

Abstract
There is discussion of expanding newborn screening (NBS) through the use of genomic sequence data; yet, challenges remain in the interpretation of DNA variants. Population-level DNA variant databases are available, and it is possible to estimate the number of newborns who would be flagged as having a risk for a genetic disease (including rare variants of unknown significance, VUS) via next-generation sequencing (NGS) positive. Estimates of the number of newborns screened as NGS positive for monogenic recessive diseases were obtained by analysis of the Genome Aggregation Database (gnomAD). For a collection of diseases for which there is interest in NBS, we provided 2 estimates for the expected number of newborns screened as NGS positive. For a set of lysosomal storage diseases, we estimated that 100 to approximately 600 NGS screen positives would be found per disease per year in a large NBS laboratory (California), and this figure may be expected to rise to a limit of about 1000 if we account for the fact that gnomAD does not contain all worldwide variants. The number of positives would drop 2.5- to 10-fold if the 10 VUS with highest allele frequency were biochemically annotated as benign. It is proposed that a second-tier biochemical assay using the same dried blood spot could be carried out as a filter and as part of NBS to reduce the number of high-risk NGS positive newborns to a manageable number.
Keywords: newborn screening, tandem mass spectrometry, next generation sequencing, genomics, dried blood spots, inborn errors of metabolism
Introduction
Next-generation sequencing (NGS) at the exome and genome level is a single assay that provides fairly comprehensive assessment of single-nucleotide variants, insertions, and deletions and can be assessed for copy number variants. This makes it appealing as a first-tier method for newborn screening (NBS) for a large collection of treatable neonatal diseases. NGS is offset by the challenges that exist in the prediction of disease phenotypes from genotypes because of the occurrence of DNA variants of uncertain pathogenic significance (VUS) and the difficulty with integration of multiple variants that partially reduce the function of the encoded protein.
For recessive diseases, one approach is to report out only cases where at least 2 variants are found (either 2 heterozygous variants or 1 homozygous variant) that are well understood to be pathogenic. These include predicted loss-of-function variants (pLoF) and missense variants for which pathogenicity is understood from clinical reports, ignoring VUS. Phasing of heterozygous variants could be carried out by NGS (or Sanger sequencing) of parents and will be aided by complementary data, such as variant co-occurrence look-up, that is available for variants in reference population databases, such as the Genome Aggregation Database (gnomAD)1. Ignoring VUS will significantly increase the false negative rates in NBS compared to those obtained with biochemical assays. In the current study, we consider the option of including VUS and passing all NGS results of concern to a biochemical assay for the disease that we term “NGS>Biochem” (NGS before biochemical investigation). In this study, we estimate the number of newborns that would be passed to biochemical testing. Notably, in silico methods used to predict the pathogenicity of missense variants in already-symptomatic newborns, despite showing success,2 are generally thought to lack the high-level accuracy needed for population based NBS in which most newborns are healthy.
An additional concern about first-tier NGS NBS are cases in which the newborn has a disease, but the responsible DNA signature (variants) is not found. Two recent studies show a DNA-based diagnostic yield of 64% and 88% when exome data were analyzed among patients already diagnosed with a newborn disease.3,4 The diagnostic yield would presumably go up if more biochemical data are used to help annotate VUS as pathogenic.
In this report we discuss the approach of NGS>Biochem. We focus on a collection of lysosomal storage diseases (LSDs) which are recessive monogenic diseases for which treatments are available for many subtypes. NBS for LSDs has started over the past decade, and further condition expansion is under discussion. We also include the bile acid disorder cerebrotendinous xanthomatosis for which NBS is being discussed.5
Materials and Methods
Detailed protocols for analysis of the gnomAD v2 database are given in the Supplemental Materials. Approach 1: individuals in the gnomAD v2 exome data set (n = 125,748) with 2 rare (popmax and global allele frequency ≤1%) pLoF and/or missense variants were counted, excluding variants that are common (>5% allele frequency) in any population that is not included in popmax and also variants that are benign (B) or likely benign (LB) in ClinVar with multiple entries in agreement (2-stars review status). Approach 2: the allele frequencies in gnomAD v2 of variants selected using the same criteria as in approach 1 were summed, and, using the Hardy-Weinberg equilibrium, q was calculated and 2q was taken as the probability of the mother or the father being a heterozygote (only 1 variant). We took q2 as the estimated number of newborns carrying variants from both parents. Both approaches assume that the US population is similar in ancestry distribution to the individuals represented in the gnomAD v2 exomes (7% African American, 14% Latino/ Admixed American, 7% East Asian, 9% South Asian, and 58% European), which is not entirely accurate but can still be used for a rough estimate.
Results
Expected LSD NGS screen positives based on observed numbers in gnomAD
Using approach 1, we estimated the number of first-tier NGS NBS positives for several LSDs using individuals in gnomAD as a representative population. We included all rare pLoF and missense variants, many of which are not classified in ClinVar, acknowledging that most of the missense variants would be VUS. We excluded all B/LB variants in ClinVar associated with a review status of 2-stars or more. For the 5 LSDs, we found 33 to 150 individuals per gene who would be NGS screen positive (~1:800 to 1:4000). It is worth noting that gnomAD is depleted for individuals with rare disease; yet, this approach has value in evaluating the expected false positive rate of the NGS screening. The true positive rate, based on the prevalence of the condition, could be added to these estimates. After scaling the number to account for the birth rate per NBS laboratory, we arrive at the estimates in Table 1, which give a reasonable accurate estimate of frequency at which VUS will be found in the general population. Supplemental Table 1 displays the counts by variant type and Supplemental Table 2 displays the screen-positive individuals when also including the ClinVar B/LB variants in the analysis.
Table 1.
Estimated number of NGS NBS screen positive newborns per year based on variants found in gnomAD, excluding all ClinVar B/LB variants with 2-star review status and above (see Methods).
| Disease (protein) | Estimated number of First-Tier NGS Screen Positives Per Year | ||||
|---|---|---|---|---|---|
| gnomAD (125,748 exomes) |
CA (500,000 births/yr) |
WA (85,000 births/yr) |
USA (4,000,000 births/yr) |
USA with 10 most common VUS removed (4,000,000 births/yr) |
|
| MLD (ARSA) | 49 | 195 | 33 | 1,559 | 159 |
| CTX (CYP27A1) | 33 | 131 | 22 | 1,050 | 318 |
| Krabbe (GALC) | 54 | 215 | 37 | 1,718 | 318 |
| MPS-I (IDUA) | 37 | 147 | 25 | 1,177 | 414 |
| Pompe (GAA) | 150 | 596 | 101 | 4,771 | 1,559 |
ARSA, arylsulfatase A; B/LB, benign or likely benign; CA, California; CTX, cerebrotendinous xanthomatosis; GAA, acid-alpha-glucosidase; GALC, galactosylceramidase; gnomAD, Genome Aggregation Database; IDUA, alpha-iduronidase; MPS-I, mucopolysaccharidosis type I, NGS, next-generation sequencing; USA, United States; VUS, variants of uncertain significance; WA, Washington; y, year.
Expected LSD NGS screen positives based on Hardy-Weinberg estimates
In a second approach, we used a Hardy-Weinberg equilibrium analysis. This assumes that the DNA variants are distributed randomly throughout the population and that the US population ancestry is similar to gnomAD. Estimates of the number of NGS screen positives, excluding all B/LB variants in ClinVar associated with a review status of 2-stars or more, are given in Supplemental Table 3. Results show that the number of NGS screen positives are up to approximately 5-fold lower than the number observed in gnomAD (see above). These approaches give different estimates because the first approach is a sampling/count in individuals depleted for rare disease (false positive rate) and the second approach extrapolates the allele frequencies from the sampling to make an estimate of the screen positive rate. In any case, the differences are relatively small and we include this approach because it is currently accessible to any gnomAD user.
Reduction in the number of VUS by variant annotation
The number of NGS NBS positives will go down over time as more VUS become annotated as benign. Figure 1 shows how the number of NGS NBS positives (using approach 1) would drop if the highest frequency VUS were further investigated and subsequently annotated as benign. As an example, the number of NGS NBS positive newborns in the US for mucopolysaccharidosis type I would drop from 1177 to 414 per year if the 10 most frequent VUS were reannotated as benign (for example, by expression of the iduronidase enzyme in human cells and measurement of its enzymatic activity) (Table 1). A systematic approach to evaluate variant function has been applied on a small set of genes including BRCA1, ARSA, and ADA2, and international research efforts are further expanding this through the Atlas of Variant Effects Alliance.6–9
Figure 1.
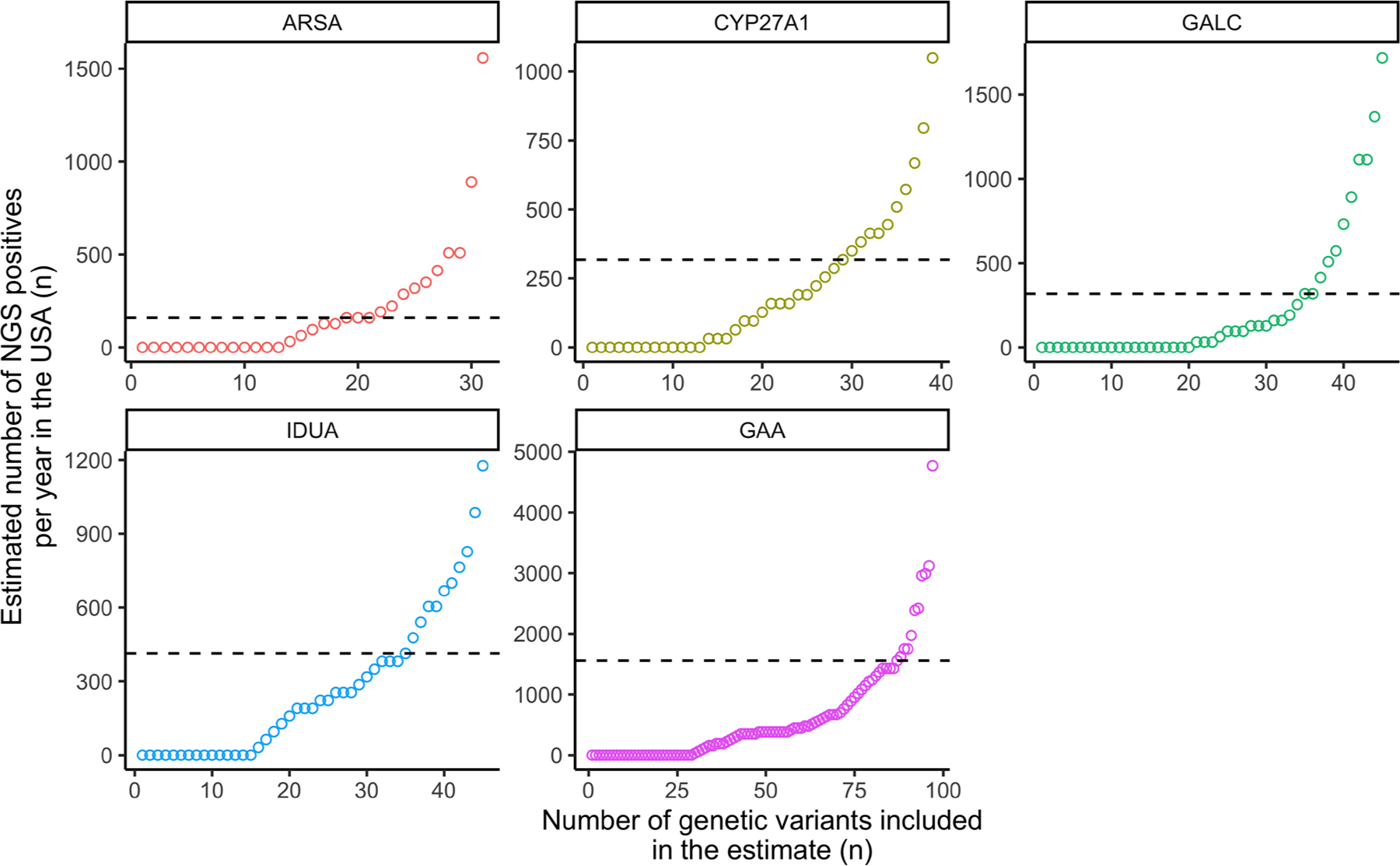
Number of NGS NBS positives estimated per year in NBS laboratories in the USA based on the number of individuals in gnomAD carrying 2 rare pLoF and/or missense variants (heterozygous or homozygous), excluding all ClinVar B/LB variants with 2-star review status and above (see Methods).
The number of genetic variants (x-axis) included in the NGS positive estimate (y-axis) is shown. By sequentially removing the variant with the highest impact on the NBS positive estimate (far right), we see the resultant reduction in the number of NGS positives. The estimated number after removing 10 variants is indicated (dashed line). B/LB, benign or likely benign; NBS, newborn screening; NGS, next-generation sequencing.
Discussion
The current report provides reasonably accurate estimates of the number of NGS screen positives that would be encountered for a set of monogenic diseases if rare VUS are included. Including a second-tier biochemical analysis using the same dried blood spot (DBS) and as part of NBS may be feasible for diseases in which relatively simple biomarker analyses exists. For diseases in which no DBS biomarker is available, the only practical option may be to ignore rare VUS, but this will come with a high false negative rate. For NBS of LSDs, second-tier with or without third-tier biochemical DBS assays reduces the false positive rates to close to zero.10–12
The number of VUS that will be encountered is not known but does have an upper limit. Of the variants that can be created by a single nucleotide change, only 11.5% of synonymous, 7.3% of missense, and 3.7% of nonsense variants are represented in gnomAD v2; therefore, there are still a large number of non-represented coding variants that may yet be identified by NBS. The majority of these variants will be very rare (though some populations are still underrepresented in reference databases). Although in silico predictors are improving,13,14 they are not of sufficient sensitivity for use in NBS. To aid in understanding the rate at which rare predicted damaging variants co-occur in reference populations, counts of individuals by gene have recently been released in gnomAD. Here, though calculated from the same data, the counts are slightly lower than those displayed in the gnomAD browser. This is due to our exclusion of 2-star review status ClinVar B/LB variants and because of counting individuals with 2 rare heterozygous and/or homozygous variants together to prevent double counting individuals with both.
Functional modeling or extensive clinical observation data are needed to resolve VUS. Because many of these variants will be benign, particularly the most common VUS, resolving these will drop the number of NGS-positive NBS (Figure 1, Table 1 right column). This raises the option that before NBS of a specific set of diseases by first-tier NGS, one could express, for instance, approximately 100 of the most frequent VUS in human cells (for example, HEK-293 cells, as was done recently for the ARSA and ADA2 genes7,8) to determine their functional impact. This could be carried out in a single laboratory in a few months and at a cost of <$10,000 per gene. In the case of ARSA, all VUS observed in gnomAD (~300) were expressed in HEK-293 cells, and enzymatic activity was measured by mass spectrometry, reducing the number of NGS NBS screen positives to a few per year per state laboratory.7 Investment in functional characterization of variants in the genes included in NGS NBS is likely to be cost effective given the large impact on decreasing the rate of sending samples for biochemical second tier screening. Ignoring all VUS after NGS NBS could lead to a significant burden of false negatives. For example, in the case of ARSA, about 25% of the missense VUS in gnomAD reduce the enzymatic activity to <3%7 and are thus expected to lead to metachromatic leukodystrophy when found in combination with a pathogenic variant.
The first-tier NGS NBS followed by second-tier biochemical assay approach will not be possible for cases in which a biochemical biomarker for the disease using the DBS is not available. However, even for diseases for which there are no good assays of the protein function, quantitative proteomic assays can be useful to measure protein abundance. One example is Wilson disease, caused by variants in the copper-transporter ATP7B. NBS for Wilson disease by biochemical screening is possible by measuring the abundance of the Wilson protein in DBS using immuno-capture of a trypsin-generated target peptide followed by detection by liquid chromatography-tandem mass spectrometry.15 A proteomics-based NBS assay for a set of primary immunodeficiencies has recently been developed using the same technology as for Wilson disease.16 Approaches such as these can be highly multiplexed and used for second-tier, biochemical NBS.
First-tier NGS followed by second-tier biochemical analysis all using DBS as part of NBS is an attractive scenario for the expansion of NBS programs to include an ever-increasing number of diseases for which early intervention provides the best clinical outcome. The use of NGS in NBS is a rapidly evolving area with several pilot programs now underway (examples include4,17,18). Experts are well aware of the likely large increase in the number of new treatments for neonatal diseases and more and more it is becoming apparent that current NBS programs will be pressured to keep pace. The current practices in NBS laboratories seem unsustainable. D. Bailey et al, at RTI International recently conducted a survey of the major groups of experts in NBS (NBS assay developers, clinical researchers, federal and state NBS advisory boards, patient advocacy groups, industry associated with NBS, and government NBS labs), and considerable consensus was reached to increasingly involve NGS in NBS and to centralize some of the steps through private-public partnerships.19 In one scenario, existing state NBS programs could take on population-level NGS, whereas second-tier biochemical assays could be centralized because it is inefficient for each NBS program to individually setup a set of about 100 biochemical NBS assays that are each infrequently run at the state level. An ideal scenario would be to create national laboratories that can receive DBS from different state NBS laboratories in which each carries out a panel of biochemical assays for 20 to 30 diseases. This would require movement of DBS across state lines.
More therapies for rare disease are expected in the coming decade and will include many that need to be initiated presymptomatically or very early in the disease process; therefore, it will be critical that NBS is able to expand to meet this demand. As a field, we have the tools needed to prepare for this future and it will require changes to the way NBS is implemented in our country to realize the potential of precision medicine.
Supplementary Material
Acknowledgments
The authors would like to thank the individuals whose data are in gnomAD for their contributions to research and the investigators who produced and shared the data. The authors thank Laurent Francioli, Michael Guo, Konrad Karczewski, and Kaitlin Samocha for contributions to the heterozygous variant analysis approach and Samantha Baxter for contributions to the Hardy-Weinberg analysis. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Funding
M.H.G. is supported National Institutes of Health NIDDK grant R01DK067859. A.P. and A.O-L are supported by NIH NHGRI awards for AnVIL U24HG010262 and gnomAD U24HG011450. S.L.S. is supported by a Manton Center Rare Disease Research Fellowship.
Footnotes
Ethics Declaration
We have complied with all relevant ethical regulations. None of the elements of our manuscript make use of human subjects. The DNA analyzed in approach 2 is publicly available on the gnomAD website. There is no human subject research in our study.
Conflict of Interest
Michael H. Gelb is a founding member of GelbChem LLC, which sells reagents for biochemical genetics assays. Anthony Philippakis is a Venture Partner at GV (formerly known as Google Ventures), a subsidiary of Alphabet corporation and an Observer on the Board of Directors at Genome Medical. Anne O’Donnell-Luria is a member of a Clinical Advisory Board for Tome Biosciences.
Websites
gnomAD v2.1.1 data set was used in this study: https://gnomad.broadinstitute.org/. Accessed October 26, 2022.
Additional Information
The online version of this article (https://doi.org/10.1016/j.gimo.2023.100821) contains supplemental material, which is available to authorized users.
Data Availability
Authors agree to share all data in this article without embargo to those who request it.
References
- 1.Guo MH, Francioli LC, Stenton SL, Goodrich JK, Watts NA, Singer-Berk M, Groopman E, Darnowsky PW, Solomonson M, Baxter S; gnomAD Project Consortium; Tiao G, Neale BM, Hirschhorn JN, Rehm HL, Daly MJ, O’Donnell-Luria A, Karczewski KJ, MacArthur DG, Samocha KE. Inferring compound heterozygosity from large-scale exome sequencing data. Nat Genet. 2024. Jan;56(1):152–161. 10.1038/s41588-023-01608-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De La Vega FM, Chowdhury S, Moore B, et al. Artificial intelligence enables comprehensive genome interpretation and nomination of candidate diagnoses for rare genetic diseases. Genome Med. 2021;13(1):153. 10.1186/s13073-021-00965-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adhikari AN, Gallagher RC, Wang Y, et al. The role of exome sequencing in newborn screening for inborn errors of metabolism. Nat Med. 2020;26(9):1392–1397. 10.1038/s41591-020-0966-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roman TS, Crowley SB, Roche MI, et al. Genomic sequencing for newborn screening: results of the NC NEXUS project. Am J Hum Genet. 2020;107(4):596–611. 10.1016/j.ajhg.2020.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaz FM, Bootsma AH, Kulik W, et al. A newborn screening method for cerebrotendinous xanthomatosis using bile alcohol glucuronides and metabolite ratios. J Lipid Res. 2017;58(5):1002–1007. 10.1194/jlr.P075051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Findlay GM, Daza RM, Martin B, et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature. 2018;562(7726):217–222. 10.1038/s41586-018-0461-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trinidad M, Hong X, Froelich S, et al. Predicting disease severity in metachromatic leukodystrophy using protein activity and a patient phenotype matrix. Genome Biol. 2023;24:172. 10.1186/s13059-023-03001-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jee H, Huang Z, Baxter S, et al. Comprehensive analysis of ADA2 genetic variants and estimation of carrier frequency driven by a function-based approach. J Allergy Clin Immunol. 2022;149(1):379–387. 10.1016/j.jaci.2021.04.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atlas of Variant Effects Alliance. Atlas of Variant Effects Alliance. Accessed November 27, 2022. https://www.varianteffect.org/
- 10.Peck DS, Lacey JM, White AL, et al. Incorporation of second-tier biomarker testing improves the specificity of newborn screening for mucopolysaccharidosis Type I. Int J Neonatal Screen. 2020;6(1):10. 10.3390/ijns6010010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herbst ZM, Urdaneta L, Klein T, et al. Evaluation of two methods for quantification of glycosaminoglycan biomarkers in newborn dried blood spots from patients with severe and attenuated mucopolysaccharidosis Type II. Int J Neonatal Screen. 2022;8(1):9. 10.3390/ijns8010009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guenzel AJ, Turgeon CT, Nickander KK, et al. The critical role of psychosine in screening, diagnosis, and monitoring of Krabbe disease. Genet Med. 2020;13:240–211. 10.1038/s41436-020-0764-y [DOI] [PubMed] [Google Scholar]
- 13.Pejaver V, Byrne AB, Feng BJ, et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet. 2022;109(12):2163–2177. 10.1016/j.ajhg.2022.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99(4):877–885. 10.1016/j.ajhg.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collins CJ, Yi F, Dayuha R, et al. Direct measurement of ATP7B peptides is highly effective in the diagnosis of Wilson disease. Gastroenterology. 2021;160(7):2367–2382.e1. 10.1053/j.gastro.2021.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins CJ, Chang IJ, Jung S, et al. Rapid multiplexed proteomic screening for primary immunodeficiency disorders from dried blood spots. Front Immunol. 2018;9:2756. 10.3389/fimmu.2018.02756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ceyhan-Birsoy O, Murry JB, Machini K, et al. Interpretation of genomic sequencing results in healthy and ill newborns: results from the BabySeq project. Am J Hum Genet. 2019;104(1):76–93. 10.1016/j.ajhg.2018.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bick D, Jones M, Taylor SL, Taft RJ, Belmont J. Case for genome sequencing in infants and children with rare, undiagnosed or genetic diseases. J Med Genet. 2019;56(12):783–791. 10.1136/jmedgenet-2019-106111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailey DB Jr, Porter KA, Andrews SM, Raspa M, Gwaltney AY, Peay HL. Expert evaluation of strategies to modernize newborn screening in the United States. JAMA Netw Open. 2021;4(12): e2140998. 10.1001/jamanetworkopen.2021.40998 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Authors agree to share all data in this article without embargo to those who request it.