

Abstract
Mitochondrial respiration is a critical energy-generating pathway in all cells, especially retinal photoreceptors that possess a highly active metabolism. In addition, photoreceptors also exhibit high aerobic glycolysis like cancer cells. Precise measurements of these metabolic activities can provide valuable insights into cellular homeostasis under physiological conditions and in disease states. High throughput microplate-based assays have been developed to measure mitochondrial respiration and various metabolic activities in live cells. However, a vast majority of these are developed for cultured cells and have not been optimized for intact tissue samples and for application ex vivo. Described here is a detailed step-by-step protocol, using microplate-based fluorescence technology, to directly measure oxygen consumption rate (OCR) as an indicator of mitochondrial respiration, as well as extracellular acidification rate (ECAR) as an indicator of glycolysis, in intact ex vivo retinal tissue. This method has been used to successfully assess metabolic activities in adult mouse retina and demonstrate its application in investigating cellular mechanisms of aging and disease.
Introduction
Mitochondria are essential organelle that regulates cellular metabolism, signaling, homeostasis, and apoptosis by coordinating multiple crucial physiological processes1. Mitochondria serve as the powerhouse in the cell to generate adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS) and provide energy that supports almost all cellular events. The majority of cellular oxygen is metabolized in mitochondria, where it serves as the final electron acceptor in the electron transport chain (ETC) during aerobic respiration. Low amounts of ATP can also be produced from glycolysis in the cytosol, where glucose is converted to pyruvate, which can be further converted to lactate or be transported into mitochondria and oxidized to acetyl-CoA, a substrate in the tricarboxylic acid cycle (TCA cycle).
The retina is one of the most metabolically active tissues in mammals2, displaying high levels of mitochondrial respiration and extremely high oxygen consumption3. The rod and cone photoreceptors contain a high density of mitochondria4, and OXPHOS generates most ATP in the retina5. In addition, the retina also relies heavily on aerobic glycolysis6,7 by converting glucose to lactate5. Mitochondrial defects are associated with various neurodegenerative diseases8,9; and with its unique high energy demands, the retina is especially vulnerable to metabolic defects, including those affecting mitochondrial OXPHOS4 and glycolysis10. Mitochondrial dysfunction and defects in glycolysis are implicated in retinal11,12 and macular13 degenerative diseases, age-related macular degeneration10,14,15,16, and diabetic retinopathy17,18. Therefore, accurate measurements of mitochondrial respiration and glycolysis can provide important parameters for assessing the integrity and health of the retina.
Mitochondrial respiration can be measured through the determination of oxygen consumption rate (OCR). Given that the conversion of glucose to pyruvate and subsequently to lactate results in extrusion of protons into and acidification of the extracellular environment, measurements of the extracellular acidification rate (ECAR) provide an indication of glycolysis flux. As the retina is composed of multiple cell types with intimate relationships and active synergy, including the exchange of substrates6, it is imperative to analyze mitochondrial function and metabolism in the context of whole retinal tissue with intact lamination and circuitry. For the past several decades, the Clark type O2 electrodes and other oxygen microelectrodes have been used to measure oxygen consumption in the retina19,20,21.
These oxygen electrodes have major limitations in sensitivity, requirement of a large sample volume, and the need for continuous stirring of suspending sample, which usually leads to the disruption of cellular and tissue context. The protocol described here was developed using a microplate-based, fluorescence technique to measure mitochondrial energy metabolism in freshly dissected ex vivo mouse retina tissue. It allows mid-throughput real-time measurements of both OCR and ECAR simultaneously using a small sample (1 mm punch) of ex vivo retinal tissue while avoiding the need for suspension and continuous stirring.
Demonstrated here is the experimental procedure for mitochondrial stress assay and glycolytic rate assay on freshly dissected retinal punch disks. This protocol allows the measurement of mitochondria-related metabolic activities in an ex vivo tissue context. Different from the assays performed using cultured cells, the readings obtained here reflect combined energy metabolism at the tissue level and are influenced by interactions between the different cell types within the tissue. The protocol is modified from a previously published version22,23 to adapt to the new generation of the Agilent Seahorse extracellular flux 24-wells (XFe24) analyzer with Islet Capture plate. The assay medium, injection compound concentrations, and number/duration of assay cycles have also been optimized for retinal tissue. A detailed step-by-step protocol is given for the preparation of retinal punch disks. More information on the program setup and data analysis can be obtained from the manufacturer’s user guide24,25,26.
Protocol
All mouse protocols were approved by the Animal Care and Use Committee of the National Eye Institute (NEI ASP# 650). Mice were housed in 12 h light-dark conditions and cared for by following the recommendations of the Guide for the Care and Use of Laboratory Animals, the Institute of Laboratory Animal Resources, and the Public Health Service Policy on Humane Care and Use of Laboratory Animals.
1. Hydrating sensor cartridge and preparation of the assay medium
-
The day before the experiment, add 1 mL of the calibration medium to each well of the utility plate. Place the Hydro-Booster cover on the top and lower the sensor cartridge through the opening on the cover. Check to ensure that the sensor is submerged in the calibration medium. Incubate the sensor cartridge overnight in a CO2-free incubator at 37 °C to activate the fluorophores.
NOTE: To prevent evaporation, the incubator is humidified by keeping a tray of water inside, and the sensor cartridge cassette is wrapped with clear plastic wrap.
Prepare the assay medium by reconstituting the Seahorse DMEM medium with the addition of glucose, pyruvate, and glutamine to the desired concentrations. In the assays reported in this article, the final concentration of substrates in the assay medium are: 6 mM of glucose, 0.12 mM of pyruvate, and 0.5 mM of glutamine. For each assay plate, 40 mL of the assay medium is prepared fresh on the day of the experiment.
Set up the assay program in the analyzer following the manufacturer’s instruction26. In the assay demonstrated here, the protocol is set as follows: 5 cycles of measurements for baseline, then inject port A, followed by 4 cycles of measurements, then inject port B and followed by 4 cycles of measurements. Each cycle is composed of mix (3 min), wait (2 min) and measure (3 min).
2. Coating mesh inserts of islet capture microplate
Prepare the coating mix by combining 20 μL of the cell attachment medium (e.g., Cell-Tak) with 171 μL of 0.1 M sodium bicarbonate and 9 μL of 1 M NaOH.
Open the lid of the cassette containing mesh inserts. Pipette 8 μL of the coating mix to each mesh inserts. Use a pipette tip to gently smear/spread the droplet around to distribute the coating mix equally throughout the mesh insert.
Close the cassette and allow the mesh inserts to incubate at room temperature for at least 25 min for adsorption.
Wash the mesh insert by pipetting 4 mL of the assay medium directly onto the mesh inserts. Gently shake the cassette to ensure all mesh inserts are washed with the assay medium.
Keep the mesh insert aside. It is ready to use.
3. Preparing injection compounds
-
Take out stock aliquots of Bam15 (10 mM), Rotenone (10 mM), Antimycin A (10 mM) and 2-DG (500 mM) from −80 °C freezer and thaw at room temperature.
NOTE: The 2-DG stock is ready to use. The other drugs need to be diluted to working stock.
Warm up 10 mL of the assay medium in a 37 °C water bath.
Dilute 10 mM Bam15 stock to 50 μM working stock using a two-step dilution procedure: mix 20 μL of 10 mM stock with 20 μL of DMSO to get 5 mM intermediate stock. Then mix 10 μL of the above 5 mM intermediate stock with 990 μL of pre-warmed assay medium to get the final 50 μM working stock.
Dilute and combine 10 mM Rotenone and 10 mM Antimycin A stock to 10 μM Rotenone/Antimycin A (Rot/AA) working stock by two steps of dilutions: mix 10 μL each of 10 mM Rotenone and 10 mM Antimycin A stock with 80 μL of DMSO to get 1 mM Rot/AA intermediate stock. Then mix 10 μL of the above 1 mM intermediate stock with 990 μL of pre-warmed assay medium to get the final 10 μM Rot/AA working stock.
Freshly prepare the above-mentioned working stocks of injection compounds on the day of the experiment and set them aside at room temperature until loading into injection ports of the sensor cartridge.
4. Retinal dissection and retinal punch preparation
-
Euthanize a mouse by CO2 asphyxiation following AVMA Guidelines on Euthanasia27.
NOTE: Do not leave the animal in a CO2 chamber longer than the time needed for euthanasia.
Enucleate eyes and place into ice-old 1x PBS buffer in a Petri-dish and then place it under a dissection microscope.
Carefully remove, by cutting with microscissors, the extra rectus muscles attached outside the eyeball and cut off the optic nerve.
Use a 30 G needle to punch a hole at the edge of the cornea (limbus); this serves as the insertion site for the microscissors. Then, use a fine dissection microscissors to make a circular cut along the edge of the cornea, separating it from the posterior eye cup.
Use sharp dissection forceps to remove the cornea, lens, and the vitreous humor away from the eye cup.
Use fine dissection microscissors to make several small cuts on the scleral layer at the rim of eye cup. Avoid cutting the retina layer. Use two sharp dissection forceps to hold on to the scleral tissue at each side of the cut and very carefully pull on the scleral layer to remove it from the neural retina. Repeat this around the eye cup until all sclera is removed and an intact retinal cup is obtained.
-
Use dissection microscissors and make radial cuts on the retinal cup to flatten it and generate several distinct sections.
NOTE: Depending on the person’s dissection skills and experience in handling fresh retinal tissue, the retinal cup can be cut to generate 3 to 5 distinct sections.
-
Use 1 mm diameter biopsy puncher to cut one retinal disk from each section of the flattened retinal cup.
NOTE: Care should be taken to get the retinal disks punched at equal distance from the optic nerve head.
-
Use forceps to transfer the pre-coated mesh inserts into the dissecting petri-dish. With the help of two superfine eyelash brushes, place the retinal punch disk onto the mesh insert. The retinal punch disk is placed at the center of the mesh insert with ganglion cell layer side down touching the mesh and photoreceptor layer facing up.
NOTE: Frequently, some RPE cells remain attached to the photoreceptors, and the pigmentation of these cells can be used as an indicator of the retinal punch disk orientation.
5. Loading the sensor cartridge injection ports and calibration
Take the hydrated sensor cartridge plate cassette out of the 37 °C incubator. Remove the Hydro-Booster cover and place the sensor cartridge back on the utility plate.
Load the desired volume of injection compound working stock solutions into appropriate ports. Hold the pipette tip at 45 ° angle. Insert the pipette tip halfway into an injection port with the bevel of the tip against the opposite wall of the injection port and gently load the compound into each port. Avoid introducing air bubbles.
Refer to the instrument user guide for the volume of the compound loaded in each injection port for a specific assay. In the experiments presented in this paper, 68 μL of 50 μM Bam15 working stock (for mitochondrial stress assay) or 68 μL of 10 μM Rot/AA working stock (for glycolytic rate assay) is loaded into port A; 75 μL of 10 μM Rot/AA working stock (for mitochondrial stress assay) or 75 μL of 500 mM 2-DG working stock (for glycolytic rate assay) is loaded into port B.
Load injection ports of all wells of the plate including background correction wells and blank wells to ensure proper injection. Load the respective compound solution in each port for the background correction wells. Assay medium can be substituted, instead of the compound solution, in each of the ports of the bank wells.
Place the loaded sensor cartridge plate, with lid off, into the analyzer machine to start calibration prior to the assay run. After the calibration is over, the program will automatically pause, waiting for the replacement of the utility plate with the islet capture plate containing retinal punches.
6. Loading the islet capture plate and start assay run
Add 607 μL of the assay medium to each well of the islet capture plate
Use forceps to grab the rim of the mesh insert containing retinal punch disks on top and take it out from the Petri-dish. Lightly tap the bottom of the mesh insert on an absorbing wipe tissue to remove extra liquid and put it into the well of the islet capture plate. Repeat this step until all mesh inserts with retinal punches are placed into the islet capture plate. Fill background correction wells and blank wells with empty mesh inserts.
Use two Graefe forceps to carefully and gently press the rim of each mesh insert and make sure that these are securely inserted at the bottom of the islet capture plate.
Place the loaded islet capture plate into a 37 °C incubator for 5 min to warm up.
Eject the utility plate after the calibration is complete and replace it with an and replace it with the islet capture plate, with lid off, containing retinal punches.
Resume the assay run.
7. Run termination and data storage
After the run is complete, eject the sensor cartridge and islet capture plate containing retinal punches. The data is automatically saved as .asyr file.
Use the associated data analysis software to view and analyze the data following the manufacturer’s user guide26.
Use the Export function to export .xslx file of the data, which can be viewed and analyzed using spreadsheet software.
8. Saving the retinal punch sample
After the assay, take out the plate from the machine, remove the sensor cartridge and gently remove the assay medium from each well using a pipette.
Apply the cover back on and seal the sides of the plate with the parafilm strip.
Store at −80 °C.
For normalization, quantify the total DNA or protein content of the punch in each well.
9. Data analysis
1. Mitochondrial stress assay
NOTE: The measured OCR value (totalOCR) represents total oxygen consumption by the tissue. After Bam15 (uncoupler) injection, OCR increases from the basal level (totalOCRbasal) to the maximum level (totalOCRmax) and goes down following the Rot/AA injection. The residual OCR value after Rot/AA injection (totalOCRRot/AA) represents non-mitochondrial oxygen consumption.
- Calculate mitochondria-related oxygen consumption as:
(Eq. 1)28 -
Calculate the mitochondrial reserve capacity (MRC) as:
(Eq. 2)29 NOTE: The last reading among the 5 measurements before Bam15 injection is taken as the “basal” value (for totalOCRbasal and mitoOCRbasal). The highest reading among the 4 measurements following Bam15 injection is used as “max” value (for totalOCRmax and mitoOCRmax). The lowest reading among the 4 measurements following the Rot/AA injection is used as totalOCRRot/AA.
2. Glycolytic rate assay
NOTE: The measured ECAR value (totalECAR) represents the total acidification of the medium by the tissue’s metabolic activity. In general, acidification of the extracellular micro-environment results mainly by extrusion of the glycolytic product, lactate. Catabolism of substrates in mitochondrial TCA cycle results in the production of CO2, which also acidifies the extracellular medium through hydration to bicarbonate.
-
Substract mitochondrial contributed medium acidification (mitoECAR) from totalECAR to obtain the glycoECAR.
(Eq. 3)28 NOTE: Mitochondrial respiration and TCA cycle are strongly coupled processes. Production of CO2 from mitochondria is a function of the rate of OXPHOS, which is measurable by mitoOCR.
-
Calculate the mitoECAR as:
where, the CCF (CO2 Contribution Factor) is an empirically calculated ratio value, representing the amount of H+ contribution from CO2-mediated acidification vs each O2 consumption from OXPHOS. CCF for this system is pre-determined to be 0.6028. Accurate measurement of medium acidification is determined by the buffer capacity of the medium, the sensitivity of instrument pH sensor, and the effective measurement chamber capacity. Here, the BF (Buffer Factor) is a parameter of the in situ experimental buffer capacity, representing the amount of H+ or OH− added to the effective measurement chamber to change the pH level by 1 unit. When customized assay medium is used, the BF can be determined by titrating known amounts of acid into the assay medium following the Buffer Factor protocol30. The Seahorse DMEM medium pH 7.4 used in this protocol has a pre-determined BF of 2.60 mmol H+/L/pH. The islet capture plate used in this protocol has a Volmicrochamber = 16.6 μL31. The volume scaling factor, Kvol, is an empirically determined constant. Kvol value is not available for the islet capture plate but can be calculated from the value of the microplate28, accounting for the volume difference in their microchambers, to be 0.41.(Eq. 4)28 NOTE: Injection of the Rot/AA shuts down mitochondrial respiration and forces the tissue to switch to glycolysis for ATP production, leading to higher lactate extrusion and an increase in ECAR measurement. Glycolysis is ceased with 2-DG injection, and the residual ECAR measurement reveals non-glycolytic and non-mitochondrial acidification of medium.
- Calculate the glycolytic reserve capacity (GRC) as:
where, the last reading among the 5 measurements before Rot/AA injection is taken as the “basal” value (). The highest reading among the 4 measurements following Rot/AA injection is used as “max” value (). The lowest reading among the 4 measurements following 2-DG injection is used as glycoECAR2-DG.(Eq. 5)32
3. Normalization
NOTE: Normalization is essential when comparing the readings from retinal tissues of different age groups or between wild-type and pathological/degenerative samples, which might differ in cell numbers.
Use commerically available kits to assess the DNA content in each retinal punch disk33,34.
-
Alternatively, use radioimmunoprecipitation assay buffer (RIPA buffer) to extract total protein from the retinal punch and use the protein content for normalization.
NOTE: The surface area of an adult mouse retina has been previously determined to be around 20 mm2, and each retina contains ~6.5 million cells35. Hence, each 1 mm diameter retinal punch is ~1/25 of a single retina and contains ~260K cells. One can refer to these numbers when comparing the data from a retinal punch to those from other tissue samples or cultured cells,
Representative Results
The data reported here are representative mitochondrial stress assay showing OCR trace (Figure 1) and glycolytic rate assay showing OCR trace and ECAR trace (Figure 2), which were performed using freshly dissected 1 mm retinal punch disks from 4 months old transgenic Nrl-L-EGFP mice36 (C57B/L6 background). These mice express GFP specifically in rod photoreceptors without altering normal retinal development, histology, and physiology and have been widely used as wild-type controls in retinal research. Two Nrl-L-GFP littermate mice were used were used in the assays presented here. GFP expressed in the Nrl-L-GFP mice does not interfere with the measurements of OCR and ECAR in this protocol. Five retinal punches were taken from each retina. Ten of the retinal punches were used for mitochondrial stress assay and the other 10 were used for glycolytic rate assay. Seahorse XF DMEM medium, pH 7.4 (constituted with 6 mM glucose, 0.12 mM pyruvate, and 0.5 mM glutamine) and Seahorse XFe24 Islet Capture plates were used in the experiments. The representative data presented here were obtained using the same 1 mm diameter puncher but was not normalized with respect to the DNA/protein content.
Figure 1: Mitochondrial stress assay.
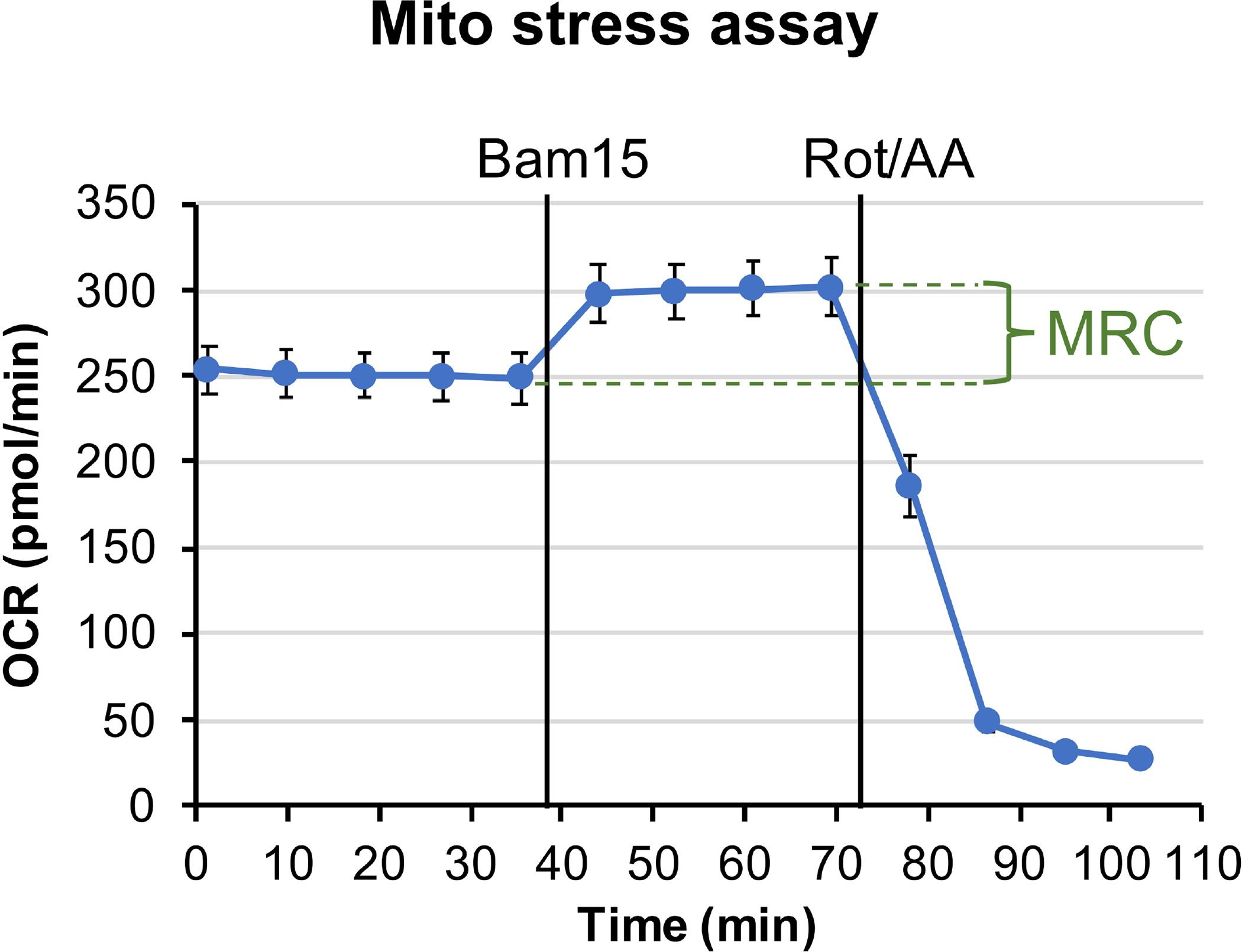
The plotted graph shows OCR trace from 1 mm retinal punch disks in Seahorse XF DMEM buffer, supplemented with 6 mM of glucose, 0.12 mM of pyruvate and 0.5 mM of glutamine. Each data point represents the average of measurements from 10 wells. Error bar = standard error. MRC is calculated to be 19.2%±3.4%.
Figure 2: Glycolytic rate assay.
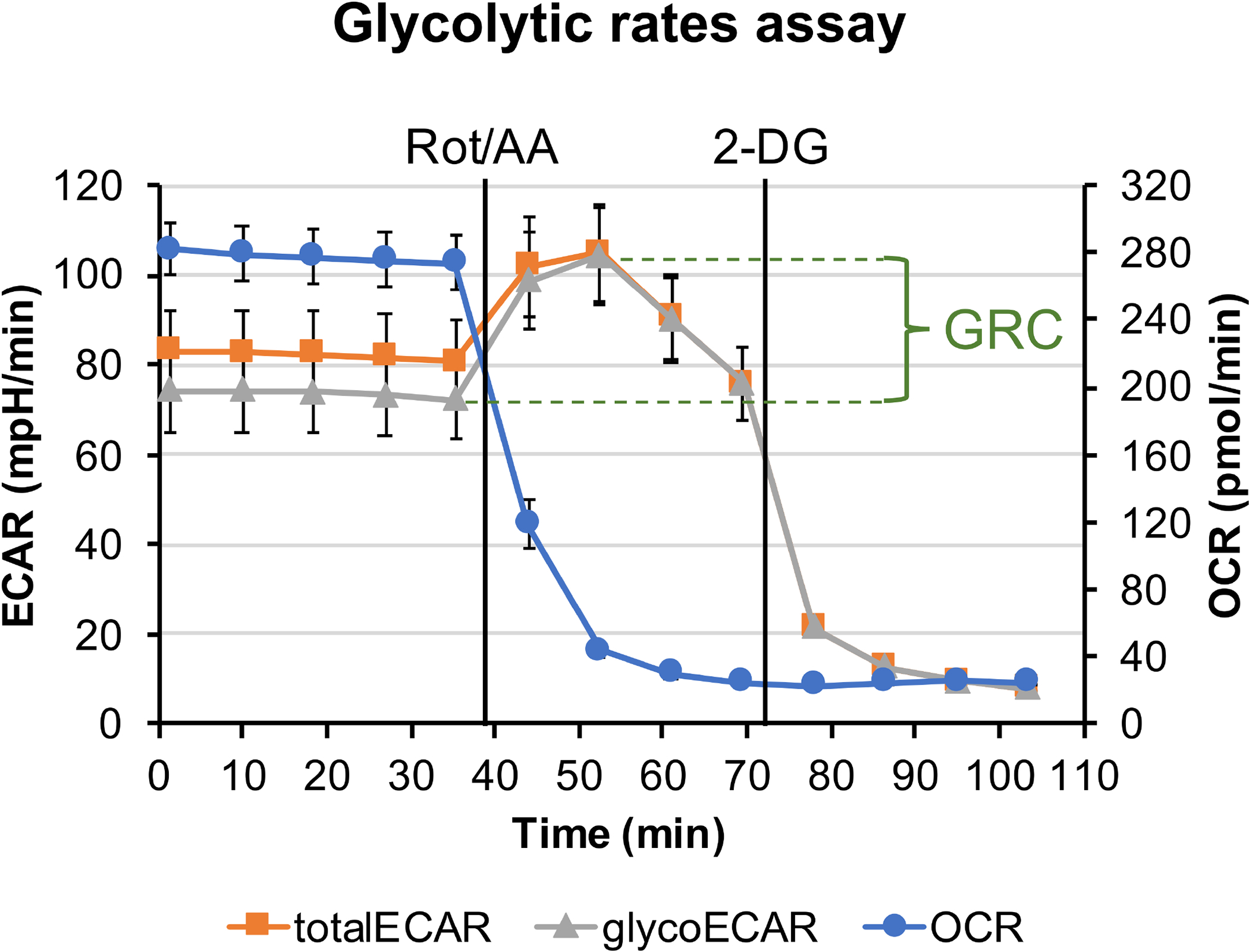
The plotted graph shows the measured OCR trace, ECAR trace (totalECAR), and the calculated glycolysis contributed ECAR (glycoECAR) from 1 mm retinal punch disks in Seahorse XF DMEM buffer supplemented with 6 mM of glucose, 0.12 mM of pyruvate, and 0.5 mM of glutamine. Each data point represents the average of measurements from 10 wells. Error bar = standard error. GRC is calculated to be 35.7%±3.4%
In mitochondrial stress assay, the uncoupler Bam1537 was injected after establishing the OCR baseline, leading to enhanced OCR to the maximal level. Rotenone and Antimycin A were injected to inhibit mitochondria respiration at complex I and complex III, respectively, resulting in OCR to drop to the minimal level (Figure 1). The difference between the maximal level of OCR and the last measurement of the basal OCR level reflects mitochondrial reserve capacity (MRC). The MRC is calculated to be 19.2%±3.4% using Eq. 2, consistent with previously measured MRC values in retinas of ~3 months old Nrl-L-EGFP mice using the previous generation Seahorse XF24 analyzer22,38.
In the glycolytic rate assay, Rotenone and Antimycin A were injected after establishing the baseline for the total ECAR. With the production of ATP from OXPHOS halted, the tissue is forced to rely on glycolysis for energy, and an increase in the extracellular release of lactate drives ECAR to the maximal level. Glycolysis is ceased by injection of 2-DG, which competes with glucose for hexokinase binding, causing ECAR to drop to the minimal level (Figure 2). Mitochondria contributed ECAR (mitoECAR) can be calculated from the mitoOCR value (Eq. 4). Glycolysis contributed ECAR glycoECAR is calculated and plotted by subtracting mitoECAR from totalECAR. The difference between maximal level of glycoECAR and the last measurement of glycoECAR basal level reflects the glycolysis reserve capacity (GRC). Here, the GRC is calculated to be 35.7% ± 3.4% using Eq. 5.
As a highly glycolytic tissue, lactate production from the retina accounts for a major source of extracellular acidification, as revealed by the small difference of glycoECAR from the totalECAR. Interestingly, ECAR measurement does not plateau immediately following the Rot/AA injection but drops after the second measurement. The retinal punch disk is an intact ex vivo system composed of different cell types, including the Müller glia cells, which are known to receive lactate (glycolysis end product) released from the photoreceptors6. Hence, a drop in ECAR measurement following the Rot/AA injection is likely explained by increased removal of lactate from the intercellular space, slowing down/preventing its release into the medium.
Discussion
Provided here are detailed instructions for performing microplate-based assays of mitochondrial respiration and glycolysis activity using ex vivo, freshly dissected retinal punch disks. The protocol has been optimized to: 1) ensure the use of a suitable assay medium for ex vivo retinal tissue; 2) employ proper size of retinal punch disks to obtain OCR and ECAR readings that fall within the machine’s optimal detecting range; 3) coating mesh inserts to enhance the adhesiveness of retinal punch for stable reading during the measuring cycle; 4) use of optimal concentration of each injected drug compounds; and 5) ensure altered cycle length to reach a plateau of mitochondrial states at each step. The reagents and protocol have been modified from a previously published version23 to adapt to the new generation Seahorse XFe24 machine. Instead of the Ames’ buffer used in the previous protocol23, a basic Seahorse DMEM medium is used here to allow the custom constitution of fuel source by adding glucose, glutamine, and pyruvate separately. This also makes it possible to perform various assays where a specific fuel substrate is supplied or deprived from the medium. In the assays presented here, the medium was constituted to the same concentration of glucose (6 mM), glutamine (0.5 mM), and pyruvate (0.12 mM) as in Ames’ buffer, which are proven suitable for retinal tissue. Another advantage of this medium (with 5 mM HEPES) over the Ames’ buffer (with 22.6 mM NaHCO3) is its low buffer capacity, which ensures sensitive and accurate measurement of ECAR28.
Both mitochondrial stress and glycolytic rate assays can be performed following the protocol described here with high precision, as evidenced by the tight standard error values between replicating wells. However, it is worthwhile to note the factors that can contribute to data variability. Avoid cell death in retinal tissue. The entire dissection process should be performed in ice-cold 1x PBS, and the process from enucleation of eyes to putting islet capture plate containing the retinal punch into the machine should not exceed 2 hours. Caution should be taken during the dissection of the retina cup to avoid any damage to the retinal tissue, and punches should not be taken from areas damaged by dissection. New, sharp biopsy puncher should be used in each experiment, and change the puncher when the edge is dull or bent to ensure consistency and accuracy in cutting retinal punches at 1 mm diameter. Try to get the retinal disks punched at equidistant from the optic nerve head to avoid regional variations (center versus peripheral). After the assay, check each well for any sign of the retinal punch being detached from the mesh insert. When a retinal punch has poor adhesion on mesh insert or detaches during measurement, the distance from senser probe to the tissue will change, affecting the readings. Omit the data from such wells with detached retinal punch.
Measurement of the real-time mitochondrial metabolism in intact retinal tissue has broad applications and can provide useful information for various studies. These assays have been used to measure mitochondrial respiration in retinal tissues from mice of different genetic backgrounds to reveal their intrinsic difference in mitochondrial activity39,40. It was also used to study changes in mitochondrial energy metabolism during aging of the retina38. By providing different fuel substrates and utilizing various inhibitors targeting different metabolic pathways, it provides insights to the preference of the cell/tissue on certain fuel sources22,38. Furthermore, comparison on OCR and MRC between wild-type mouse and mouse models of inherited retinal degeneration can provide evidence of mitochondrial defects in degenerating retina22.
There are limitations of this technique. The islet capture plate used in these assays only contains 24 wells; hence, it is only able to provide mid-throughput analysis. The data quality from this method is contingent upon the quality of retinal punch disks and viability of cells. Also, retinal dissection and retinal punch disks preparation is a time-consuming process, rendering it less feasible to high-throughput analysis on live ex vivo retinal tissues even when 96-well plates are available. Compared to a monolayer of cultured cells, penetration of drug compound into the retinal tissue also affects data readout. In addition, the measured OCR and ECAR values represent the total performance of the entire tissue, which is composed by many different cell types; hence, one needs to consider the relationship and interactions among different neuronal and glial cells in the retina while interpreting the data. Specific experimental designs should be implemented by tailoring to each project. It is recommended that one includes 3 to 5 retinal punches (from same eye or same mouse) as technical replicates and use samples from 3 or more mice as biological replicates.
Supplementary Material
Acknowledgments
This work is supported by the Intramural Research Program of the National Eye Institute (ZIAEY000450 and ZIAEY000546).
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/62914.
Disclosures
The authors have nothing to disclose.
References
- 1.Nunnari J, Suomalainen A Mitochondria: In sickness and in health. Cell. 148 (6), 1145–1159, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wong-Riley MT Energy metabolism of the visual system. Eye Brain. 2 99–116, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu DY, Cringle SJ Oxygen distribution and consumption within the retina in vascularised and avascular retinas and in animal models of retinal disease. Progress in Retina and Eye Research. 20 (2), 175–208 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Barot M, Gokulgandhi MR, Mitra AK Mitochondrial dysfunction in retinal diseases. Current Eye Research. 36 (12), 1069–1077 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joyal JS, Gantner ML, Smith LEH. Retinal energy demands control vascular supply of the retina in development and disease: The role of neuronal lipid and glucose metabolism. Progress in Retina and Eye Research. 64 131–156 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hurley JB, Lindsay KJ, Du J Glucose, lactate, and shuttling of metabolites in vertebrate retinas. Journal of Neuroscience Research. 93 (7), 1079–1092 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haydinger CD, Kittipassorn T, Peet DJ Power to see-Drivers of aerobic glycolysis in the mammalian retina: A review. Clinical and Experimental Ophthalmology. 48 (8), 1057–1071 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Wright AF et al. Lifespan and mitochondrial control of neurodegeneration. Nature Genetics. 36 (11), 1153–1158 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Bossy-Wetzel E, Schwarzenbacher R, Lipton SA Molecular pathways to neurodegeneration. Nature Medicine. 10 Suppl S2–9 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Leveillard T, Philp NJ, Sennlaub F Is retinal metabolic dysfunction at the center of the pathogenesis of age-related macular degeneration? International Journal of Molecular Sciences. 20 (3), (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vlachantoni D et al. Evidence of severe mitochondrial oxidative stress and a protective effect of low oxygen in mouse models of inherited photoreceptor degeneration. Human Molecular Genetics. 20 (2), 322–335 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Grenell A et al. Loss of MPC1 reprograms retinal metabolism to impair visual function. Proceedings of the National Academy of Science U. S. A. 116 (9), 3530–3535 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wright AF, Chakarova CF, Abd El-Aziz MM, Bhattacharya SS Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nature Reviews in Genetics. 11 (4), 273–284 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Jarrett SG, Boulton ME Consequences of oxidative stress in age-related macular degeneration. Molecular Aspects of Medicine. 33 (4), 399–417 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rozing MP et al. Age-related macular degeneration: A two-level model hypothesis. Progress in Retina Eye Research. 76, 100825 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Yokosako K et al. Glycolysis in patients with age-related macular degeneration. Open Ophthalmology Journal. 8, 39–47 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bek T Mitochondrial dysfunction and diabetic retinopathy. Mitochondrion. 36, 4–6 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Yumnamcha T, Guerra M, Singh LP, Ibrahim AS Metabolic dysregulation and neurovascular dysfunction in diabetic retinopathy. Antioxidants (Basel). 9 (12), (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Futterman S, Kinoshita JH Metabolism of the retina. I. Respiration of cattle retina. Journal of Biological Chemistry. 234 (4), 723–726 (1959). [PubMed] [Google Scholar]
- 20.Linsenmeier RA Effects of light and darkness on oxygen distribution and consumption in the cat retina. Journal of General Physiology. 88 (4), 521–542 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Medrano CJ, Fox DA Oxygen consumption in the rat outer and inner retina: light- and pharmacologically-induced inhibition. Experiments in Eye Research. 61 (3), 273–284, (1995). [DOI] [PubMed] [Google Scholar]
- 22.Kooragayala K et al. Quantification of oxygen consumption in retina ex vivo demonstrates limited reserve capacity of photoreceptor mitochondria. Investigative Ophthalmology and Visual Science. 56 (13), 8428–8436 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adlakha YK, Swaroop A Determination of mitochondrial oxygen consumption in the retina ex vivo: applications for retinal disease. Methods in Molecular Biology. 1753, 167–177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agilent Mitocondrial stress test user guide. https://www.agilent.com/cs/library/usermanuals/public/XF_Cell_Mito_Stress_Test_Kit_User_Guide.pdf. (2021). [Google Scholar]
- 25.Agilent Glycolytic rate assay user guide. https://www.agilent.com/cs/library/usermanuals/public/103344-400.pdf. (2021). [Google Scholar]
- 26.Agilent wave 2.6 user guide. https://www.agilent.com/cs/library/usermanuals/public/S7894-10000_Rev_C_Wave_2_6_User_Guide.pdf. (2021). [Google Scholar]
- 27.AVMA Guidelines for the Euthanasia of Animals. https://www.avma.org/sites/default/files/2020-01/2020-Euthanasia-Final-1-17-20.pdf. (2021). [Google Scholar]
- 28.Improving Quantification of Cellular Glycolytic Rate Using Agilent Seahorse XF Technology. https://www.agilent.com/cs/library/whitepaper/public/whitepaper-improve-quantification-of-cellular-glycolytic-rate-cell-analysis-5991-7894en-agilent.pdf. (2021). [Google Scholar]
- 29.Report Generator User Guide. https://www.agilent.com/cs/library/usermanuals/public/Report_Generator_User_Guide_Seahorse_XF_Cell_Mito_Stress_Test_Single_File.pdf [Google Scholar]
- 30.Agilent Seahorse XF Cell Mito Stress Test. https://www.agilent.com/cs/library/usermanuals/public/usermanual-xf-buffer-factor-protocol-cell-analysis-S7888-10010en-agilent.pdf. (2021). [Google Scholar]
- 31.Agilent Seahorse XF Buffer Factor Protocol. https://www.agilent.com/cs/library/brochures/5991-8657EN_seahorse_plastics_brochure.pdf. (2021. [Google Scholar]
- 32.Agilent sensor cartridges and cell culture microplates Agilent Seahorse XF Glycolysis Stress Test Kit User Guide. https://www.agilent.com/cs/library/usermanuals/public/XF_Glycolysis_Stress_Test_Kit_User_Guide.pdf. (2021). [Google Scholar]
- 33.Fan YY et al. A bioassay to measure energy metabolism in mouse colonic crypts, organoids, and sorted stem cells. American Journal of Physiology-Gastrointestinal and Liver Physiology. 309 (1), G1–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang L et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nature Medicine. 21 (11), 1364–1371 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeon CJ, Strettoi E, Masland RH The major cell populations of the mouse retina. Journal of Neuroscience. 18 (21), 8936–8946 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akimoto M et al. Targeting of GFP to newborn rods by Nrl promoter and temporal expression profiling of flow-sorted photoreceptors. Proceedings of the National Academy of Science U. S. A. 103 (10), 3890–3895, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kenwood BM et al. Identification of a novel mitochondrial uncoupler that does not depolarize the plasma membrane. Molecular Metabolism. 3 (2), 114–123 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corso-Diaz X et al. Genome-wide profiling identifies DNA methylation signatures of aging in rod photoreceptors associated with alterations in energy metabolism. Cell Reports. 31 (3), 107525 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berkowitz BA et al. Mitochondrial respiration in outer retina contributes to light-evoked increase in hydration in vivo. Investigative Ophthalmology and Visual Science. 59 (15), 5957–5964 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joyal JS et al. Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor Ffar1. Nature Medicine. 22 (4), 439–445 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.