

Abstract
The genus Cryptococcus comprises two major fungal species that cause clinical infections in humans: C. gattii and C. neoformans. To establish invasive human disease, inhaled Cryptococci must penetrate the lung tissue and reproduce. Each year, about 1 million cases of Cryptococcus infection are reported worldwide, and the infection’s mortality rate ranges from 20% to 70%. Many HIV+/AIDS patients are affected by Cryptococcus infections, with 220,000 cases of cryptococcal meningitis reported worldwide in this population every year (C neoformans infection statistics, CDC). To escape from host immune cell attack, Cryptococcus covers itself in a sugar-based capsule composed primarily of glucuronoxylomannan (GXM). To evade phagocytosis, the yeast cells increase to a >45-μm perimeter then referred to as titan or giant cells. Cryptococci virulence is directly proportional to the percentage of titan/giant cells present during Cryptococcus infection. To combat cryptococcosis, we proposed the redirection of CD8+ T cells to target the GXM in the capsule, via expression of a GXM-specific chimeric antigen receptor (GXMR-CAR). GXMR-CAR has an anti-GXM single-chain variable fragment followed by an IgG4 stalk in the extracellular domain, a CD28 transmembrane domain, and CD28 and CD3-ς signaling domains. After lentiviral transduction of human T cells with the GXMR-CAR construct, flow cytometry demonstrated that 82.4% of the cells expressed GXMR-CAR on their surface. To determine whether the GXMR-CAR+ T cells exhibited GXM-specific recognition, these cells were incubated with GXM for 24 h and examined with use of bright-field microscopy. Large clusters of proliferating GXMR-CAR+ T cells were observed in GXM-treated cells, whereas no clusters were observed in control cells. Moreover, the interaction of GXM with GXMR-CAR+ T cells was detected via flow cytometry by using a GXM-specific antibody and the recognition of GXM by GXMR-CAR T cells triggered the secretion of granzyme and IFN-γ. The ability of GXMR-CAR T cells to bind to the yeast form of C. neoformans was detected by fluorescent microscopy, but no binding was detected in mock-transduced control T cells (NoDNA T cells). Moreover, lung tissue sections were stained with Gomori methenamine-silver (GMS) and evaluated by Nanoozomer, revealing a significantly lower number of titan cells with perimeters ranging from 50 to 130 μm and giant cells >130 μm compared with other groups. Therefore, we have validated our hypothesis by the redirection of GXMR-CAR+ T cells to target GXM which induces the secretion of cytotoxic granules and IFN-γ that will aid in the control the cryptococcosis. Thus, these findings revealed that GXMR-CAR+ T cells can target Cryptococcus neoformans. Future studies will be focused on determining the therapeutic efficacy of GXMR-CAR+ T cells in an animal model of cryptococcosis.
Keywords: Chimeric antigen receptor T-cell therapy, Cryptococcus, glucuronoxylomannan, fungal therapy, GXMR-CAR T cells, cell therapy, immunotherapy
Introduction
Cryptococcus gattii and Cryptococcus neoformans are the two major Cryptococcus species that cause clinical infections in humans [1] due to the ability of basidiomycetous yeasts to grow at 37°C through asexual yeast budding in human and animal hosts. Upon inhalation of infectious propagules, Cryptococcus spp. penetrate the lung tissue and reproduce, causing pulmonary cryptococcosis, leading to the possibility of dissemination to other organs, most commonly the brain [2]. C. gattii infections occur in both immunocompetent and immunocompromised individuals, whereas C. neoformans infections are more common in immunosuppressed patients, such as those who have received organ transplants or have HIV infection/AIDS or hematological malignancies [3]. In mice infected with C. neoformans, CD4+ T-helper 1 (Th1) cells play an important role in fighting infection [4, 5] by secreting proinflammatory cytokines to boost the host immune system, as demonstrated by the fact that interferon (IFN)-γ and interleukin (IL)-12–knockout mice had a higher mortality index than did IFN-γ and IL-12 wild- type mice [6–9]. In addition, mice infected with C. gattii displayed reduced dendritic cell– mediated Th1/Th17 immune responses [10]. Several fungal vaccine studies have demonstrated an essential role for CD8+ T cells in destroying host cells harboring intracellular fungus [11]. In addition, CD8+ T cells have been shown to mediate direct killing of extracellular fungus by releasing cell lytic granules [12–14]. NKp30, a natural killer (NK) cell receptor, directly recognizes Cryptococcus spp., causing NK cells to release cell lytic granules. This suggests that when the fungal burden overwhelms cytotoxic T cells, NK cells will participate in clearing the fungal infection [15, 16].
Our group was the first to explore the direct killing of extracellular fungi through the redirection of CD8+ T cells via expression of an engineered chimeric antigen receptor (CAR) to target a carbohydrate expressed on fungal cell walls [17, 18]. CARs are usually composed of four domains: 1) an extracellular antigen-specific binding domain or domains, 2) a hinge or spacer region, 3) a transmembrane region, and 4) a cytoplasmic signaling region [19, 20]. Interest in using CAR+ T cells to treat diseases such as cancer has increased in recent years because CAR-based recognition enables T cells to identify proteins, glycoproteins, or glycolipids on the cell surface and release cytotoxic granules that destroy the target cells in a manner that does not depend on the peptide-major histocompatibility complex (MHC) [21]. CAR-dependent T-cell activation is achieved through the CAR endodomain, which is composed of cytoplasmic signaling domains from CD28 or CD137 and CD3-ς [22, 23]. In most studies using CARs, researchers have administered CD19-specific CARs for the treatment of B-cell malignancies. Recently, the U.S. Food and Drug Administration approved CAR+ T-cell therapies for leukemia and lymphoma [21, 24–28]. We demonstrated that T cells expressing a CAR containing the extracellular portion of Dectin-1 (D-CAR) targeted germinating fungal hyphae and inhibited hyphal growth of Aspergillus spp. [18]. However, the Cryptococcus-killing activity of CD8+ CAR T cells has yet to be explored in the development of immunotherapy for cryptococcosis.
Fungal cell wall glycans and exopolysaccharides are the first point of physical contact in fungal-host interactions, and these polysaccharides are common to multiple fungi [29]. Pattern recognition receptors present on innate immune cells recognize these glycans and eliminate fungal spores, germinating hyphae, and yeast. However, the Cryptococcus spp. has exopolysaccharides that can mask these glycans that are recognized by immune cells. Disrupting the glucuronoxylomannan (GXM) capsule may aid the host immune system by increasing glycan recognition and fungal clearance. In the present study, we generated a CAR to redirect engineered CD8+ T cells to target the polysaccharide capsule of Cryptococcus spp. The capsule of Cryptococcus spp. is a major virulence factor primarily composed of GXM, with lesser amounts of galactoxylomannan (GalXM) and mannoproteins [30–32]. One of the functions of the capsule is to provide protection by preventing recognition and phagocytosis by the host innate immune cells, inhibiting the migration of phagocytes, and suppressing the proliferation of T cells [33]. Casadevall et al. [34] developed and characterized the murine monoclonal antibody (mAb) 18B7 that binds to Cryptococcus spp., promoting antibody- dependent cellular toxicity mediated by innate immune cells such as macrophages. Phase I clinical studies were performed by using 18B7 to treat cryptococcosis in HIV+ patients. Titers of serum cryptococcal antigen decreased by a median of 2- to 3-fold in patients treated with of 1–2 mg/kg at 1–2 weeks post-infusion, and the treatment was well tolerated [34, 35].
The GXM-specific chimeric antigen receptor (GXMR-CAR) was designed to recognize GXM by using a single-chain variable fragment (scFv) originating from 18B7. We hypothesized that GXMR-CAR T cells would display cytotoxic activity against fungi expressing GXM on the cell wall. Our study demonstrates that engineered human T cells expressing GXMR-CAR bound to GXM extract in solution and interacted with heat-killed C. neoformans yeast. The GXMR-CAR+ T cell–treated NOD Scid gamma (NSG) mice displayed significantly lower C. neoformans (giant cell) burden than did mock-transduced control T cell–treated mice.
Materials and Methods
Ethics statement
All animal experiments were conducted after the study was approved by the MD Anderson Cancer Center Institutional Animal Care and Use Committee (IACUC). Our IACUC-approved protocol number is 00000555-RN01. MD Anderson’s IACUC is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). The IACUC was formed to meet the standards of the Public Health Service Policy on the Humane Care and Use of Laboratory Animals.
Blood samples (buffy coats) were obtained from heathy donors at the MD Anderson Blood Bank, and all donors provided written informed consent. The MD Anderson Institutional Review Board (IRB) approved protocol number LAB07-0296 for obtaining peripheral blood from healthy donors.
Mice and C. neoformans
Eight-week-old female NSG mice were acquired from the Department of Experimental Radiation Oncology mouse colony at The University of Texas MD Anderson Cancer Center. The mice were maintained in a sterile biohazard facility at MD Anderson.
The C. neoformans yeast strain H99 (ATCC, cat. no. 208821) was allowed to grow in YPD medium until the late logarithmic phase, washed one time with phosphate-buffered saline (PBS), followed by suspension in PBS [36]. The yeast concentration was determined by using a hemocytometer, and 20 μl of a suspension of 2 × 106 yeast cells/ml was used to perform intranasal inoculation of the mice [37].
Construction of GXMR-CAR targeting Cryptococcus spp.
The DNA sequence encoding the light and heavy chains of the anti-GXM mAb 18B7 clone was obtained from Casadevall et al. [34] and was used to design the targeting domain of GXMR-CAR. The GXMR-CAR is a second-generation CAR in which the anti-GXM extracellular domain was fused to modified human IgG4 hinge and Fc regions [38] followed by the transmembrane and cytoplasmic domains of human CD28 and CD3-ς chains [22]. Full-length GXMR-CAR was subcloned into a lentiviral (LV) vector (Addgene; cat. no. 61422) containing the green fluorescent protein (GFP) sequence. The LV-GXMR-CAR construct was sequenced and verified at the Sequencing and Microarray Facility at MD Anderson.
Generation of GXMR-CAR+ viral particles
To generate GXMR-CAR+ LV particles, GXMR-CAR containing the LV vectors, pMD2.G (VSV-G envelope-expressing plasmid; Addgene; cat. no. 12259) and psPAX2 (second-generation LV packaging plasmid; Addgene; cat. no. 12260) were transfected into HEK-293FT cells (ThermoFisher; cat. no. R70007) by using Lipofectamine 3000 reagent (Thermo Fisher Scientific; cat. no. L3000008) according to the manufacturer’s instructions. The viral supernatant of LV-CAR–transfected HEK-293FT cells (green cells in Fig. 1B) was collected every day for 3 days, and the pool of viral particles was concentrated by using the Lenti-X concentrator (Clontech Laboratories; cat. no. 631231). The titer of the GXMR-CAR viral particles was determined by using HEK-293FT cells, and LV stocks were aliquoted and maintained at −80°C.
Fig 1. Construction of a GXMR-CAR that targets Cryptococcus spp.

(A) DNA sequence (left) and schematic representation (right) of the GXMR-CAR targeting Cryptococcus spp. has a scFv portion derived from the anti-GXM mAb 18B7. The DNA encodes four domains: the signaling domains of CD3-ς and CD28 (yellow and green regions, respectively), the transmembrane (Tm) domain of CD28 (green region), the constant region of IgG4 (black line), a scFv portion from 18B7 (blue region), and an enhanced GFP (EGFP) portion for detection via fluorescence (white region). The DNA sequence was subcloned into a lentiviral (LV) vector backbone. The LV-GXMR-CAR construct was used for transfection and transduction. Ec, extracellular binding domain; cyto, cytoplasmic signaling region. (B) HEK-293FT cells (1 × 106 cells/ml) were used to make GXMR-CAR+ viral particles via transfection with all three plasmids combined: LV-GXMR-CAR, pMD2.G, and psPAX2, using Lipofectamine. Mock- transduced HEK-293FT cells (NoDNA) were used as negative controls. HEK-293FT cells were visualized by using fluorescence microscopy in bright-field (BF) and green (GFP) channels at 100× and 200× magnification 24 h after transfection. (C) GXMR-CAR+ viral particles were used to transduce HEK-293FT cells. HEK-293FT cells were evaluated via flow cytometry and expressed in a histogram (left). HEK-293FT cells were visualized by using fluorescence microscopy in brightfield and GFP channels at 100× magnification (right) 3 days after transduction. Mock-transduced negative control cells (NoDNA) did not receive the GXMR-CAR construct.
Transduction of cell lines and peripheral blood mononuclear cells
HEK-293FT cells were cultured in a six-well plate at a concentration of 5 × 105 cells/ml. GXMR-CAR+ viral particles were added to the cells in 1 ml of RPMI 1640 medium (HyClone Laboratories) supplemented with 2 mM GlutaMAX-1 (Life Technologies; cat. no. 35050-061) and 10% heat-inactivated fetal bovine serum (HyClone Laboratories). After 24 h of incubation at 37°C, the cells were fed growth media and maintained for 2 days to evaluate the transduction efficiency via quantification of GFP expression using fluorescence microscopy and flow cytometry.
Blood samples (buffy coats) obtained from heathy donors at the MD Anderson Blood Bank were used for peripheral blood mononuclear cell (PBMC) isolation with Ficoll-Paque PLUS solution (GE Healthcare Life Sciences; cat. no. 17144002) according to the manufacturer’s instructions. Transduction of PBMCs with LV-GXM-CAR constructs was performed with use of RetroNectin reagent (Takara Bio USA; cat. no. T100A/B) according to the manufacturer’s instructions. After viral transduction, human T cells were maintained in the presence of IL-2 (50 U/ml) plus IL-21 (20 ng/ml) [18]. Ten days after transduction, LV-CAR–infected T cells expressing GFP were sorted at the South Campus Flow Cytometry Facility at MD Anderson. LV-CAR T cells were expanded by stimulation with an antibody cocktail (anti-CD3/CD28; STEMCELL Technologies; cat. no. 10971) combined with IL-2 (50 U/ml) and IL-21 (20 ng/ml).
Detection of GXMR-CAR expression and identification of the memory-phenotype of GXMR-CAR+ T cells by flow cytometry
The GXMR-CAR construct expresses GFP (CAR-GFP) and a modified human IgG4 hinge region combined with an Fc region that can be detected with an anti-Fc antibody. The expression of GXMR-CAR on the surface of human T cells was analyzed via flow cytometry (MACSQuant; Miltenyi Biotech) by using a goat anti-human IgG antibody (Fc-γ fragment-specific, 1:100 dilution; Jackson ImmunoResearch Laboratories; cat. no. 109-606-098). Human T cells were identified with use of flow cytometry with an anti-human CD3-PE antibody (1:100 dilution; Miltenyi Biotech; cat. no. 130-091-374). The cells that were double-positive for an anti-human IgG antibody and CAR-GFP represented GXMR-CAR expression on the surface of human T cells. NoDNA T cells (mock-transduced) were used as negative controls.
GXMR-CAR+ T cells were expanded in vitro for 30 days (ImmunoCult human CD3/CD28 T-cell activator), and the memory phenotype was determined by flow cytometry. All antibodies for this analysis were obtained from Miltenyi Biotec. Memory cell subsets were determined by using a mixture of anti-CD3-PE (cat. no. 130-091-374), anti-CD8-PerCP (cat. no. 130-113-160), anti-CCR7-APC (cat. no. 130-099-363), and anti-CD45RA-PE-Vio770 (cat. no. 130-108-716). Gates were set up by using PBMCs, and these parameters were applied to GXMR-CAR+ T cells. CD3+CD8+ T cells were gated to evaluate the memory cell subsets using anti-CCR7 and anti-CD45RA antibodies as follows: TCM, central memory T cells (CCR7+/CD45-); TEM, effector memory T cells (CCR7-/CD45-); TEFF, effector T cells (CCR7-/CD45+); Tnaive/SCM, stem cell–like memory T cells (CCR7+/CD45+).
Detection of interaction between GXMR-CAR+ T cells and the sugar moiety GXM
GXMR-CAR+ and NoDNA T cells were incubated with GXM (C. neoformans polysaccharide antigen; ZeptoMetrix, ) at a dilution of 1:100. An ImmunoCult human CD3/CD28 T-cell activator (STEMCELL Technologies; cat. no. 10971) was used to activate positive control T cells according to the manufacturer’s instructions. GXMR-CAR+ and NoDNA T cells incubated with medium alone were considered as negative controls. These cells were imaged via bright-field microscopy (Leica DMI6000B) at 400× magnification. GXMR-CAR+ T cells that bound to GXM were identified with use of an anti-GXM antibody and analyzed by flow cytometry. To conduct this study, the cells were incubated for 30 min with or without C. neoformans polysaccharide antigens at 1:100 dilution. After T cells were washed with PBS, the cells were incubated with a murine anti-GXM IgG mAb for 1 h (EMD Millipore; cat. no. MABF2069), washed with PBS, incubated with a goat anti-mouse IgG phycoerythrin-conjugated secondary antibody (Jackson ImmunoResearch Laboratories; cat. no. 115-116-071), and analyzed by using flow cytometry. T cells without the addition of GXM served as negative controls.
In vitro assay demonstrating that GXMR-CAR+ T cells target C. neoformans yeast
Fluorescence microscopy was used to visualize the interaction between GXMR-CAR+ T cells (GFP+) and heat-killed C. neoformans yeast (ATCC, cat. no. 208821). The yeast was labeled with Calcofluor-white (Thermo Fisher Scientific; cat. no. L7009) according to the manufacturer’s instructions. GXMR-CAR+ T cells and NoDNA T cells (mock-transduced cells) labeled with carboxyfluorescein succinimidyl ester (Thermo Fisher Scientific; cat. no. C1157) were cultured at a concentration of 1 × 106 cells/ml and incubated with yeast (1 × 105 cells/ml) at 37°C. After 24 h, the interaction between GXMR-CAR+ T cells (green) or NoDNA T cells (green) with C. neoformans yeast (blue) was viewed under a fluorescence microscope (Leica DMI6000B) at 100× magnification.
Secretion of granzyme and IFN-γ by GXMR-CAR T cells incubated with soluble GXM
GXMR-CAR+ T cells (0.5 × 106/well) were incubated with GXM (C. neoformans polysaccharide antigen; ZeptoMetrix) at a dilution of 1:100. ImmunoCult human CD3/CD28 T-cell activator (STEMCELL Technologies; cat. no. 10971) was used according to manufacturer’s instructions to activate positive control T cells and negative controls were incubated with medium alone. Golgi-stop was added according to manufacturer’s instructions (Cat#554715), and after 3 h the GXMR-CAR+ T cells were fixed/permeabilized to perform the detection of granzyme B antibody (cat#561016, BD Biosciences) and IFN-γ (anti- IFN-γ antibody; Cat#51-20685z). The cells were analyzed by flow cytometry.
Detection of Cryptococcus spp. in lung tissue by GMS staining
Hematoxylin and eosin (H&E) staining and Gomori methenamine-silver (GMS) staining were performed with use of 5-μm thick lung tissue sections collected from PBS, GXMR-CAR+ T cell or NoDNA T cell treated groups. H&E staining was performed by the Research Histology Core Lab at MD Anderson. Brightfield images of H&E-stained slides were captured on a Nikon Ti Eclipse. GMS staining was performed by the Histology Core Lab at the Department of Pediatrics at MD Anderson. The GMS slides were scanned with Hamamatsu’s Nanozoomer at 40×, and images were analyzed by using NDP.view software to measure the number, perimeter, and area of C. neoformans yeast. This analysis categorized the C. neoformans cells by size, ranging from 4 to 50 μm, 51 to 90 μm, and <91 μm to compare all parameters between groups; Pearson’s chi-square test was used to determine whether there were statistically significant differences between the expected frequencies and the observed frequencies in one or more categories of a contingency table based on the perimeter data [39].
Cytokine profiling
Plasma was isolated from mouse blood and stored at −20°C. Plasma samples were thawed on ice and diluted 1:4 in buffer; duplicates were analyzed by using the Bio-Plex Pro Human Th17 Cytokine 15plex Assay and Bio-Plex 200 (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions.
Results
Construction of GXMR-CAR to target Cryptococcus spp.
CAR+ T cells that recognize tumor antigen undergo activation in a non–MHC-restricted manner that triggers an immune response against cancer cells. We adopted this clinically translatable approach to generate CAR+ T cells that target Cryptococcus spp., which causes morbidity and mortality in immunocompromised (C. neoformans) and immunocompetent (C. gattii) individuals. GXM is the major virulence factor in the Cryptococcus spp. capsule and protects the yeast from host immune cell attack. The light and heavy chains in the Fab domain of the murine monoclonal antibody 18B7 [34] were selected to generate the scFv domain of the GXMR-CAR that specifically recognizes GXM present in the Cryptococcus spp. capsule.
To generate the GXMR-CAR construct, the DNA sequence of the GXM-specific scFv (scFv-18B7, targeting domain) was fused to modified Fc regions of the human IgG4 (hinge domain), along with the transmembrane and co-stimulatory domains of CD28, and the signaling domain of CD3-ς (Fig. 1A) [18]. HEK-293FT cells were used to make GXMR-CAR+ viral particles by transfecting them with three plasmids: pMD2.G for the envelope, psPAX2 for packaging, and GXMR-CAR, according to the manufacturer’s instructions as described in the Materials and Methods section. The success of transfection was determined by analyzing GFP expression after 24 h (Fig. 1B) at 100× and 200× magnification.
After the viral particles containing GXMR-CAR were generated, HEK-293FT cells were infected with the particles to evaluate the GXMR-CAR transduction efficiency. After 3 days in culture, the GFP-positive cells were identified with use of fluorescence microscopy (Fig. 1C, right), and the GFP expression in transduced cells was quantified by using flow cytometry (Fig. 1C, left). A high efficiency of transduction of HEK-293FT cells with GXMR-CAR+ viral particles was observed, as 67% of the cells were GFP+ (Fig. 1C).
GXMR-CAR expression on human T cells
After GXMR-CAR was successfully expressed in HEK-293FT cells, the same protocol was used for the transduction of human T cells. Initially, PBMCs were stimulated with an antibody cocktail (anti-CD3/CD28) combined with IL-2 for 3 days. Activated human T cells were transduced with GXMR-CAR+ viral particles; 3 days post-transduction, GXMR-CAR+ T cells were enriched by sorting based on GFP expression, and expansion of GXMR-CAR+ T cells was performed as described in the Materials and Methods section. After 3 days of expansion, 82.4% of the human GXMR-CAR+ T cells expressed GFP, as determined by flow cytometry (Fig. 2A). To demonstrate expression of GXMR-CAR on the surface of T cells, an anti-human Fc antibody (Fc-γ fragment-specific) was used to bind to the spacer region of IgG4 (shown in Fig. 1A) localized on their surface. The results show that 77.8% of the GXMR-CAR+ T cells were positive for the antibody (Fig. 2B), confirming the expression of GXMR-CAR on the T-cell surface. Expanded GXMR-CAR+ T cells were evaluated by flow cytometry to determine the memory phenotypes. Naive/stem cell like memory, central memory, and effector memory T-cell subsets, which are critical for long-term immune memory, were prevalent in GXMR-CAR+ T cells (Fig. 2C, 2D).
Fig. 2. Expression of GXMR-CAR on the surface of human T cells.

(A) Flow cytometry plots show anti-CD3 antibody-positive T cells on the y-axis and GXMR-CAR-GFP–expressing T cells on the x-axis. The cells double-positive for the antibody and GFP (82.4%) are GXMR- CAR+ GFP-expressing T cells. (B) Flow cytometry plots show T cells positive for an anti- human Fc antibody on the y-axis and GXMR-CAR-GFP–expressing cells on the x-axis. The cells double-positive for the antibody and CAR-GFP (77.8%) were considered GXMR-CAR+ T cells. Mock-transduced negative control T cells (NoDNA) did not receive the GXMR-CAR construct. Phenotypic analysis of GXMR-CAR+ T cells (C, D): This analysis of GXMR- CAR+ T cells stimulated by using an ImmunoCult human CD3/CD28 T-cell activator was performed after 30 days of in vitro expansion of the cells. (C) PBMCs and (D) GXMR-CAR+ T cells were stained with a mixture of anti-CD3-PE, anti-CD8-PerCP, anti-CCR7-APC, and anti-CD45RA-PE-Vio770 antibodies, and CAR+ T cells were evaluated according to GFP expression. CD3+CD8+ T cells were gated to evaluate the memory cell subsets using anti-CCR7 memory T cells; TEM, effector memory T cells; TEFF, effector T cells; TSCM, stem cell memory
GXMR-CAR+ T cells interact with soluble GXM from the Cryptococcus spp. capsule and heat killed Cryptococcus spp. yeast
Since GXMR-CAR+ T cells were designed to target GXM, experiments were conducted to investigate whether GXMR-CAR+ T cells bind to C. neoformans yeast and to GXM. First, the presence of GXM on the C. neoformans capsule was evaluated in both live and heat-killed yeast. The heat-killed and live yeast forms were incubated with anti-GXM antibody (18B7 clone), followed by detection with a secondary antibody conjugated to FITC. The FITC labeled yeast cells were analyzed by flow cytometry and the mean fluorescence intensity was similar between live and heat-killed yeast (Fig. S1). These results suggests that heat-killed C. neoformans yeast has a GXM capsule on its surface. To determine whether GXMR-CAR T cells target heat-killed C. neoformans yeast, heat-killed C. neoformans yeast was stained with Calcofluor-white (blue) and incubated with GXMR-CAR+ T cells (green) or NoDNA T cells (negative control; green), and their interaction was visualized by using fluorescence microscopy. The results showed that GXMR-CAR+ T cells co-localized with C. neoformans yeast (Fig. 3A, right), whereas the NoDNA T cells did not interact with the yeast (Fig. 3A, left). Moreover, in GXMR-CAR+ T cell–treated wells C. neoformans yeast was trapped inside T-cell clusters, whereas in wells with NoDNA T cells, the cells did not interact with the yeast, and no cell clusters were observed. These results showed that GXMR-CAR+ T cells can recognize Cryptococcus spp. yeast and form complexes with it. To corroborate these findings, additional experiments were conducted to determine the specificity of GXMR-CAR+ T cells for GXM. Polysaccharide antigens from the capsule of Cryptococcus spp. were incubated with GXMR-CAR+ T cells, and an anti-GXM mAb 18B7 was added to detect GXM on the surface of GXMR-CAR+ T cells by using flow cytometry (Fig. 3B, right). The recognition of soluble GXM by GXMR-CAR+ T cells was demonstrated by cells that were double-positive for CAR-GFP and the anti-GXM mAb 18B7 clone (Fig. 3B), whereas NoDNA T cells did not recognize GXM. In addition, after 24 h of incubation with polysaccharide antigens from Cryptococcus spp., cell clusters were observed by bright-field microscopy in wells treated with GXMR-CAR T cells, whereas no cell clusters were detected upon incubation of NoDNA T cells (mock-transduced cells) with the polysaccharide antigen (Fig. 4A). Similar types of cell clusters were observed in the anti-CD3/CD28 antibody–treated positive control, whereas no cell clusters were observed in the medium-alone negative control wells (Fig. 4A). These findings confirmed that the GXMR-CAR–expressing T cells targeted GXM, a major compound in the Cryptococcus spp. capsule.
Fig 3. Recognition of GXM from Cryptococcus spp. by GXMR-CAR+ T cells.

(A) GXMR- CAR+ T cells and NoDNA T cells (mock-transduced, negative controls) were labeled with carboxyfluorescein succinimidyl ester and incubated with C. neoformans yeast labeled with Calcofluor-white. The interaction of GXMR-CAR+ T cells (green) and NoDNA T cells (green) with C. neoformans yeast (blue) was evaluated by using fluorescence microscopy. The bottom right image shows the cell clusters formed by co-localization of GXMR-CAR+ T cells and C. neoformans yeast. NoDNA T cells (top right) did not display co-localization. (B) GXMR- CAR+ and NoDNA T cells were assayed with polysaccharide antigens from the capsule of Cryptococcus spp., and the interaction of GXM with the cell surface was evaluated via flow cytometry by using an anti-GXM mAb 18B7 clone stained with a phycoerythrin-conjugated secondary antibody. The cells double-positive for 18B7 and GXMR-CAR-GFP demonstrated the interaction between GXMR-CAR+ T cells and GXM from Cryptococcus spp. The NoDNA T cells and GXMR-CAR+ T cells not incubated with polysaccharide antigens from the capsule of Cryptococcus spp. (without GXM) were used to demonstrate the absence of nonspecific binding of 18B7 to cells
Fig 4. Activation of GXMR-CAR+ T cells after recognition of GXM from Cryptococcus spp..
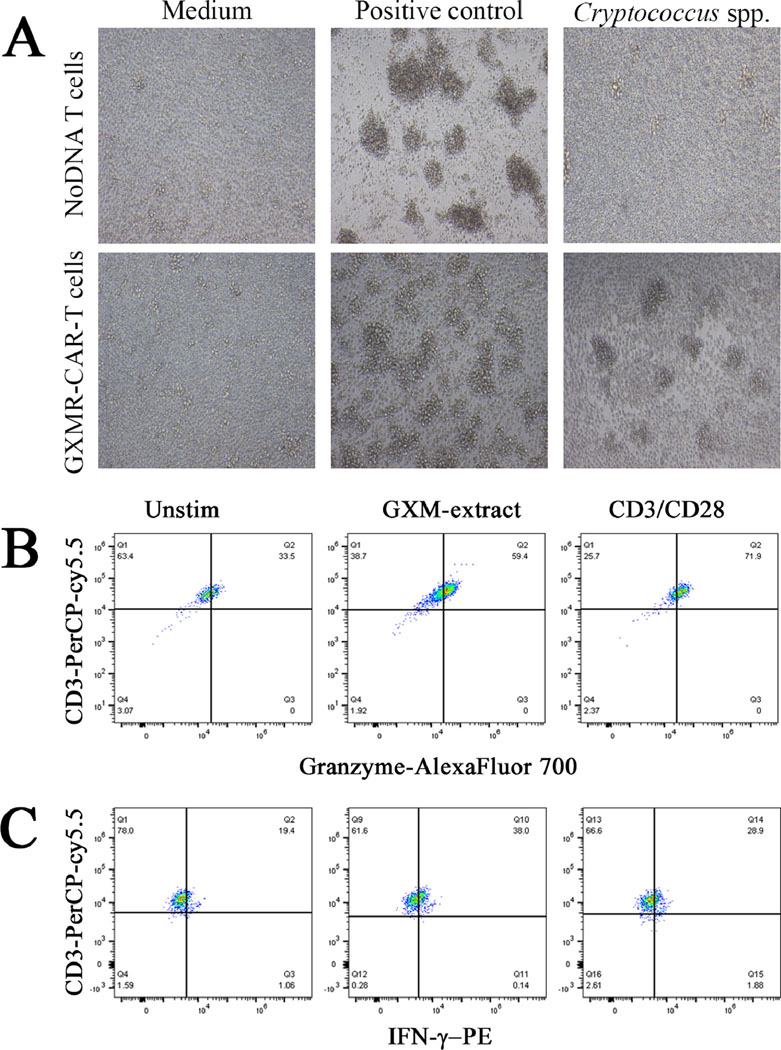
(A) GXMR-CAR+ and NoDNA T cells were incubated with a preparation of polysaccharide antigens from the capsule of Cryptococcus spp. at a 1:100 dilution. After 24 h, the images were acquired by bright-field microscopy at 400x magnification. Positive control T cells were activated by using an ImmunoCult human CD3/CD28 T-cell activator (STEMCELL Technologies) according to the manufacturer’s instructions. T cells incubated with medium alone were used as negative control cells. (B and C) GXMR-CAR+ T cells were incubated with soluble GXM from C. neoformans at a dilution of 1:100 and Golgi-stop was added as recommended by manufacturer’s instructions. After 3 h the GXMR-CAR+ T cells were used for detection of granzyme (B) and IFN-γ (C) by flow cytometry. An ImmunoCult human CD3/CD28 T-cell activator (anti-CD3/CD28 stimulus) was used as positive control and medium alone were considered as negative controls.
GXMR-CAR+ T cells can be activated by soluble GXM from Cryptococcus spp. capsule
To evaluate whether GXMR-CAR+ receptor induces cell signaling upon recognition of GXM polysaccharide from Cryptococcus spp., GXMR-CAR+ T cells were incubated with a preparation of polysaccharide antigens from the capsule of Cryptococcus spp. In this context, GXMR-CAR+ T cells were incubated with soluble GXM for 3 h and granzyme and perforin levels in the cytoplasm were measured by flow cytometry using specific antibodies. This assay demonstrated that GXMR-CAR+ T cells showed increased amounts of granzyme after exposure to soluble GXM, compared to GXMR-CAR+ T cells in the absence of GXM (Figure 4B). Moreover, the recognition of soluble GXM by GXMR-CAR+ T cells resulted in an increased percentage of IFN-γ positive cells in comparison to unstimulated GXMR-CAR+ T cells (Figure 4C). These results suggest that GXMR-CAR+ T cells interact with polysaccharide antigens in the capsule of Cryptococcus spp., promoting the clustering and activation of GXMR-CAR+ T cells.
Targeting efficacy of GXMR-CAR+ T cells in a pulmonary Cryptococcus infection model
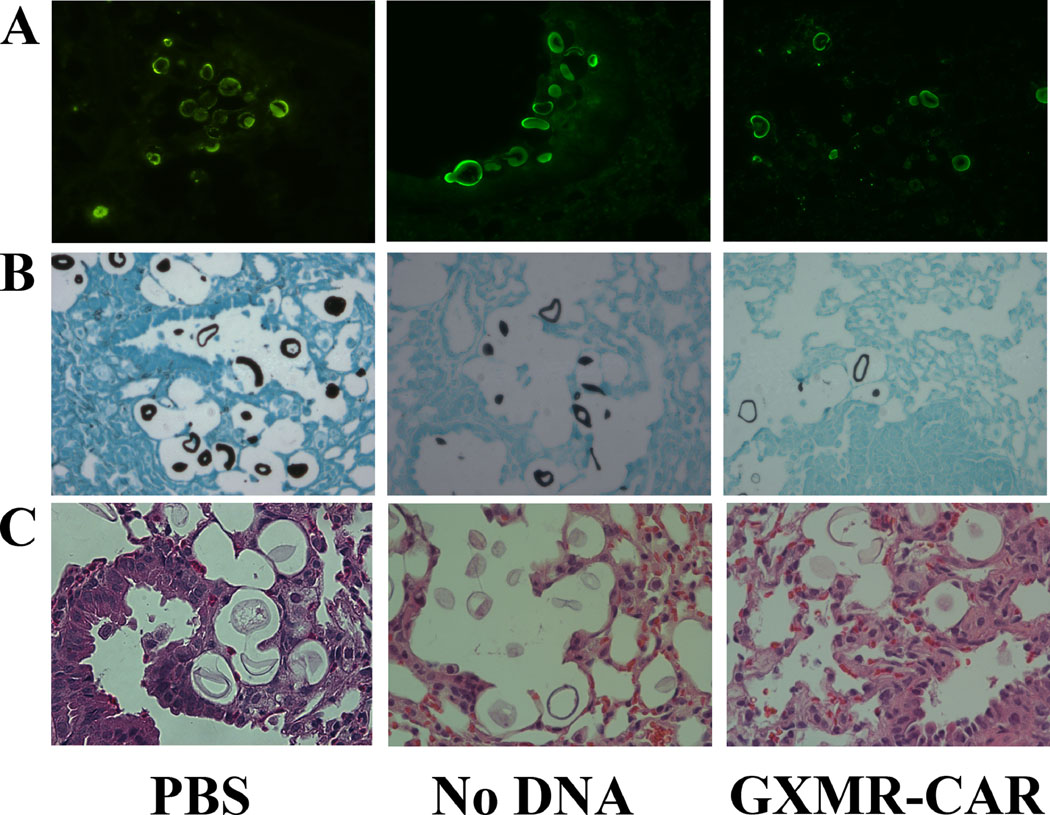
NSG mice were used for these studies because they are deficient in mature B, T, and functional NK cells. Therefore, they are unable to target and destroy infused GXMR-CAR+ T cells. Pulmonary fungal infection was established in the mice by infecting their lungs with C. neoformans yeast (1 × 105/mouse) via intranasal infusion [40]. The mice were placed into 3 groups according to the infusions they received: GXMR-CAR+ T cells, NoDNA T cells, and PBS. The mice were infused with 5 million GXMR-CAR+ T cells, NoDNA T cells, or PBS alone via tail vein injection on the first and fourth day after infection. Eight days after infection, the mice were humanely euthanized, and entire sections of lung tissue were evaluated for fungal burden by GMS. GMS staining was used to measure perimeter (length) of the yeast categorized in distinct cell types (yeast, titan, and giant cells) according to size: <50 μm or >51 μm [41, 42]. There were no significant differences in length among the distinct cell types of C. neoformans yeasts in the treated and untreated groups (Fig. 5A, 5B). However, Pearson’s chi- square test analysis demonstrated that there are significant differences in the perimeter size distribution of cell types of C. neoformans, such as yeast and titan cells, among the 3 groups (Fig. 5C). The C. neoformans cells range in size from 51 to 130 μm, and those >130 μm are indicative of titanization, which is critical for pathogenicity. Interestingly, the GXMR-CAR+ T-cell group had significantly lower numbers of both cell sizes compared with other groups (Fig. 5C). Nanozoomer scanning analysis of GMS stained lung sections revealed that GXMR- CAR T cell target C. neoformans in the lungs, resulting in decreased titanization compared to the NoDNA and PBS groups. Elevated levels of plasma IFN-γ was observed in GXMR-CAR T cell treated group when compared to NoDNA T cell treated group (Fig. 5D). In addition, more mononuclear cells were observed in GXMR-CAR T cells treated group than in the NoDNA-treated group (Fig. 6C). Corroborating our in vitro results, these data demonstrate that intravenously infused GXMR-CAR+ T cells directly target C. neoformans and may control its spread in the lungs of infected mice.
Fig 5. Measuring Cryptococcus perimeter size and distribution of cell type.
Nanozoomer software was used to measure the Cryptococcus yeast size from GMS-stained slides and categorized into 3 cell types (yeast): <50 μm, ≥51 to <90 μm (titan cell), and >91 μm (giant cell) in the PBS, NoDNA, and GXMR-CAR T cell–treated groups. (A) The perimeter size <50 μm was plotted in the upper panel and >50 μm in the lower panel. (B) Frequency distributions of cell types categorized into <50 μm; >51 μm to >90 μm, and >91 μm in PBS-treated, NoDNA T cell–treated, and GXMR-CAR T cell–treated groups are shown in (C). Distribution co- efficient shows significant differences between the cell types in the GXMR-CAR T cell–treated group; giant cells were not observed in the group. (D) Plasma of mice infected with C. neoformans and treated with PBS, NoDNA cells or GXMR-CAR T cells were obtained after infection and the levels of IFN-γ were measured using a Bio-Plex Human Cytokine Assay (Bio- Rad, Hercules, CA).
Fig. 6. GXMR-CAR T cell recruitment at the Cryptococcus infection site.

Immunofluorescent GXM-staining (A), Gomori methenamine-silver (GMS) staining (B), and Hematoxylin and eosin (H&E) staining (C) were performed with use of 5-μm thick lung tissue sections collected from PBS, GXMR-CAR+ T cell or NoDNA T cell treated groups, as described in the Materials and Methods section.
Discussion
CAR T-cell therapy has been successfully used to treat hematological malignancies, with an overall success rate of >80% [43]. In 2017, the U.S. Food and Drug Administration approved two CD19-targeting CARs (tisagenlecleucel [Kymriah] and axicabtagene ciloleucel [Yescarta]) for treatment of chronic lymphoblastic leukemia [27], pediatric relapsed or refractory acute lymphoblastic leukemia, and relapsed or refractory large B-cell lymphoma [43]. In addition, CAR T cells targeting HIV have been designed to reduce the viral burden [44]. The redirection of T cells by a CAR targeting a carbohydrate expressed on fungal cell walls was first reported by Kumaresan et al. [18]. In the present study, we developed CAR+ T cells targeting the yeast virulence factor GXM, a carbohydrate present in the Cryptococcus spp. capsule. To generate GXMR-CAR+ T cells, the extracellular domain of GXMR-CAR+ T cells was derived from the murine mAb 18B7, which binds to GXM and has a high affinity for Cryptococcus spp. In a phase I dose escalation trial, researchers tested treatment of cryptococcal meningitis in HIV+ patients with 18B7 [34, 35]. Transient decreases in serum cryptococcal antigen levels were observed in patients receiving 18B7 doses of 1–2 mg/kg, and doses up to 1 mg/kg were well tolerated. However, the use of CAR T-cell therapy for treating cryptococcosis had not been explored.
We designed a second-generation GXMR-CAR with a CD28 costimulatory domain and a CD3γ activation domain. The LV vector that was used for genomic insertion of CAR constructs was selected because (1) it has been used in clinical trials, (2) large quantities of CAR T cells can be produced within 7–10 days of transduction, and (3) more memory cells are produced (TCM and TEM subtypes) compared with non-viral methods of CAR T-cell production.
GXMR-CAR T cells were able to bind to GXM after incubation with C. neoformans polysaccharide antigens and to recognize C. neoformans yeast in both in vitro and in vivo studies. Image scanning analysis revealed that the perimeters of the titan cells were greatly reduced in the GXMR-CAR T cell–treated group compared with perimeters in the PBS and NoDNA T cell–treated groups. Titan cells are a major contributing factor for the virulence and pathogenesis of the Cryptococcus-infected host [41, 42]. We hypothesize that the destruction of titan cells would substantially improve cryptococcosis treatment. Our findings demonstrated that the scFv domain of the GXMR-CAR+ T cells retained its targeting potential against the GXM present in the capsule of C. neoformans.
The currently available antifungal drugs used to combat cryptococcosis consist of a combination of amphotericin B deoxycholate or liposomal amphotericin B and 5- fluorocytosine administered for 2 weeks. Fluconazole is recommended for use during the consolidation and maintenance phases, but this antifungal drug requires a long treatment course, which reduces patient compliance and/or tolerability [45, 46]. The rising number of antibiotic-resistant strains of fungi, such as C. gattii [47] and Candida auris [48], poses a significant threat to health. From 2004 to 2007, there were 83 cases of C. gattii infection in Washington and Oregon, with a mortality rate of 33%. Antibiotic resistance may have contributed to the poor response rate to antibiotic therapy in these cases [47]. To treat these deadly diseases, other treatment options such as fungal immunotherapy are needed. To improve the currently available treatment options for cryptococcosis, combining immunomodulators with existing antifungal drugs may be beneficial. Along these lines, researchers evaluated the effects of administering adjuvant recombinant IFN-γ in 75 HIV+ patients with acute cryptococcal meningitis, which led to more rapid sterilization of cerebrospinal fluid in 36% of the treated group compared with 13% of the placebo controls. Moreover, the addition of recombinant IFN-γ to amphotericin B in HIV+ patients with cryptococcal meningitis caused a significant reduction in Cryptococcus spp. in cerebrospinal fluid [49]. Recently, a research group used a vaccination strategy to control cryptococcosis by using glucan particles as a delivery system to carry protective protein antigens from C. neoformans or C. gattii. This approach induced CD4+ T-cell responses in the lungs of vaccinated mice, providing partial protection after challenge with C. neoformans or C. gattii [50]. These studies demonstrated that polarization of adaptive immune cells is critical to an appropriate immune response against cryptococcosis.
Another immunomodulatory treatment strategy is adoptive T-cell therapy, which can induce cell-mediated immunity against invasive fungal infections. Like CD4+ T cells, CD8+ T cells differentiate into Tc1 and Tc17 cells to recruit innate immune cells to fight fungal infections [11, 51]. Tc1 and Tc17 cells also activate B cells, resulting in the secretion of antigen-specific antibodies against fungi [12]. The major mechanism that CD8+ T cells use to control fungal infections is the targeted release of cytotoxic factors that act directly on fungi, such as perforin, granulysin, and granzyme [12, 17]. Similar mechanisms have also been shown in NK cell–mediated fungal yeast killing. NKp30, a NK cell receptor, was shown to directly recognize beta-glucan present in the Cryptococcus spp. cell wall and to activate NK cells to release cytolytic granules, such as granzyme and perforin, to lyse Cryptococcus yeast [15, 16]. The antifungal activity of adoptive T-cell therapy can be improved by redirecting T-cell specificity with use of a CAR that recognizes specific fungal antigens. In this context, Kumaresan et al. [18] demonstrated that engineered CAR+ T cells containing an extracellular domain of the carbohydrate recognition domain of Dectin-1 (D-CAR–expressing T cells) were capable of targeting β-glucan–expressing fungi. In vitro and in vivo studies showed that D- CAR–expressing T cells target germinating Aspergillus hyphae and inhibit aspergillosis dissemination by using the cytolytic machinery of the genetically modified T cells [18].
These findings led us to develop CAR+ T cells expressing an scFv from the mAb 18B7 to target Cryptococcus spp. that contain a polysaccharide capsule predominantly made up of GXM [52]. Because the compounds in the Cryptococcus spp. capsule impair recognition of pathogen-associated molecular patterns by pattern-recognition receptors, the β-glucans, mannoproteins, and chitin are poorly recognized by the immune system. GXMR-CAR expressed on human T cells recognizes the soluble form of GXM and targets C. neoformans yeast, demonstrating the specificity of GXMR-CAR to this pathogen. The major fungi responsible for invasive fungal infections, Candida and Aspergillus species, lack GXM and have different polysaccharide compositions in the outer cell wall [29], whereas GXMR-CAR specifically targets only fungi that have GXM in the outer cell wall. Our in vivo studies demonstrated that GXMR-CAR+ T cells efficiently reduced the size and number of titan cells in the lungs of mice infected with C. neoformans compared with the size and number of these cells in the negative control group, as demonstrated by GMS staining and GXM- immunofluorescence (Fig. 4 and Fig. S2, respectively). In addition, C. neoformans mitogen can directly activate CD8+ T cells to secrete cytolytic granules, especially granulysin, to kill Cryptococcus spp. [12–14]. When the fungal burden is high, scavenger receptors such as CD5 expressed on T-cell surfaces can modulate T-cell function to reduce the effect of fungal sepsis [15, 16, 53]. In this context, mock-transduced negative control T cells (NoDNA) in the current work, a small population of T cells may recognize an antigen present on the capsule of C. neoformans in the lungs [14, 54, 55]. In spite of that, the size of the titan cells was greatly reduced in the GXMR-CAR+ T cell–treated group, suggesting that GXMR-CAR+ T cells can directly control GXM–mediated virulence in cryptococcosis (Fig. 4).
GXMR-CAR+ T cells may reduce the severity of Cryptococcus spp. infection in the lungs (Fig. 6) through the following mechanisms: 1) production of proinflammatory cytokines such as IFN-γ, which augments the response of antigen-presenting cells (APCs) and neutrophils against Cryptococcus spp.; 2) release of granzyme, granulysin, and perforin by GXMR-CAR+ T cells, which may cause degradation of Cryptococcus spp. cell walls, and 3) reduction of GXM-induced virulence. Our new approach to targeting Cryptococcus spp. with CAR+ T cells using an anti-GXM mAb may be extended to other invasive fungal infections that use monoclonal antibodies specific to other cell wall proteins.
Cytokine release syndrome and macrophage activating syndrome are the major limiting factors for CAR cell–mediated immunotherapy. Clinicians have developed strategies to address cytokine release syndrome, such as treatment with tocilizumab, an antibody to IL6R that blocks binding to the IL-6 receptor, which has been successfully used in clinics. At present we are not sure whether any off-target toxicities are associated with GXMR-CAR T cell therapy. One of the challenges for GXMR-CAR T cells to overcome is the potential unwanted targeting of soluble GXM that is shed into the blood and cerebrospinal fluid during Cryptococcus spp. infection [56]. One way to overcome this unwanted targeting of soluble GXM is by treating patients with anti-GXM antibodies to reduce the GXM in the circulation before infusing with GXMR-CAR T cells [35].
Since immunodeficient mouse models like NSG have been used to evaluate targeting efficacy of CAR T cells in cancer models, we adapted this model to evaluate the functional efficacy of GXMR-CAR T cells. Future studies will be conducted using a murine version of GXMR-CAR T cells to study the efficacy in immunocompetent mouse models to provide a wealth of information about the therapeutic efficacy of GXMR-CAR T cells in participation with the host innate immune system.
Supplementary Material
Fig. 7. Approach to targeting C. neoformans with CAR+ T cells.

Schematic shows that T cells engineered to express GXMR-CAR via lentiviral transduction can target C. neoformans. In vitro and in vivo studies demonstrated that GXMR-CAR+ T cells interact with C. neoformans yeast and that GXMR-CAR+ T cells infused in NSG mice infected with C. neoformans promoted a reduction in the number of titan cells in the lungs. We suggest that GXMR-CAR+ T cells use two major mechanisms to control cryptococcosis: 1) activation of neutrophils and APCs by proinflammatory mediators released by GXMR-CAR+ T cells and 2) production of granzyme, perforin, and granulysin by GXMR-CAR+ T cells acting on the C. neoformans.
Acknowledgments
This research was financially supported in part by the São Paulo Research Foundation, Brazil (grant numbers 2016/23044-1, 2016/04877-2, and 2018/18538-0). Additional funding was provided by the Department of Pediatrics Research, MD Anderson, and by the National Institute of Allergy and Infectious Disease (R21 grant AI127381-02).
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Harris J, Lockhart S, Chiller T, Cryptococcus gattii: where do we go from here?, Med Mycol 50(2) (2012) 113–29. [DOI] [PubMed] [Google Scholar]
- [2].Skolnik K, Huston S, Mody CH, Cryptococcal Lung Infections, Clin Chest Med 38(3) (2017) 451–464. [DOI] [PubMed] [Google Scholar]
- [3].Rajasingham R, Smith RM, Park BJ, Jarvis JN, Govender NP, Chiller TM, Denning DW, Loyse A, Boulware DR, Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis, Lancet Infect Dis 17(8) (2017) 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lindell DM, Ballinger MN, McDonald RA, Toews GB, Huffnagle GB, Diversity of the T-cell response to pulmonary Cryptococcus neoformans infection, Infect Immun 74(8) (2006) 4538–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Murdock BJ, Huffnagle GB, Olszewski MA, Osterholzer JJ, Interleukin-17A enhances host defense against cryptococcal lung infection through effects mediated by leukocyte recruitment, activation, and gamma interferon production, Infect Immun 82(3) (2014) 937–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zaragoza O, Alvarez M, Telzak A, Rivera J, Casadevall A, The relative susceptibility of mouse strains to pulmonary Cryptococcus neoformans infection is associated with pleiotropic differences in the immune response, Infect Immun 75(6) (2007) 2729–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hoag KA, Lipscomb MF, Izzo AA, Street NE, IL-12 and IFN-gamma are required for initiating the protective Th1 response to pulmonary cryptococcosis in resistant C.B-17 mice, Am J Respir Cell Mol Biol 17(6) (1997) 733–9. [DOI] [PubMed] [Google Scholar]
- [8].Chen GH, McDonald RA, Wells JC, Huffnagle GB, Lukacs NW, Toews GB, The gamma interferon receptor is required for the protective pulmonary inflammatory response to Cryptococcus neoformans, Infect Immun 73(3) (2005) 1788–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Coelho C, Bocca AL, Casadevall A, The intracellular life of Cryptococcus neoformans, Annu Rev Pathol 9 (2014) 219–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Angkasekwinai P, Sringkarin N, Supasorn O, Fungkrajai M, Wang YH, Chayakulkeeree M, Ngamskulrungroj P, Angkasekwinai N, Pattanapanyasat K, Cryptococcus gattii infection dampens Th1 and Th17 responses by attenuating dendritic cell function and pulmonary chemokine expression in the immunocompetent hosts, Infect Immun 82(9) (2014) 3880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wuthrich M, Deepe GS Jr., Klein B, Adaptive immunity to fungi, Annu Rev Immunol 30 (2012) 115–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Oykhman P, Mody CH, Direct microbicidal activity of cytotoxic T-lymphocytes, J Biomed Biotechnol 2010 (2010) 249482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Levitz SM, Dupont MP, Smail EH, Direct activity of human T lymphocytes and natural killer cells against Cryptococcus neoformans, Infect Immun 62(1) (1994) 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mody CH, Wood CJ, Syme RM, Spurrell JC, The cell wall and membrane of Cryptococcus neoformans possess a mitogen for human T lymphocytes, Infect Immun 67(2) (1999) 936–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li SS, Ogbomo H, Mansour MK, Xiang RF, Szabo L, Munro F, Mukherjee P, Mariuzza RA, Amrein M, Vyas JM, Robbins SM, Mody CH, Identification of the fungal ligand triggering cytotoxic PRR-mediated NK cell killing of Cryptococcus and Candida, Nat Commun 9(1) (2018) 751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Qiu Y, Dayrit JK, Davis MJ, Carolan JF, Osterholzer JJ, Curtis JL, Olszewski MA, Scavenger receptor A modulates the immune response to pulmonary Cryptococcus neoformans infection, J Immunol 191(1) (2013) 238–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kumaresan PR, da Silva TA, Kontoyiannis DP, Methods of Controlling Invasive Fungal Infections Using CD8(+) T Cells, Front Immunol 8 (2018) 1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kumaresan PR, Manuri PR, Albert ND, Maiti S, Singh H, Mi T, Roszik J, Rabinovich B, Olivares S, Krishnamurthy J, Zhang L, Najjar AM, Huls MH, Lee DA, Champlin RE, Kontoyiannis DP, Cooper LJ, Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection, Proc Natl Acad Sci U S A 111(29) (2014) 10660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dotti G, Gottschalk S, Savoldo B, Brenner MK, Design and development of therapies using chimeric antigen receptor-expressing T cells, Immunol Rev 257(1) (2014) 107–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sadelain M, Brentjens R, Riviere I, The basic principles of chimeric antigen receptor design, Cancer Discov 3(4) (2013) 388–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gill S, Maus MV, Porter DL, Chimeric antigen receptor T cell therapy: 25years in the making, Blood Rev 30(3) (2016) 157–67. [DOI] [PubMed] [Google Scholar]
- [22].Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, Smith DD, Forman SJ, Jensen MC, Cooper LJ, CD28 costimulation provided through a CD19- specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells, Cancer Res 66(22) (2006) 10995–1004. [DOI] [PubMed] [Google Scholar]
- [23].Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M, Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor, Nat Biotechnol 20(1) (2002) 70–5. [DOI] [PubMed] [Google Scholar]
- [24].Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, Steinberg SM, Stroncek D, Tschernia N, Yuan C, Zhang H, Zhang L, Rosenberg SA, Wayne AS, Mackall CL, T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial, Lancet 385(9967) (2015) 517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA, Chimeric antigen receptor T cells for sustained remissions in leukemia, N Engl J Med 371(16) (2014) 1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, Yang JC, Kammula US, Devillier L, Carpenter R, Nathan DA, Morgan RA, Laurencot C, Rosenberg SA, B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells, Blood 119(12) (2012) 2709–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Porter DL, Levine BL, Kalos M, Bagg A, June CH, Chimeric antigen receptor- modified T cells in chronic lymphoid leukemia, N Engl J Med 365(8) (2011) 725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, Olszewska M, Bernal Y, Pegram H, Przybylowski M, Hollyman D, Usachenko Y, Pirraglia D, Hosey J, Santos E, Halton E, Maslak P, Scheinberg D, Jurcic J, Heaney M, Heller G, Frattini M, Sadelain M, Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias, Blood 118(18) (2011) 4817–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Snarr BD, Qureshi ST, Sheppard DC, Immune Recognition of Fungal Polysaccharides, J Fungi (Basel) 3(3) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ma H, Hagen F, Stekel DJ, Johnston SA, Sionov E, Falk R, Polacheck I, Boekhout T, May RC, The fatal fungal outbreak on Vancouver Island is characterized by enhanced intracellular parasitism driven by mitochondrial regulation, Proc Natl Acad Sci U S A 106(31) (2009) 12980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ma H, May RC, Virulence in Cryptococcus species, Adv Appl Microbiol 67 (2009) 131–90. [DOI] [PubMed] [Google Scholar]
- [32].Cherniak R, Sundstrom JB, Polysaccharide antigens of the capsule of Cryptococcus neoformans, Infect Immun 62(5) (1994) 1507–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yang CL, Wang J, Zou LL, Innate immune evasion strategies against Cryptococcal meningitis caused by Cryptococcus neoformans, Exp Ther Med 14(6) (2017) 5243–5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Casadevall A, Cleare W, Feldmesser M, Glatman-Freedman A, Goldman DL, Kozel TR, Lendvai N, Mukherjee J, Pirofski LA, Rivera J, Rosas AL, Scharff MD, Valadon P, Westin K, Zhong Z, Characterization of a murine monoclonal antibody to Cryptococcus neoformans polysaccharide that is a candidate for human therapeutic studies, Antimicrob Agents Chemother 42(6) (1998) 1437–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Larsen RA, Pappas PG, Perfect J, Aberg JA, Casadevall A, Cloud GA, James R, Filler S, Dismukes WE, Phase I evaluation of the safety and pharmacokinetics of murine- derived anticryptococcal antibody 18B7 in subjects with treated cryptococcal meningitis, Antimicrob Agents Chemother 49(3) (2005) 952–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hommel B, Sturny-Leclere A, Volant S, Veluppillai N, Duchateau M, Yu CH, Hourdel V, Varet H, Matondo M, Perfect JR, Casadevall A, Dromer F, Alanio A, Cryptococcus neoformans resists to drastic conditions by switching to viable but non- culturable cell phenotype, PLoS Pathog 15(7) (2019) e1007945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Oliveira-Brito PKM, Rezende CP, Almeida F, Roque-Barreira MC, da Silva TA, iNOS/Arginase-1 expression in the pulmonary tissue over time during Cryptococcus gattii infection, Innate Immun 26(2) (2019) 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cooper LJ, Topp MS, Serrano LM, Gonzalez S, Chang WC, Naranjo A, Wright C, L. 633 Popplewell, A. Raubitschek, S.J. Forman, M.C. Jensen, T-cell clones can be rendered 634 specific for CD19: toward the selective augmentation of the graft-versus-B-lineage 635 leukemia effect, Blood 101(4) (2003) 1637–44. [DOI] [PubMed] [Google Scholar]
- [39].R.C. Team, A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria, 2018. [Google Scholar]
- [40].Almeida F, Wolf JM, da Silva TA, DeLeon-Rodriguez CM, Rezende CP, Pessoni AM, Fernandes FF, Silva-Rocha R, Martinez R, Rodrigues ML, Roque-Barreira MC, Casadevall A, Galectin-3 impacts Cryptococcus neoformans infection through direct antifungal effects, Nat Commun 8(1) (2017) 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zaragoza O, Garcia-Rodas R, Nosanchuk JD, Cuenca-Estrella M, Rodriguez- Tudela JL, Casadevall A, Fungal cell gigantism during mammalian infection, PLoS Pathog 6(6) (2010) e1000945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hommel B, Mukaremera L, Cordero RJB, Coelho C, Desjardins CA, Sturny- Leclere A, Janbon G, Perfect JR, Fraser JA, Casadevall A, Cuomo CA, Dromer F, Nielsen K, Alanio A, Titan cells formation in Cryptococcus neoformans is finely tuned by environmental conditions and modulated by positive and negative genetic regulators, PLoS Pathog 14(5) (2018) e1006982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Boyiadzis MM, Dhodapkar MV, Brentjens RJ, Kochenderfer JN, Neelapu SS, Maus MV, Porter DL, Maloney DG, Grupp SA, Mackall CL, June CH, Bishop MR, Chimeric antigen receptor (CAR) T therapies for the treatment of hematologic malignancies: clinical perspective and significance, J Immunother Cancer 6(1) (2018) 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Beck SE, Blankson JN, Replacing cART with CAR-T Cells: Using Immunotherapy to Cure HIV, Mol Ther (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Firacative C, Lizarazo J, Illnait-Zaragozi MT, Castaneda E, Latin G. American Cryptococcal Study, The status of cryptococcosis in Latin America, Mem Inst Oswaldo Cruz 113(7) (2018) e170554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Perfect JR, The antifungal pipeline: a reality check, Nat Rev Drug Discov 16(9) (2017) 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gast CE, Basso LR Jr., Bruzual I, Wong B, Azole resistance in Cryptococcus gattii from the Pacific Northwest: Investigation of the role of ERG11, Antimicrob Agents Chemother 57(11) (2013) 5478–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kordalewska M, Lee A, Park S, Berrio I, Chowdhary A, Zhao Y, Perlin DS, Understanding Echinocandin Resistance in the Emerging Pathogen Candida auris, Antimicrob Agents Chemother 62(6) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Antachopoulos C, Walsh TJ, Immunotherapy of Cryptococcus infections, Clin Microbiol Infect 18(2) (2012) 126–33. [DOI] [PubMed] [Google Scholar]
- [50].Specht CA, Lee CK, Huang H, Hester MM, Liu J, Luckie BA, Torres Santana MA, Mirza Z, Khoshkenar P, Abraham A, Shen ZT, Lodge JK, Akalin A, Homan J, Ostroff GR, Levitz SM, Vaccination with Recombinant Cryptococcus Proteins in Glucan Particles Protects Mice against Cryptococcosis in a Manner Dependent upon Mouse Strain and Cryptococcal Species, MBio 8(6) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].LeibundGut-Landmann S, Wuthrich M, Hohl TM, Immunity to fungi, Curr Opin Immunol 24(4) (2012) 449–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zaragoza O, Rodrigues ML, De Jesus M, Frases S, Dadachova E, Casadevall A, The capsule of the fungal pathogen Cryptococcus neoformans, Adv Appl Microbiol 68 (2009) 133–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Vera J, Fenutria R, Canadas O, Figueras M, Mota R, Sarrias MR, Williams DL, Casals C, Yelamos J, Lozano F, The CD5 ectodomain interacts with conserved fungal cell wall components and protects from zymosan-induced septic shock-like syndrome, Proc Natl Acad Sci U S A 106(5) (2009) 1506–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Avci FY, Li X, Tsuji M, Kasper DL, Carbohydrates and T cells: a sweet twosome, Semin Immunol 25(2) (2013) 146–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Carbone FR, Gleeson PA, Carbohydrates and antigen recognition by T cells, Glycobiology 7(6) (1997) 725–30. [DOI] [PubMed] [Google Scholar]
- [56].Powderly WG, Cloud GA, Dismukes WE, Saag MS, Measurement of cryptococcal antigen in serum and cerebrospinal fluid: value in the management of AIDS-associated cryptococcal meningitis, Clin Infect Dis 18(5) (1994) 789–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.