

Abstract
Small molecule drug conjugates are an emerging targeted therapy for cancer treatment. Building upon the overexpressed prostate-specific membrane antigen (PSMA) in prostate cancer, we herein report the design and synthesis of a novel PSMA–PI3K small molecule drug conjugate 1. Conjugate 1 demonstrates potent inhibition against PI3K with an IC50 value of 0.40 nM and simultaneously targets PSMA, giving rise to selective growth inhibition activity for PSMA-positive cancer cells. Conjugate 1 also potently inhibits the phosphorylation of PI3K main downstream effectors and arrests the cell cycle in the G0/G1 phase in PSMA-positive 22Rv1 prostate cancer cells. Further in vivo evaluation shows that conjugate 1 has favorable efficacy and tolerability in a 22Rv1 xenograft model, demonstrating its potential application in targeted cancer therapy.
A novel PSMA–PI3K small molecule drug conjugate has been prepared, highlighting its potential in targeted cancer therapy.
Introduction
Conventional chemotherapeutic approaches predominantly depend upon cytotoxic agents, which frequently exhibit a lack of selective accumulation within tumor tissues, leading to increased drug dosage and undesirable off-target toxicities.1 Targeted therapy with reduced side effects offers a viable option for precision medicine. Among various targeted approaches, antibody drug conjugates (ADCs) and small molecule drug conjugates (SMDCs) have gained considerable attention over the past several decades. These conjugated drugs typically consist of three key components: a targeting module for the desired receptor/cell, a cytotoxic payload, and a stable or enzymatically/chemically cleavable linker bridging the two entities.2,3 In contrast to ADCs, SMDCs demonstrate several advantages with their small size, non-immunogenic nature, controllable synthesis, and ease of storage and transportation,2,4 representing an encouraging targeted cancer therapy with great potential.
The phosphoinositide 3-kinase (PI3K) pathway, an extensively studied target for cancer treatment, plays a critical function in cell growth, proliferation, survival, metabolism, and angiogenesis.5,6 It is well documented that over-activation of PI3K signaling and mutations in related genes occur in many cancer types.7,8 Hence, the exploitation of PI3K inhibitors is of great value as an effective therapeutic approach for cancer treatment. To date, six PI3K inhibitors have been approved by the US Food and Drug Administration.9,10 However, due to their inevitable on-target systemic toxicity and subsequent unwanted side effects, PI3K inhibitors generally face challenges and obstacles in clinical development.11–13 Therefore, improving the accumulation of PI3K inhibitors in the lesion area could be an attractive approach to facilitate their development.
Prostate-specific membrane antigen (PSMA), also known as glutamate carboxypeptidase II, is one of the most attractive biomarkers for selective delivery of anti-tumor drugs for diagnostic or therapeutic purposes. Several small molecule ligand-targeted drugs have been approved for clinical use, such as [68Ga]PSMA-11 and [18F]DCFPyL for imaging of prostate cancer.14–16 Recently, [177Lu]PSMA-617 was approved by the FDA as the first targeted radioligand therapy for the treatment of PSMA-positive metastatic castration-resistant prostate cancer (mCRPC).17,18 Moreover, PSMA-targeted ligands conjugated with small molecule drugs are also actively explored, such as conjugates with doxorubicin,19 paclitaxel,20,21 docetaxel,22 DM1,23 MMAE,4,24,25 SERCA,26 and topoisomerase inhibitors.27
Herein, we report the discovery of a PSMA–PI3K small molecule drug conjugate. The conjugate is composed of a PI3K inhibitor, a PSMA-targeting motif and a linker connecting the two moieties. In such a design, we use a hybridization strategy that combines key features of our earlier disclosed 4-methylquinazoline based PI3K inhibitor and PSMA-617 (Fig. 1).28 In particular, the commonly used Glu-Urea-Lys dipeptide with high affinity to PSMA is then introduced to the 8 position of the quinazoline scaffold to occupy the solvent-exposed region of the PI3K ATP-binding pocket with a suitable linker. We provide a simple method to synthesize conjugates and evaluate them biologically.
Fig. 1. Design strategy of the novel PSMA–PI3K small molecule drug conjugate 1.

Results and discussion
Chemistry
The synthetic route of the PI3K inhibitor is shown in Scheme 1. Notably, the starting material tert-butyl 4-((6-bromo-4-methylquinazolin-8-yl)oxy)butanoate 2 was prepared according to our previous work.29 It was reacted with 2,4-difluoro-N-(2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)benzenesulfonamide through Suzuki coupling to produce 3, and finally hydrolyzed to obtain the acid 4.
Scheme 1. Synthesis of PI3K inhibitor 4. Reagents and conditions: (a) 2,4-difluoro-N-(2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)benzenesulfonamide prepared according to the literature,30 PdCl2(dppf), 2 M K2CO3, dioxane, 100 °C, 6 h, 85%; (b) trifluoroacetic acid, dichloromethane, rt, 3 h.

The synthetic route of the PSMA–PI3K conjugate 1 is shown in Scheme 2. Initially, the commercially available material 5 was used to react with triphosgene to produce the intermediate isocyanate, which was then coupled with l-glutamic acid dibenzyl ester to yield 6. After removal of Boc protection using trifluoroacetic acid, the substituted lysine was condensed with naphthylalanine in the presence of HATU to yield 7. The Boc protection of 7 was then removed to react with Boc-trans-4-(aminomethyl)-cyclohexane-1-carboxylic acid to afford 8, which was further condensed with intermediate 4 to generate 9 after the removal of the Boc protection. Finally, compound 9 was hydrogenated to give the desired conjugate 1 in a good yield.
Scheme 2. Synthesis of PSMA–PI3K conjugate 1. Reagents and conditions: (a) triphosgene, DIPEA, dibenzyl l-glutamate, dichloromethane, 0 °C to rt, 12 h, 71%; (b) trifluoroacetic acid, dichloromethane, rt, 0.5 h; (S)-2-((tert-butoxycarbonyl)amino)-3-(naphthalen-2-yl)propanoic acid, HATU, DIPEA, dichloromethane, 78%; (c) trifluoroacetic acid, dichloromethane, rt, 0.5 h; (1r,4r)-4-(((tert-butoxycarbonyl)amino)methyl)cyclohexane-1-carboxylic acid, HATU, DIPEA, dichloromethane, 82%; (d) trifluoroacetic acid, dichloromethane, rt, 0.5 h; intermediate 4, HATU, DIPEA, dichloromethane, 85%; (e) 10% Pd/C, methanol, rt, 12 h, 77%.

Biological evaluation
The PI3Kα inhibitory potency of conjugate 1 was first evaluated to show an IC50 value of 0.4 nM, displaying higher potency compared to the free 4-methylquinazoline PI3K inhibitor (IC50 = 1.67 nM).31 This validates our design, demonstrating that the inhibitory potency could be maintained or enhanced by introducing the PSMA targeting moiety to the 8-position of the quinazoline scaffold through a suitable linker. In addition, a cell binding assay also indicated that conjugate 1 occupied the PSMA receptors in cancer cells (Fig. 2).
Fig. 2. (A) Inhibitory potency of conjugate 1 against PI3Kα; (B) PSMA binding of conjugate 1 in the 22Rv1 cell line.

Molecular docking studies were also performed to study the binding modes of conjugate 1 with PI3K α and PSMA respectively. As depicted in Fig. 3A, conjugate 1 could form a hydrogen bond with Val851 and a water bridge with Asp810 and Tyr836. Moreover, a charge interaction existed between the deprotonated sulfonamide and Lys802. Docking simulations also revealed that conjugate 1 established hydrogen bonds with Arg210, Asn257, Tyr552, Lys699, Tyr700 and active-site water molecules of PSMA. Notably, the ureido nitrogen atom of conjugate 1 interacted with the main chain carbonyl of Glu424, while its carbonyl oxygen established contacts with the catalytic zinc atom, Tyr552, and His553. Moreover, conjugate 1 is held in place by hydrogen bonding to an arginine-rich patch (Arg534 and Arg536) along with Asn519 (Fig. 3B).
Fig. 3. Predicted binding modes of conjugate 1 with PI3Kα (PDB ID: 4JPS,32 A) and PSMA (PDB ID: 5O5T, B). Conjugate 1 (yellow) and residues (smudge) are shown in stick representation, while the zinc ions and water ions in the binding site are labeled as light blue and red spheres. Hydrogen bonds are shown as black dashed lines.

Next, the antiproliferative activities of conjugate 1 were assessed on both PSMA-positive (22Rv1 and LNCaP) and PSMA-negative (PC3) cells, resulting in IC50 values of 9.68 μM, 1.08 μM, and 57.78 μM, respectively. Notably, its growth inhibitory activities against PSMA-positive LNCaP and 22Rv1 cells were 54- and 6-fold higher, relative to PSMA-negative PC3 cells (Table 1). In comparison, the free 4-methylquinazoline PI3K inhibitor and the PSMA ligand (Lys-Urea-Glu) were also tested against LNCaP cells and showed IC50 values of 1.18 μM and >100 μM. These findings indicated that our rationally designed PSMA–PI3K conjugate 1 can selectively target PSMA overexpressed cancer cells and may improve the safety profile of PI3K inhibitors.
Antiproliferative activity of conjugate 1 against prostate cancer cellsa.
| Cell lines | PSMA+ | PSMA− | |
|---|---|---|---|
| 22Rv1 | LNCaP | PC3 | |
| IC50 (μM) | 9.68 | 1.08 | 57.78 |
Data are expressed as the mean value (n = 3).
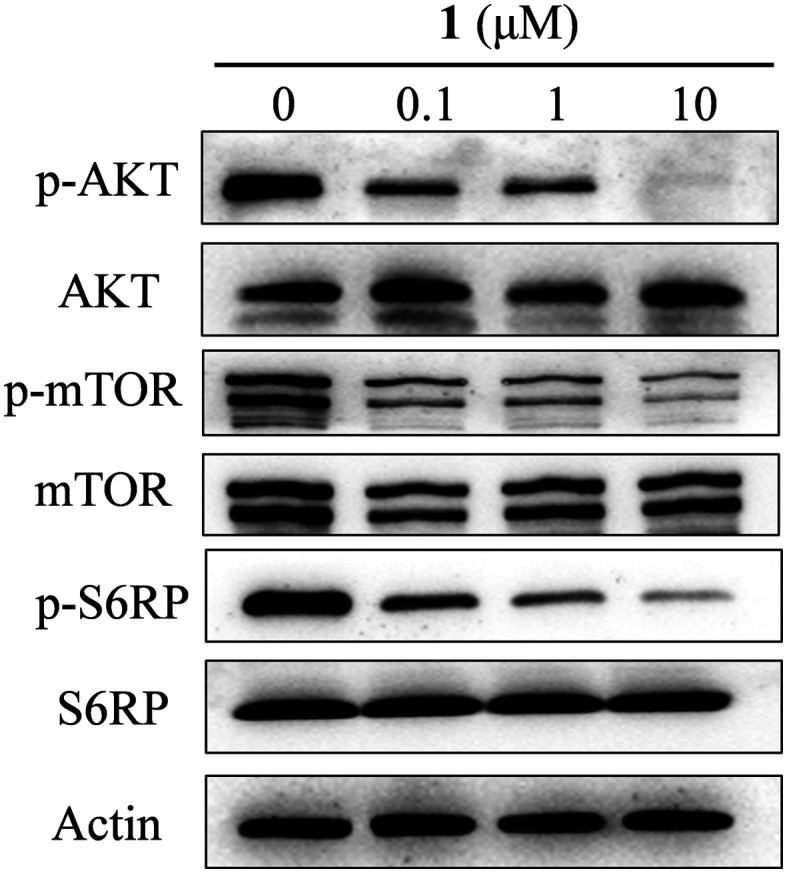
To explore the anti-cancer mechanism triggered by conjugate 1, its inhibitory activity against PI3K downstream effectors in 22Rv1 cells was analyzed by Western blotting. As shown in Fig. 4, conjugate 1 suppressed the expression levels of phosphorylated-AKT (S473 site), mTOR and S6RP in a dose-dependent manner. These immunoblotting results were consistent with the aforementioned kinase inhibitory activity and cellular antiproliferative activity exerted by conjugate 1.
Fig. 4. Conjugate 1 efficiently inhibited the PI3K pathway in 22Rv1 cells. The cells were treated with the indicated concentrations for 3 h.
To further elucidate its anticancer mechanism, cell cycle analysis was performed with flow cytometry. As shown in Fig. 5, conjugate 1 dose-dependently induced cell cycle arrest at the G0/G1 phase in 22Rv1 cells.
Fig. 5. Conjugate 1 dose-dependently induced cell cycle arrest in 22Rv1 cells. (A) Flow cytometry analysis of the cell cycle distribution following treatment of 22Rv1 cells with 1 at indicated concentrations for 24 h; (B) percentage of 22Rv1 cells in different phases of the cell cycle.

Subsequently, conjugate 1 underwent in vivo antitumor assessment in a 22Rv1 prostate cancer xenograft model. As depicted in Fig. 6, conjugate 1 demonstrated in vivo antitumor effectiveness in a dose-dependent manner. Particularly, the intraperitoneal (IP) administration of 20 mg kg−1 led to 53% tumor growth inhibition (TGI). Throughout the treatment regimen, the model mice exhibited overall good tolerance, as evidenced by stable body weight profiles.
Fig. 6. In vivo antitumor efficacy of conjugate 1 in a 22Rv1 prostate cancer xenograft model. (A) Tumor volume changes after treatment with 1 in the xenograft model; (B) tumor weight changes after treatment with 1 in the xenograft model; (C) changes in anatomical sections of tumors in xenograft models after treatment with 1; (D) body weight changes after treatment with 1 in the xenograft model.

Conclusion
In summary, a novel PSMA–PI3K small molecule drug conjugate 1 was rationally designed and synthesized through a hybridization strategy. Conjugate 1 not only maintains potent inhibition against PI3K but also demonstrates significant binding affinity with PSMA in prostate cancer cells, thereby enabling the simultaneous targeting of both PI3K and PSMA. Notably, conjugate 1 exhibited greater antiproliferative activities on PSMA-positive cancer cells compared to PSMA-negative counterparts. Furthermore, the conjugate potently suppressed the phosphorylation of PI3K main downstream effectors and induced cell cycle arrest. Impressively, conjugate 1 exhibited dose-dependent anticancer efficacy in a 22Rv1 xenograft model. Overall, this work offers valuable insights into the design of small molecule drug conjugates using PI3K inhibitors. Moreover, it presents a promising chemical scaffold that can be further refined for the creation of targeted anticancer therapeutics.
Experimental section
General information
All starting materials, solvents and reagents are from commercial sources and are usable without further purification. NMR spectra were recorded on a JEOL 400 MHz NMR or Bruker 500 MHz NMR spectrometer. NMR spectra were obtained with dimethyl sulfoxide (DMSO-d6) or chloroform (CDCl3) as the solvent and tetramethylsilane (TMS) as the internal standard. Chemical shifts are given in δ units of ppm (in NMR description, s = singlet, d = doublet, t = triplet, q = quartet and m = multiple). HRMS spectra were obtained in positive ion mode using a Thermo LCQ Deca XP Maz mass spectrometer with electrospray ionization (ESI). For the HRMS analysis of the bromine-containing compounds, their isotopes were measured as 79. The purity of end compounds was confirmed to be >95% by UPLC analysis (Waters ACQUITY UPLC ICLASS QDA), where the method follows: water with 0.1% formic acid as mobile phase A, acetonitrile as mobile phase B, Waters C18 plus column (2.7 μm, 4.6 mm × 50 mm), 25 °C, 5% acetonitrile at 0 min, 95% acetonitrile at 4 min and lasted for 2.0 min, 5% acetonitrile at 6.5 min and lasted for 1.0 min, 0.3 mL min−1, UV detector at 254 nm.
Synthesis of the PSMA–PI3K conjugate
tert-Butyl 4-((6-(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-4-methylquinazolin-8-yl)oxy)butanoate (3)
Compound 2 (1.2 g, 1.0 eq.), 2,4-difluoro-N-(2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)benzenesulfonamide (1.61 g, 1.2 eq.) and 2 M K2CO3 aqueous solution (3.0 eq.) in dioxane were degassed with Ar, followed by the addition of PdCl2(dppf) (231 mg, 0.10 eq.). The mixture was degassed again and backfilled with argon (three cycles) and then stirred at 100 °C for 5 h under Ar atmosphere. The reaction mixture was cooled to rt, diluted with water and EtOAc, and acidified with 2 M HCl solution to a mixture pH of 5–6. The two layers were separated and the aqueous layer was extracted with EtOAc (30 mL × 2). The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by column chromatography (silica gel, DCM/MeOH = 50 : 1, v/v) to give the desired product 3 as a brown solid (1.54 g, 81% yield). 1H NMR (500 MHz, CDCl3) δ 9.21 (s, 1H), 8.18 (d, J = 2.3 Hz, 1H), 8.03 (d, J = 2.2 Hz, 1H), 7.92–7.84 (m, 1H), 7.65 (d, J = 1.7 Hz, 1H), 7.37–7.30 (m, 2H), 6.96 (t, J = 8.3 Hz, 2H), 4.33 (t, J = 6.5 Hz, 2H), 3.96 (s, 3H), 2.97 (s, 3H), 2.54 (t, J = 7.2 Hz, 2H), 2.35–2.26 (m, 2H), 1.44 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 172.51, 166.39 (dd, JC–F = 259, 11.5 Hz), 165.30, 159.84 (dd, JC–F = 259, 12.1 Hz), 155.18, 154.65, 153.97, 141.60, 141.04, 136.87, 132.59 (d, JC–F = 10 Hz), 130.41, 127.20, 125.68, 123.54 (dd, JC–F = 13.6, 3.8 Hz), 120.66, 114.14, 112.09 (dd, JC–F = 22.3, 3.5 Hz), 105.95 (t, JC–F = 25.0 Hz), 80.71, 68.51, 54.33, 31.95, 28.24, 24.42, 22.42. HRMS (ESI): m/z [M + H]+ calcd for C29H31O6N4F2S, 601.1927; found 601.1929, Δ 0.30 ppm.
4-((6-(5-((2,4-Difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-4-methylquinazolin-8-yl)oxy)butanoic acid (4)
Compound 3 (1.0 g, 0.9 eq.) was dissolved in DCM, and TFA (428 μL, 3 eq.) was added and stirred at room temperature for 3–4 h. After the reaction was completed, the reaction mixture was concentrated under vacuum to remove the vast majority of TFA. The mixture was diluted with water and EtOAc, and saturated sodium bicarbonate solution was added to achieve a mixture pH of 5–6. The two layers were separated and the mixture was extracted with EtOAc (30 mL × 2) to extract the aqueous layer. The combined organic layers were washed with water and brine, dried with anhydrous Na2SO4, filtered and concentrated to give the pale-yellow product 4 (0.92 g, 93% yield). 1H NMR (500 MHz, DMSO-d6) δ 12.20 (s, 1H), 10.35 (s, 1H), 9.08 (s, 1H), 8.57 (d, J = 2.4 Hz, 1H), 8.12 (d, J = 2.4 Hz, 1H), 7.92 (d, J = 1.7 Hz, 1H), 7.76 (td, J = 8.5, 6.2 Hz, 1H), 7.69–7.54 (m, 2H), 7.25–7.19 (m, 1H), 4.31 (t, J = 6.4 Hz, 2H), 3.65 (s, 3H), 3.17 (d, J = 4.2 Hz, 2H), 2.95 (s, 3H), 2.14–2.05 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 174.16, 168.17, 165.09 (dd, JC–F = 254, 12.1 Hz), 159.45 (dd, JC–F = 258, 13.8 Hz), 158.11, 154.56, 153.21, 143.51, 140.64, 135.76, 135.32, 131.88 (d, JC–F = 10.4 Hz), 129.15, 125.25 (dd, JC–F = 14.5, 3.8 Hz), 124.95, 119.54, 113.98, 111.87 (dd, JC–F = 22.8, 3.3 Hz), 105.84 (t, JC–F = 26.2), 67.86, 53.42, 30.11, 24.18, 22.06. HRMS (ESI): m/z [M + H]+ calcd for C25H23O6N4F2S, 545.1301; found 545.1296, Δ −0.86 ppm.
Tribenzyl (10S,14S)-2,2-dimethyl-4,12-dioxo-3-oxa-5,11,13-triazahexadecane-10,14,16-tricarboxylate (6)
Triphosgene (557 mg, 0.35 eq.) was placed in a reaction vessel cooled to 0 °C under Ar atmosphere and dissolved in DCM (80 mL). Benzyl N6-(tert-butoxycarbonyl)-l-lysinate (2.0 g, 1.0 eq.) was dissolved in DCM (20 mL) and DIPEA (2.3 mL, 2.5 eq.) was added and dissolved by shaking. The mixture was added dropwise to a stirred solution of triphosgene over 0.5 h. The reaction system was brought to room temperature and then stirred for 10 min. The prepared mixture of dibenzyl l-glutamate (1.95 g, 1.0 eq.) and DIPEA (2.3 mL, 2.5 eq.) in DCM (20 mL) was then added to the reaction vessel over 10 min at 0 °C. The reaction was stirred for an additional 0.5 h and monitored by LC-MS at 0.5 h intervals. Upon completion of the reaction, the reaction solution was concentrated by rotary evaporation, EtOAc and H2O extraction was performed, and the product was recovered from the organic layer. Purification was performed by rapid chromatography on silica gel using a 10–60% EtOAc/DCM gradient. The pure fractions were mixed, rotary evaporated and dried under high vacuum to the white product 6 (2.9 g, 71% yield). 1H NMR (400 MHz, CDCl3) δ 7.37–7.28 (m, 15H), 5.39 (s, 2H), 5.19–5.07 (m, 6H), 4.68 (s, 1H), 4.56 (dd, J = 8.2, 5.0 Hz, 1H), 4.49 (dd, J = 7.6, 5.0 Hz, 1H), 3.07–2.97 (m, 2H), 2.50–2.34 (m, 2H), 2.19 (dtd, J = 13.6, 6.9, 5.0 Hz, 1H), 1.96 (dtd, J = 14.6, 8.4, 6.4 Hz, 1H), 1.84–1.71 (m, 1H), 1.71–1.59 (m, 1H), 1.48–1.37 (m, 12H), 1.36–1.15 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 173.32, 172.96, 172.87, 157.04, 156.34, 135.93, 135.59, 135.40, 128.71, 128.70, 128.67, 128.52, 128.50, 128.39, 128.35, 128.33, 79.27, 77.41, 77.16, 76.91, 67.35, 67.10, 66.56, 52.98, 52.64, 40.16, 32.37, 30.43, 29.51, 28.56, 28.05, 22.39. HRMS (ESI): m/z [M + H]+ calcd for C38H48O9N3, 690.3385; found 690.3381, Δ −0.60 ppm.
Dibenzyl (((S)-1-(benzyloxy)-6-((S)-2-((tert-butoxycarbonyl)amino)-3-(naphthalen-2-yl)propanamido)-1-oxohexan-2-yl)carbamoyl)-l-glutamate (7)
Compound 6 (1.5 g, 1.0 eq.) was dissolved in a mixed solution of TFA/DCM = 1/1 and stirred at room temperature for 0.5 h. The reaction mixture was vacuum concentrated to give the white intermediate. To a solution of the intermediate (1 g, 1.0 eq.) in DCM (20 mL) at room temperature were added (S)-2-((tert-butoxycarbonyl)amino)-3-(naphthalen-2-yl)propanoic acid (589 mg, 1.1 eq.), 2-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU, 968 mg, 1.5 eq.) and DIPEA (1.2 mL, 4.0 eq.), and the mixture was stirred for 12 hours at room temperature. The reaction mixture was concentrated under vacuum, diluted with water and EtOAc, the two layers were separated and the aqueous layer was extracted with EtOAc (30 mL × 2). The combined organic layers were washed with water and brine, dried with anhydrous Na2SO4, filtered and concentrated. The residue was purified by column chromatography (silica gel, DCM/MeOH = 50 : 1, v/v) to give the light yellow product 7 (1.23 g, 82% yield). 1H NMR (400 MHz, CDCl3) δ 7.71 (s, 1H), 7.68 (d, J = 8.2 Hz, 1H), 7.55 (d, J = 8.2 Hz, 1H), 7.44 (ddd, J = 8.1, 6.8, 1.3 Hz, 1H), 7.39–7.29 (m, 17H), 7.28–7.17 (m, 2H), 6.81 (s, 1H), 6.46 (s, 2H), 5.27–5.18 (m, 3H), 5.13–5.05 (m, 3H), 4.87–4.74 (m, 2H), 4.54 (td, J = 7.8, 3.8 Hz, 1H), 3.50–3.39 (m, 1H), 3.21 (dd, J = 13.9, 4.3 Hz, 1H), 3.13–2.99 (m, 2H), 2.63–2.39 (m, 2H), 2.35–2.22 (m, 1H), 2.10–1.96 (m, 1H), 1.70 (tt, J = 12.3, 4.3 Hz, 1H), 1.59 (tt, J = 10.1, 4.1 Hz, 1H), 1.37–1.22 (m, 3H), 1.19 (s, 9H), 1.15–1.06 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 174.57, 174.31, 173.10, 172.84, 157.28, 156.82, 136.07, 136.05, 135.20, 134.84, 133.51, 132.33, 128.73, 128.61, 128.58, 128.53, 128.31, 128.29, 128.24, 128.17, 128.13, 127.76, 127.59, 127.04, 126.02, 125.55, 79.91, 77.42, 77.16, 76.91, 67.44, 66.64, 66.39, 55.83, 52.75, 52.13, 39.94, 39.48, 32.36, 30.15, 29.81, 29.28, 28.57, 28.31, 23.27. HRMS (ESI): m/z [M + H]+ calcd for C51H59O10N4, 887.4226; found 887.4220, Δ −0.63 ppm.
Dibenzyl (((S)-1-(benzyloxy)-6-((S)-2-((1r,4S)-4-(((tert-butoxycarbonyl)amino)methyl)cyclohexane-1-carboxamido)-3-(naphthalen-2-yl)propanamido)-1-oxohexan-2-yl)carbamoyl)-l-glutamate (8)
Compound 7 (1 g, 1.0 eq.) was combined with (1r,4r)-4-(((tert-butoxycarbonyl)amino)methyl)cyclohexane-1-carboxylic acid (360 mg, 1.1 eq.) to obtain a light yellow product 8 (1.1 g, 85% yield) using the same method as compound 7. 1H NMR (500 MHz, CDCl3) δ 7.71 (d, J = 8.2 Hz, 1H), 7.63 (s, 1H), 7.58 (d, J = 8.2 Hz, 1H), 7.45 (t, J = 7.5 Hz, 1H), 7.41–7.20 (m, 17H), 7.14 (d, J = 8.4 Hz, 1H), 7.07 (s, 1H), 6.14 (d, J = 7.8 Hz, 1H), 5.29–5.03 (m, 6H), 4.80 (d, J = 5.8 Hz, 1H), 4.49 (t, J = 6.1 Hz, 1H), 4.43 (td, J = 10.0, 3.6 Hz, 1H), 3.40–3.30 (m, 1H), 3.19 (dd, J = 13.8, 5.3 Hz, 1H), 3.09–2.98 (m, 1H), 2.93–2.83 (m, 1H), 2.82–2.79 (m, 6H), 2.59–2.43 (m, 2H), 2.27 (td, J = 8.9, 5.7 Hz, 1H), 2.01 (td, J = 8.3, 5.7 Hz, 1H), 1.79 (t, J = 13.8 Hz, 2H), 1.62 (d, J = 12.5 Hz, 2H), 1.58–1.46 (m, 2H), 1.44–1.41 (m, 9H), 1.31–1.23 (m, 3H), 1.22–0.93 (m, 2H), 0.90–0.80 (m, 1H), 0.76–0.53 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 177.16, 175.21, 174.09, 173.05, 172.74, 157.52, 156.06, 136.03, 136.01, 135.22, 134.77, 133.44, 132.33, 128.87, 128.75, 128.62, 128.57, 128.30, 128.27, 128.25, 128.13, 128.05, 127.83, 127.49, 127.22, 126.08, 125.59, 79.14, 77.42, 77.16, 76.91, 67.50, 66.63, 66.44, 54.61, 52.80, 51.85, 46.62, 44.53, 39.80, 39.41, 37.41, 32.46, 30.28, 29.95, 29.56, 29.51, 29.06, 28.87, 28.53, 27.98, 23.36. HRMS (ESI): m/z [M + H]+ calcd for C59H72O11N5, 1026.5223; found 1026.5231, Δ 0.78 ppm.
Dibenzyl (((2S)-1-(benzyloxy)-6-((2S)-2-(4-((4-((6-(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-4-methylquinazolin-8-yl)oxy)butanamido)methyl)cyclohexane-1-carboxamido)-3-(naphthalen-2-yl)propanamido)-1-oxohexan-2-yl)carbamoyl)-l-glutamate (9)
Compound 8 (1.0 g, 1.1 eq.) and 4 (534 mg, 1.0 eq.) obtained previously were subjected to the same method as compound 7 to give a light yellow product 9 (1.1 g, 72% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.08 (s, 1H), 8.56 (d, J = 2.3 Hz, 1H), 8.11 (d, J = 2.4 Hz, 1H), 7.91 (d, J = 1.6 Hz, 2H), 7.89 (s, 1H), 7.85–7.79 (m, 2H), 7.79–7.73 (m, 3H), 7.67 (s, 1H), 7.61 (d, J = 1.8 Hz, 1H), 7.61–7.55 (m, 1H), 7.46–7.40 (m, 2H), 7.40–7.36 (m, 1H), 7.36–7.28 (m, 15H), 7.22 (td, J = 8.3, 2.2 Hz, 1H), 6.47 (dd, J = 23.1, 8.2 Hz, 2H), 5.16–5.03 (m, 6H), 4.53 (td, J = 8.1, 5.2 Hz, 1H), 4.33–4.21 (m, 3H), 4.12 (td, J = 8.0, 5.5 Hz, 1H), 3.66 (s, 3H), 3.16–3.12 (m, 1H), 3.09 (dd, J = 10.4, 3.2 Hz, 1H), 3.05–2.96 (m, 1H), 2.95 (s, 3H), 2.93–2.88 (m, 1H), 2.88–2.83 (m, 1H), 2.45–2.32 (m, 4H), 2.14–2.04 (m, 2H), 2.04–1.95 (m, 1H), 1.82 (td, J = 14.6, 8.3 Hz, 1H), 1.69–1.56 (m, 1H), 1.56–1.44 (m, 2H), 1.33–1.14 (m, 11H), 1.09–0.95 (m, 1H), 0.86–0.72 (m, 1H). 13C NMR (126 MHz, DMSO-d6) δ 174.92, 172.86, 172.39, 171.98, 171.46, 170.97, 168.17, 165.12 (dd, JC–F = 254.4, 12.2 Hz), 159.27 (dd, JC–F = 258.1, 13.5 Hz), 158.08, 157.19, 154.63, 153.15, 143.48, 140.65, 136.10, 135.97, 135.90, 135.77, 135.72, 135.27, 132.87, 131.85 (d, JC–F = 10.8 Hz), 131.76, 129.15, 128.40, 128.04, 128.00, 127.97, 127.89, 127.87, 127.80, 127.78, 127.43, 127.41, 127.28, 127.24, 125.89, 125.30 (dd, JC–F = 14.7, 3.5 Hz), 124.95, 119.53, 113.93, 111.94, 111.82 (dd, JC–F = 22.3, 3.1 Hz), 105.81 (t, JC–F = 26.2 Hz), 68.34, 65.97, 65.77, 65.55, 53.67, 53.40, 52.59, 51.81, 44.72, 43.66, 38.20, 38.17, 36.97, 31.75, 31.30, 29.69, 29.53, 28.76, 28.57, 28.32, 27.09, 24.87, 22.45, 22.04. HRMS (ESI): m/z [M + H]+ calcd for C79H84O14N9F2S, 1452.5821; found 1452.5824, Δ 0.21 ppm.
(((S)-1-Carboxy-5-((S)-2-((1s,4R)-4-((4-((6-(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)-4-methylquinazolin-8-yl)oxy)butanamido)methyl)cyclohexane-1-carboxamido)-3-(naphthalen-2-yl)propanamido)pentyl)carbamoyl)-l-glutamic acid (1)
A mixture of compound 9 (500 mg, 1.0 eq.) and 10% Pd/C (2.0 g, 5.0 eq.) in methanol (20 mL) was stirred in a test tube equipped with a H2 balloon at 25 °C. After 24 h, the reaction system was filtered and the catalyst on the filter paper was washed with methanol (5 mL × 3) and DMSO (1 mL × 3). The combined filtrates were concentrated under vacuum. The mixture was purified by preparative HPLC (MeCN/H2O = 1 : 1, v/v) to afford product 1 as a bronze solid (203 mg, 50% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.03 (s, 1H), 8.46 (d, J = 2.3 Hz, 1H), 8.02 (d, J = 2.3 Hz, 1H), 7.93–7.86 (m, 2H), 7.85 (d, J = 1.7 Hz, 1H), 7.81–7.78 (m, 2H), 7.76–7.70 (m, 3H), 7.63 (d, J = 1.7 Hz, 1H), 7.55 (d, J = 1.7 Hz, 1H), 7.52 (ddd, J = 10.3, 9.1, 2.5 Hz, 1H), 7.43–7.36 (m, 2H), 7.35 (dd, J = 8.5, 1.7 Hz, 1H), 7.17 (td, J = 8.7, 2.9 Hz, 1H), 6.26 (dd, J = 10.9, 8.2 Hz, 2H), 4.48 (td, J = 8.9, 5.2 Hz, 1H), 4.25 (t, J = 6.3 Hz, 2H), 4.06 (td, J = 8.2, 5.4 Hz, 1H), 3.98 (td, J = 8.1, 5.1 Hz, 1H), 3.63 (s, 3H), 3.06 (dd, J = 13.6, 5.0 Hz, 1H), 3.03–2.97 (m, 1H), 2.94 (dd, J = 7.4, 5.8 Hz, 1H), 2.91 (s, 3H), 2.89–2.81 (m, 3H), 2.31 (t, J = 7.5 Hz, 2H), 2.26–2.13 (m, 2H), 2.08–2.00 (m, 3H), 1.92–1.81 (m, 1H), 1.73–1.65 (m, 1H), 1.65–1.52 (m, 3H), 1.47–1.39 (m, 2H), 1.31–1.23 (m, 2H), 1.24–1.10 (m, 5H), 1.06–0.94 (m, 1H), 0.85–0.70 (m, 3H).13C NMR (126 MHz, DMSO-d6) δ 175.48, 175.19, 174.90, 174.51, 172.14, 171.39, 168.48, 165.00 (dd, JC–F = 253.7, 12.0 Hz), 159.63 (dd, JC–F = 257.4, 13.3 Hz), 158.16, 157.70, 154.74, 153.24, 140.70, 136.57, 135.85, 133.11, 132.08, 132.01, 129.23, 128.09, 127.69, 127.60, 127.51, 126.46, 126.20, 125.63, 125.19, 113.92, 112.12, 111.87 (dd, JC–F = 22.1, 12.0 Hz), 105.87 (t, JC–F = 26.4 Hz), 68.48, 53.99, 53.59, 52.80, 52.41, 44.98, 43.94, 38.64, 38.30, 37.13, 32.04, 31.93, 30.61, 29.84, 29.69, 28.97, 28.89, 28.48, 28.23, 25.15, 22.76, 22.20. HRMS (ESI): m/z [M + H]+ calcd for C58H66O14N9F2S, 1182.4413; found 1182.4424, Δ 1.01 ppm.
Cell viability assay
Human prostate cancer cell lines 22Rv1, LNCaP and PC3 were purchased from China Infrastructure of Cell Line Resources (Beijing, China). All of the cells were cultured in RPMI 1640 medium with 10% fetal bovine serum. Cell viability was assessed by CellTiter-Glo Luminescent Cell Viability Assay (Promega, USA). The cells were plated in 96-well culture plates with 3 × 103 cells per well and incubated overnight. Then the cells were treated with the test compound at different concentrations. After 72 h incubation, 30 μl of CellTiter Glo reagent was added into each well. The contents were mixed on an orbital shaker for 2 minutes to induce cell lysis, and the plate was incubated at room temperature for 10 minutes. The luminescence was recorded by a BioTek Synergy H1 Plate Reader. The half maximum inhibitory concentration (IC50) was then calculated by GraphPad Prism 8.1 (GraphPad Software, San Diego, USA).
Molecular docking study
The X-ray crystal structures of PI3Kα (PDB ID: 4JPS) and PSMA (PDB ID: 5O5T) were retrieved from the Protein Data Bank (PDB). Molecular docking of conjugate 1 was carried out using the Schrödinger Maestro 2021-2. The binding-sites of PI3Kα and PSMA with conjugate 1 were analyzed and the high-resolution image files were produced using PyMOL.
Immunoblotting
The cells were harvested and lysed in RIPA lysis buffer supplemented with a protease inhibitor cocktail and phosphatase inhibitor (Topscience, Shanghai, China). The protein concentrations were measured by using a BCA Protein Assay Kit (Lablead Biotech, China). A total of 25 μg of protein was resolved by via SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. Anti-phosphor-mTOR, anti-mTOR, anti-PMSA, anti-phosphor-AKT (Ser473), anti-AKT, anti-phosphor-S6RP, anti-S6RP, anti-β-actin were incubated overnight at 4 °C. Then, the membranes were incubated with peroxidase-conjugated anti-rabbit or mouse IgG secondary antibody. Finally, the protein bands were visualized with the chemiluminescence method (Pierce™ ECL Western blotting Substrate, Thermo Fisher Scientific, USA).
Cell cycle analysis
A total of 100 000 22Rv1 cells were treated with various concentrations of conjugate 1 for 24 h. Then the cells were harvested, fixed with ice-cold 70% ethanol and stained with PI/RNase solution (Thermo Fisher Scientific). Data were acquired by flow cytometry (BD Verse, BD Biosciences, San Jose) with a laser at 488 nm wavelength and analyzed by FlowJo software (Three Star, Ashland).
Cell binding assay
The 22Rv1 cells were treated with conjugate 1 for 6 h. Then the cells were harvested and washed with PBS 3 times. The cells were incubated with anti-PMSA (Proteintech Inc, Rosemont, USA) or IgG (1 : 100) for 1 h at 4 °C. After being washed with PBS, the cells were incubated with secondary antibody (Alexa Fluor 488 donkey anti-rabbit IgG, Invitrogen) for 30 min at 4 °C. Then the cells were washed with PBS and tested by flow cytometry.
Animal study
Male Balb/c athymic nude mice of 8–10 weeks old (SPF Biotech, Beijing, China) were subcutaneously implanted with 1 × 107 22Rv1 cells in 0.2 mL Matrigel solution in the right flank. When the tumor volume reached 1000 mm3, tumor issues were harvested sterilely, and tumor cells were extracted from the tissue homogenate. Then, the mice were implanted with 2 × 106 tumor cells each in the right flank. Six days later when the average tumor volume reached 100 mm3, the mice were randomized and received treatment. The test compound, which was dissolved in saline with 1% PEG400, was intraperitoneally administered once per day and taxol was intraperitoneally injected every week for 3 doses. The vehicle saline with 1% PEG400 was also intraperitoneally administered. All the groups were administered with 0.2 mL per 20 g of mice weight. Tumor volume and body weight were measured twice a week. Tumor volume was calculated as V = 1/2 × L × W2, where L is the maximum length of tumor, and W is the maximum width of tumor. Data including tumor volume and body weight were collected and values were analyzed by GraphPad Prism 8.
All procedures were approved by the Ethics Committee for Animal Experiments of the Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College and conducted under the Guidelines for Animal Experiments of Peking Union Medical College (Beijing, China).
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There is no conflict of interest to declare.
Supplementary Material
Acknowledgments
Financial support from the CAMS Innovation Fund for Medical Sciences (CIFMS) (2021-I2M-1-026, 2022-I2M-1-013 and 2022-I2M-1-014) is gratefully acknowledged. We thank Xinhua Liu for his help on molecular modeling.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00246f
References
- Patel T. K. Adhikari N. Amin S. A. Biswas S. Jha T. Ghosh B. Small Molecule Drug Conjugates (SMDCs): An Emerging Strategy for Anticancer Drug Design and Discovery. New J. Chem. 2021;45(12):5291–5321. doi: 10.1039/D0NJ04134C. [DOI] [Google Scholar]
- Zhuang C. Guan X. Ma H. Cong H. Zhang W. Miao Z. Small Molecule-Drug Conjugates: A Novel Strategy for Cancer-Targeted Treatment. Eur. J. Med. Chem. 2019;163:883–895. doi: 10.1016/j.ejmech.2018.12.035. [DOI] [PubMed] [Google Scholar]
- Srinivasarao M. Low P. S. Ligand-Targeted Drug Delivery. Chem. Rev. 2017;117(19):12133–12164. doi: 10.1021/acs.chemrev.7b00013. [DOI] [PubMed] [Google Scholar]
- Amin T. U. Emara R. Pal A. Aldawod H. Jiang G. Liang D. Haque Tuhin M. T. Balgoname A. Patel A. D. Alhamadsheh M. M. Enhancing the Safety and Efficacy of PSMA-Based Small-Molecule Drug Conjugates by Linker Stabilization and Conjugation to Transthyretin Binding Ligand. J. Med. Chem. 2022;65(22):15473–15486. doi: 10.1021/acs.jmedchem.2c01423. [DOI] [PubMed] [Google Scholar]
- Engelman J. A. Luo J. Cantley L. C. The Evolution of Phosphatidylinositol 3-Kinases as Regulators of Growth and Metabolism. Nat. Rev. Genet. 2006;7(8):606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- Janku F. Yap T. A. Meric-Bernstam F. Targeting the PI3K Pathway in Cancer: Are We Making Headway? Nat. Rev. Clin. Oncol. 2018;15(5):273–291. doi: 10.1038/nrclinonc.2018.28. [DOI] [PubMed] [Google Scholar]
- Thorpe L. M. Yuzugullu H. Zhao J. J. PI3K in Cancer: Divergent Roles of Isoforms, Modes of Activation and Therapeutic Targeting. Nat. Rev. Cancer. 2015;15(1):7–24. doi: 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counihan J. L. Grossman E. A. Nomura D. K. Cancer Metabolism: Current Understanding and Therapies. Chem. Rev. 2018;118(14):6893–6923. doi: 10.1021/acs.chemrev.7b00775. [DOI] [PubMed] [Google Scholar]
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/217759s000lbl.pdf (accessed March 24, 2023)
- Peng Y. Wang Y. Zhou C. Mei W. Zeng C. PI3K/Akt/mTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front. Oncol. 2022;12:819128. doi: 10.3389/fonc.2022.819128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curigliano G. Shah R. R. Safety and Tolerability of Phosphatidylinositol-3-Kinase (PI3K) Inhibitors in Oncology. Drug Saf. 2019;42(2):247–262. doi: 10.1007/s40264-018-0778-4. [DOI] [PubMed] [Google Scholar]
- Dreyling M. Santoro A. Mollica L. Leppä S. Follows G. Lenz G. Kim W. S. Nagler A. Dimou M. Demeter J. Özcan M. Kosinova M. Bouabdallah K. Morschhauser F. Stevens D. A. Trevarthen D. Munoz J. Rodrigues L. Hiemeyer F. Miriyala A. Garcia-Vargas J. Childs B. H. Zinzani P. L. Long-Term Safety and Efficacy of the PI3K Inhibitor Copanlisib in Patients with Relapsed or Refractory Indolent Lymphoma: 2-Year Follow-up of the CHRONOS-1 Study. Am. J. Hematol. 2020;95(4):362–371. doi: 10.1002/ajh.25711. [DOI] [PubMed] [Google Scholar]
- Esposito A. Viale G. Curigliano G. Safety, Tolerability, and Management of Toxic Effects of Phosphatidylinositol 3-Kinase Inhibitor Treatment in Patients With Cancer: A Review. JAMA Oncol. 2019;5(9):1347. doi: 10.1001/jamaoncol.2019.0034. [DOI] [PubMed] [Google Scholar]
- Werner R. A. Derlin T. Lapa C. Sheikbahaei S. Higuchi T. Giesel F. L. Behr S. Drzezga A. Kimura H. Buck A. K. Bengel F. M. Pomper M. G. Gorin M. A. Rowe S. P. 18F-Labeled, PSMA-Targeted Radiotracers: Leveraging the Advantages of Radiofluorination for Prostate Cancer Molecular Imaging. Theranostics. 2020;10(1):1–16. doi: 10.7150/thno.37894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. Cai P. Feng Y. Sun Z. Wang Y. Chen Y. Zhang W. Liu N. Zhou Z. In Vitro and in Vivo Comparative Study of a Novel 68Ga-Labeled PSMA-Targeted Inhibitor and 68Ga-PSMA-11. Sci. Rep. 2021;11(1):19122. doi: 10.1038/s41598-021-98555-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroenke M. Schweiger L. Horn T. Haller B. Schwamborn K. Wurzer A. Maurer T. Wester H.-J. Eiber M. Rauscher I. Validation of 18F-rhPSMA-7 and 18F-rhPSMA-7.3 PET Imaging Results with Histopathology from Salvage Surgery in Patients with Biochemical Recurrence of Prostate Cancer. J. Nucl. Med. 2022;63(12):1809–1814. doi: 10.2967/jnumed.121.263707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennrich U. Eder M. [177Lu]Lu-PSMA-617 (PluvictoTM): The First FDA-Approved Radiotherapeutical for Treatment of Prostate Cancer. Pharmaceuticals. 2022;15(10):1292. doi: 10.3390/ph15101292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor O. de Bono J. Chi K. N. Fizazi K. Herrmann K. Rahbar K. Tagawa S. T. Nordquist L. T. Vaishampayan N. El-Haddad G. Park C. H. Beer T. M. Armour A. Pérez-Contreras W. J. DeSilvio M. Kpamegan E. Gericke G. Messmann R. A. Morris M. J. Krause B. J. Lutetium-177–PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021;385(12):1091–1103. doi: 10.1056/NEJMoa2107322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanenkov Y. A. Machulkin A. E. Garanina A. S. Skvortsov D. A. Uspenskaya A. A. Deyneka E. V. Trofimenko A. V. Beloglazkina E. K. Zyk N. V. Koteliansky V. E. Bezrukov D. S. Aladinskaya A. V. Vorobyeva N. S. Puchinina M. M. Riabykh G. K. Sofronova A. A. Malyshev A. S. Majouga A. G. Synthesis and Biological Evaluation of Doxorubicin-Containing Conjugate Targeting PSMA. Bioorg. Med. Chem. Lett. 2019;29(10):1246–1255. doi: 10.1016/j.bmcl.2019.01.040. [DOI] [PubMed] [Google Scholar]
- Machulkin A. E. Skvortsov D. A. Ivanenkov Y. A. Ber A. P. Kavalchuk M. V. Aladinskaya A. V. Uspenskaya A. A. Shafikov R. R. Plotnikova E. A. Yakubovskaya R. I. Nimenko E. A. Zyk N. U. Beloglazkina E. K. Zyk N. V. Koteliansky V. E. Majouga A. G. Synthesis and Biological Evaluation of PSMA-Targeting Paclitaxel Conjugates. Bioorg. Med. Chem. Lett. 2019;29(16):2229–2235. doi: 10.1016/j.bmcl.2019.06.035. [DOI] [PubMed] [Google Scholar]
- Lv Q. Yang J. Zhang R. Yang Z. Yang Z. Wang Y. Xu Y. He Z. Prostate-Specific Membrane Antigen Targeted Therapy of Prostate Cancer Using a DUPA–Paclitaxel Conjugate. Mol. Pharmaceutics. 2018;15(5):1842–1852. doi: 10.1021/acs.molpharmaceut.8b00026. [DOI] [PubMed] [Google Scholar]
- Machulkin A. E. Uspenskaya A. A. Zyk N. Y. Nimenko E. A. Ber A. P. Petrov S. A. Shafikov R. R. Skvortsov D. A. Smirnova G. B. Borisova Y. A. Pokrovsky V. S. Kolmogorov V. S. Vaneev A. N. Ivanenkov Y. A. Khudyakov A. D. Kovalev S. V. Erofeev A. S. Gorelkin P. V. Beloglazkina E. K. Zyk N. V. Khazanova E. S. Majouga A. G. PSMA-Targeted Small-Molecule Docetaxel Conjugate: Synthesis and Preclinical Evaluation. Eur. J. Med. Chem. 2022;227:113936. doi: 10.1016/j.ejmech.2021.113936. [DOI] [PubMed] [Google Scholar]
- Murce E. Spaan E. Beekman S. van den Brink L. Handula M. Stuurman D. de Ridder C. Dalm S. U. Seimbille Y. Synthesis and Evaluation of ePSMA-DM1: A New Theranostic Small-Molecule Drug Conjugate (T-SMDC) for Prostate Cancer. Pharmaceuticals. 2023;16(8):1072. doi: 10.3390/ph16081072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olatunji F. P. Pun M. Herman J. W. Romero O. Maniatopoulos M. Latoche J. D. Parise R. A. Guo J. Beumer J. H. Anderson C. J. Berkman C. E. Modular Smart Molecules for PSMA-Targeted Chemotherapy. Mol. Cancer Ther. 2022;21(11):1701–1709. doi: 10.1158/1535-7163.MCT-22-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. Shirke A. Walker E. Sun R. Ramamurthy G. Wang J. Shan L. Mangadlao J. Dong Z. Li J. Wang Z. Schluchter M. Luo D. Wang Y. Stauffer S. Brady-Kalnay S. Hoimes C. Lee Z. Basilion J. P. Small Molecule-Based Prodrug Targeting Prostate Specific Membrane Antigen for the Treatment of Prostate Cancer. Cancers. 2021;13(3):417. doi: 10.3390/cancers13030417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denmeade S. R. Mhaka A. M. Rosen D. M. Brennen W. N. Dalrymple S. Dach I. Olesen C. Gurel B. DeMarzo A. M. Wilding G. Carducci M. A. Dionne C. A. Møller J. V. Nissen P. Christensen S. B. Isaacs J. T. Engineering a Prostate-Specific Membrane Antigen–Activated Tumor Endothelial Cell Prodrug for Cancer Therapy. Sci. Transl. Med. 2012;4(140):140ra86. doi: 10.1126/scitranslmed.3003886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy J. Nguyen T. X. Kanduluru A. K. Venkatesh C. Lv W. Reddy P. V. N. Low P. S. Cushman M. DUPA Conjugation of a Cytotoxic Indenoisoquinoline Topoisomerase I Inhibitor for Selective Prostate Cancer Cell Targeting. J. Med. Chem. 2015;58(7):3094–3103. doi: 10.1021/jm5018384. [DOI] [PubMed] [Google Scholar]
- Lin S. Jin J. Liu Y. Tian H. Zhang Y. Fu R. Zhang J. Wang M. Du T. Ji M. Wu D. Zhang K. Sheng L. Li Y. Chen X. Xu H. Discovery of 4-Methylquinazoline Based PI3K Inhibitors for the Potential Treatment of Idiopathic Pulmonary Fibrosis. J. Med. Chem. 2019;62(19):8873–8879. doi: 10.1021/acs.jmedchem.9b00969. [DOI] [PubMed] [Google Scholar]
- Zhang K. Huang R. Ji M. Lin S. Lai F. Wu D. Tian H. Bi J. Peng S. Hu J. Sheng L. Li Y. Chen X. Xu H. Rational Design and Optimization of Novel 4-Methyl Quinazoline Derivatives as PI3K/HDAC Dual Inhibitors with Benzamide as Zinc Binding Moiety for the Treatment of Acute Myeloid Leukemia. Eur. J. Med. Chem. 2024;264:116015. doi: 10.1016/j.ejmech.2023.116015. [DOI] [PubMed] [Google Scholar]
- Lin S. Wang C. Ji M. Wu D. Lv Y. Zhang K. Dong Y. Jin J. Chen J. Zhang J. Sheng L. Li Y. Chen X. Xu H. Discovery and Optimization of 2-Amino-4-Methylquinazoline Derivatives as Highly Potent Phosphatidylinositol 3-Kinase Inhibitors for Cancer Treatment. J. Med. Chem. 2018;61(14):6087–6109. doi: 10.1021/acs.jmedchem.8b00416. [DOI] [PubMed] [Google Scholar]
- Xia L. Jiang L. Du T. Lin S. Xiong T. Peng S. Tian H. Zhang K. Wu D. Sheng L. Ji M. Chen X. Xu H. Design, Synthesis, and Biological Evaluation of Novel Bivalent PI3K Inhibitors for the Potential Treatment of Cancer. Bioorg. Chem. 2023;140:106814. doi: 10.1016/j.bioorg.2023.106814. [DOI] [PubMed] [Google Scholar]
- Furet P. Guagnano V. Fairhurst R. A. Imbach-Weese P. Bruce I. Knapp M. Fritsch C. Blasco F. Blanz J. Aichholz R. Hamon J. Fabbro D. Caravatti G. Discovery of NVP-BYL719 a Potent and Selective Phosphatidylinositol-3 Kinase Alpha Inhibitor Selected for Clinical Evaluation. Bioorg. Med. Chem. Lett. 2013;23(13):3741–3748. doi: 10.1016/j.bmcl.2013.05.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting this article have been included as part of the ESI.†