

Abstract
The unprecedented habitat fragmentation or loss has threatened the existence of many species. Therefore, it is essential to understand whether and how these species can pace with the environmental changes. Recent advantages in landscape genomics enabled us to identify molecular signatures of adaptation and predict how populations will respond to changing environments, providing new insights into the conservation of species. Here, we investigated the pattern of neutral and putative adaptive genetic variation and its response to changing environments in a tertiary relict tree species, Taxus cuspidata Sieb. et Zucc, which is distributed in northeast China and adjacent regions. We investigated the pattern of genetic diversity and differentiation using restriction site‐associated DNA sequencing (RAD‐seq) and seven nuclear microsatellites (nSSRs) datasets. We further explored the endangered mechanism, predicted its vulnerability in the future, and provided guidelines for the conservation and management of this species. RAD‐seq identified 16,087 single nucleotide polymorphisms (SNPs) in natural populations. Both the SNPs and nSSRs datasets showed high levels of genetic diversity and low genetic differentiation in T. cuspidata. Outlier detection by F ST outlier analysis and genotype‐environment associations (GEAs) revealed 598 outlier SNPs as putative adaptive SNPs. Linear redundancy analysis (RDA) and nonlinear gradient forest (GF) showed that the contribution of climate to genetic variation was greater than that of geography, and precipitation played an important role in putative adaptive genetic variation. Furthermore, the genetic offset and risk of non‐adaptedness (RONA) suggested that the species at the northeast edge may be more vulnerable in the future. These results suggest that although the species has maintained high current genetic diversity in the face of recent habitat loss and fragmentation, future climate change is likely to threaten the survival of the species. Temperature (Bio03) and precipitation (Prec05) variables can be potentially used as predictors of response of T. cuspidata under future climate. Together, this study provides a theoretical framework for conservation and management strategies for wildlife species in the context of future climate change.
Keywords: climate change, conservation, genetic diversity, genotype‐environment associations, habitat fragmentation, local adaptation
1. INTRODUCTION
Habitat loss or fragmentation due to climate change or human activity is recognized as a major threat to global biodiversity (Pykälä, 2019; Rands et al., 2010). Among the various forms of biodiversity, genetic diversity is particularly important in conservation biology, as it provides the raw material for evolutionary change and thus has the potential to adapt to changing environments (Laikre et al., 2010; Nielsen et al., 2022; O'Brien et al., 2022). Normally, genetic diversity might decrease because of increased random genetic drift, inbreeding, and reductions in gene flow due to habitat loss or fragmentation of species (Aguilar et al., 2008; Chung et al., 2023; Lowe et al., 2005; Young et al., 1996). However, habitat loss or fragmentation can also lead to highly specific genetic consequences (e.g., increased genetic divergence, genetic bottleneck and reduced effective population size) for plants because of their different life histories, particularly for tree species with long generation times (e.g., González et al., 2020; Petit & Hampe, 2006; Piotti, 2009).
Forest tree species are largely undomesticated and exhibit local adaptation across heterogeneous environments (Bonan, 2008; Litkowiec et al., 2018). Neutral molecular markers, such as microsatellite genotyping, chloroplast DNA (cpDNA) and mitochondrial DNA (mtDNA) fragments, have been used to reveal the genetic dynamics of forest tree species especially endangered tree species. In general, low genetic diversity and strong genetic differentiation were revealed in endangered or critically endangered tree species with small population size because of the enhanced genetic drift effect and inbreeding (e.g., Frankham, 2005; Yang et al., 2020), such as T. yunnanensis (Miao et al., 2016), Abies ziyuanensis (Tang et al., 2008), Cathaya argyrophylla (Wang & Ge, 2006). However, high levels of genetic diversity have also been detected in some endangered tree species with current small or scattered population size (e.g., de Sousa et al., 2020; Gitzendanner et al., 2012; Yousefzadeh et al., 2018).
To date, neutral molecular markers are the most common tools for conservation genetic studies, however, genetic variation at neutral loci cannot provide direct information on adaptive selective processes involving the interactions between individuals and their environment (Hoffmann & Willi, 2008; Holderegger et al., 2006). Recently, with the advent of high‐throughput sequencing technologies, adaptive genetic markers have become available even for tree species with large and complex genomes (Feng & Du, 2022; Mackay et al., 2012; Neale & Kremer, 2011). In addition, emerging landscape genomics aims to detect adaptive variation along the landscape by integrating geographic and environmental information to provide unprecedented insights into the evolutionary mechanisms of local adaptation (Borrell et al., 2020; Du et al., 2020; Sork et al., 2013; Wang et al., 2021). F ST outlier analysis and genotype‐environment associations (GEAs) in landscape genomics have been suggested as promising tools for detecting adaptive genetic variation in conservation practices (Ahrens et al., 2018; Chung et al., 2023; Schoville et al., 2012). In recent years researchers have increasingly employed these methods to study how spatial heterogeneity promotes population genetic dynamics, and how genomic variation promotes adaptive evolution in forest tree species, particularly conifers (Jia et al., 2020; Mayol et al., 2020; Zhao et al., 2020).
Taxus cuspidata Sieb. et Zucc., a tertiary relict tree species, is a keystone‐dominant coniferous long‐lived wind‐pollinated tree species with a discontinuous distribution in Japan, Korea, Northeast (NE) China and Far Eastern Russia (Kunashir Island, Sakhalin and Primorye) (Chung et al., 1999; Kitamura & Murata, 1987). In China, the species is mainly located at altitudes ranging from 600 m to 1200 m in the Changbai Mountains and neighboring areas. The number of natural populations of T. cuspidata has drastically decreased in the last century, partially because of human disturbance. Therefore, T. cuspidata is considered as “Plant Species with Extremely Small Populations (PSESP)” in China (Wade et al., 2016), however, it was a Least Concern (LC) species in the International Union for Conservation of Nature (IUCN) (Katsuki & Luscombe, 2013). Previous studies based on paternal inherited mtDNA and cpDNA fragments have shown high degrees of genetic diversity in T. cuspidata (Cheng et al., 2015; Kozyrenko et al., 2017; Su et al., 2018). However, the contributions of geography and climate to genetic variation in T. cuspidata remain unclear.
Here, we sampled T. cuspidata natural populations throughout the species' distribution range and combined their neutral and non‐neutral genetic variation using nSSRs and SNPs. We aimed to (1) estimate the genetic diversity and genetic differentiation within and among populations, (2) understand the contribution of geographic and climatic factors to genetic variation and (3) predict future adaptability of the species using landscape genomic tools under a scenario of future climate change. Our study lays the groundwork for understanding the molecular mechanisms of local adaptation, and provides a theoretical foundation for the conservation and management of important conifer tree species in East Asia.
2. MATERIALS AND METHODS
2.1. Field sampling
In our study, we collected leaf material in natural populations of T. cuspidata in the Changbai Mountains and adjacent areas. The sampling points were strategically distributed across the area, spanning from the southwest to the northeast of Changbai Mountains and adjacent areas. A total of 200 individuals of T. cuspidata were sampled from 19 natural populations and each population was separated by at least 30 km. The detail sampling information of each population was listed in Table S1. Leaf samples were labeled and stored in plastic bags with silica gel for DNA extraction. Voucher specimens of each population were deposited at Beijing Forestry University.
2.2. DNA isolation and microsatellite genotyping
Total genomic DNA was extracted from leaf tissue using Plant Genomic DNA Kit (Tiangen, Beijing, China) following the manufacturer's protocol. The DNA quality was initially checked by 1% agarose electrophoresis gel and then the concentration was measured by an ultramicro‐spectrophotometer (Thermo Fisher, USA). Seven polymorphic nuclear microsatellite (nSSR) loci were used for genotyping all 200 individuals after initial screening of 24 nSSR primers that had been already applicable to T. cuspidata (Cheng et al., 2015; Dubreuil et al., 2008; Kondo, 2016) (see Table S2 for primer details). The PCR conditions followed Du et al., 2017 and the PCR products were analyzed using an ABI PRISM 3730 Genetic Analyzer (Applied Biosystems, USA). Subsequently, the alleles were scored using GENEMARKER v. 2.2 (Softgenetics, USA) and the genotype was checked twice.
2.3. Double‐digest RADseq‐derived SNP dataset
A subset of samples (130 out of 200 individuals in 19 populations) was used for SNP dataset collection by a double‐digestion restriction fragment‐based procedure (ddRAD‐seq) (Peterson et al., 2012) (Table S3). The subset of sampling by RAD‐seq covered almost the entire distribution range to capture the vast majority of diversity. For each sample, the genomic DNA was processed for library construction and sequencing. Briefly, DNA was double‐digested with restriction enzyme TaqI and MseI, followed by the ligation of Illumina adaptors. Ligation products were size‐selected about 500 bp and amplified by kapa HotStart ReadyMix (cat. no. KK2601; KAPA Biosystems) with 13 cycles. Paired‐end sequencing (2 × 150 bp) was performed using an Illumina HiSeq2000 platform at Majorbio Pharm Technology Co., Ltd., Shanghai, China.
The data quality control was assessed by FastQC v0.11.7 (Andrews, 2010). We removed adapter sequences and low‐quality bases (Phred quality <20) from raw data using Trimmomatic 0.36 (Bolger et al., 2014). Then the reads were all trimmed to 120 bp and designed for SNP genotyping using Stacks v1.48 (Catchen et al., 2011, 2013). In detail, ustacks was used to assemble our sequences into de novo, cstacks was used to build a catalog of loci, sstcaks was used to match loci against the catalog and the Populations was used to output SNPs. A strict criteria was used to filter SNPs using VCFtools (Danecek et al., 2011): first, we only kept individuals that represented at least 60% of the SNPs; second, a filtering parameter of 0.8 was used to avoid the influence of missing data, i.e., more than 80% of individuals in a population were required to process a locus; third, a minor allele frequency (MAF) < 0.1 was used to filter the data and to reduce the likelihood of false‐positive results due to spurious correlations.
2.4. Genetic diversity and structure
We estimated genetic diversity including mean effective number of alleles (N E), mean observed heterozygosity (H O), mean expected heterozygosity (H E) and mean number of different alleles (N A) for each population by GenAlEx 6.5 for nSSR dataset (Peakall & Smouse, 2006). We defined two types for ddRAD‐seq dataset: neutral genetic variation based on neutral SNPs and non‐neutral (putatively adaptive) genetic variation based on outlier SNPs (i.e., outliers were detected by F ST and GEA analysis; see section outlier detection below). We estimated the mean frequency of the most frequent allele at each locus (P), mean nucleotide diversity (π), H O and H E for all, neutral, F ST‐outlier and GEA‐outlier SNPs by Populations module in Stacks (Catchen et al., 2013).
We conducted a principal component analysis (PCA) to produce a lower dimensional subspace that captured most of the variation in each of three data types. For nSSRs, “adegenet” (Jombart & Ahmed, 2011) was used for the PCA in R 3.6.1 (R Core Team, 2019). PLINK v.1.07 was used for the PCA in all, neutral and outlier SNPs (Purcell et al., 2007; Zheng et al., 2012).
Population structure was also accessed using Bayesian clustering. For nSSRs, we identified population genetic structure through the Bayesian Markov chain Monte Carlo (MCMC) clustering method implemented in STRUCTURE v 2.3.4 (Pritchard et al., 2000). Twenty independent runs were performed for each value of K (1–10) using 200,000 generations for the MCMC cycles and 100,000 generations for the burn‐in by STRUCTURE HARVESTER (Earl & vonHoldt, 2012). The most likely number of clusters (K) was determined using ΔK and LnP(K) statistics, according to Evanno et al. (2005). CLUMPP (Jakobsson & Rosenberg, 2007) and DISTRUCT v1.1 (Rosenberg, 2004) were used to aligned independent runs and visualize the bar plots of the individual's probabilities of population membership.
ADMIXTURE v1.3.0 (Alexander et al., 2009; Alexander & Lange, 2011), which can deal with large data with fast speed, was applied to assess population structure of genetic variation for all, neutral and outlier SNPs. We ran ADMIXTURE with default 5‐fold cross‐validation (−cv = 5) to select the optimal K from one to ten, the best K exhibits low cross‐validation error (CV error) opposed to others (Alexander & Lange, 2011).
We conducted a hierarchical analysis of molecular variance (AMOVA) to quantify the degree of genetic divergence among and within populations for our nSSRs, all SNPs, neutral SNPs, F ST‐outlier SNPs and GEA‐outlier SNPs by Arlequin 3.5 (Excoffier & Lischer, 2010). The significance of genetic differentiation differences was evaluated using 10,000 permutations.
2.5. Climatic variables
Bioclimatic variables of current climate (representative of 1960–1990) and future climate (2070: average for 2061–2080) were downloaded from the WorldClim Version2 (http://worldclim.org/version2, Fick & Hijmans, 2017) at spatial resolutions of 30 s (~1 km2). A total of 31 climate variables were extracted by longitudes and latitudes of population sites based on raster layers by ArcMap10.2. To avoid biased estimates of model coefficients and spurious significance levels resulting from multicollinearity, we excluded highly correlated climate variables with the threshold values of 0.7 using a variance inflation factor (VIF) test in “usdm” R package (Marquaridt, 1970; Naimi et al., 2014; R Core Team, 2019). Six climate variables were finally retained: isothermality (bio03), maximum temperature of warmest month (bio05), mean temperature of driest quarter (bio09), precipitation seasonality (bio15), precipitation in May (prec05) and precipitation in October (prec10). We also carried out Shapiro–Wilk and Levene test (package car, Fox & Weisberg, 2019) to explore each variable's data distribution and homogeneity. We found that the data for each variable was not normally distributed nor homogeneous; therefore, we performed the Kruskal‐Wallis rank sum test to explore the significance level for each variable.
2.6. Outlier detection
Three algorithms were used to detect potentially adaptive loci, including one F ST outlier analysis approach and two genotype‐environment associations (GEAs) outlier detection approaches.
For the F ST outlier analysis, we use Bayesian approach in BayeScan 2.0 to directly estimate the posterior probability of a given locus under selection (Foll & Gaggiotti, 2008). We used the following parameter values: sample size of 5000, 20 pilot runs with 5000 run length, 50,000 burn‐in iterations and thinning interval of 10. Prior odds for neutral model were set to 10 and SNPs with q < 0.01 (−log10 q > 2) were considered as F ST‐outliers (Puebla et al., 2014).
For genotype‐environment associations (GEAs) outlier detection approaches, we firstly applied latent factor mixed models (LFMM) by package LEA in R (R Core Team, 2019). We use a hierarchical Bayesian mixed modelling approach to identify allele–environment correlations, while modelling residual population structure with ‘latent factors’ (Frichot et al., 2013; Frichot & François, 2015). In LFMM, environmental variables were tested separately and introduced into each model as fixed effects, and the number of latent factors (K = 3) was selected to account for genetic structure by sparse non‐negative matrix factorization (SNMF) (Figure S7). The parameters were set as follows: 100,000 iterations with a burn‐in of 50,000 iterations and ten replicate runs. Significant outliers were determined as SNPs with p‐values of p < 10−3 or –log10 (p‐value) > 3. We secondly used Bayesian generalized linear mixed models (BayEnv) to detect the correlation between SNPs and environmental variables with a neutral dataset generated by BayeScan as a null model (Günther & Coop, 2013). We initially computed a null covariance matrix of relatedness between populations, over 100,000 iterations and five independent runs. We then tested all SNPs (including those initially identified by BayeScan) under an alternative model where allele frequencies are determined by a combination of the covariance matrix and an environmental variable. We performed our analysis independently across six environmental variables, and using Bayes factor (BF) to evaluate the posterior probability that each SNP is under selection across independent environmental variable. We also performed nonparametric Spearman's rank correlation as alternative tests to the BF and detect the correlation between ranks of SNP allele frequencies and environmental variables. Therefore, we considered the SNPs in the top 1% of BF values (BF > 3) and top 10% of the absolute value of Spearman rank correlation coefficients (ρ) as significant outliers.
2.7. Multivariate relationship between genetic variation and environmental gradients
To estimate how the genomic variation is influenced by climate or geographic variables, we firstly performed redundancy analyses (RDAs) and partial redundancy analyses (pRDAs) to detect linear relationships between genetic variations and multivariate climatic gradients, using “vegan” R package (Oksanen et al., 2019). We constructed two pRDAs models to differentiate the independent effects of climate and geography by reciprocally constraining one of the two factors. Briefly, climatic effects were conditioned on the effects of geography (pRDAenv to extract pure effects of the climate) and vice versa (pRDAgeo for pure effects of geography). All SNPs, neutral and non‐neutral genetic variation with the six climate variables and geographic variables (longitude and latitude) were considered as predictor variables. Statistical significance was evaluated from 999 permutations.
We then employed gradient forest (GF) analyses to explore the nonlinear relationships between environmental variables and their contribution to genetic variation using R package GradientForest (Ellis et al., 2012). Gradient forest is a machine learning, regression tree approach that allows for exploration of nonlinear associations of spatial, environmental and allelic variables (Ellis et al., 2012; Fitzpatrick & Keller, 2015). We conducted a GF analysis on six climate variables to assess the relative importance of each predictor variable using weighted R2 value, split importance; Ellis et al. (2012). Split importance, a measure of the amount of variation explained, is high in positions along the gradient where allelic change is large. We ran a gradient forest with 500 regression trees per SNP, maxLevel = log2(0.368n)/2, and variable correlation threshold of 0.5 to calculate conditional variable importance as recommended (Ellis et al., 2012; Fitzpatrick & Keller, 2015). The default values of the other parameters were carried out for each GF model.
2.8. Adaptation potential of Taxus cuspidata
We employed two different methods to estimate the adaptive potential of T. cuspidata. We selected the Global Climate Model BCC‐CSM1‐1 (IPCC, 2014) under two contrasting representative concentration pathways (RCPs), including a low‐emission scenario (RCP 2.6) and a high‐emission scenario (RCP 8.5) in 2070 years for future climate scenarios. We first used GF to estimate the genetic offset based on all SNPs, F ST‐outlier SNPs and GEA‐outlier SNPs under future climatic conditions. Genetic offset, a measure of the magnitude of genetic change required between present and future climate (Fitzpatrick & Keller, 2015) was used to predict genetic variation across grid cells of NE China by “GradientForest” package (Ellis et al., 2012) in R (R Core Team, 2019). For each grid cell, the Euclidian distances between the current and future genetic compositions were calculated and served as the metric for genetic offset (Ellis et al., 2012).
We also performed a risk of non‐adaptedness (RONA) to predict the adaptive potential of T. cuspidata to future local climate using default settings in PYRONA v0.3.6 (Pina‐Martins et al., 2019; Rellstab et al., 2016). PYRONA ranked the environment factors by the number of associations and provided an average RONA value weighted by the regression R2 value for each predictor factors. The “RONA value” here indicates the mean difference between current and future expected allele frequencies, which was used to estimate the expected allele frequencies in future based on the present allele frequencies and environmental variables. The inferred linear model was used to predict expected allele frequencies in future environmental gradients in 2070 according to the Global Climate Model BCC‐CSM1‐1 (IPCC, 2014) under RCP 2.6 and RCP 8.5.
3. RESULTS
3.1. RAD‐based SNP data
We attained a total of 373,894,701 loci with the mean depth of coverage for filtered SNPs at 4.89 (range: 3.58–7.47) and mean proportion of loci with missing data per sample is 0.46 (range: 0.07–0.98) (Table S3). A total of 37 individuals with high proportion missing data were discarded, and the retaining 93 individuals is at least four individuals per population (Table 1). Stacks initially recovered 144,575 SNPs and 16,087 high‐quality SNPs were retained after stringent quality control.
TABLE 1.
Genetic diversity of Taxus cuspidata based on neutral and non‐neutral genetic variation.
| Neutral genetic variation | Non‐neutral genetic variation | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| nSSRs | Neutral SNPs | F ST‐outlier SNPs | GEA‐outlier SNPs | ||||||||||||||
| Population | N | N A | N E | H o | H E | P | H o | H E | π | P | H o | H E | π | P | H o | H E | π |
| MDJHCH | 15 (8) | 4.14 | 2.48 | 0.39 | 0.48 | 0.79 | 0.27 | 0.28 | 0.30 | 0.86 | 0.18 | 0.19 | 0.21 | 0.77 | 0.27 | 0.32 | 0.34 |
| MDJS | 15 (8) | 4.14 | 2.34 | 0.36 | 0.44 | 0.78 | 0.25 | 0.30 | 0.32 | 0.77 | 0.16 | 0.30 | 0.33 | 0.77 | 0.25 | 0.31 | 0.33 |
| HCM | 15 (8) | 3.43 | 1.90 | 0.29 | 0.40 | 0.80 | 0.26 | 0.27 | 0.30 | 0.81 | 0.31 | 0.25 | 0.27 | 0.75 | 0.29 | 0.33 | 0.35 |
| MDJHP | 15 (7) | 3.57 | 2.17 | 0.38 | 0.43 | 0.80 | 0.24 | 0.26 | 0.29 | 0.87 | 0.15 | 0.17 | 0.18 | 0.78 | 0.25 | 0.29 | 0.31 |
| YBD | 15 (8) | 3.57 | 2.13 | 0.42 | 0.46 | 0.79 | 0.28 | 0.29 | 0.31 | 0.80 | 0.29 | 0.29 | 0.31 | 0.76 | 0.31 | 0.32 | 0.34 |
| YBHG | 15 (8) | 3.00 | 1.90 | 0.29 | 0.34 | 0.78 | 0.31 | 0.30 | 0.32 | 0.84 | 0.23 | 0.22 | 0.24 | 0.76 | 0.34 | 0.32 | 0.35 |
| YBJ | 5 (4) | 2.29 | 1.71 | 0.46 | 0.34 | 0.81 | 0.31 | 0.26 | 0.30 | 0.88 | 0.19 | 0.17 | 0.20 | 0.79 | 0.33 | 0.28 | 0.33 |
| YJX | 15 (8) | 2.57 | 1.58 | 0.26 | 0.29 | 0.78 | 0.30 | 0.30 | 0.32 | 0.83 | 0.23 | 0.23 | 0.25 | 0.76 | 0.31 | 0.32 | 0.35 |
| YBHS | 4 (4) | 2.14 | 1.73 | 0.39 | 0.34 | 0.79 | 0.28 | 0.27 | 0.33 | 0.84 | 0.20 | 0.21 | 0.26 | 0.77 | 0.30 | 0.29 | 0.35 |
| YBHSP | 6 (6) | 3.14 | 2.21 | 0.36 | 0.39 | 0.80 | 0.28 | 0.27 | 0.31 | 0.75 | 0.26 | 0.32 | 0.35 | 0.76 | 0.32 | 0.31 | 0.36 |
| LJD | 7 (7) | 2.71 | 2.23 | 0.31 | 0.47 | 0.79 | 0.26 | 0.29 | 0.31 | 0.77 | 0.29 | 0.32 | 0.34 | 0.77 | 0.27 | 0.31 | 0.34 |
| LJB | 5 (4) | 2.43 | 1.71 | 0.43 | 0.34 | 0.79 | 0.32 | 0.28 | 0.32 | 0.79 | 0.30 | 0.29 | 0.33 | 0.77 | 0.37 | 0.31 | 0.36 |
| BSSCZ | 5 (5) | 2.29 | 1.94 | 0.26 | 0.28 | 0.81 | 0.27 | 0.26 | 0.29 | 0.85 | 0.21 | 0.21 | 0.24 | 0.77 | 0.33 | 0.31 | 0.35 |
| THL | 10 (8) | 2.57 | 1.72 | 0.30 | 0.34 | 0.78 | 0.30 | 0.30 | 0.33 | 0.71 | 0.34 | 0.38 | 0.40 | 0.75 | 0.30 | 0.34 | 0.36 |
| Mean | 2.99 | 1.98 | 0.35 | 0.38 | 0.79 | 0.28 | 0.28 | 0.31 | 0.81 | 0.24 | 0.25 | 0.28 | 0.77 | 0.30 | 0.31 | 0.34 | |
Abbreviations: N, number of individuals used for nSSRs and RAD‐seq in parentheses; N A, No. of different alleles and N E, effective number of alleles; H o and H E, observed and expected heterozygosity; P, mean frequency of the most frequent allele at each locus; π, mean nucleotide diversity.
3.2. Genetic diversity and differentiation based on nSSRs and SNP datasets
For nSSRs, observed and expected heterozygosity estimated per population ranged from 0.26 to 0.46 and 0.28 to 0.48, respectively (Table 1, Table S5). The expected heterozygosity (H E) and nucleotide diversity (π) ranged from 0.26 to 0.30 and 0.29 to 0.33 in all (Table S4) and neutral SNPs (Table 1). For F ST‐outlier SNPs, H E and π ranged from 0.17 to 0.38 and 0.18 to 0.40, and for GEA‐outlier SNPs, H E and π ranged from 0.28 to 0.34 and 0.31 to 0.36 (Table 1).
PCA (Figures S1, S2) based on both nSSRs and RAD‐seq datasets were consistent with Bayesian clustering analysis, which failed to detect clear population genetic structure in all populations. The most likely number of clusters (K) was two for the nSSRs (Figure 1a, Figures S3, S4), one for all and neutral SNPs (Figure 5b, S5a), and three for outlier SNPs (Figure 1b, Figures S5c, S6). These results showed a mixed genetic makeup of clusters in all populations.
FIGURE 1.
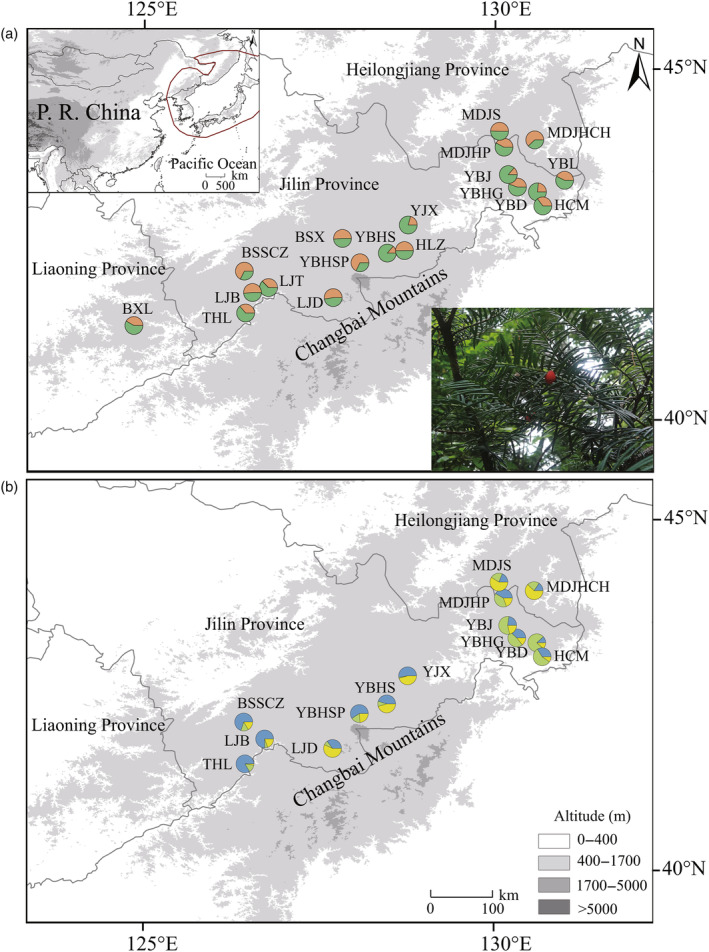
Geographic distribution and sampling sites of Taxus cuspidata. The pie chart shows the genetic clustering of each population based on (a) neutral genetic variation by microsatellites (SSRs) and (b) non‐neutral genetic variation by outlier SNPs. The red line indicates the distribution range of T. cuspidata. Orange, green, blue and yellow represent genetic cluster assignments; the details of the cluster assignments were showed in Table S1.
The results of AMOVA indicated that a large proportion of genetic variation occurred within populations, with low levels of genetic differentiation (F ST = 0.07) in nSSRs, all SNPs and neutral SNPs; For outlier SNPs, high F ST values (F ST = 0.32) in F ST‐outlier SNPs and moderate F ST values (F ST = 0.1) in GEA‐outlier SNPs (Table 2).
TABLE 2.
Hierarchical analyses of molecular variance (AMOVA) of Taxus cuspidata populations based on all SNPs, neutral and non‐neutral genetic variation.
| Source of variation | df | Percentage of variation (%) | Fixation indices |
|---|---|---|---|
| All SNPs | |||
| Among groups | 13 | 6.6 | |
| Within populations | 172 | 93.4 | F ST = 0.07 |
| Neutral genetic variation | |||
| nSSRs | |||
| Among groups | 18 | 7.5 | |
| Within populations | 381 | 92.5 | F ST = 0.07 |
| Neutral SNPs | |||
| Among groups | 13 | 7.2 | |
| Within populations | 172 | 92.8 | F ST = 0.07 |
| Non‐neutral genetic variation | |||
| F ST‐outlier SNPs | |||
| Among groups | 13 | 38.5 | |
| Within populations | 172 | 79.1 | F ST = 0.32 |
| GEA‐outlier SNPs | |||
| Among groups | 13 | 10.3 | |
| Within populations | 172 | 89.7 | F ST = 0.10 |
Note: Significance tests (1000 permutations) showed all fixation indices were significant (p < 0.05).
Abbreviation: df, degree of freedom.
3.3. Outlier detection and environmental association
We identified 63 SNPs as putative outliers and did not detect any significantly low outlier F ST values that would be indicative of balancing or purifying selection using BayeScan (Figure S8). According to the significance test, each climate variable was significantly different among the populations (Kruskal–Wallis chi‐squared = 92, p < 0.001). We identified 279 and 286 putative adaptive SNPs that were significantly associated with at least one climatic variable using LFMM (Figure S9) and BayEnv (Figure S10), respectively, with five common SNPs (Figure 2c). More putative adaptive SNPs were significantly associated with precipitation variables than temperature variables (Table S6). A total of 598 outlier SNPs were identified as non‐neutral genetic variations and 15,489 neutral SNPs as neutral genetic variations.
FIGURE 2.
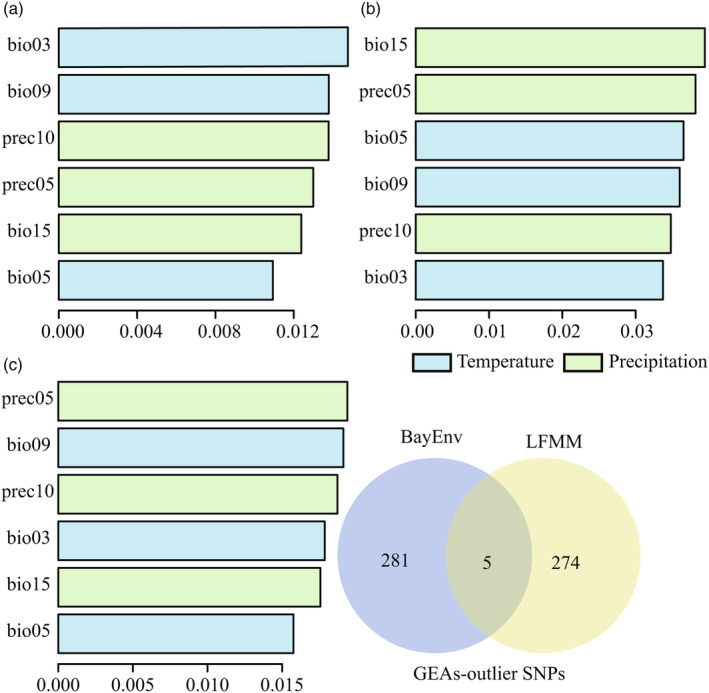
R 2‐weighted importance of environmental variables explaining genetic gradients for all (a), F ST‐outlier (b), and GEA‐outlier (c) SNPs in Taxus cuspidata using GF analysis. A venn diagram showing the overlap of outliers between all SNPs associated with at least one climatic variable, as identified by LFMM and BayEnv.
3.4. Environmental associations with genetic variation
RDA revealed that a large proportion of genetic variation among populations was associated with the six climate variables. The RDA results were similar to that of pRDA (Tables S7 and S8; Figures S11a, S12); here, we explained the results of pRDA (Table 3, Figures S11b, S13). For nSSRs, genetic variation was found to be mainly associated with climate variables: 5% of the nSSRs variance was explained by climate, while geography only accounted for 1% (Table 3). For all SNPs and neutral SNPs, 10% of the explained variance was explained by climate and 3% by geography (Table 3, Table S8). For outlier SNPs, 21% of the F ST‐outlier SNPs variance was explained by climate and 4% by geography; 13% of the GEA‐outlier SNPs variance was explained by climate and 3% by geography (Table 3).
TABLE 3.
Summary and partitioning of the variance associated with climate and geographical variables based on pRDA in neutral and non‐neutral genetic variation.
| Neutral genetic variation | Non‐neutral genetic variation | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| nSSRs | Neutral SNPs | F ST‐outlier SNPs | GEA‐outlier SNPs | |||||||||
| PVE | Eigenvalue | p | PVE | Eigenvalue | p | PVE | Eigenvalue | p | PVE | Eigenvalue | p | |
| Geography | 1.41 | 1.10 | 0.373 | 3.45 | 1.69 | 0.001 | 4.44 | 2.6 | 0.002 | 2.89 | 1.53 | 0.001 |
| Climate | 4.97 | 1.69 | 0.004 | 9.96 | 1.62 | 0.001 | 20.96 | 4.10 | 0.001 | 13.41 | 2.37 | 0.001 |
| bio03 | 1.22 | 2.49 | 0.014 | 1.78 | 1.74 | 0.001 | 1.75 | 2.06 | 0.047 | 3.17 | 3.37 | 0.001 |
| bio05 | 0.66 | 1.35 | 0.222 | 1.35 | 1.32 | 0.004 | 4.4 | 5.16 | 0.002 | 1.71 | 1.81 | 0.004 |
| bio09 | 0.96 | 1.96 | 0.055 | 1.63 | 1.59 | 0.001 | 2.79 | 3.28 | 0.003 | 1.99 | 2.12 | 0.001 |
| bio15 | 0.87 | 1.78 | 0.082 | 1.60 | 1.56 | 0.001 | 2.28 | 2.68 | 0.012 | 2.07 | 2.20 | 0.001 |
| prec05 | 0.61 | 1.21 | 0.268 | 1.89 | 1.85 | 0.001 | 7.63 | 8.95 | 0.001 | 2.68 | 2.85 | 0.001 |
| prec10 | 0.65 | 1.33 | 0.244 | 1.72 | 1.68 | 0.001 | 2.11 | 2.46 | 0.025 | 1.79 | 1.90 | 0.001 |
Abbreviation: PVE, percentage of explained variance.
For all SNPs, the GF indicated that isothermality (bio03) was the most important climate response factor (Figure 2a). However, for outlier SNPs, precipitation in May (prec05) and precipitation seasonality (bio15) were the most important climate response factors, respectively (Figure 2b,c).
3.5. Prediction of population vulnerability under future climate change
In the three GF models by all SNPs, F ST‐outlier and GEA‐outlier SNPs, the predicted turnover in allele frequencies across the landscape followed a southwest to northeast direction: northeastern populations are expected to have higher genetic offsets in the Changbai Mountains and adjacent areas in the future (Figure 3a–f), the degree of genetic offset is slightly higher under RCP 8.5 than that of RCP 2.6 (Table S9). The RONA revealed that the most represented environmental variables were prec05, bio03, and prec10 (Table S10). Under the combined effect of the three most represented climate variables, population THL showed the highest RONA value for RCP 2.6 (Figure 4). MDJS, HCM and YBJ populations showed the highest RONA value for RCP 8.5 in 2070 (Figure 4). The THL and MDJS populations had a lower adaptive potential for prec10 by 2070.
FIGURE 3.
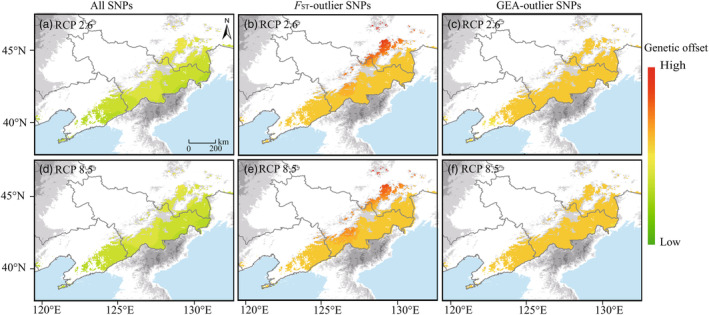
Genetic offset to future climate change predicted for all (a, d), F ST‐outlier (b, e) and GEA‐outlier SNPs (c, f) of Taxus cuspidata in 2070. (a–c), RCP 2.6; (d–f), RCP 8.5. Red and green indicate high and low genetic offset, respectively. The details of the genetic offset values were showed in Table S9.
FIGURE 4.
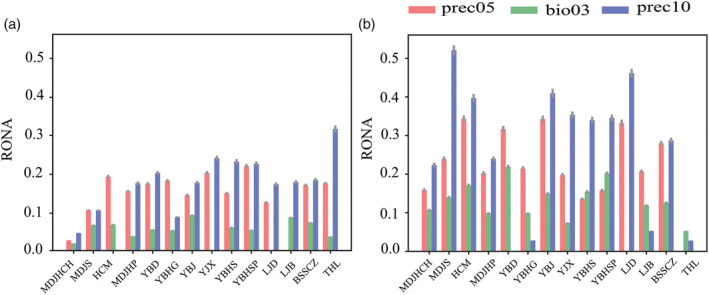
RONA of Taxus cuspidata under RCP 2.6 (a) and RCP 8.5 (b) prediction models for 2070. Bars represent weighted means (by R 2 value), and grey error bars represent the standard error.
4. DISCUSSION
Our study integrated population genetics and landscape genomic methods to explore neutral and non‐neutral genetic variations in T. cuspidata populations in the Changbai Mountains. The neutral genetic variation dataset revealed high genetic diversity and low genetic differentiation, whereas non‐neutral genetic variation showed high genetic differentiation. GEAs revealed that the contribution of climate to genetic variation was greater than that of geography, and precipitation played an important role in the putative adaptive genetic variation in response to climate change. We found that populations in the northeast range will be more vulnerable to future climate change as suggested by putative adaptive genetic variation. Our study provides insight into how neutral and putative adaptive genetic variation interacts with the environment, which is essential for future conservation and management of natural populations.
4.1. High genetic diversity was maintained in present fragmented populations
We integrated the patterns of neutral and non‐neutral genetic variation in T. cuspidata using nSSRs and RAD‐seq datasets. In this study, high current genetic diversity and low genetic differentiation were observed across the extant geographical range in China. The present results are in agreement with previous T. cuspidata genetic studies by cpDNA fragments in Russian and South Korea (Kozyrenko et al., 2017), cpDNA and mtDNA fragments in China (Su et al., 2018), and were similar to that of T. baccata from Poland by microsatellites (Litkowiec et al., 2018). In addition, admixture analysis revealed substantial gene flow among the populations. For fragmented and isolated T. cuspidata populations, substantial gene flow may compensate for the genetic barrier caused by fragmented habitats (Browne & Karubian, 2018; Hu et al., 2008; Petit et al., 2005).
Theoretically, fragmentation of species usually has a negative effect on genetic diversity compared to intact populations (Nybom, 2004; Vranckx et al., 2012). One possibility for the high genetic diversity and gene flow might be the outcrossing nature of the species. T. cuspidata is a wind pollination and typical bird‐dispersed plant. Gene flow may be accomplished by both seeds and pollen, and pollen often disperses over longer distances than seeds (Jordano, 2017; Kremer et al., 2012). However, this seems to be difficult, particularly for long‐distance intervals among the studied populations. For yew, a multi‐model approach indicated that 95% of the seed and pollen dispersal distances were <109 m and 704 m, respectively (Chybicki & Oleksa, 2018). An alternative explanation for high genetic diversity is the delayed sexual maturity of the species. The unbalanced ratio of females to males of T. cuspidata (1:2.3) was found in this region (Long et al., 2021). The sex ratio in dioecious plants might significantly affect genetic diversity (Rosche et al., 2018), e.g., the dioecy may increase genetic diversity due to obligate outcrossing, which has been found in Pherospheara hookeriana (Worth et al., 2021). Isolation occurred mainly throughout the last few decades and was probably not long enough to impact current genetic diversity (Münzbergová et al., 2013; Rosche et al., 2018). Therefore, the short duration of fragmentation tends to maintain the standing variations in long‐lived plants, which may delay adverse effect in the progenies of T. cuspidata (Aguilar et al., 2008; Vranckx et al., 2012).
High genetic differentiation was also observed in the putative adaptive genetic variation, the high F ST values may be due to the fixation of different alleles in the local populations. This might also be caused by natural selection in response to changing environments, and adaptive processes may contribute to high F ST values. Therefore, natural populations of T. cuspidata primarily stem from intense selective pressure imposed by environmental factors.
4.2. Signature of natural selection in T. cuspidata
Outlier detection test is an effective approach to identify the presence of putatively adaptive genetic variation. Although outlier detection tests can produce false outliers due to confounding factors (Schoville et al., 2012), tests with various demographic hypotheses can be utilized and compared to solve this issue, and common loci detected in consensus are more likely to be actual targets of selection (Ahrens et al., 2018). In this study, we applied three outlier methods to detect different sets of outlier SNPs that showed signatures of selection. The proportion of F ST outlier (0.4%) aligns closely with the findings in other conifers, such as Pinus taeda (0.2%) (De La Torre et al., 2019), Keteleeria davidiana (0.2%) (Shih et al., 2018) and Cupressus gigantea (0.4%) (Yang et al., 2022). The proportion of GEA‐outlier SNPs (ranging from 1.7% to 1.8%) was similar to those of T. baccata (2.6% to 3.0%) (Mayol et al., 2020) and Pinus albicaulis (2.9%) (van Mantgem et al., 2023). In the present study, more GEA‐outlier SNPs associated with climatic variables were correlated with precipitation variables than temperature variables. These outlier SNPs of T. cuspidata suggested that signatures of selection may be involved in local adaptation processes in response to selective pressure. However, due to the short RAD reads and no annotated reference genome, we failed to further annotate any genes associated with putatively adaptive SNPs.
4.3. Genetic variation associated with adaptation to climate change
Precipitation and temperature are main climatic factors influencing plant's distribution and survival (Cuervo‐Alarcon et al., 2018). Temperature is a key factor influencing the growth and phenology of tree species (Schmiege et al., 2021; Vitasse et al., 2009). RDA and GF indicated that temperature was found to be associated with our nSSRs, neutral SNPs and all SNPs sets. An explanation to this temperature‐driven genetic variation is that temperature could affect the connection between populations (i.e., gene flow), which has been observed in yews in the northern Italy (Mercuri et al., 2013). Yews are also highly sensitive to extreme temperature changes, including T. globosa (Antúnez, 2021) and T. baccata (Mayol et al., 2020; Moir, 1999). Low temperatures can encourage the rooting of yews during the early growth stage (Muñoz‐Gutiérrez et al., 2009). Therefore, the maximum temperature of warmest month (bio05) and the mean temperature of driest quarter (bio09) might affect the growth and distribution of T. cuspidata.
We also found that precipitation was an important variable associated with putative adaptive genetic variation. Water availability may be an important selection factor for yew species, especially during the last interglacial period, as indicated by the association study in the common yew, T. baccata (Mayol et al., 2020). A previous study using ecological niche modeling has also found that the best suitable distribution time for T. cuspidata is during the last glacial maximum and the most restricted distribution time for the species is during the last interglacial period (c. 130 ka), suggesting the cold‐tolerant and wet‐sensitive features of the species (Su et al., 2018). Therefore, water availability will be a challenge for T. cuspidata when facing the changing environments, as suggested by other yew species (Linares, 2013).
4.4. Species vulnerability and conservation strategies
Assessing the vulnerability of species to climate change can provide insight into the potential risk of species persistence in future climate scenarios (Capblancq et al., 2020; Feng & Du, 2022). Genetic prediction of adaptation to future climate indicated that populations (e.g., MDJHP, YJX and MDJS) in the northeast area (Heilongjiang and Jilin Provinces) exhibited a higher risk of genetic maladaptation than other populations. These populations showed low potential to adapt to changing environments in small or isolated habitats.
For GF genetic offset, the predictive power of genomic offset estimates on fitness effects is increasingly being assessed through experimental and simulation studies, showing promising results (Láruson et al., 2022). The GF offset results in our study differed not much between the two future climate scenarios. An explanation for the phenomenon is that T. cuspidata has a long lifecycle with long generation intervals, in which the rates of emergence and spread of novel adaptive alleles in populations through de novo mutations are likely to be too slow to respond to climate change.
For all SNPs and putatively adaptive SNPs, northeastern populations showed higher genetic offsets and vulnerabilities than the other populations. Su et al. (2018) found that ecological niche modeling showed a contraction trend in the distribution of T. cuspidata by 2070, with northeastern populations located in a contracting area. Láruson et al. (2022) showed that higher genetic offset may be caused by genetics drift at small population rather than a selection‐driven response. Negative associations between GF offset and population size have also been found in previous bird studies (Bay et al., 2018; Ruegg et al., 2018). Therefore, the high genetic offset of T. cuspidata may also be caused by natural selection and/or small population size.
Based on our study and previous reports, we came to the conclusion that T. cuspidata is at risk in China with number of challenges, such as fragmentation and habitat loss, climate change, disturbance and the unbalanced ratio of females to males (Long et al., 2021; Su et al., 2018; Wang et al., 2019). Our study specifically recommends adopting in situ conservation strategies for THL, LJD and YBHSP populations with high adaptive genetic diversity but vulnerable in the future. In addition, populations displaying low adaptive variation, including MDJHCH, MDJHP and YBJ, could benefit from ex situ conservation strategies. Furthermore, for small or isolated populations characterized by limited genetic diversity and high vulnerability (e.g., BSSCZ, YBJ), genetic rescue and assisted gene flow techniques should be considered to facilitate their adaptation to climate change. Previous studies have demonstrated the effectiveness of such approaches (Aitken & Bemmels, 2016; Aitken & Whitlock, 2013; Whiteley et al., 2015).
It is worth noting that our study represents a pioneering effort to integrate neutral and adaptive genetic variation to the conservation of a threatened tree species. However, to gain deeper insights into the molecular mechanisms underlying the threats faced by T. cuspidata, future research endeavors should include comprehensive investigations encompassing whole‐genome or transcriptome sequencing, along with the inclusion of phenotypic and fine‐scale environmental data.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
Supporting information
Figure S1.
Table S1.
ACKNOWLEDGEMENTS
Funding: This work was supported by grants from the Fundamental Research Funds for the Central Universities, P. R. China (2021ZY80), the Special Program for the Institute of National Parks, Chinese Academy, P. R. China Sciences (KFJ‐STS‐ZDTP‐2022‐001) and National Key Research and Development Plan (Grant No. 2016YFC0503106). We thank Prof. Jingwen Li from BFU for discussing the sexual ratio of yews and Prof. Erli Pang from BNU for bioinformatics analysis.
Luo, Y. , Qin, W. , Yan, Y. , Yin, K. , Zang, R. , & Du, F. K. (2024). Climate change vulnerability and conservation strategies for tertiary relict tree species: Insights from landscape genomics of Taxus cuspidata . Evolutionary Applications, 17, e13686. 10.1111/eva.13686
Yanjun Luo and Wei Qin contributed equally to this work.
Contributor Information
Kangquan Yin, Email: yinkq@bjfu.edu.cn.
Fang K. Du, Email: dufang325@bjfu.edu.cn.
DATA AVAILABILITY STATEMENT
Data for this study are available at 10.6084/m9.figshare.23515398.
REFERENCES
- Aguilar, R. , Quesada, M. , Ashworth, L. , Herrerias‐Diego, Y. V. O. N. N. E. , & Lobo, J. (2008). Genetic consequences of habitat fragmentation in plant populations: Susceptible signals in plant traits and methodological approaches. Molecular Ecology, 17(24), 5177–5188. 10.1111/j.1365-294X.2008.03971.x [DOI] [PubMed] [Google Scholar]
- Ahrens, C. W. , Rymer, P. D. , Stow, A. , Bragg, J. , Dillon, S. , Umbers, K. D. , & Dudaniec, R. Y. (2018). The search for loci under selection: Trends, biases and progress. Molecular Ecology, 27(6), 1342–1356. 10.1111/mec.14549 [DOI] [PubMed] [Google Scholar]
- Aitken, S. N. , & Bemmels, J. B. (2016). Time to get moving: Assisted gene flow of forest trees. Evolutionary Applications, 9(1), 271–290. 10.1111/eva.12293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken, S. N. , & Whitlock, M. C. (2013). Assisted gene flow to facilitate local adaptation to climate change. Annual Review of Ecology, Evolution, and Systematics, 44, 367–388. 10.1146/annurev-ecolsys-110512-135747 [DOI] [Google Scholar]
- Alexander, D. H. , & Lange, K. (2011). Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinformatics, 12, 1–6. 10.1186/1471-2105-12-246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, D. H. , Novembre, J. , & Lange, K. (2009). Fast model‐based estimation of ancestry in unrelated individuals. Genome Research, 19(9), 1655–1664. 10.1101/gr.094052.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Antúnez, P. (2021). Influence of physiography, soil and climate on Taxus globosa . Nordic Journal of Botany, 39(3), e03058. 10.1111/njb.03241 [DOI] [Google Scholar]
- Bay, R. A. , Harrigan, R. J. , Underwood, V. L. , Gibbs, H. L. , Smith, T. B. , & Ruegg, K. (2018). Genomic signals of selection predict climate‐driven population declines in a migratory bird. Science, 359(6371), 83–86. 10.1126/science.aan438 [DOI] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonan, G. B. (2008). Forests and climate change: Forcings, feedbacks, and the climate benefits of forests. Science, 320(5882), 1444–1449. 10.1126/science.1155121 [DOI] [PubMed] [Google Scholar]
- Borrell, J. S. , Zohren, J. , Nichols, R. A. , & Buggs, R. J. (2020). Genomic assessment of local adaptation in dwarf birch to inform assisted gene flow. Evolutionary Applications, 13(1), 161–175. 10.1111/eva.12883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne, L. , & Karubian, J. (2018). Habitat loss and fragmentation reduce effective gene flow by disrupting seed dispersal in a neotropical palm. Molecular Ecology, 27(15), 3055–3069. 10.1111/mec.14765 [DOI] [PubMed] [Google Scholar]
- Capblancq, T. , Fitzpatrick, M. C. , Bay, R. A. , Exposito‐Alonso, M. , & Keller, S. R. (2020). Genomic prediction of (mal) adaptation across current and future climatic landscapes. Annual Review of Ecology, Evolution, and Systematics, 51, 245–269. 10.1146/annurev-ecolsys-020720-042553 [DOI] [Google Scholar]
- Catchen, J. , Hohenlohe, P. A. , Bassham, S. , Amores, A. , & Cresko, W. A. (2013). Stacks: An analysis tool set for population genomics. Molecular Ecology, 22(11), 3124–3140. 10.1111/mec.12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen, J. M. , Amores, A. , Hohenlohe, P. , Cresko, W. , & Postlethwait, J. H. (2011). Stacks: Building and genotyping loci de novo from short‐read sequences. G3: Genes, Genomes, Genetics, 1(3), 171–182. 10.1534/g3.111.000240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, B. B. , Zheng, Y. Q. , & Sun, Q. W. (2015). Genetic diversity and population structure of Taxus cuspidata in the Changbai Mountains assessed by chloroplast DNA sequences and microsatellite markers. Biochemical Systematics and Ecology, 63, 157–164. 10.1016/j.bse.2015.10.009 [DOI] [Google Scholar]
- Chung, M. G. , Oh, G. S. , & Chung, J. M. (1999). Allozyme variation in Korean populations of Taxus cuspidata (Taxaceae). Scandinavian Journal of Forest Research, 14(2), 103–110. 10.1080/02827589950152827 [DOI] [Google Scholar]
- Chung, M. Y. , Merilä, J. , Li, J. , Mao, K. , López‐Pujol, J. , Tsumura, Y. , & Chung, M. G. (2023). Neutral and adaptive genetic diversity in plants: An overview. Frontiers in Ecology and Evolution, 11, 1116814. 10.3389/fevo.2023.1116814 [DOI] [Google Scholar]
- Chybicki, I. J. , & Oleksa, A. (2018). Seed and pollen gene dispersal in Taxus baccata, a dioecious conifer in the face of strong population fragmentation. Annals of Botany, 122(3), 409–421. 10.1093/aob/mcy081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo‐Alarcon, L. , Arend, M. , Müller, M. , Sperisen, C. , Finkeldey, R. , & Krutovsky, K. V. (2018). Genetic variation and signatures of natural selection in populations of European beech (Fagus sylvatica L.) along precipitation gradients. Tree Genetics & Genomes, 14, 1–21. 10.1007/s11295-018-1297-2 [DOI] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C. A. , Banks, E. , DePristo, M. A. , Handsaker, R. E. , Lunter, G. , Marth, G. T. , Sherry, S. T. , McVean, G. , & Durbin, R . (2011). The variant call format and VCFtools. Bioinformatics, 27(15), 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Torre, A. R. , Wilhite, B. , & Neale, D. B. (2019). Environmental genome‐wide association reveals climate adaptation is shaped by subtle to moderate allele frequency shifts in loblolly pine. Genome Biology and Evolution, 11(10), 2976–2989. 10.1093/gbe/evz220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sousa, V. A. , Reeves, P. A. , Reilley, A. , de Aguiar, A. V. , Stefenon, V. M. , & Richards, C. M. (2020). Genetic diversity and biogeographic determinants of population structure in Araucaria angustifolia (Bert.) O. Ktze. Conservation Genetics, 21(2), 217–229. 10.1007/s10592-019-01242-9 [DOI] [Google Scholar]
- Du, F. K. , Hou, M. , Wang, W. , Mao, K. , & Hampe, A. (2017). Phylogeography of Quercus aquifolioides provides novel insights into the Neogene history of a major global hotspot of plant diversity in south‐west China. Journal of Biogeography, 44(2), 294–307. 10.1111/jbi.12836 [DOI] [Google Scholar]
- Du, F. K. , Wang, T. , Wang, Y. , Ueno, S. , & de Lafontaine, G. (2020). Contrasted patterns of local adaptation to climate change across the range of an evergreen oak, Quercus aquifolioides . Evolutionary Applications, 13(9), 2377–2391. 10.1111/eva.13030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil, M. , Sebastiani, F. , Mayol, M. , González‐Martínez, S. C. , Riba, M. , & Vendramin, G. G. (2008). Isolation and characterization of polymorphic nuclear microsatellite loci in Taxus baccata L. Conservation Genetics, 9, 1665–1668. 10.1007/s10592-008-9515-3 [DOI] [Google Scholar]
- Earl, D. A. , & VonHoldt, B. M. (2012). STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetics Resources, 4, 359–361. 10.1007/s12686-011-9548-7 [DOI] [Google Scholar]
- Ellis, N. , Smith, S. J. , & Pitcher, C. R. (2012). Gradient forests: Calculating importance gradients on physical predictors. Ecology, 93(1), 156–168. 10.1890/11-0252.1 [DOI] [PubMed] [Google Scholar]
- Evanno, G. , Regnaut, S. , & Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Molecular Ecology, 14(8), 2611–2620. 10.1111/j.1365-294X.2005.02553.x [DOI] [PubMed] [Google Scholar]
- Excoffier, L. , & Lischer, H. E. (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and windows. Molecular Ecology Resources, 10(3), 564–567. 10.1111/j.1755-0998.2010.02847.x [DOI] [PubMed] [Google Scholar]
- Feng, L. , & Du, F. K. (2022). Landscape genomics in tree conservation under a changing environment. Frontiers in Plant Science, 13(822), 217. 10.3389/fpls.2022.822217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fick, S. E. , & Hijmans, R. J. (2017). WorldClim 2: New 1‐km spatial resolution climate surfaces for global land areas. International Journal of Climatology, 37(12), 4302–4315. 10.1002/joc.5086 [DOI] [Google Scholar]
- Fitzpatrick, M. C. , & Keller, S. R. (2015). Ecological genomics meets community‐level modelling of biodiversity: Mapping the genomic landscape of current and future environmental adaptation. Ecology Letters, 18(1), 1–16. 10.1111/ele.12376 [DOI] [PubMed] [Google Scholar]
- Foll, M. , & Gaggiotti, O. (2008). A genome‐scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics, 180(2), 977–993. 10.1534/genetics.108.092221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox, J. , & Weisberg, S. (2019). An R companion to applied regression (Third ed.). Sage. https://socialsciences.mcmaster.ca/jfox/Books/Companion/ [Google Scholar]
- Frankham, R. (2005). Conservation biology: Ecosystem recovery enhanced by genotypic diversity. Heredity, 95(3), 183. 10.1038/sj.hdy.6800706 [DOI] [PubMed] [Google Scholar]
- Frichot, E. , & François, O. (2015). LEA: An R package for landscape and ecological association studies. Methods in Ecology and Evolution, 6(8), 925–929. 10.1111/2041-210x.12382 [DOI] [Google Scholar]
- Frichot, E. , Schoville, S. D. , Bouchard, G. , & François, O. (2013). Testing for associations between loci and environmental gradients using latent factor mixed models. Molecular Biology and Evolution, 30, 1687–1699. 10.1093/molbev/mst063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitzendanner, M. A. , Weekley, C. W. , Germain‐Aubrey, C. C. , Soltis, D. E. , & Soltis, P. S. (2012). Microsatellite evidence for high clonality and limited genetic diversity in Ziziphus celata (Rhamnaceae), an endangered, self‐incompatible shrub endemic to the Lake Wales ridge, Florida, USA. Conservation Genetics, 13, 223–234. 10.1007/s10592-011-0287-9 [DOI] [Google Scholar]
- González, A. V. , Gómez‐Silva, V. , Ramírez, M. J. , & Fontúrbel, F. E. (2020). Meta‐analysis of the differential effects of habitat fragmentation and degradation on plant genetic diversity. Conservation Biology, 34(3), 711–720. 10.1111/cobi.13422 [DOI] [PubMed] [Google Scholar]
- Günther, T. , & Coop, G. (2013). Robust identification of local adaptation from allele frequencies. Genetics, 195(1), 205–220. 10.1534/genetics.113.152462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, A. A. , & Willi, Y. (2008). Detecting genetic responses to environmental change. Nature Reviews Genetics, 9(6), 421–432. 10.1038/nrg2339 [DOI] [PubMed] [Google Scholar]
- Holderegger, R. , Kamm, U. , & Gugerli, F. (2006). Adaptive vs. neutral genetic diversity: Implications for landscape genetics. Landscape Ecology, 21, 797–807. 10.1007/s10980-005-5245-9 [DOI] [Google Scholar]
- Hu, L. J. , Uchiyama, K. , Shen, H. L. , Saito, Y. , Tsuda, Y. , & Ide, Y. (2008). Nuclear DNA microsatellites reveal genetic variation but a lack of phylogeographical structure in an endangered species, Fraxinus mandshurica, across north‐east China. Annals of Botany, 102(2), 195–205. 10.1093/aob/mcn074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- IPCC . (2014). Climate change 2014: Synthesis report. Contribution of working groups I. II and III to the fifth assessment report of the intergovernmental panel on climate change 151(10.1017). IPCC. [Google Scholar]
- Jakobsson, M. , & Rosenberg, N. A. (2007). CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics, 23(14), 1801–1806. 10.1093/bioinformatics/btm233 [DOI] [PubMed] [Google Scholar]
- Jia, K. H. , Zhao, W. , Maier, P. A. , Hu, X. G. , Jin, Y. , Zhou, S. S. , … Mao, J. F. (2020). Landscape genomics predicts climate change‐related genetic offset for the widespread Platycladus orientalis (Cupressaceae). Evolutionary Applications, 13(4), 665–676. 10.1111/eva.12891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart, T. , & Ahmed, I. (2011). Adegenet 1.3–1: New tools for the analysis of genome‐wide SNP data. Bioinformatics, 27(21), 3070–3071. 10.1093/bioinformatics/btr521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordano, P. (2017). What is long‐distance dispersal? And a taxonomy of dispersal events. Journal of Ecology, 105(1), 75–84. 10.1111/1365-2745.12690 [DOI] [Google Scholar]
- Katsuki, T. , & Luscombe, D. (2013). Taxus cuspidata. The IUCN red list of threatened species. 2013: E.T42549A2987373. 10.2305/IUCN.UK.2013-1 [DOI]
- Kitamura, S. , & Murata, G. (1987). Colored illustrations of woody plants of Japan (Vol. II, p. 545). Hoikusha Publ. Co., Ltd. [Google Scholar]
- Kondo, T. (2016). Development of polymorphic microsatellite markers for Japanese yew, Taxus cuspidata, and T. Cuspidata var. nana (Taxaceae). Applications in Plant Sciences, 4(7), 1600020. 10.3732/apps.1600020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozyrenko, M. M. , Artyukova, E. V. , & Chubar, E. A. (2017). Genetic diversity and population structure of Taxus cuspidata Sieb. Et Zucc. Ex Endl. (Taxaceae) in Russia according to data of the nucleotide polymorphism of intergenic spacers of the chloroplast genome. Russian Journal of Genetics, 53, 865–874. 10.1134/S1022795417070079 [DOI] [Google Scholar]
- Kremer, A. , Ronce, O. , Robledo‐Arnuncio, J. J. , Guillaume, F. , Bohrer, G. , Nathan, R. , … Schueler, S. (2012). Long‐distance gene flow and adaptation of forest trees to rapid climate change. Ecology Letters, 15(4), 378–392. 10.1111/j.1461-0248.2012.01746.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laikre, L. , Allendorf, F. W. , Aroner, L. C. , Baker, C. S. , Gregovich, D. P. , Hansen, M. M. , … Waples, R. S. (2010). Neglect of genetic diversity in implementation of the convention of biological diversity. Conservation Biology, 24(1), 86–88. 10.1111/j.1523-1739.2009.01425.x [DOI] [PubMed] [Google Scholar]
- Láruson, Á. J. , Fitzpatrick, M. C. , Keller, S. R. , Haller, B. C. , & Lotterhos, K. E. (2022). Seeing the forest for the trees: Assessing genetic offset predictions from gradient forest. Evolutionary Applications, 15(3), 403–416. 10.1111/eva.13354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares, J. C. (2013). Shifting limiting factors for population dynamics and conservation status of the endangered English yew (Taxus baccata L., Taxaceae). Forest Ecology and Management, 291, 119–127. 10.1016/j.foreco.2012.11.009 [DOI] [Google Scholar]
- Litkowiec, M. , Lewandowski, A. , & Wachowiak, W. (2018). Genetic variation in Taxus baccata L.: A case study supporting Poland's protection and restoration program. Forest Ecology and Management, 409, 148–160. 10.1016/j.foreco.2017.11.026 [DOI] [Google Scholar]
- Long, T. , Wu, X. , Wang, Y. , Chen, J. , Xu, C. , Li, J. , … Zang, R. (2021). The population status and threats of Taxus cuspidata, a plant species with extremely small populations in China. Global Ecology and Conservation, 26, e01495. 10.1016/j.gecco.2021.e01495 [DOI] [Google Scholar]
- Lowe, A. J. , Boshier, D. , Ward, M. , Bacles, C. F. E. , & Navarro, C. (2005). Genetic resource impacts of habitat loss and degradation; reconciling empirical evidence and predicted theory for neotropical trees. Heredity, 95(4), 255–273. 10.1038/sj.hdy.6800725 [DOI] [PubMed] [Google Scholar]
- Mackay, J. , Dean, J. F. , Plomion, C. , Peterson, D. G. , Cánovas, F. M. , Pavy, N. , … Cervera, M. T. (2012). Towards decoding the conifer giga‐genome. Plant Molecular Biology, 80, 555–569. 10.1007/s11103-012-9961-7 [DOI] [PubMed] [Google Scholar]
- Marquaridt, D. W. (1970). Generalized inverses, ridge regression, biased linear estimation, and nonlinear estimation. Technometrics, 12(3), 591–612. 10.1080/00401706.1970.10488699 [DOI] [Google Scholar]
- Mayol, M. , Riba, M. , Cavers, S. , Grivet, D. , Vincenot, L. , Cattonaro, F. , … González‐Martínez, S. C. (2020). A multiscale approach to detect selection in nonmodel tree species: Widespread adaptation despite population decline in Taxus baccata L. Evolutionary Applications, 13(1), 143–160. 10.1111/eva.12838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri, A. M. , Torri, P. , Casini, E. , & Olmi, L. (2013). Climate warming and the decline of Taxus airborne pollen in urban pollen rain (Emilia Romagna, northern Italy). Plant Biology, 15, 70–82. 10.1111/j.1438-8677.2012.00624.x [DOI] [PubMed] [Google Scholar]
- Miao, Y. C. , Zhang, Z. J. , & Su, J. R. (2016). Low genetic diversity in the endangered Taxus yunnanensis following a population bottleneck, a low effective population size and increased inbreeding. Silvae Genetica, 65, 59–66. 10.1515/sg-2016-0008 [DOI] [Google Scholar]
- Moir, A. K. (1999). The dendrochronological potential of modern yew (Taxus baccata) with special reference to yew from Hampton court palace. UK. The New Phytologist, 144(3), 479–488. 10.1046/j.1469-8137.1999.00545.x [DOI] [PubMed] [Google Scholar]
- Muñoz‐Gutiérrez, L. , Vargas‐Hernández, J. J. , López‐Upton, J. , & Soto‐Hernández, M. (2009). Effect of cutting age and substrate temperature on rooting of Taxus globosa . New Forests, 38, 187–196. 10.1007/s11056-009-9139-6 [DOI] [Google Scholar]
- Münzbergová, Z. , Cousins, S. A. , Herben, T. , Plačková, I. , Mildén, M. , & Ehrlén, J. (2013). Historical habitat connectivity affects current genetic structure in a grassland species. Plant Biology, 15(1), 195–202. 10.1111/j.1438-8677.2012.00601.x [DOI] [PubMed] [Google Scholar]
- Naimi, B. , Hamm, N. A. , Groen, T. A. , Skidmore, A. K. , & Toxopeus, A. G. (2014). Where is positional uncertainty a problem for species distribution modelling? Ecography, 37(2), 191–203. 10.1111/j.1600-0587.2013.00205.x [DOI] [Google Scholar]
- Neale, D. B. , & Kremer, A. (2011). Forest tree genomics: Growing resources and applications. Nature Reviews Genetics, 12(2), 111–122. 10.1038/nrg2931 [DOI] [PubMed] [Google Scholar]
- Nielsen, E. S. , Hanson, J. O. , Carvalho, S. B. , Beger, M. , Henriques, R. , Kershaw, F. , & Von der Heyden, S. (2022). Molecular ecology meets systematic conservation planning. Trends in Ecology & Evolution, 38, 143–155. 10.1016/j.tree.2022.09.006 [DOI] [PubMed] [Google Scholar]
- Nybom, H. (2004). Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Molecular Ecology, 13(5), 1143–1155. 10.1111/j.1365-294X.2004.02141.x [DOI] [PubMed] [Google Scholar]
- O'Brien, D. , Laikre, L. , Hoban, S. , Bruford, M. W. , Ekblom, R. , Fischer, M. C. , … MacDonald, A. J. (2022). Bringing together approaches to reporting on within species genetic diversity. Journal of Applied Ecology, 59(9), 2227–2233. 10.1111/1365-2664.14 [DOI] [Google Scholar]
- Oksanen, J. , Blanchet, F. G. , Friendly, M. , Kindt, R. , Legendre, P. , McGlinn, D. , … Imports, M. A. S. S. (2019). Package ‘vegan’. Community Ecology Package, Version 2.9. https://CRAN.R‐project.org/package=vegan
- Peakall, R. O. D. , & Smouse, P. E. (2006). GENALEX 6: Genetic analysis in excel. Population genetic software for teaching and research. Molecular Ecology Notes, 6(1), 288–295. 10.1093/bioinformatics/bts460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, B. K. , Weber, J. N. , Kay, E. H. , Fisher, H. S. , & Hoekstra, H. E. (2012). Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non‐model species. PLoS One, 7(5), e37135. 10.1371/journal.pone.0037135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit, J. R. , Duminil, J. , Fineschi, S. , Hampe, A. , Salvini, D. , & Ven‐dramin, G. G. (2005). Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Molecular Ecology, 14, 689–701. 10.1111/j.1365-294X.2004.02410.x [DOI] [PubMed] [Google Scholar]
- Petit, R. J. , & Hampe, A. (2006). Some evolutionary consequences of being a tree. Annual Review of Ecology, Evolution, and Systematics, 37, 187–214. 10.1146/annurev.ecolsys.37.091305.1102 [DOI] [Google Scholar]
- Pina‐Martins, F. , Baptista, J. , Pappas, G., Jr. , & Paulo, O. S. (2019). New insights into adaptation and population structure of cork oak using genotyping by sequencing. Global Change Biology, 25(1), 337–350. 10.1111/gcb.14497 [DOI] [PubMed] [Google Scholar]
- Piotti, A. (2009). The genetic consequences of habitat fragmentation: The case of forests. iForest – Biogeosciences and Forestry, 2(3), 75–76. 10.3832/ifor0496-002 [DOI] [Google Scholar]
- Pritchard, J. K. , Stephens, M. , & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155(2), 945–959. 10.1093/genetics/155.2.945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puebla, O. , Bermingham, E. , & McMillan, W. O. (2014). Genomic atolls of differentiation in coral reef fishes (Hypoplectrus spp., Serranidae). Molecular Ecology, 23(21), 5291–5303. 10.1111/mec.12926 [DOI] [PubMed] [Google Scholar]
- Purcell, S. , Neale, B. , Todd‐Brown, K. , Thomas, L. , Ferreira, M. A. , Bender, D. , … Sham, P. C. (2007). PLINK: A tool set for whole‐genome association and population‐based linkage analyses. The American Journal of Human Genetics, 81(3), 559–575. 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pykälä, J. (2019). Habitat loss and deterioration explain the disappearance of populations of threatened vascular plants, bryophytes and lichens in a hemiboreal landscape. Global Ecology and Conservation, 18, e00610. 10.1016/j.gecco.2019.e00610 [DOI] [Google Scholar]
- R Core Team . (2019). R: A language and environment for statistical computing. R Foundation for Statistical Computing. https://www.R‐project.org/ [Google Scholar]
- Rands, M. R. , Adams, W. M. , Bennun, L. , Butchart, S. H. , Clements, A. , Coomes, D. , … Vira, B. (2010). Biodiversity conservation: Challenges beyond 2010. Science, 329(5997), 1298–1303. 10.1126/science.1189138 [DOI] [PubMed] [Google Scholar]
- Rellstab, C. , Zoller, S. , Walthert, L. , Lesur, I. , Pluess, A. R. , Graf, R. , … Gugerli, F. (2016). Signatures of local adaptation in candidate genes of oaks (Quercus spp.) with respect to present and future climatic conditions. Molecular Ecology, 25(23), 5907–5924. 10.1111/mec.13889 [DOI] [PubMed] [Google Scholar]
- Rosche, C. , Schrieber, K. , Lachmuth, S. , Durka, W. , Hirsch, H. , Wagner, V. , … Hensen, I. (2018). Sex ratio rather than population size affects genetic diversity in Antennaria dioica . Plant Biology, 20(4), 789–796. 10.1111/plb.12716 [DOI] [PubMed] [Google Scholar]
- Rosenberg, N. A. (2004). DISTRUCT: A program for the graphical display of population structure. Molecular Ecology Notes, 4(1), 137–138. 10.1046/j.1471-8286.2003.00566.x [DOI] [Google Scholar]
- Ruegg, K. , Bay, R. A. , Anderson, E. C. , Saracco, J. F. , Harrigan, R. J. , Whitfield, M. , … Smith, T. B. (2018). Ecological genomics predicts climate vulnerability in an endangered southwestern songbird. Ecology Letters, 21(7), 1085–1096. 10.1111/ele.12977 [DOI] [PubMed] [Google Scholar]
- Schmiege, S. C. , Buckley, B. M. , Stevenson, D. W. , Heskel, M. A. , Cuong, T. Q. , Nam, L. C. , & Griffin, K. L. (2021). Respiratory temperature responses of tropical conifers differ with leaf morphology. Functional Ecology, 35(7), 1408–1423. 10.1111/1365-2435.13814 [DOI] [Google Scholar]
- Schoville, S. D. , Bonin, A. , François, O. , Lobreaux, S. , Melodelima, C. , & Manel, S. (2012). Adaptive genetic variation on the landscape: Methods and cases. Annual Review of Ecology, Evolution, and Systematics, 43, 23–43. 10.1146/annurev-ecolsys-110411-160248 [DOI] [Google Scholar]
- Shih, K. M. , Chang, C. T. , Chung, J. D. , Chiang, Y. C. , & Hwang, S. Y. (2018). Adaptive genetic divergence despite significant isolation‐by‐distance in populations of Taiwan cow‐tail fir (Keteleeria davidiana var. formosana). Frontiers Plant Science, 9, 92. 10.3389/fpls.2018.00092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sork, V. L. , Aitken, S. N. , Dyer, R. J. , Eckert, A. J. , Legendre, P. , & Neale, D. B. (2013). Putting the landscape into the genomics of trees: Approaches for understanding local adaptation and population responses to changing climate. Tree Genetics & Genomes, 9, 901–911. 10.1007/s11295-013-0596-x [DOI] [Google Scholar]
- Su, J. , Yan, Y. , Song, J. , Li, J. , Mao, J. , Wang, N. , … Du, F. K. (2018). Recent fragmentation may not alter genetic patterns in endangered long‐lived species: Evidence from Taxus cuspidata . Frontiers in Plant Science, 9, 1571. 10.3389/fpls.2018.01571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, S. , Dai, W. , Li, M. , Zhang, Y. , Geng, Y. , Tang, L. , & Zhong, Y. (2008). Genetic diversity of relictual and endangered plant Abies ziyuanensis (Pinaceae) revealed by AFLP and SSR markers. Genetica, 133, 21–30. 10.1007/s10709-007-9178-x [DOI] [PubMed] [Google Scholar]
- van Mantgem, P. J. , Milano, E. R. , Dudney, J. , Nesmith, J. C. , Vandergast, A. G. , & Zald, H. S. (2023). Growth, drought response, and climate‐associated genomic structure in whitebark pine in the Sierra Nevada of California. Ecology and Evolution, 13(5), e10072. 10.1002/ece3.10072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitasse, Y. , Delzon, S. , Bresson, C. C. , Michalet, R. , & Kremer, A. (2009). Altitudinal differentiation in growth and phenology among populations of temperate‐zone tree species growing in a common garden. Canadian Journal of Forest Research, 39(7), 1259–1269. 10.1139/X09-054 [DOI] [Google Scholar]
- Vranckx, G. U. Y. , Jacquemyn, H. , Muys, B. , & Honnay, O. (2012). Meta‐analysis of susceptibility of woody plants to loss of genetic diversity through habitat fragmentation. Conservation Biology, 26(2), 228–237. 10.1111/j.1523-1739.2011.01778.x [DOI] [PubMed] [Google Scholar]
- Wade, E. M. , Nadarajan, J. , Yang, X. , Ballesteros, D. , Sun, W. , & Pritchard, H. W. (2016). Plant species with extremely small populations (PSESP) in China: A seed and spore biology perspective. Plant Diversity, 38(5), 209–220. 10.1016/j.pld.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. W. , & Ge, S. (2006). Phylogeography of the endangered Cathaya argyrophylla (Pinaceae) inferred from sequence variation of mitochondrial and nuclear DNA. Molecular Ecology, 15(13), 4109–4122. 10.1111/j.1365-294X.2006.03086.x [DOI] [PubMed] [Google Scholar]
- Wang, J. , Wang, Y. , Feng, J. , Chen, C. , Chen, J. , Long, T. , … Li, J. (2019). Differential responses to climate and land‐use changes in threatened Chinese Taxus species. Forests, 10(9), 766. 10.3390/f10090766 [DOI] [Google Scholar]
- Wang, T. , Feng, L. , & Du, F. K. (2021). New approaches for ecological adaptation study: From population genetics to landscape genomics. Scientia Sinica Vitae, 51, 167–178. 10.1360/SSV-2020-0265 [DOI] [Google Scholar]
- Whiteley, A. R. , Fitzpatrick, S. W. , Funk, W. C. , & Tallmon, D. A. (2015). Genetic rescue to the rescue. Trends in Ecology & Evolution, 30(1), 42–49. 10.1016/j.tree.2014.10.009 [DOI] [PubMed] [Google Scholar]
- Worth, J. R. , Marthick, J. R. , Harrison, P. A. , Sakaguchi, S. , & Jordan, G. J. (2021). The palaeoendemic conifer Pherosphaera hookeriana (Podocarpaceae) exhibits high genetic diversity despite quaternary range contraction and post glacial bottlenecking. Conservation Genetics, 22, 307–321. 10.1007/s10592-021-01338-1 [DOI] [Google Scholar]
- Yang, H. , Li, J. , Milne, R. I. , Tao, W. , Wang, Y. , Miao, J. , … Mao, K. (2022). Genomic insights into the genotype–environment mismatch and conservation units of a Qinghai–Tibet plateau endemic cypress under climate change. Evolutionary Applications, 15(6), 919–933. 10.1111/eva.13377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Cai, L. , Liu, D. , Chen, G. , Gratzfeld, J. , & Sun, W. (2020). China's conservation program on plant species with extremely small populations (PSESP): Progress and perspectives. Biological Conservation, 244(108), 535. 10.1016/j.biocon.2020.108535 [DOI] [Google Scholar]
- Young, A. , Boyle, T. , & Brown, T. (1996). The population genetic consequences of habitat fragmentation for plants. Trends in Ecology & Evolution, 11(10), 413–418. 10.1016/0169-5347(96)10045-8 [DOI] [PubMed] [Google Scholar]
- Yousefzadeh, H. , Rajaei, R. , Jasińska, A. , Walas, Ł. , Fragnière, Y. , & Kozlowski, G. (2018). Genetic diversity and differentiation of the riparian relict tree Pterocarya fraxinifolia (Juglandaceae) along altitudinal gradients in the Hyrcanian forest (Iran). Silva Fennica, 52(5), 10000. 10.14214/sf.10000 [DOI] [Google Scholar]
- Zhao, W. , Sun, Y. Q. , Pan, J. , Sullivan, A. R. , Arnold, M. L. , Mao, J. F. , & Wang, X. R. (2020). Effects of landscapes and range expansion on population structure and local adaptation. New Phytologist, 228(1), 330–343. 10.1111/nph.16619 [DOI] [PubMed] [Google Scholar]
- Zheng, X. , Levine, D. , Shen, J. , Gogarten, S. M. , Laurie, C. , & Weir, B. S. (2012). A high‐performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics, 28(24), 3326–3328. 10.1093/bioinformatics/bts606 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Table S1.
Data Availability Statement
Data for this study are available at 10.6084/m9.figshare.23515398.