das Risiko für suizidales Verhalten, weshalb das Vorhandensein psychischer Symptome sorgfältig überwacht werden sollte [26] und depressiven Symptomen besondere Aufmerksamkeit zu schenken ist. Es wurde festgestellt, dass nur 55 % aller Patienten mit mittelschweren bis schweren Depressionen im Zusammenhang mit HK Antidepressiva einnahmen, was auf eine Unterbehandlung
Unterbehandlung
hindeutet [20].
Angststörungen
Angststörungen treten bei bis zu 50 % der Betroffenen in allen Stadien der Krankheit auf [25] und beginnen oft schon vor dem Auftreten motorischer Symptome. Neben Angstsymptomen, die durch die psychische Belastung durch das eigene Schicksal, aber auch durch die Erlebnisse in der Familie bedingt sind und in Form konkreter Ängste
konkreter Ängste
in Erscheinung treten, gibt es auch unspezifische und diffuse Ängste
diffuse Ängste
, insbesondere durch die Veränderungen z. B. der Amygdala, des Striatums und des präfrontalen Kortex [15, 28]. Störungen insbesondere des serotonergen und dopaminergen Systems
serotonergen und dopaminergen Systems
werden bei der Entstehung von Angst eine wesentliche Rolle beigemessen [28].
Bei der HK können sich Ängste als Panikattacken
Panikattacken
, generalisierte Angststörung, isolierte Phobien und soziale Phobien manifestieren [22, 29]. Zudem kann die Selbstwahrnehmung von Symptomen Ängste auslösen. Im fortgeschrittenen Stadium können in Ermangelung verbaler Kommunikationsmöglichkeiten Ängste auch als eine vermehrte Unruhe in Erscheinung treten [22].
Antriebsstörung und Apathie
Antriebstörung sowie Apathie äußern sich in einem Mangel an Interesse
Mangel an Interesse
und/oder Motivation für Aktivitäten, die früher Spaß gemacht haben, sowie für das tägliche Leben und soziale Interaktionen. Es handelt sich um ein sehr häufiges neuropsychiatrisches Symptom der HK, mit zunehmender Prävalenz im Krankheitsverlauf [25, 30, 31]. Als Ursache der Apathie werden kognitive, emotionale und autoaktivierende Defizite
autoaktivierende Defizite
diskutiert [32, 33], die mit verschiedenen Veränderungen frontokortikaler Trakte der weißen Substanz einhergehen [34]. Die Apathie kann sich bereits im Prodromalstadium
Prodromalstadium
der HK, korrelierend mit einer Volumenminderung im Putamen und Kaudatum, manifestieren [35]. Bei der HK muss die Apathie von den Symptomen einer Depression unterschieden werden, was klinisch in der Regel gut möglich ist, und eine gezielte Behandlung der Symptome ermöglicht. Die Apathie korreliert direkt mit der Krankheitsprogression bei HK und ist ein Zeichen für die kortikostriatale Beeinträchtigung
kortikostriatale Beeinträchtigung
[31]. Apathie ist mit schlechter kognitiver Leistungsfähigkeit und einer Zunahme motorischer und psychiatrischer Beeinträchtigungen assoziiert [36].
Zwangsähnliche Verhaltensmuster und Perseveration
Mit dem Fortschreiten der HK können zwangsähnliche Verhaltensmuster auftreten, die Ausdruck einer zunehmenden orbitofrontalen und striatalen Dysfunktion
orbitofrontalen und striatalen Dysfunktion
sind [37]. Eine Differenzierung zwischen Zwangsstörungen (mit Zwängen) und Perseveration (das „Haftenbleiben“ an Gedanken, aber auch „repetitives“ Verhalten), die auf Veränderungen der exekutiven Funktionen zurückzuführen sind, ist klinisch wichtig [37]. Obwohl diese Verhaltensmuster auf den ersten Blick ähnlich erscheinen (repetitives Verhalten), unterscheiden sich die möglichen Behandlungswege je nach Ursache erheblich.
Perseveration lässt sich wahrscheinlich am besten von Zwangsstörungen unterscheiden, da sie auftritt, ohne dass sich der Betroffene der Problematik bewusst ist und sie meist nicht als störend empfindet [37]. Perseverationen stellen eine Herausforderung für die Familie
Herausforderung für die Familie
und das Pflegepersonal dar, da der Betroffene das gleiche Verhalten (z. B. Wiederholung von Fragen, auf die Toilette gehen, rauchen, etwas trinken wollen) viele Male hintereinander, manchmal über Stunden, wiederholt. Im Gegensatz zu Perseveration treten Zwangsstörungen bei HK nicht signifikant häufiger als in der Allgemeinbevölkerung auf [38].
Reizbarkeit, Impulsivität und Aggressivität
Es wird angenommen, dass Reizbarkeit, Impulsivität und Aggressivität bei der HK aus der komplexen Beziehung zwischen den neurobiologischen Veränderungen, die im Krankheitsverlauf auftreten, und den psychischen Reaktionen auf die subjektiv von den Betroffenen wahrgenommenen Veränderungen resultieren [39, 40, 41]. Impulsivität als Handeln ohne Voraussicht
Handeln ohne Voraussicht
oder Rücksicht auf mögliche Konsequenzen und Reizbarkeit als Zustand der Ungeduld und Intoleranz treten bei der HK häufig gemeinsam auf. Sie können aber auch isoliert voneinander auftreten und zu aggressiven Ausbrüchen
aggressiven Ausbrüchen
führen [42], in den meisten Fällen in Verbindung mit anderen neuropsychiatrischen Symptomen wie kognitive Beeinträchtigung, Apathie oder Perseveration [30].
Angehörige, Betreuer und Pflegepersonal sind in der Regel am meisten von diesen Verhaltensmustern betroffen, da sie in engem Kontakt mit den Betroffenen stehen und sie im Alltag betreuen [8]. Aggressive Episoden können durch die geringste Provokation
geringste Provokation
ausgelöst werden und zu wütendem oder im Extremfall gewalttätigem Verhalten führen, das stundenlang anhalten kann. Manche Erkrankte zeigen nach diesen Ausbrüchen Einsicht, sind über das Ausmaß überrascht und fühlen sich schuldig [43]. Es ist jedoch nicht ungewöhnlich, dass Erkrankte während oder nach aggressiven Ausbrüchen uneinsichtig
uneinsichtig
oder sich ihrer Handlungen nicht bewusst sind, was die Gesamtsituation und die Betreuung zusätzlich erschweren kann [43].
Es ist wichtig zu betonen, dass auch Hunger, Schmerzen oder andere belastende Empfindungen bei erschwerter Kommunikation
erschwerter Kommunikation
zu Erregung und Reizbarkeit beitragen können, was durch entsprechende Überprüfung/Untersuchung ausgeschlossen werden sollte [42].
Psychose
Psychotische Symptome (Wahnvorstellungen, Halluzinationen) werden in der Literatur zur HK meist mit einer Prävalenz von ca. 10 % angegeben, wobei Wahnvorstellungen häufiger als Halluzinationen zu beobachten sind [25, 44]. Bei den Halluzinationen
Halluzinationen
überwiegen akustische Halluzinationen. Psychotische Symptome treten häufiger bei manifest erkrankten Personen auf [25]. Die Wahnvorstellungen
Wahnvorstellungen
können Verfolgungswahn, Eifersuchtswahn, überwertige, aber auch nihilistische Gedanken beinhalten [25, 44]. Psychotische Symptome betreffen aber auch alle anderen möglichen Inhalte wie Zönästhesien
Zönästhesien
wie z. B. „Kribbeln und Jucken“ als Ausdruck von „Parasitenbefall“, bis hin zu „Folie à deux“
„Folie à deux“
, bei der Wahnvorstellungen einer anderen Person übernommen werden [44]. Es sollte differenzialdiagnostisch eine drogen- und/oder medikamenteninduzierte psychotische Symptomatik ausgeschlossen werden [22].
Mit Zunahme der kognitiven Einschränkungen lässt die Intensität der psychotischen Symptomatik häufig nach. Psychotische Symptome sind manchmal schwer zu behandeln [45] und sind dann ein häufiger Grund für stationäre Behandlungen
stationäre Behandlungen
und ggf. Unterbringung in einem Pflegeheim [8].
Merke
Psychiatrische Symptome können lange vor dem Auftreten typischer motorischer Symptome bei der HK auftreten und stellen eine enorme Belastung für die Betroffenen und ihre Familien dar.
Die Patienten weisen ein erhöhtes Risiko für Suizidgedanken, Suizidversuche und Suizide auf.
Diagnostik
Klinische Diagnose
Ein Krankheitsbild mit choreatischen Bewegungsstörungen,
choreatischen Bewegungsstörungen,
kognitivem Abbau und psychiatrischen Störungen sowie (falls erfragbar) einer positiven Familienanamnese impliziert klinisch die Verdachtsdiagnose einer HK. Sie ist die häufigste Ursache „choreatischer Bewegungsstörungen“ und wird bei ca. 90 % der Betroffenen durch einen Gentest bestätigt [46]. Differenzialdiagnostisch sind andere seltene neurologische Erkrankungen in Erwägung zu ziehen, die der HK ähneln und auch als „HK-Phänokopien“
„HK-Phänokopien“
bezeichnet werden. Ebenso können im Rahmen von Autoimmun‑, Stoffwechsel- und neurodegenerativen Prozessen HK-ähnliche Symptome auftreten (siehe dazu [46, 47]).
Eine genaue Anamnese mit vollständiger Familienanamnese
Familienanamnese
ist unerlässlich, um den Krankheitsverlauf zu beschreiben und den autosomal-dominanten Erbgang zu erkennen. Die klinische Beurteilung sollte die Erhebung des psychopathologischen Befundes, die neurologische und internistische Untersuchung sowie die Erfassung von Komorbiditäten umfassen. Standardisierte klinische Bewertungsskalen
klinische Bewertungsskalen
(wie die Unified Huntington’s Disease Rating Scale [UHDRS]) sollten angewendet werden, da sie alle von der HK betroffenen Bereiche (motorisch, psychiatrisch und kognitiv) erfassen und zur Beurteilung des Funktionsniveaus und der Aktivitäten des täglichen Lebens sowie des Krankheitsverlaufs nützlich sind [48]. Eine zerebrale Bildgebung
zerebrale Bildgebung
sollte die Diagnostik der HK vervollständigen, wobei der striatale Volumenverlust das empfindlichste Maß für krankheitsbedingte Veränderungen ist [49, 50].
Molekulargenetische Diagnostik
Die genetische Beratung
genetische Beratung
entsprechend dem „Gendiagnostik-Gesetz“ (GenDG) ist sowohl im Falle einer genetischen Untersuchung bei Symptomträgern
Symptomträgern
als auch bei einer prädiktiven Untersuchung sog. „Risikopersonen“
„Risikopersonen“
essenziell. Sie ist wichtig, um den Betroffenen und ihren Familien informierte Entscheidungen zu ermöglichen und die möglichen Implikationen eines Mutationsnachweises zu verstehen. In der S2k-Leitlinie Chorea/Morbus Huntington (2023; [51]) wird bei symptomatischen Patienten mit typischem klinischen Bild eine molekulargenetische Untersuchung zur Diagnosesicherung empfohlen. Gemäß GenDG [52] kann diese differenzialdiagnostische Abklärung bei symptomatischen Patienten von jedem Arzt durchgeführt werden. Bei asymptomatischen Personen hingegen ist gemäß § 10 des GenDG eine genetische Untersuchung nur durch Fachärzte für Humangenetik oder andere Ärzte zulässig, die sich durch den Erwerb einer Facharzt‑, Schwerpunkt- oder Zusatzbezeichnung für genetische Untersuchungen qualifiziert haben.
Es wird grundsätzlich empfohlen, vor einer genetischen Untersuchung und bei der Ergebnismitteilung eine genetische Beratung, ggf. auch eine psychologische Begleitung
psychologische Begleitung
, anzubieten. Während des Beratungsprozesses ist es unerlässlich, auf psychische Symptome, insbesondere auf Depressivität und Suizidalität, zu achten. In Einzelfällen kann es sinnvoll sein, den Prozess durch eine ambulante Psychotherapie
ambulante Psychotherapie
zu begleiten.
Über das GenDG hinaus haben sich die z. B. im Europäischen Huntington-Netzwerk (EHDN
EHDN
) zusammengeschlossenen Zentren auf Empfehlungen zur Durchführung der prädiktiven genetischen Untersuchung geeinigt [53], deren verbindliche Anwendung auch von der Deutschen Huntington-Hilfe (DHH
DHH
e. V.) empfohlen wird.
Merke
Vor einer genetischen Untersuchung (Gentest) muss eine genetische Beratung erfolgen
Behandlungsoptionen und ethische Aspekte
In Ermangelung einer kausalen Therapie konzentriert sich das derzeitige Management der HK auf die symptomatische Behandlung
symptomatische Behandlung
. Die Behandlung von Symptomen im Zusammenhang mit der HK umfasst nicht nur die pharmakologische neurologische und psychiatrische Behandlung, sondern auch nichtpharmakologische Maßnahmen, einschließlich psychotherapeutischer und sozialmedizinischer Begleitung zur Bewältigung psychischer Probleme, Unterstützungsdienste für Menschen mit Behinderungen zur Förderung der Alltagsbewältigung und schließlich Palliativmedizin [6, 12].
Die maßgeschnittene Behandlung
maßgeschnittene Behandlung
umfasst ein multidisziplinäres Team (Infobox), das sich sowohl um den Betroffenen selbst als auch um die Aufklärung und Betreuung der Angehörigen kümmert [54]. Das multidisziplinäre Team
multidisziplinäre Team
setzt sich aus verschiedenen Berufsgruppen zusammen, die eng zusammenarbeiten. Wichtig ist auch, dass für jeden Betroffenen (inkl. Familie) ein individueller Pflege- und Behandlungsplan erstellt wird.
Infobox Das multidisziplinäre Team zur Behandlung von Patienten mit Huntington-Krankheit setzt sich aus verschiedenen Berufsgruppen zusammen, die eng zusammenarbeiten
Ärztliche Versorgung
Hausarzt: erste Anlaufstelle, wohnortnah
Neurologe/Psychiater/Nervenarzt: Diagnostik und Behandlung (pharmakologisch, psychotherapeutisch), nichtmedikamentös (Verordnung von Physio‑, Logo‑, Ergotherapie)
Humangenetiker: genetische Beratung und molekulargenetische Testung (auch bei Fachärzten mit Qualifikation zur fachgebundenen genetischen Beratung in Huntington-Zentren und Ambulanzen möglich)
Zahnarzt: Sicherstellung einer adäquaten zahnärztlichen Versorgung
Weitere Fachärzte je nach Bedarf und Fragestellung (z. B. Palliativmediziner, Internist)
Psychologische/psychotherapeutische Betreuung
Psychologe/Psychotherapeut: psychologische Beurteilung, Beratung und Unterstützung (für Patienten und Angehörige), Psychotherapie
Neuropsychologe: kognitive Abklärung und Beratung
Spezifische therapeutische Betreuung
Ergotherapeut: Beurteilung der Alltagsfunktionen, kognitives Training, Hilfsmittel
Logopäde: Beurteilung von Sprache und Kommunikation; Dysphagiediagnostik und -beratung
Ernährungstherapeut: Beurteilung und Beratung in Ernährungsfragen
Physiotherapeut: Beurteilung des Gangbildes; Übungsprogramm; Hilfsmittel
Pflegeberatung/Case-Management: Unterstützung des Patienten und der Angehörigen
Sozialmedizinische Betreuung
Sozialarbeiter: sozialmedizinische Beratung (z. B. Finanzen, Wiedereingliederung, Rente, Pflegestufe, Heimunterbringung, Hilfsmittel)
Forschung
Spezialambulanzen und -zentren: Einbeziehung von Patienten und Angehörigen in die Forschung
Selbsthilfegruppen
Kontakt zu regionalen Vertretern von Selbsthilfegruppen, Peer-to-Peer-Beratung, Kontakt zu Angehörigen und Unterstützung
Pharmakologische Ansätze
Es gibt es bis heute nur wenig Evidenz
wenig Evidenz
aus kontrollierten randomisierten Studien für die Wirksamkeit von Psychopharmaka bei psychischen Symptomen
psychischen Symptomen
und Verhaltensstörungen im Rahmen der HK [30]. Die erste Studie dieser Art erbrachte keinen Nachweis der Wirksamkeit von Bupropion
Bupropion
bei Apathie durch HK [55]. Obwohl die Behandlung der Chorea nicht Gegenstand dieses Artikels ist, ist es wichtig zu erwähnen, dass eine symptomatische Behandlung Auswirkungen auf die psychiatrische Symptomatik haben kann. Nachdem für Tiaprid
Tiaprid
, das bereits seit den 1970er-Jahren zur Behandlung von Bewegungsstörungen (auch bei der HK) eingesetzt wurde, keine heutigen Qualitätsstandards entsprechenden Studien, dafür aber langjährige klinische Erfahrung vorliegen und es außerhalb Europas auch nicht verfügbar ist, empfehlen internationale Leitlinien Tetrabenazin
Tetrabenazin
als einziges Medikament, das für die Behandlung der Chorea bei HK zugelassen ist. Tetrabenazin hat aber ein bezüglich psychischer Symptome ungünstigeres Nebenwirkungsprofil
ungünstigeres Nebenwirkungsprofil
als Tiaprid. Es kann jedoch laut der Sk2-Leitlinie der Deutschen Gesellschaft für Neurologie [51] erwogen werden, eine antihyperkinetische Therapie zunächst mit Tiaprid einzuleiten und Tetrabenazin (in Kombination oder als Monotherapie) dann einzusetzen, wenn die Behandlungsmöglichkeiten mit Tiaprid (Wirkung, Verträglichkeit) ausgereizt sind [56]. Als Inhibitor des vesikulären Monoamintransporters 2 (VMAT2) reduziert Tetrabenazin die Aufnahme von Monoaminen (einschließlich Dopamin) in die synaptischen Vesikel, was zu Nebenwirkungen im Sinne von Depressionen, suizidalen Gedanken sowie Sedierung führen kann und bei der Verordnung bei HK-Patienten mit affektiven Störungen berücksichtigt werden muss [22, 57].
Zur Behandlung der motorischen Symptome
motorischen Symptome
der HK werden Antipsychotika
Antipsychotika
wegen ihrer antagonistischen Wirkung auf Dopaminrezeptoren eingesetzt, zeigen aber auch positive Effekte bei gleichzeitigem Vorliegen von Verhaltensauffälligkeiten, Depressionen oder Psychosen [22, 57]. Andere Antipsychotika wie Olanzapin, Risperidon, Sulpirid, Amisulprid, Aripiprazol, Quetiapin, Haloperidol können ebenfalls die Chorea reduzieren [22, 57]. Die Evidenz hierfür stammt jedoch nur aus kleinen, meist offenen Studien und es gibt keine großen direkten Vergleiche zwischen den verschiedenen Medikamenten [58]. Wenn ein Medikament nicht wirkt oder schlecht vertragen wird, ist es sinnvoll, ein anderes Antipsychotikum in Betracht zu ziehen. Als Nebenwirkungen von Antipsychotika können extrapyramidale Symptome
extrapyramidale Symptome
(EPS), Herzrhythmusstörungen (Verlängerung der QTc-Zeit) sowie ein metabolisches Syndrom auftreten.
Affektive Störungen und Angststörung
Depressionen und Angstzustände treten bei der HK häufig auf. Zur Behandlung können selektive Serotoninwiederaufnahmehemmer
selektive Serotoninwiederaufnahmehemmer
(SSRI) wie Sertralin, Escitalopram oder Citalopram und Serotonin-Noradrenalin-Wiederaufnahmehemmer (SNRI) wie Venlafaxin oder Duloxetin eingesetzt werden [22, 59]. Auch Mirtazapin kann hilfreich sein, insbesondere bei Schlafstörungen. Antidepressiva mit ausgeprägten anticholinergen Effekten sollten vermieden werden, da sie choreatische Bewegungsstörungen verstärken können.
Bei Angststörungen
Angststörungen
sind Antipsychotika eine wertvolle therapeutische Alternative, wenn andere Behandlungen erfolglos bleiben [12]. Der Einsatz von Benzodiazepinen, z. B. Lorazepam oder Diazepam, sollte zurückhaltend und nur auf der Grundlage einer Risiko-Nutzen-Abwägung in Betracht gezogen werden [45].
Antriebsstörung und Apathie
Es ist wichtig, die Angehörigen und das Pflegepersonal über die verschiedenen Aspekte und Ursachen der Apathie aufzuklären [22]. Wenn möglich, werden aktivierende Maßnahmen
aktivierende Maßnahmen
, die Einführung von Routinen und ein strukturiertes Programm von Aktivitäten empfohlen. Depressionen können Apathie verstärken. Bei Verdacht auf Depression sollte ein SSRI
SSRI
erwogen werden [60]. Bupropion erwies sich in einer randomisierten kontrollierten klinischen Studie ähnlich wie der Alzheimer-Krankheit als nicht wirksam [55]. Sedativa und Antipsychotika können die Apathie verstärken, daher wird empfohlen, bei der Verschreibung darauf zu achten.
Zwangsähnliche Verhaltensmuster und Perseveration
Für die Behandlung von Zwangsstörungen sind SSRI
SSRI
in der Regel die erste Wahl, wobei auch Komorbiditäten eine Rolle bei der Therapiewahl spielen [22]. Bei mangelndem Ansprechen auf die Therapie wird entweder ein Wechsel auf Clomipramin oder eine Augmentation mit Stimmungsstabilisatoren
Stimmungsstabilisatoren
und Antipsychotika bevorzugt, während Benzodiazepine häufiger eingesetzt werden, wenn Angststörungen als Komorbidität vorliegen [61]. Eine Kombination aus medikamentöser Therapie (z. B. mit SSRI) und psychotherapeutischen und psychoedukativen Maßnahmen kann möglicherweise bessere Effekte erzielen [12, 37].
Reizbarkeit, Impulsivität und Aggressivität
Bei der Behandlung von Impulsivität und Aggression ist es wichtig, die verhaltenstherapeutischen Maßnahmen
verhaltenstherapeutischen Maßnahmen
mit Medikamenten zu kombinieren. Bei akuter Erregung, die nicht auf verhaltenstherapeutische Strategien anspricht, sind Benzodiazepine
Benzodiazepine
oder Antipsychotika die bevorzugten pharmakologischen Optionen [45]. Bei chronischer Erregung, die durch wiederkehrende und andauernde Beschwerden gekennzeichnet ist oder bei der die Gefahr besteht, dass der Betroffene sich selbst oder anderen Schaden zufügt, kann man entweder ein Antipsychotikum
Antipsychotikum
oder ein stimmungsstabilisierendes Antiepileptikum (z. B. Valproat) einsetzen. Reizbarkeit kann auf SSRIs ansprechen, erfordert aber oft höhere Dosen.
Psychose
Ein Antipsychotikum ist die erste Wahl bei der pharmakologischen Behandlung von Psychosen bei der HK, wobei Antipsychotika der zweiten Generation
Antipsychotika der zweiten Generation
aufgrund ihres besseren Nebenwirkungsprofils in der Regel bevorzugt werden [45, 62]. Bestehen gleichzeitig ausgeprägte choreatische Bewegungsstörungen, kann der Einsatz eines „klassischen“ Antipsychotikums (z. B. Haloperidol) erwogen werden, dessen dopaminrezeptorantagonistische Wirkungen dann gleichzeitig „antihyperkinetisch“ wirken können.
Eine Umstellung sollte erfolgen, wenn die psychotischen Symptome mit dem ersten Medikament nicht ausreichend kontrolliert werden konnten oder Nebenwirkungen aufgetreten sind. Die Kombination von Antipsychotika
Kombination von Antipsychotika
wird nicht empfohlen und sollte nur bei schweren Formen der Psychose eingesetzt werden. Clozapin sollte in Erwägung gezogen werden, wenn die psychotischen Symptome nicht ausreichend auf andere Antipsychotika ansprechen, sofern regelmäßige Blutbildkontrollen möglich sind [22]. Die Herausforderung bei der Behandlung besteht darin, die Nebenwirkungen
Nebenwirkungen
der Antipsychotika zu erkennen, da sie schwer von Symptomen der HK im Krankheitsverlauf zu unterscheiden sind.
Im Allgemeinen ist es bei der Behandlung psychiatrischer Symptome der HK wichtig, auf komorbide Erkrankungen
komorbide Erkrankungen
zu achten, die den Allgemeinzustand des Patienten verschlechtern können, wie Infektionskrankheiten, Stoffwechselerkrankungen, Drogenkonsum oder andere Ursachen, die zu akuter Agitation oder psychotischen oder deliranten Zuständen führen können. Ebenso ist es wichtig, auf Schlafhygiene
Schlafhygiene
als Präventionsstrategie gegen Agitation zu achten und Schlafstörungen zu vermeiden [22].
Bei der Pflege von HK-Patienten im fortgeschrittenen Stadium sollte eine reizarme Umgebung
reizarme Umgebung
hergestellt werden. Die Erkrankten profitieren von einem geregelten Tagesablauf
geregelten Tagesablauf
und Routinen. Es ist wichtig, die Angehörigen und das Pflegepersonal über die Verhaltenssymptome aufzuklären und sie über Strategien im Umgang mit Erregungszuständen, Perseveration, Apathie und auch über aktivierende, ressourcenorientierte Pflege zu informieren ([54]. Die pharmakologischen Behandlungsmöglichkeiten der Huntington-Krankheit sind in Tab. 1 dargestellt.
Tab. 1.
Pharmakologische Behandlungsmöglichkeiten bei der Huntington-Krankheita
| Symptom | Medikament | Gruppe | Startdosis | Empfehlungsdosis (allg.) |
|---|---|---|---|---|
| Affektive Störungen/Angst | Sertralin | Antidepressivum (SSRI) | 25 mg | 75–150 mg |
| Escitalopram | Antidepressivum (SSRI) | 5 mg | 10–20 mg | |
| Citalopram | Antidepressivum (SSRI) | 10 mg | 20–40 mg | |
| Venlafaxin | Antidepressivum (SSNRI) | 37,5–75 mg | 150–225 mg | |
| Duloxetin | Antidepressivum (SSNRI) | 30 mg | 60–90 mg | |
| Mirtazapin | Antidepressivum (NaSSA) | 7,5–15 mg | 30 mg | |
| Bupropion | Antidepressivum (SNDRI) | 150 mg | 300 mg | |
| Olanzapin | Antipsychotikum 2. Gen | 2,5–5 mg | 10–20 mg | |
| Risperidon | Antipsychotikum 2. Gen | 0,5–1 mg | 2–4 mg | |
| Zwänge/Perseveration | Sertralin | Antidepressivum (SSRI) | 25–50 mg | 75–150 mg |
| Paroxetin | Antidepressivum (SSRI) | 10 mg | 20–40 mg | |
| Clomipramin | Antidepressivum (TCA) | 50 mg | 100–150 mg | |
| Olanzapin | Antipsychotikum 2. Gen | 2,5–5 mg | 10–20 mg | |
| Impulsivität/Aggressivität | Olanzapin | Antipsychotikum 2. Gen | 2,5–5 mg | 10–20 mg |
| Sulpirid/Amisulpirid | Antipsychotikum 2. Gen | 50–100 mg | 200–400 mg | |
| Risperidon | Antipsychotikum 2. Gen | 0,5–1 mg | 2–4 mg | |
| Zuclopenthixol | Antipsychotikum 1. Gen | 5 mg | 10–20 mg | |
| Haloperidol | Antipsychotikum 1. Gen | 0,5–1 mg | 2–4 mg | |
| Valproat | Stimmungsstabilisator | 150–300 mg | 600–900 mg | |
| Clonazepam | Benzodiazepine | 0,25–0,5 mg | 1–2 mg | |
| Diazepam | Benzodiazepine | 2,5–5,0 mg | 5–10 mg | |
| Lorazepam | Benzodiazepine | 0,5 mg | 1–2 mg | |
| Psychose | Olanzapin | Antipsychotikum 2. Gen | 2,5–5 mg | 10–20 mg |
| Quetiapin | Antipsychotikum 2. Gen | 25 mg | 200–400 mg | |
| Sulpirid/Amisulpirid | Antipsychotikum 2. Gen | 50–100 mg | 200–400 mg | |
| Aripiprazol | Antipsychotikum 3. Gen | 2,5–5 mg | 10–20 mg | |
| Risperidon | Antipsychotikum 2. Gen | 0,5–1 mg | 2–4 mg | |
| Clozapin | Antipsychotikum 2. Gen | 12,5 mg | 100–200 mg | |
| Zuclopenthixol | Antipsychotikum 1. Gen | 5 mg | 10–20 mg | |
| Haloperidol | Antipsychotikum 1. Gen | 0,5–1 mg | 2–4 mg | |
| Chorea/Hyperkinesienb | Tiaprid | Antipsychotikum 1. Gen | 50–100 mg | 400–600 mg |
| Tetrabenazine | Antihyperkinetikum | 12,5 mg | 25–75 mg |
SSNRI selektive Serotonin-Noradrenalin-Wiederaufnahmehemmer, SSRI selektive Serotoninwiederaufnahmehemmer, NaSSA noraadrenerge und spezifisch serotonerge Antidepressiva, SNDRI selektive Noradrenalin-Dopamin-Wiederaufnahmehemmer, TCA trizyklische Antidepressiva, Gen Generation
aEs handelt sich um Empfehlungen, die auf klinischer Erfahrung, Expertenmeinungen und Leitlinien basieren [12, 22, 57, 59, 62]. Die Dosierungen sollten individuell angepasst und evaluiert werden, eine regelmäßige Überprüfung ist erforderlich
bDie antichoreatische Medikation ist nicht Hauptbestandteil des Artikels, daher werden hier nur die am häufigsten verordneten Medikamente zur Übersicht aufgeführt
Psychotherapie
Psychotherapie (PT) hat sich bei einer Vielzahl HK-bezogener psychischer und Verhaltensproblemen als wertvoll erwiesen. Beispiele hierfür sind die Unterstützung
Unterstützung
bei der Entscheidungsfindung vor einer genetischen Untersuchung, die Unterstützung nach einer Befund- oder Diagnosemitteilung, die Krankheitsbewältigung und die Behandlung psychiatrischer Symptome wie z. B. Depression, Angst und Zwang, solange die kognitiven Ressourcen
kognitiven Ressourcen
ausreichend sind [63, 64]. Neben einer Symptomlinderung steht die Verbesserung der Lebensqualität
Verbesserung der Lebensqualität
im Vordergrund. Auch Familienangehörige
Familienangehörige
sind oft stark belastet oder zeigen sogar krankheitsrelevante Symptome, weshalb auch sie von einer PT profitieren können. Da die Pflege von HK-Patienten eine hohe Belastung darstellt, ist es wichtig, auch ihnen Unterstützung anzubieten.
Trotz des Potenzials von PT bei der HK gibt es jedoch nur begrenzte wissenschaftlich fundierte Daten hierzu. Die ohnehin bestehende Komplexität der PT-Forschung wird durch die gleichzeitige Präsenz meist mehrerer psychiatrischer Symptomkomplexe und die Vielfalt der verfügbaren Therapieverfahren bei der HK noch verstärkt, was es schwierig macht, qualitativ hochwertige Wirksamkeitsnachweise zu erbringen. Einige kleine Studien haben einen potenziellen Nutzen gezeigt, z. B. die Mindfulness-Based Cognitive Therapy
Mindfulness-Based Cognitive Therapy
(MBCT) für prämanifeste Mutationsträger [64]. Die British Psychological Society hat 2021 Therapieempfehlungen herausgegeben, die für die HK insbesondere kognitive Verhaltenstherapie
kognitive Verhaltenstherapie
(Cognitive Behavioral Therapy, CBT), MBCT und Akzeptanz-und-Commitment-Therapie
Akzeptanz-und-Commitment-Therapie
(ACT) empfehlen [65].
Bei der HK müssen wegen der fortschreitenden Natur der Erkrankung u. U. die Therapieansätze angepasst werden, um den veränderten Bedürfnissen und Fähigkeiten der Betroffenen gerecht werden zu können. Die Therapieempfehlungen für psychiatrische Symptome bei der HK (z. B. auch in den deutschen Leitlinien) konzentrieren sich häufig auf medikamentöse Ansätze und erwähnen Psychotherapie nur unter „Weitere nicht medikamentöse Therapieoptionen“, wobei angenommen werden kann, dass der Grund hierfür fehlende wissenschaftliche Wirksamkeitsnachweise
fehlende wissenschaftliche Wirksamkeitsnachweise
bei der HK sind. In einigen Fällen wird auch eine „psychologische“ oder „psychosoziale Beratung“ empfohlen, wobei es jedoch schwierig sein kann, zwischen Beratungsbedarf und einer behandlungsbedürftigen psychischen Störung zu unterscheiden. Aspekte wie Verfügbarkeit, Finanzierung und versicherungsrechtliche Fragen könnten ebenfalls eine Rolle spielen. Die PT-Forschung entwickelt inzwischen neue Ansätze für eine evidenz- und prozessbasierte modulare Psychotherapie, die in Zukunft auch für die Versorgung von Patienten mit einer HK hilfreich sein könnten. Psychotherapeutische Interventionen können sowohl in Einzel- als auch in Paarsitzungen sinnvoll sein. In den Niederlanden wurde z. B. ein Programm für Paare (Hold Me Tight Program) entwickelt und wissenschaftlich mit 15 Paaren begleitet [66].
Nichtpharmakologische Ansätze
Physiotherapie, Ergotherapie und Logopädie gehören in der Behandlung der HK mittlerweile zum etablierten Standard, insbesondere zur Verbesserung und Stabilisierung zahlreicher neurologisch-motorischer Symptome [54]. Neben diesen unmittelbaren Wirkungen sind in vielen Studien auch positive Wirkungen auf kognitive Defizite und psychische Symptomkomplexe nachgewiesen worden [67] Zudem helfen regelmäßige ambulante Termine auch bei der Tagesstrukturierung
Tagesstrukturierung
und können manchmal auch bei der Überwindung der sonst medikamentös schwer beeinflussbaren Apathie helfen. Bei HK-Betroffenen kommt es regelmäßig zu einer Reduktion der sozialen Kontakte
sozialen Kontakte
, sodass auch hier diese unterstützenden Behandlungen zu einer Verbesserung führen können, weil die Therapiekontakte wesentliche Sozialkontakte bedeuten können. Die Entscheidung sollte jedoch individuell und in sorgfältiger Absprache und unter Berücksichtigung der Motivation erfolgen, da diese Therapieverfahren die Betroffenen auch mit den bestehenden Symptomen, Einschränkungen und Defiziten konfrontieren und im Einzelfall dadurch auch zu einer Verschlechterung von Ängsten und depressiven Symptomen führen können [67].
Ethische Aspekte
Bei der HK gibt es sowohl für Betroffene als auch für Angehörige zahlreiche ethisch-moralische Aspekte, die zu beachten sind. Diese umfassen beispielsweise sowohl subjektive als auch institutionelle ethische Herausforderungen im Zusammenhang mit einer genetischen Untersuchung
genetischen Untersuchung
. Beispiele hierfür sind die präzise Aussage zu einer Mutationsträgerschaft bei fehlender kausaler Therapieoption oder Situationen, in denen sich ein (volljähriges) Kind genetisch untersuchen lassen möchte, der mutmaßlich betroffene Elternteil jedoch nicht. In solchen Fällen könnte die Mutationsträgerschaft
Mutationsträgerschaft
des Kindes einen unerwünschten Rückschluss auf den Genstatus des Elternteils zulassen. Ein weiteres Dilemma kann bei eineiigen Zwillingen
eineiigen Zwillingen
auftreten, wenn nur einer von ihnen eine genetische Untersuchung durchführen lassen möchte. Die Möglichkeit einer prädiktiven genetischen Untersuchung kann schwerwiegende psychologische Auswirkungen haben und Fragen zu Autonomie, Verantwortlichkeit, Fürsorge, Respekt vor den Wünschen anderer und Stigmatisierung aufwerfen.
Weitere ethische Fragen ergeben sich im Zusammenhang mit Kinderwunsch
Kinderwunsch
und der Option einer Präimplantationsdiagnostik (PID) oder Pränataldiagnostik (PND). Darüber hinaus bestehen relevante Themen im Zusammenhang mit lebensverlängernden Maßnahmen
lebensverlängernden Maßnahmen
, wie die Verwendung einer Ernährungssonde oder Trachealkanüle, Fragen zu Therapielimitationen und einer Palliativversorgung. Die frühzeitige Abfassung von Patientenverfügungen ist zu empfehlen (möglichst gemeinsam mit den behandelnden Ärzten) mit konkreten Festlegungen (z. B. ob und wann – wenn gewünscht – die Anlage einer perkutanen endoskopischen Gastrostomie [PEG] erfolgen soll).
Palliative Aspekte
Um eine angemessene Palliativversorgung zu gewährleisten, müssen sowohl allgemeine als auch HK-spezifische Symptome und Probleme berücksichtigt werden [68]. Eine bedarfsgerechte Palliativversorgung sollte rechtzeitig eingeleitet werden. Die häufigsten Todesursachen
häufigsten Todesursachen
sind rezidivierende Aspirationspneumonien, Herz-Kreislauf-Erkrankungen und Kachexie. Die HK geht mit einer hohen psychischen Belastung einher, z. B. Suizidgedanken, Hoffnungslosigkeit, aber auch Sorge um den Tod und das Sterben [69].
Die „Angst vor dem Sterben“
„Angst vor dem Sterben“
stellt eine große Herausforderung dar, wobei die Betroffenen explizit über Angst vor dem Sterbeprozess, Grübeln über den Tod und Angst vor der Zukunft und den Auswirkungen der Erkrankung darauf berichten [69]. Es wird daher empfohlen, psychologische, psychotherapeutische und pharmakologische Maßnahmen zu kombinieren und in die neurologische oder psychiatrische Palliativversorgung einzubeziehen.
Weitere Probleme, die es zu berücksichtigen gilt, sind Schluckstörungen, Kommunikationsschwierigkeiten, Dystonie, Schmerzen sowie kognitive und psychiatrische Symptome [68].
Merke
Das multidisziplinäre Team spielt eine wichtige Rolle bei der Betreuung und Behandlung von HK-Patienten, deren Angehörigen und Betreuern.
Wichtige Links.
Netzwerk für Ärzte, Therapeuten, Forscher und Patienten: European Huntington’s Disease Network (EHDN) https://ehdn.org/
Deutsche Patientenorganisation: Deutsche Huntington-Hilfe e. V. (DHH) https://www.dhh-ev.de/
Europäische Patientenorganisation: European Huntington Assoziation (EHA) https://eurohuntington.org/
Neuigkeiten aus der Huntington-Forschung: HD Buzz https://de.hdbuzz.net/
Fazit für die Praxis
Psychiatrische Symptome sind bei der Huntington-Krankheit (HK) häufig und vielfältig. Sie können viele Jahre vor ersten motorischen Symptomen auftreten.
Bei der HK sind Depressivität und Suizidalität besonders zu beachten.
Bei fortschreitender Erkrankung können Apathie, aggressive Verhaltensweisen und psychotische Symptome zu gravierenden Problemen führen, die neben einer stationären Behandlung auch eine Heimunterbringung erforderlich machen können.
In Ermangelung eines kausalen Therapieansatzes erfolgt die Behandlung der psychiatrischen Symptome symptomatisch. Es werden pharmakologische Ansätze, Psychotherapie und weitere nichtpharmakologische Maßnahmen inkl. sozialmedizinische Begleitung eingesetzt. Die pharmakologische und psychotherapeutische Behandlung erfolgt nach den Leitlinien für die Behandlung der jeweiligen Störungsbilder, wobei einige Besonderheiten der HK zu berücksichtigen sind.
Die Behandlung sollte durch ein multidisziplinäres Team erfolgen und die Beratung der Familienangehörigen einschließen.
CME-Fragebogen
Was könnte der 38-jährigen Patientin aus dem Fallbeispiel am Anfang des Artikels gegen die Reizbarkeit und das Aufbrausen, was zu vielen Problemen in der Familie geführt hat, empfohlen werden?
Ketamininfusionen im Rahmen eines stationären Aufenthaltes
Methylphenidat und Mediation
MAO(Monoaminoxidase)-Hemmer und systemische Psychotherapie
Selektive Serotoninwiederaufnahmehemmer (SSRI), ggf. atypisches Neuroleptikum und Psychotherapie
Dopaminagonist und Neurofeedback
Durch eine Trias welcher Symptome zeichnet sich die Huntington-Krankheit aus?
Periphere, zentrale und vegetative Symptome
Neurologische, endokrine und metabolische Symptome
Motorisch-neurologische, kognitive und psychiatrische Symptome
Muskuläre, koordinative und sensible Symptome
Schmerzen, Lähmungen und Fatigue
Die Beteiligung welcher Hirnareale erklärt die Vielfalt und Komplexität der klinischen Symptomatik der Huntington-Krankheit?
Basalganglien, frontaler Kortex, Amygdala und Striatum
Kleinhirn, Pons, Subthalamus und Gyrus cinguli
Hirnstamm, Thalamus, Tegmentum und Corpora mamillaria
Hippokampus, limbisches System, Tectum und visueller Kortex
Corpus callosum, Hypothalamus, Vermis cerebelli und Capsula interna
Ein 62-jähriger Patient mit fortgeschrittener Huntington-Krankheit wird wegen einer Sepsis nach Aspirationspneumonie mit bekannten Schluckstörungen und Kachexie stationär aufgenommen. Es liegt eine Patientenverfügung vor, nach der lebensverlängernde Maßnahmen sowie eine PEG(perkutane endoskopische Gastrostomie)-Anlage nicht gewünscht werden. Welche Maßnahme erscheint am sinnvollsten?
Stabilisierung auf einer Intensivstation
Anlage einer PEG-Sonde
Symptombezogene und palliative Behandlung
Vorübergehende parenterale Ernährung
Vorübergehende transnasale Ernährung
Wie häufig sind Suizidversuche bei der Huntington-Erkrankung?
1–2 %
7–10 %
23–25 %
27–29 %
70–80 %
Welcher Befund sollte bei Huntington-Patienten durch weitere Diagnostik abgeklärt werden, weil er untypisch für den Verlauf ist?
Gewichtsverlust
Neurologisch-motorische Symptome
Nierenfunktionsstörungen
Psychische Symptome
Kognitive Einschränkungen und Demenz
In welche Stadien wird die Huntington-Krankheit klinisch deskriptiv eingeteilt?
Asymptomatisch – prodromal – manifest
Akut und chronisch
Langsam progredient – progredient – schubförmig
Psychiatrisch – neurologisch – neuropsychiatrisch
Neurotisch – psychotisch – motorisch
Welche Symptome werden Jahre vor dem Auftreten erster choreatischer Symptome bei der Huntington-Krankheit beobachtet?
Psychiatrische Symptome
Ataxie
Chronisches Schmerzsyndrom
Elektrolytstörungen
Epileptische Anfälle
In Ihrer Praxis stellt sich ein 24-jähriger Mann mit einer positiven Familienanamnese für die Huntington-Krankheit vor, der weder neurologische noch psychiatrische Symptome zeigt und eine genetische Untersuchung auf das Vorliegen einer Mutation für die Huntington-Krankheit wünscht. Wie sollten Sie vorgehen?
Den Patienten darüber aufklären, dass sich bei positiver Familienanamnese eine genetische Untersuchung erübrigt
Eine umgehende Blutentnahme für die molekulargenetische Diagnostik anbieten
Eine genetische Beratung nach dem Gendiagnostikgesetz bei einem hierfür zertifizierten Arzt empfehlen
Den Patienten informieren, dass eine genetische Untersuchung nur beim Vorliegen neurologischer Symptome sinnvoll ist
Dem Patienten in dieser Situation von einer genetischen Untersuchung abraten
Worauf ist besonders zu achten, wenn bei Patienten eine ausgeprägte Chorea mit Tetrabenazin behandelt wird?
Es können Depressionen und suizidale Gedanken ausgelöst werden.
Der Serumkaliumspiegel kann bedrohlich ansteigen.
Eine hormonelle Kontrazeption kann durch Enzyminduktion beeinträchtigt werden.
Es können psychotische und/oder maniforme Episoden ausgelöst werden.
Der Tetrabenazinspiegel ist regelmäßig zu kontrollieren.
Einhaltung ethischer Richtlinien
Interessenkonflikt
Gemäß den Richtlinien des Springer Medizin Verlags werden Autoren und Wissenschaftliche Leitung im Rahmen der Manuskripterstellung und Manuskriptfreigabe aufgefordert, eine vollständige Erklärung zu ihren finanziellen und nichtfinanziellen Interessen abzugeben.
Autoren
P. Brieger: A. Finanzielle Interessen: P. Brieger gibt an, dass kein finanzieller Interessenkonflikt besteht. – B. Nichtfinanzielle Interessen: Ärztlicher Direktor des kbo-Isar-Amper-Klinikums, München | Mitgliedschaft: DGPPN. M. Dose: A. Finanzielle Interessen: Reisekosten für Leitung von Gruppen bei Jahrestagungen der Deutschen Huntington-Hilfe (DHH e.V.). – kbo (Kliniken des Bezirks Oberbayern) Fachberater für Autismus-Spektrum-Störungen bei Erwachsenen und Huntington-Krankheit. – B. Nichtfinanzielle Interessen: kbo (Kliniken des Bezirks Oberbayern) Fachberater für Autismus-Spektrum-Störungen bei Erwachsenen und Huntington-Krankheit | Mitglied des wissenschaftlichen Beirats der Deutschen-Huntington-Hilfe/DHH e.V. und von Autismus Deutschland, Mitglied des Europäischen Huntington Netzwerkes (EHDN) und der DGPPN. R. Hoffmann: A. Finanzielle Interessen: Forschungsförderung zur persönlichen Verfügung: Prüfer in Studien für Hoffmann-LaRoche, Wave, SOM-Tech, Prilenia und CHDI. – B. Nichtfinanzielle Interessen: Angestellter Oberarzt Huntington-Zentrum-Süd, kbo-Isar-Amper-Klinikum, Region München, Taufkirchen (Vils). M. Marziniak: A. Finanzielle Interessen: M. Marziniak gibt an, dass kein finanzieller Interessenkonflikt besteht. – B. Nichtfinanzielle Interessen: Angestellt als Chefarzt der Klinik für Neurologie am Isar-Amper-Klinikum, 85540 Haar seit 2013 | Mitgliedschaften: DGN, DMSG, DGKN, DSG, DGSS | Mitgliedschaft und bis Ende 2021 2. Vizepräsident der DMKG. A. Mühlbäck: A. Finanzielle Interessen: Meine Institution wurde für meine Rolle als Hauptprüferin in Studien für Hoffmann- LaRoche, Wave, SOM-Tech, Prilenia und die Cure Huntington’s Disease Initiative Foundation (CHDI) bezahlt. Bis 2023 arbeitete ich am Huntington-Zentrum der Neurologischen Klinik der Universität Ulm, diese Einrichtung erhielt Zuwendungen für klinische Studien von Hoffmann-LaRoche, Ionis-Pharma, Prilenia, Novartis, TEVA und PTC-Therapeutics. Ich habe an der Universität Ulm (unter der Leitung von Prof. Landwehrmeyer) an EU-Forschungsprojekten (Horizon 2020, JPND 2018) teilgenommen, die vom Bundesministerium für Bildung und Forschung (BMBF) finanziert wurden. – Ich habe Reise- und Übernachtungskosten erstattet bekommen von European Huntington Association (EHA), Europäischen Huntington Netzwerk (EHDN), der Deutschen Huntington Hilfe (DHH), der Deutschen Heredoataxie Gesellschaft (DHAG), der Deutschen Parkinson Gesellschaft (DPG) und der Firma Gen.Orph erstattet. – B. Nichtfinanzielle Interessen: FÄ, Huntington-Zentrum, Neurologie, Uniklinik Ulm, Ulm (Teilzeit 2017 -2023); OÄ, Huntington-Zentrum Süd, kbo-Isar-Amper-Klinikum, Klinik Taufkirchen, Taufkirchen (Teilzeit 2020 bis dato ); FÄ, Psychiatrie, Klinikum re. d. Isar, TUM, München (Teilzeit 03/2024 bis dato ) | Mitgliedschaften: European Huntington Association (EHA), Deutsche Parkinson Gesellschaft (DPG), Deutsche Gesellschaft für Psychiatrie und Psychotherapie, Psychosomatik und Nervenheilkunde (DGPPN) | Mitglied des Wissenschaftlichen Beirats - Deutsche Huntington Hilfe (DHH), Vorstandsmitglied - Europäischen Huntington Netzwerk (EHDN), Vorstandsmitglied - Deutsche Heredoataxie Gesellschaft (DHAG). N. Pozzi: A. Finanzielle Interessen: N. Pozzi wurde gefördert von Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project-ID 424778381—TRR 295. – B. Nichtfinanzielle Interessen: Angestellter Arzt in Weiterbildung im Huntington-Zentrum-Süd, kbo-Isar-Amper-Klinikum, Region München, Taufkirchen (Vils) und in Neurologische Klinik und Poliklinik, Universitätsklinikum Würzburg, Würzburg. J. Priller: A. Finanzielle Interessen: Studienteilnahme: EHDN, CHDI, Ionis/Roche, Ergomed. – Reisekosten: EHDN, CHDI. – Kein Interessenkonflikt durch wissenschaftliche Zusammenarbeit mit Neurimmune und Beratertätigkeit bei Axon. – Patent auf EPO-Varianten. – B. Nichtfinanzielle Interessen: Ärztlicher Direktor, Klinik und Poliklinik für Psychiatrie und Psychotherapie, Klinikum rechts der Isar, School of Medicine and Health, TU München, München; Leiter, Labor für Mol. Psychiatrie, Neuropsychiatrie, Charité - Universitätsmedizin Berlin; PI, DZNE Berlin und DZPG München; Chair of Brain Inflammation and Repair, University of Edinburgh and UK DRI | Kein direkter Interessenkonflikt durch Mitgliedschaften in der Deutschen Gesellschaft für Psychiatrie und Psychotherapie, Psychosomatik und Nervenheilkunde (DGPPN), der Deutschen Gesellschaft für Gerontopsychiatrie und -psychotherapie (DGGPP), der Deutschen Gesellschaft für Biologische Psychiatrie (DGBP), dem Deutschen Zentrum für Neurodegenerative Erkrankungen (DZNE) und Deutschen Zentrum für Psychische Gesundheit (DZPG), dem Deutschen Netzwerk Gedächtnisambulanzen (DNG), Generate e.V. und der International Group for The Study of Lithium Treated Patients (IGSLI).
Wissenschaftliche Leitung
Die vollständige Erklärung zum Interessenkonflikt der Wissenschaftlichen Leitung finden Sie am Kurs der zertifizierten Fortbildung auf www.springermedizin.de/cme.
Der Verlag
erklärt, dass für die Publikation dieser CME-Fortbildung keine Sponsorengelder an den Verlag fließen.
Für diesen Beitrag wurden von den Autor/-innen keine Studien an Menschen oder Tieren durchgeführt. Für die aufgeführten Studien gelten die jeweils dort angegebenen ethischen Richtlinien.
Footnotes

QR-Code scannen & Beitrag online lesen
Hinweis des Verlags
Der Verlag bleibt in Hinblick auf geografische Zuordnungen und Gebietsbezeichnungen in veröffentlichten Karten und Institutsadressen neutral.
Literatur
- 1.Huntington G, Chorea O (1872) The Medical and Surgical Reporter. S.W. Butler, Philadelphia [Google Scholar]
- 2.Huntington’s disease Collaborative Research, G. (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72(6):971–983 10.1016/0092-8674(93)90585-E [DOI] [PubMed] [Google Scholar]
- 3.Lee JM et al (2012) CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology 78(10):690–695 10.1212/WNL.0b013e318249f683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gardiner SL et al (2019) Prevalence of Carriers of Intermediate and Pathological Polyglutamine Disease–Associated Alleles Among Large Population-Based Cohorts. JAMA Neurol 76(6):650 10.1001/jamaneurol.2019.0423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pringsheim T et al (2012) The incidence and prevalence of Huntington’s disease: A systematic review and meta-analysis. Mov Disord 27(9):1083–1091 10.1002/mds.25075 [DOI] [PubMed] [Google Scholar]
- 6.Bates GP et al (2015) Huntington disease. Nat Rev Dis Primers 1(1):15005 10.1038/nrdp.2015.5 [DOI] [PubMed] [Google Scholar]
- 7.Tabrizi SJ et al (2022) A biological classification of Huntington’s disease: the Integrated Staging System. Lancet Neurol 21(7):632–644 10.1016/S1474-4422(22)00120-X [DOI] [PubMed] [Google Scholar]
- 8.Paoli R et al (2017) Neuropsychiatric Burden in Huntington’s Disease. Brain Sci 7(12):67 10.3390/brainsci7060067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paulsen et al (2014) Clinical and Biomarker Changes in Premanifest Huntington Disease Show Trial Feasibility: A Decade of the PREDICT-HD Study. Front Aging Neurosci 6:78 10.3389/fnagi.2014.00078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stout JC et al (2012) Evaluation of longitudinal 12 and 24 month cognitive outcomes in premanifest and early Huntington’s disease. J Neurol Neurosurg Psychiatry 83(7):687–694 10.1136/jnnp-2011-301940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Burg JMM et al (2021) Effect of Body Weight on Age at Onset in Huntington Disease. Neurol Genet 7(4):e603 10.1212/NXG.0000000000000603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bachoud-Lévi AC et al (2019) International Guidelines for the Treatment of Huntington’s Disease. Front Neurol 10:710 10.3389/fneur.2019.00710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosas HD et al (2008) Cerebral cortex and the clinical expression of Huntington’s disease: complexity and heterogeneity. Brain 131(Pt 4):1057–1068 10.1093/brain/awn025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vonsattel J‑P et al (1985) Neuropathological Classification of Huntington’s Disease. J Neuropathol Exp Neurol 44(6):559–577 10.1097/00005072-198511000-00003 [DOI] [PubMed] [Google Scholar]
- 15.Thu DC et al (2010) Cell loss in the motor and cingulate cortex correlates with symptomatology in Huntington’s disease. Brain 133(4):1094–1110 10.1093/brain/awq047 [DOI] [PubMed] [Google Scholar]
- 16.Martínez-Horta S et al (2018) Structural and metabolic brain correlates of apathy in Huntington’s disease. Mov Disord 33(7):1151–1159 10.1002/mds.27395 [DOI] [PubMed] [Google Scholar]
- 17.van Wamelen DJ et al (2013) Suprachiasmatic nucleus neuropeptide expression in patients with Huntington’s Disease. Sleep 36(1):117–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh-Bains MK et al (2019) Cerebellar degeneration correlates with motor symptoms in Huntington disease. Ann Neurol 85(3):396–405 10.1002/ana.25413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dorsey ER (2013) Natural History of Huntington Disease. JAMA Neurol [DOI] [PubMed]
- 20.van Duijn et al (2014) Neuropsychiatric symptoms in a European Huntington’s disease cohort (REGISTRY). J Neurol Neurosurg Psychiatry 85(12):1411–1418 10.1136/jnnp-2013-307343 [DOI] [PubMed] [Google Scholar]
- 21.Orth M et al (2010) Observing Huntington’s Disease: the European Huntington’s Disease Network’s REGISTRY. PLoS Curr 2:Rrn1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson KE et al (2018) Clinical Management of Neuropsychiatric Symptoms of Huntington Disease: Expert-Based Consensus Guidelines on Agitation, Anxiety, Apathy, Psychosis and Sleep Disorders. J Huntingtons Dis 7(4):355–366 10.3233/JHD-180293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McAllister B et al (2021) Timing and Impact of Psychiatric, Cognitive, and Motor Abnormalities in Huntington Disease. Neurology 96(19):e2395–e2406 10.1212/WNL.0000000000011893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jellinger KA (2024) The pathobiology of depression in Huntington’s disease: an unresolved puzzle. J Neural Transm [DOI] [PubMed]
- 25.van Duijn E, Kingma EM, van der Mast RC (2007) Psychopathology in verified Huntington’s disease gene carriers. J Neuropsychiatry Clin Neurosci 19(4):441–448 10.1176/jnp.2007.19.4.441 [DOI] [PubMed] [Google Scholar]
- 26.Kachian ZR et al (2019) Suicidal ideation and behavior in Huntington’s disease: Systematic review and recommendations. J Affect Disord 250:319–329 10.1016/j.jad.2019.03.043 [DOI] [PubMed] [Google Scholar]
- 27.Nock MK et al (2008) Cross-national prevalence and risk factors for suicidal ideation, plans and attempts. Br J Psychiatry 192(2):98–105 10.1192/bjp.bp.107.040113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kenwood MM, Kalin NH, Barbas H (2022) The prefrontal cortex, pathological anxiety, and anxiety disorders. Neuropsychopharmacology 47(1):260–275 10.1038/s41386-021-01109-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Duijn et al (2008) Cross-sectional study on prevalences of psychiatric disorders in mutation carriers of Huntington’s disease compared with mutation-negative first-degree relatives. J Clin Psychiatry 69(11):1804–1810 10.4088/JCP.v69n1116 [DOI] [PubMed] [Google Scholar]
- 30.Eddy CM, Parkinson EG, Rickards HE (2016) Changes in mental state and behaviour in Huntington’s disease. Lancet Psychiatry 3(11):1079–1086 10.1016/S2215-0366(16)30144-4 [DOI] [PubMed] [Google Scholar]
- 31.Abdollah Zadegan S et al (2023) Frequency and Pathophysiology of Apathy in Huntington Disease: A Systematic Review and Meta-Analysis. J Neuropsychiatry Clin Neurosci 35(2):121–132 10.1176/appi.neuropsych.20220033 [DOI] [PubMed] [Google Scholar]
- 32.Levy R, Dubois B (2006) Apathy and the functional anatomy of the prefrontal cortex-basal ganglia circuits. Cereb Cortex 16(7):916–928 10.1093/cercor/bhj043 [DOI] [PubMed] [Google Scholar]
- 33.Pagonabarraga J et al (2015) Apathy in Parkinson’s disease: clinical features, neural substrates, diagnosis, and treatment. Lancet Neurol 14(5):518–531 10.1016/S1474-4422(15)00019-8 [DOI] [PubMed] [Google Scholar]
- 34.De Paepe AE et al (2021) Gray Matter Vulnerabilities Predict Longitudinal Development of Apathy in Huntington’s Disease. Mov Disord 36(9):2162–2172 10.1002/mds.28638 [DOI] [PubMed] [Google Scholar]
- 35.Misiura MB et al (2019) Apathy Is Related to Cognitive Control and Striatum Volumes in Prodromal Huntington’s Disease. J Int Neuropsychol Soc 25(5):462–469 10.1017/S1355617719000067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andrews SC et al (2021) Apathy predicts rate of cognitive decline over 24 months in premanifest Huntington’s disease. Psychol Med 51(8):1338–1344 10.1017/S0033291720000094 [DOI] [PubMed] [Google Scholar]
- 37.Oosterloo M et al (2019) Obsessive-Compulsive and Perseverative Behaviors in Huntington’s Disease. J Huntingtons Dis 8(1):1–7 10.3233/JHD-180335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoffmann R et al (2019) Obsessive-Compulsive Symptoms are Less Common in Huntington’s Disease than Reported Earlier. J Huntingtons Dis 8(4):493–500 10.3233/JHD-190351 [DOI] [PubMed] [Google Scholar]
- 39.Karagas NE, Rocha NP, Stimming EF (2020) Irritability in Huntington’s Disease. J Huntingtons Dis 9(2):107–113 10.3233/JHD-200397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simpson J et al (2019) Validity of irritability in Huntington’s disease: A scoping review. Cortex 120:353–374 10.1016/j.cortex.2019.06.012 [DOI] [PubMed] [Google Scholar]
- 41.Pardina-Torner H et al (2024) Disentangling the neurobiological bases of temporal impulsivity in Huntington’s disease. Brain Behav 14(3):e3335 10.1002/brb3.3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McLauchlan DJ, Linden DEJ, Rosser AE (2022) Excessive response to provocation rather than disinhibition mediates irritable behaviour in Huntington’s disease. Front Neurosci 16:993357 10.3389/fnins.2022.993357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCusker E, Loy CT (2014) The many facets of unawareness in huntington disease. Tremor Other Hyperkinet Mov 4:257 10.5334/tohm.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rossi M et al (2020) Nosology and Phenomenology of Psychosis in Movement Disorders. Mov Disord Clin Pract 7(2):140–153 10.1002/mdc3.12882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bonelli RM, Hofmann P (2004) A review of the treatment options for Huntington’s disease. Expert Opin Pharmacother 5(4):767–776 10.1517/14656566.5.4.767 [DOI] [PubMed] [Google Scholar]
- 46.Cardoso F (2014) Differential diagnosis of Huntington’s disease: what the clinician should know. Neurodegen Dis Manage 4(1):67–72 10.2217/nmt.13.78 [DOI] [PubMed] [Google Scholar]
- 47.Saft C et al (2023) Differential diagnosis of chorea (guidelines of the German Neurological Society). Neurol Res Pract 5(1):63 10.1186/s42466-023-00292-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huntington Study Group (1996) Unified Huntington’s disease rating scale: Reliability and consistency. Mov Disord 11(2):136–142 10.1002/mds.870110204 [DOI] [PubMed] [Google Scholar]
- 49.Mason SL et al (2018) Predicting clinical diagnosis in Huntington’s disease: An imaging polymarker. Ann Neurol 83(3):532–543 10.1002/ana.25171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeun P et al (2019) Fluid and imaging biomarkers for Huntington’s disease. Mol Cell Neurosci 97:67–80 10.1016/j.mcn.2019.02.004 [DOI] [PubMed] [Google Scholar]
- 51.Saft C et al (2022) S2k-Leitlinie Chorea/Morbus Huntington. Hrsg. Leitlinien für Diagnostik und Therapie in der Neurologie. Deutsche Gesellschaft für Neurologie (DGN) [Google Scholar]
- 52.Gendiagnostikgesetz, Gesetz über genetische Untersuchungen bei Menschen (Gendiagnostikgesetz – GenDG) vom 31. Juli 2009 (BGBl. I S. 2529, 3672), das zuletzt durch Artikel 15 Absatz 4 des Gesetzes vom 4. Mai 2021 (BGBl. I S. 882) geändert worden ist. 31.07.2009: Ein Service des Bundesministeriums der Justiz sowie des Bundesamts für Justiz – www.gesetze-im-internet.de.
- 53.Macleod R et al (2013) Recommendations for the predictive genetic test in Huntington’s disease. Clin Genet 83(3):221–231 10.1111/j.1399-0004.2012.01900.x [DOI] [PubMed] [Google Scholar]
- 54.Mühlbäck A et al (2023) What we don’t need to prove but need to do in multidisciplinary treatment and care in Huntington’s disease: a position paper. Orphanet J Rare Dis 18(1):19 10.1186/s13023-023-02622-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gelderblom H et al (2017) Bupropion for the treatment of apathy in Huntington’s disease: A multicenter, randomised, double-blind, placebo-controlled, prospective crossover trial. PLoS ONE 12(3):e173872 10.1371/journal.pone.0173872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saft C et al (2023) Symptomatic treatment options for Huntington’s disease (guidelines of the German Neurological Society). Neurol Res Pract 5(1):61 10.1186/s42466-023-00285-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stoker TB et al (2022) Huntington’s disease: diagnosis and management. Pract Neurol 22(1):32–41 10.1136/practneurol-2021-003074 [DOI] [PubMed] [Google Scholar]
- 58.Burgunder JM et al (2011) An International Survey-based Algorithm for the Pharmacologic Treatment of Chorea in Huntington’s Disease. PLoS Curr 3:Rrn1260 10.1371/currents.RRN1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Massey TH, McLauchlan DJ (2024) Huntington’s disease: A clinical primer for acute and general physicians. Clin Med 24(2):100200 10.1016/j.clinme.2024.100200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clark ML et al (2023) A systematic review and meta-analysis of depression and apathy frequency in adult-onset Huntington’s disease. Neurosci Biobehav Rev 149:105166 10.1016/j.neubiorev.2023.105166 [DOI] [PubMed] [Google Scholar]
- 61.Anderson K et al (2011) An International Survey-based Algorithm for the Pharmacologic Treatment of Obsessive-Compulsive Behaviors in Huntington’s Disease. PLoS Curr 3:Rrn1261 10.1371/currents.RRN1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saft C et al (2023) Symptomatic treatment options for Huntington’s disease (guidelines of the German Neurological Society). Neurol Res Pract 5(1):61 10.1186/s42466-023-00285-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barth J et al (2013) Comparative efficacy of seven psychotherapeutic interventions for patients with depression: a network meta-analysis. PLoS Med 10(5):e1001454 10.1371/journal.pmed.1001454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eccles FJR et al (2021) Experiences of Mindfulness-Based Cognitive Therapy for Premanifest Huntington’s Disease. J Huntingtons Dis 10(2):277–291 10.3233/JHD-210471 [DOI] [PubMed] [Google Scholar]
- 65.Simpson JEF, Zarotti N, for British Psychological Society (2021) Psychological interventions for people with Huntington’s disease, Parkinson’s disease, motor neurone disease, and multiple sclerosis: Evidence-based guidance. Lancaster University [Google Scholar]
- 66.Petzke TM, Rodriguez-Girondo M, van der Meer LB (2022) The Hold me Tight Program for Couples Facing Huntington’s Disease. J Huntingtons Dis 11(2):203–215 10.3233/JHD-210516 [DOI] [PubMed] [Google Scholar]
- 67.Jones U, Kegelmeyer DA, Kloos AD (2022) Implementing Physiotherapy Huntington’s Disease Guidelines in Clinical Practice. J Huntingtons Dis 11(3):307–311 10.3233/JHD-220532 [DOI] [PubMed] [Google Scholar]
- 68.Boersema-Wijma DJ et al (2023) Palliative care in advanced Huntington’s disease: a scoping review. BMC Palliat Care 22(1):54 10.1186/s12904-023-01171-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sokol LL et al (2023) Death Anxiety in Huntington Disease: Longitudinal Heath-Related Quality-of-Life Outcomes. J Palliat Med 26(7):907–914 10.1089/jpm.2022.0160 [DOI] [PMC free article] [PubMed] [Google Scholar]