

Summary
Pathogenic LRRK2 mutations have been implicated in Parkinson’s disease. A new study provides compelling evidence that LRRK2 kinase hyperactivation reduces the processivity of autophagosomal retrograde transport in axons through an unproductive tug-of-war between anterograde and retrograde motors, thus contributing to autophagy dysfunction and axonal degeneration.
Keywords: autophagy, autophagosome transport, dynein, kinesin, LRRK2, lysosomes, Parkinson’s disease, Rab GTPases
Neurons are highly polarized cells with an extremely long axon and extensive branches at their terminal regions. This morphological feature is particularly unique and complex in dopaminergic nigrostriatal neurons which possess widely spread and highly dense axonal arborizations. In the rat brain, they can extend to ~0.47 meters in length if all the axon fibers are summed 1. Lysosomes serve as degradation hubs for autophagic and endocytic components, thus maintaining cellular homeostasis essential for neuronal survival and function. In neurons, autophagic vacuoles (AVs) are preferentially formed at the distal axonal tip and then undergo stepwise maturation through dynein-driven retrograde trafficking en route towards the soma, where mature lysosomes are relatively enriched for autophagic degradation 2,3. Retrograde transport plays a critical role in autophagic clearance and thus the maintenance of cellular homeostasis in distal axons and synapses. Autophagic dysfunction contributes to the pathogenesis of several major neurodegenerative diseases, including Parkinson’s disease (PD) 4,5. However, the cellular mechanisms underlying impaired autophagic trafficking and maturation by the PD-linked gene mutations remain largely elusive.
Mutations in leucine-rich repeat kinase 2 (LRRK2) are the most common genetic cause of PD 6. LRRK2-G2019S is the most frequent type of autosomal dominant missense mutation that is located in the kinase domain and associated with the hyperactivity of LRRK2 kinase 7,8. Despite its pathogenic gain-of-function roles, precisely how increased LRRK2 kinase activity leads to PD-linked autophagic dysfunction remains largely unknown. In a new study published in this issue of Current Biology, Boecker et al. 9 addressed this issue by elegantly employing live neuron imaging combined with genetic manipulation, biochemical, and cellular analyses. Their study revealed that pathogenic hyperactivation of LRRK2 by the G2019S mutation plays a crucial role in recruiting an enhanced amount of the anterograde motor kinesin and its adaptor JIP4 to the AV outer membrane, resulting in an unbalanced tug-of-war between anterograde and retrograde motors, leading to decreased processivity of AV retrograde transport. Their findings fill in a missing piece of the puzzle and represent conceptual advances as to how PD-linked LRRK2 hyperactivity impairs axonal AV trafficking and maturation, thus contributing to the pathogenesis of PD.
LRRK2 has been implicated in the autophagy-lysosomal pathway10, and autophagic stress is a central problem associated with major neurodegenerative diseases 4. A critical question relevant to PD-linked LRRK2 mutations is whether its kinase hyperactivity has a direct link with impaired autophagy in neurons. To address this, Boecker et al. applied three different neuron models. In the first model by overexpressing LRRK2-G2019S in rat hippocampal neurons, they found striking alterations in axonal AV processivity. Axonal AVs undergo more frequent pauses, spend more time in a paused status per AV, and display increased reversals when compared with neurons expressing kinase-dead or WT LRRK2, thus highlighting kinase-dependent changes in AV motility. To examine whether an endogenous level of LRRK2-G2019S is sufficient to induce these phenotypes, they then used G2019S knock-in (KI) mouse neurons as a second model and consistently found similar defects in axonal AV processivity. Such a dramatic increase in reversals affects the overall processivity of AV retrograde transport, as revealed by a robust difference between the total and net run length of each AV, indicating reduced retrograde processivity due to an increase in non-processive motility. To further confirm these kinase-dependent phenotypes, they applied the LRRK2 kinase inhibitor MLi-2 and found that it effectively rescues AV transport in G2019S KI neurons. To consolidate these findings, Boecker et al. alternatively employed gene-edited human iPSC-derived LRRK2-G2019S neurons as a third model. Similarly, they discovered disrupted processivity of axonal AV transport. Altogether, these intriguing phenotypes observed in three neuron model systems provide a compelling link between LRRK2 kinase hyperactivation and impaired axonal AV processivity.
Retrograde transport is essential for maintaining autophagic maturation and degradation capacity 2,3,11. Since axonal AV maturation is linked to their retrograde transport 2,12, they further determined the maturation status of these axonal AVs using the dual-color mCherry-EGFP-tagged LC3 reporter that allows for differentiation between acidified and non-acidified AVs. Boecker et al. observed a significant reduction in the fraction of acidified AVs in the proximal axonal region of G2019S KI neurons, a phenotype that could be rescued by blocking LRRK2 kinase activity with MLi-2, thus supporting their claim that LRRK2 kinase hyperactivation impairs AV acidification and maturation.
Boecker et al. then investigated the mechanism underlying the kinase-dependent regulation of axonal AV processivity. Rab GTPases act as master regulators of multiple intracellular trafficking pathways by interacting with motor adaptors and also serve as substrates of LRRK2 13,14. Of particular interest, Rab29 is genetically linked to PD and functions as both a substrate and activator of LRRK2 kinase 15,16. In this study, Boecker et al. showed that both Rab29 and LRRK2 associate with outer AV membranes. Overexpressing Rab29 decreases the processivity of AV transport to a similar extent to neurons expressing LRRK2-G2019S. Interestingly, increased Rab29 phosphorylation was observed on AVs from G2019S KI mouse brain lysates, and the LRRK2 kinase inhibitor MLi-2 was able to rescue this phenotype, demonstrating that LRRK2 and Rab29 act within the same pathway in controlling the processivity of AVs.
Bidirectional axonal transport is driven by an integrated opposing force between the anterograde motor kinesin and the retrograde motor dynein. Boecker et al. thus hypothesized that the increased reversals and non-processive motility of AVs along axons of LRRK2-G2019S neurons may be attributed to an unregulated tug-of-war between the opposing motors as a result of the increased kinesin recruitment and activity on the AV. To test this, they focused on JIP4, a scaffolding protein that forms a complex with kinesin on trafficking cargoes 17,18. In line with recent findings that LRRK2 induces the phosphorylation of Rab GTPase including Rab10 and Rab35 and increases JIP4 recruitment to lysosomes 19, they revealed increased levels of JIP4, phospho-Rab proteins, and kinesin-1 heavy chain (KHC), but not the dynactin subunit p150Glued, on AVs from G2019S KI mouse brain lysates as compared to WT. By overexpressing JIP4 together with LC3, they further confirmed a similar phenotype with a more robust disruption in AV processivity and increased reversals. Altogether, the study suggests an attractive pathogenic tug-of-war model: PD-linked hyperactive LRRK2 kinase reduces AV retrograde transport and impairs AV maturation through the phosphorylation of Rab proteins that recruit JIP4 to the AV surface, where JIP4 in turn recruits the kinesin motor. Based on these findings, they proposed an enhanced engagement of kinesin motors with microtubules, thus leading to an unproductive tug-of-war (Figure 1).
Figure 1. A pathogenic tug-of-war model in PD-linked AV transport.
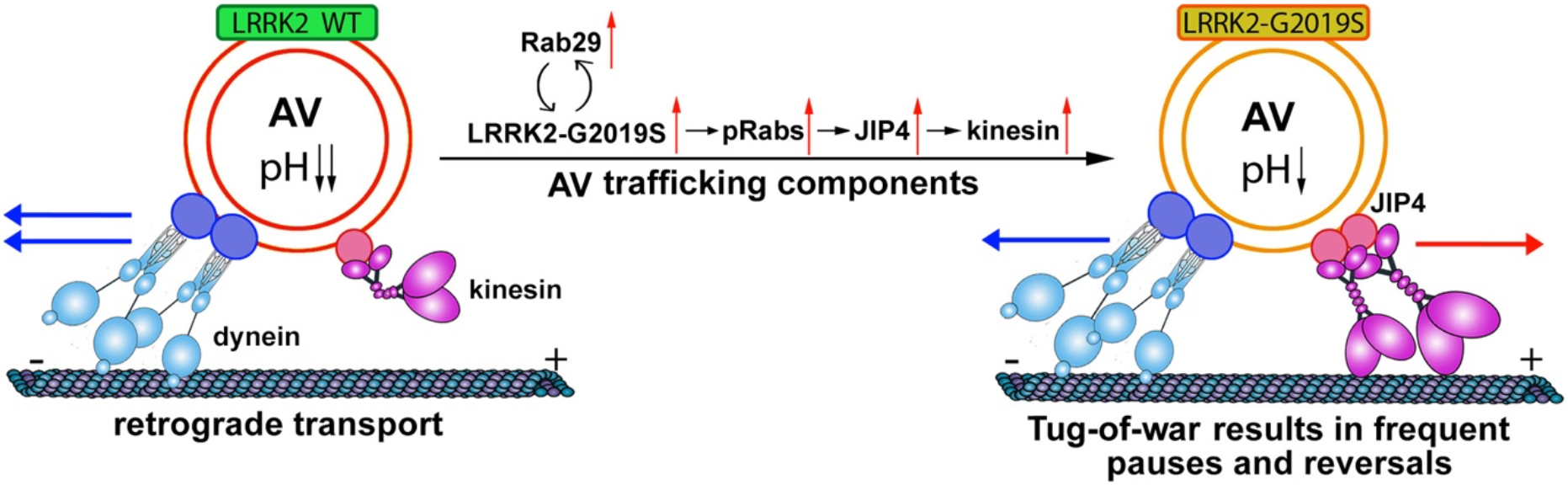
Axonal transport is driven by an integrated opposing force between the anterograde motor kinesin and the retrograde motor dynein. AVs are preferentially formed at the distal axonal tip and then undergo stepwise maturation along the dynein-driven trafficking pathway en route towards the soma for degradation. PD-linked hyperactive LRRK2 kinase reduces AV retrograde processivity and impairs their maturation through the phosphorylation of Rab proteins that recruit JIP4 to the outer membrane of AVs. JIP4 in turn recruits more kinesin motors that then engage with microtubules, thus leading to an unproductive tug-of-war. This model nicely interprets the striking phenotypes of the reduced AV processivity in LRRK2-G2019S neurons, where axonal AVs undergo more frequent pauses, spend more time in a paused status per AV, and display increased reversals.
The study advances our knowledge of the pathogenic role of LRRK2-G2019S that has been linked to the dysfunction of various organelles including endo-lysosomes, Golgi, mitochondria, and synaptic vesicles. Thus, there are several questions to be answered before we are able to fully understand pathologic contributions of LRRK2 hyperactivation. First, it is well documented that lysosomal dysfunction occurs in LRRK2-G2019S models 20. The current study provided evidence showing no significant changes in the pH and motility of LAMP1-labeled endosome/lysosome-like organelles in LRRK2-G2019S neurons. Perhaps the most direct follow-up study would be to examine whether reduced lysosomal degradation capacity or their defective fusion with AVs co-contributes to reduced AV maturation and axonal autophagic stress found in the LRRK2-G2019S models. It would be more compelling to examine whether mature lysosomal dynamics are disrupted using activity-based reporters that monitor active lysosomal enzymes and lysosomal degradative cargoes. Second, it is not known whether disrupted AV processivity is the primary contributor to axonal PD pathology, given the fact that LRRK2 mutants have also been linked to dysfunction in multiple trafficking routes through the phosphorylation of several members of the Rab family. In addition, it would be intriguing to determine which Rab protein(s) specifically recruits JIP4 to the AV surface upon their phosphorylation by LRRK2-G2019S. Despite these new questions raised, the study by the Holzbaur group established a robust mechanistic link between pathogenic LRRK2 and impaired AV transport, thus representing a conceptual tour de force for new insights into PD-associated autophagic stress.
Acknowledgments:
We thank J.C. Roney for critical reading. This work was supported by the Intramural Research Program of NINDS, NIH ZIA NS003029 and ZIA NS002946 (Z-H. Sheng).
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare no competing interests.
References:
- 1.Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, and Kaneko T (2009). Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. The Journal of neuroscience : the official journal of the Society for Neuroscience 29, 444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maday S, Wallace KE, and Holzbaur EL (2012). Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol 196, 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng XT, Zhou B, Lin MY, Cai Q, and Sheng ZH (2015). Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. The Journal of cell biology 209, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nixon RA (2013). The role of autophagy in neurodegenerative disease. Nature medicine 19, 983–997. [DOI] [PubMed] [Google Scholar]
- 5.Mizushima N, and Levine B (2020). Autophagy in Human Diseases. The New England journal of medicine 383, 1564–1576. [DOI] [PubMed] [Google Scholar]
- 6.Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, et al. (2008). Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol 7, 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, et al. (2006). Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiology of disease 23, 329–341. [DOI] [PubMed] [Google Scholar]
- 8.Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, and Ross CA (2006). Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci 9, 1231–1233. [DOI] [PubMed] [Google Scholar]
- 9.Boecker CA, Goldsmith J, Dou D,Cajka GG, and Holzbaur EL (2021). Increased LRRK2 kinase activity alters neuronal autophagy by disrupting the axonal transport of autophagosomes. Curr.Biol ??? [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giaime E, Tong Y, Wagner LK, Yuan Y, Huang G, and Shen J (2017). Age-Dependent Dopaminergic Neurodegeneration and Impairment of the Autophagy-Lysosomal Pathway in LRRK-Deficient Mice. Neuron 96, 796–807 e796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai Q, Lu L, Tian JH, Zhu YB, Qiao H, and Sheng ZH (2010). Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron 68, 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee S, Sato Y, and Nixon RA (2011). Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy. J Neurosci 31, 7817–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiral FR, Kohrs FE, Jin EJ, and Hiesinger PR (2018). Rab GTPases and Membrane Trafficking in Neurodegeneration. Current biology : CB 28, R471–R486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, et al. (2016). Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Purlyte E, Dhekne HS, Sarhan AR, Gomez R, Lis P, Wightman M, Martinez TN, Tonelli F, Pfeffer SR, and Alessi DR (2018). Rab29 activation of the Parkinson's disease-associated LRRK2 kinase. The EMBO journal 37, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, et al. (2009). Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nature genetics 41, 1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bowman AB, Kamal A, Ritchings BW, Philp AV, McGrail M, Gindhart JG, and Goldstein LS (2000). Kinesin-dependent axonal transport is mediated by the sunday driver (SYD) protein. Cell 103, 583–594. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen Q, Lee CM, Le A, and Reddy EP (2005). JLP associates with kinesin light chain 1 through a novel leucine zipper-like domain. The Journal of biological chemistry 280, 30185–30191. [DOI] [PubMed] [Google Scholar]
- 19.Bonet-Ponce L, Beilina A, Williamson CD, Lindberg E, Kluss JH, Saez-Atienzar S, Landeck N, Kumaran R, Mamais A, Bleck CKE, et al. (2020). LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci Adv 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wallings RL, Humble SW, Ward ME, and Wade-Martins R (2019). Lysosomal Dysfunction at the Centre of Parkinson's Disease and Frontotemporal Dementia/Amyotrophic Lateral Sclerosis. Trends in neurosciences 42, 899–912. [DOI] [PMC free article] [PubMed] [Google Scholar]