

Abstract
The unprecedentedly large size of the global SARS-CoV-2 phylogeny makes any computation on the tree difficult. Lineage identification (e.g. the PANGO nomenclature for SARS-CoV-2) and assignment are key to track the virus evolution. It requires annotating clade roots of lineages to unlabeled ancestral nodes in a phylogenetic tree. Then the lineage labels of descendant samples under these clade roots can be inferred to be the corresponding lineages. This is the ancestral lineage annotation problem, and matUtils (a package in pUShER) and PastML are commonly used methods. However, their computational tractability is a challenge and their accuracy needs further exploration in huge SARS-CoV-2 phylogenies. We have developed an efficient and accurate method, called “F1ALA”, that utilizes the F1-score to evaluate the confidence with which a specific ancestral node can be annotated as the clade root of a lineage, given the lineage labels of a set of taxa in a rooted tree. Compared to these methods, F1ALA achieved roughly an order of magnitude faster yet with ∼12% of their memory usage when annotating 2277 PANGO lineages in a phylogeny of 5.26 million taxa. F1ALA allows real-time lineage tracking to be performed on a laptop computer. F1ALA outperformed matUtils (pUShER) with statistical significance, and had comparable accuracy to PastML in tests on empirical and simulated data. F1ALA enables a tree refinement by pruning taxa with inconsistent labels to their closest annotation nodes and re-inserting them back to the pruned tree to improve a SARS-CoV-2 phylogeny with both higher log-likelihood and lower parsimony score. Given the ultrafast speed and high accuracy, we anticipated that F1ALA will also be useful for large phylogenies of other viruses. Codes and benchmark datasets are publicly available at https://github.com/id-bioinfo/F1ALA.
Keywords: PANGO lineages, SARS-CoV-2, ancestral reconstruction, tree refinement, F1-score
Introduction
Phylogenetics can play an important role in tracing the spread of emerging virus variants by integrating lineage information into a phylogenetic tree. During the COVID-19 pandemic, the Phylogenetic Assignment of Named Global Outbreak Lineages (PANGO lineages) has been widely utilized to categorize SARS-CoV-2 sequences into specific lineages to assist public health control measures (Rambaut et al. 2020). With clade roots of these lineages being annotated at ancestral nodes in the tree, lineage information of descendant samples can be efficiently determined while the ancestral nodes with annotations can provide the evolution history for the virus variants (McBroome et al. 2021). We call the problem of identifying and annotating the clade roots of lineages in a phylogeny to be ancestral lineage annotation (ALA) (Fig. 1).
Figure 1.
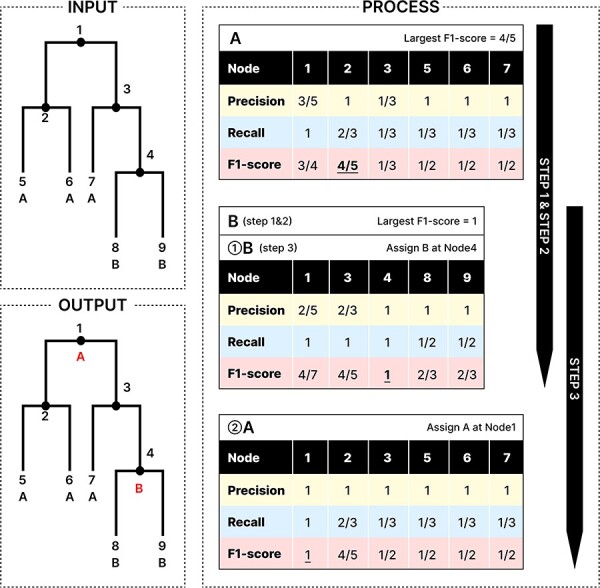
Illustration of the algorithm for ALA. Given a tree with 5 taxa (Nodes 5–9) and 4 internal nodes (Nodes 1–4) where Nodes 5–7 are labeled as lineage A and Nodes 8–9 are labeled as lineage B, the ALA is computed in three steps. Step1: Extract potential annotation nodes for lineages A (Nodes 1–3 and 5–7) and B (Nodes 1, 3–4, and 8–9) [shown in the headers (black background) of the top two tables]. Step2: Determine the order of lineages for ancestral annotation based on the annotation confidence score (the largest F1-score for each lineage, i.e. A = 4/5 and B = 1, marked by underlines in the top two tables). So lineage B is assigned first and then A, as shown by ① and ② in the bottom two tables. Step3: Assign the annotation for B at Node 4 first (middle table), then for A at Node 1. When recalculating F1-scores for potential annotation nodes of lineage A, the taxa at Nodes 8 and 9 are excluded from formulae (1–3) due to Nodes 8 and 9 having been already assigned to the confirmed annotation of Node 4 (bottom table). The F1-score tables for lineage B are the same in Step 2 and Step 3.
Ancestral character reconstruction (ACR) can be used to infer evolutionary dynamics by estimating the states of ancestral nodes for a character of interest (e.g. ecological, phenotypic, and biogeographic traits) in a phylogenetic tree when character labels are given for some or all taxa (Ishikawa et al. 2019). If lineages are the characters of interest in ALA, an ACR method, e.g. PastML (Ishikawa et al. 2019), would construct ancestral states of lineages, and subtrees with identical lineage states are considered as clusters for the annotation of corresponding lineages.
Most conventional ACR methods are not suitable for the ALA in a huge SARS-CoV-2 phylogeny. pUShER is currently the default inference pipeline for lineage assignment in SARS-CoV-2 PANGO lineage nomenclature system (pangolin) (O’Toole 2022). pUShER applies its packaged tool “matUtils” to annotate PANGO lineages of SARS-CoV-2 at ancestral nodes in a reference tree (i.e. ALA problem) (McBroome et al. 2021, Turakhia et al. 2021). For ALA, matUtils constructs consensus sequences for all SARS-CoV-2 sequences with the same lineage label on a given phylogenetic tree. It then searches for the optimal node to insert a consensus sequence to the tree for each lineage resulting in the lowest additional parsimony score by the phylogenetic placement method UShER (Turakhia et al. 2021). This optimal node is defined as the clade root of the lineage. However, occasions of multiple optimal nodes for a single consensus sequence in the ALA by matUtils were frequently observed, e.g., 1139 out of 1248 PANGO lineage members in the benchmarking 100K dataset in this scenario (“Materials and Methods” section). Multiple optimal nodes would cause uncertainty in the phylogenetic placement to determine which node should be considered as the clade root of a lineage. At the same time, the quality of consensus sequence inferred for a lineage will be affected by the quality of sequences belonging to this lineage. This observation was verified by our simulation benchmarks that the accuracy of matUtils dropped significantly when the error rate of sequences increased and the number of used sequences for ALA decreased (details in “Results” section). Its runtime and memory usage were still substantial (see Table 1 for details).
Table 1.
Runtime and memory used for ALA.
| Runtime (h:mm:ss) | Peak memory used (GB) | 100K | 660K | 5.26M |
|---|---|---|---|
| F1ALA | 0:00:07 |0.29 | 0:03:40 |1.83 | 0:12:41 |3.60 |
| PastML | 0:00:50 |0.68 | 0:25:10 |5.50 | 1:33:06 |29.00 |
| matUtils (pUShER) | 0:00:53 |0.47 | 0:28:36 |5.05 | 3:20:37 |27.17 |
Tests were run on an AMD Ryzen Threadripper PRO 5975WX server with 32-Cores and 500 GB RAM using 8 threads. 10 repetitions of each run were performed and average values are presented.
Here, we present a novel ALA approach (F1ALA) that applies the F1-score (Powers 2008) to evaluate the confidence with which ancestral nodes in a tree can be annotated as the clade roots of lineages. When compared to PastML and matUtils (pUShER) on medium, large, and huge SARS-CoV-2 phylogenies, F1ALA achieved roughly an order of magnitude faster than these methods with ∼12% of their memory usage, which is able be run on a laptop computer even for the ALA in a 5.26M-taxa tree (Table 1). F1ALA achieved high accuracies comparable to those of PastML and significantly outperformed matUtils (pUShER) in tests on empirical and simulated datasets.
The phylogenetic trees for millions of SARS-CoV-2 genome sequences in Global Initiative on Sharing All Influenza Data (GISAID; Shu and McCauley 2017) and Genome Browser (Turakhia et al. 2021) are constructed by the online tree updating method UShER (Turakhia et al. 2021), where new SARS-CoV-2 genome sequences are sequentially inserted into a backbone tree. However, as repeated sample insertions do not update the backbone tree, any error in prior insertions cannot be corrected. Hence, tree optimization is required to detect and correct potential mis-insertions using techniques, such as nearest-neighbor interchange or subtree-pruning-regrafting (SPR) which remain time-consuming (Price et al. 2010). We propose a new tree refinement method by iteratively removing all inconsistently labeled taxa relative to their closest annotation nodes, as detected by F1ALA, and reinserting them back using online tree updating tools such as UShER and TIPars (Turakhia et al. 2021, Ye et al. 2024). This achieved both larger tree log-likelihood and smaller parsimony score for the refined tree.
Materials and methods
Algorithm for ancestral lineage annotation
To trace the spread of viral lineages, ALA is to infer the clade roots (as annotation nodes) for these lineages when providing a set of taxon names for each lineage in a rooted phylogenetic tree. It should ensure that taxa under these annotation nodes remain monophyletic for all lineages (McLennan 2010). Nevertheless, because the provided taxa from pangolin are sometimes non-monophyletic in a given tree (McBroome et al. 2021), simply using the most recent common ancestor does not yield accurate inference of their clade roots. Instead, F1ALA calculates F1-score for unlabeled ancestral nodes and iteratively assigns a lineage annotation to the ancestral node with the largest F1-score (this ancestral node with lineage annotation is called “annotation node”). F1-score is a metric of predictive performance being as the harmonic mean of the precision and recall. A true positive (TP) is defined as the provided lineage label of a taxon being the same as its closest annotation node. Then, the precision is the number of TP taxa divided by the number of all taxa in subtrees of the annotation nodes, including those identified incorrectly (their given lineage labels different from those of their closest annotation nodes), and the recall is the number of TP taxa divided by the number of all taxa with provided lineage labels.
In a rooted tree 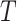 , with taxon nodes
, with taxon nodes 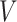 , let
, let 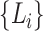 be all members of the lineages,
be all members of the lineages, 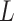 , and
, and 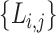 be the lineage labels given to a set of taxa
be the lineage labels given to a set of taxa 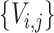 belonging to the lineage
belonging to the lineage 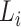 , where
, where 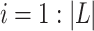 and
and 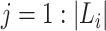 . F1ALA computes ALA in three steps; that is to determine the clade root
. F1ALA computes ALA in three steps; that is to determine the clade root 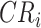 in tree T to annotate lineage
in tree T to annotate lineage 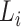 (
(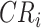 becomes an annotation node). The lineage label of any internal or external node in tree T is inferred from the lineage of its closest annotation node (Fig. 1).
becomes an annotation node). The lineage label of any internal or external node in tree T is inferred from the lineage of its closest annotation node (Fig. 1).
Step 1. Extract potential annotation nodes. A potential annotation node of a lineage must be among the ancestral nodes of the taxa belonging to this lineage. A recursive function determines all ancestral nodes, 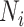 , for a lineage,
, for a lineage, 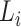 , where
, where 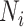 are all unique ancestral nodes for any taxon
are all unique ancestral nodes for any taxon 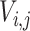 in Li (for
in Li (for 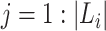 ,with lineage label
,with lineage label 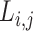 ) to the root of tree T.
) to the root of tree T.
Step 2. Determine the order of lineages for ancestral annotation. For a lineage 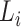 , let the subtree under any potential annotation node
, let the subtree under any potential annotation node 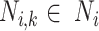 be
be 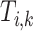 where
where 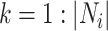 , then the taxa in subtree
, then the taxa in subtree 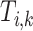 are denoted as
are denoted as 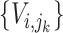 (
(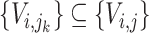 ). When annotating the clade root
). When annotating the clade root 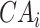 of lineage
of lineage 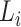 at node
at node 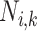 , we have
, we have
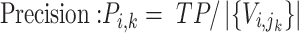 |
(1) |
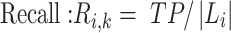 |
(2) |
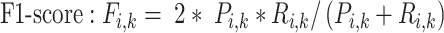 |
(3) |
where 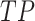 is the number of taxa within subtree
is the number of taxa within subtree 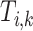 that have the lineage label
that have the lineage label 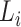 .
.
The highest F1-score 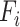 among all
among all 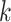 in {
in {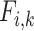 } (i.e.
} (i.e. 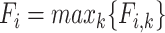 ) is referred as the annotation confidence score for lineage
) is referred as the annotation confidence score for lineage 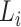 . Smaller annotation confidence score for a lineage means there is more uncertainty about its potential annotation in tree T. F1ALA computes the annotation confidence scores for all lineages
. Smaller annotation confidence score for a lineage means there is more uncertainty about its potential annotation in tree T. F1ALA computes the annotation confidence scores for all lineages 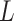 and sort them in descending order.
and sort them in descending order.
Step 3. Assign the annotation for each linage according to the order from Step 2. To compute the annotation for lineage 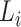 , let the taxa of subtrees of previously confirmed annotation nodes for lineages ranking in front of
, let the taxa of subtrees of previously confirmed annotation nodes for lineages ranking in front of 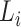 in the sorted order be
in the sorted order be 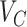 . Then F1ALA re-computes the F1-scores for any potential annotation node
. Then F1ALA re-computes the F1-scores for any potential annotation node 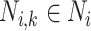 by excluding
by excluding 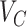 in
in 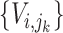 when calculating formulae (1–3), we have
when calculating formulae (1–3), we have
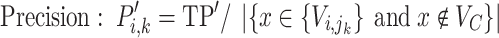 |
(4) |
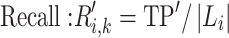 |
(5) |
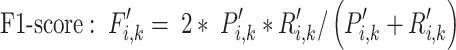 |
(6) |
where 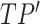 is the number of taxa in
is the number of taxa in 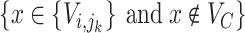 which have the lineage label
which have the lineage label 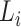 . An example of this re-computation is given in the bottom table of Fig. 1.
. An example of this re-computation is given in the bottom table of Fig. 1.
Lineage 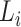 is annotated at the node with the highest F1-score (
is annotated at the node with the highest F1-score (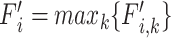 ), which is the clade root
), which is the clade root 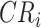 of lineage
of lineage 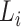 .
.
Each lineage will only be annotated at a node of tree T as a monophyletic group. This does not guarantee all taxa in a tree are assigned under the annotation nodes but the assignments are generally high quality (Supplementary Table S1). This is the same case with matUtils (McBroome et al. 2021) and PastML (Ishikawa et al. 2019).
Algorithm for tree refinement
Given a rooted phylogenetic tree, lineage labels and sequences for all or a set of taxa, the algorithm uses ancestral annotation information to refine the tree topology. After ancestral lineages are annotated by F1ALA, all taxa with labels different from their closest annotation nodes are removed from the tree. The removed taxa are sorted in ascending order by the number of ambiguous nucleotides in their sequences. An online tree updating method (e.g. TIPars or UShER) is used to re-insert them sequentially into the reduced tree. This refinement process is repeated until there is no improvement of the accuracy of ALA or a maximum iteration limit is exceeded.
Benchmark datasets and programs
Three empirical 100K-, 660K-, and 5.26M-taxa SARS-CoV-2 phylogenies were used as benchmark datasets. To form the 100K-taxa dataset, genomes were subsampled from all lineages with high sequence quality (n = 96 020; collected before January 2021) and a maximum likelihood phylogenetic tree was constructed by IQTREE2 (GTR model) (Minh et al. 2020) using the genome hCoV-19/Wuhan/WIV04/2019/EPI_ISL_402124 as root [details in Ye et al. (2024)]. The 660K-taxa tree (659 885 genomes) was downloaded from GISAID on 6 September 2021 (Shu and McCauley 2017). PANGO lineage labels, 1248 and 1181 unique members respectively, were extracted from the metadata of GISAID for the 100K and 660K datasets. For the 5.26M-taxa tree, 5 256 518 genomes and their PANGO lineage labels were taken from http://hgdownload.soe.ucsc.edu/goldenPath/wuhCor1/UShER_SARS-CoV-2/ on 19 February 2023 (giving 2277 unique PANGO lineages). All taxa in the trees were labeled with PANGO lineages and used for ALA. Nextclade labels (Aksamentov et al. 2021) were not used for benchmarking since they are not available in GISAID metadata.
Since errors in PANGO lineages labeling SARS-CoV-2 sequences are a well-known problem (O’Toole et al. 2021), 70 757 taxa from the 100K dataset that had identical lineage labels based on the annotations by F1ALA, PastML, and matUtils (Fig. 2e) were considered as a “ground truth.” A phylogenetic tree was constructed using these sequences by FastTree2 v2.1.11 (double-precision version) under the GTR GAMMA20 model using hCoV-19/Wuhan/WIV04/2019/EPI_ISL_402124 as the root and the output binary tree was collapsed to a polytomous tree using the “ape” R package (tolerance = 1.0*E-6). The accuracy of ALA was evaluated when wrong lineage labels were artificially introduced to this 70 757-taxa reference tree. PANGO lineages labeling errors (replacement of the original lineage label by a false one) was randomly applied to 5%, 10%, 20%, and 50% of the taxa in the tree with 100 replicates of these labeling “errors.” Independently lineage labels were masked for 5%, 10%, 20%, and 50% of taxa in the tree with 100 replicates.
Figure 2.
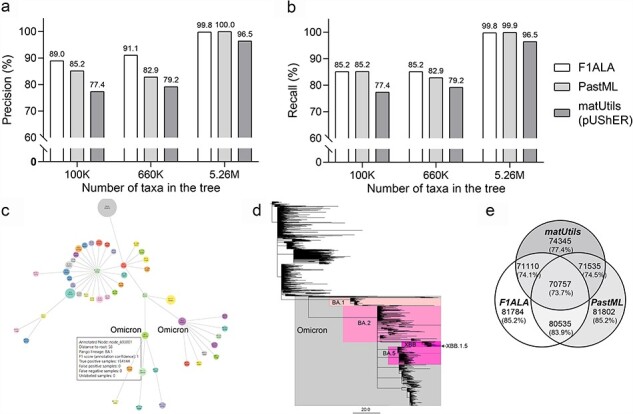
Accuracy of ALA for the 100K-, 660K-, and 5.26M-taxa SARS-CoV-2 phylogenies. (a) Precision (b) Recall for ALA by F1ALA, PastML, and matUtils (pUShER). (c) The ALA using the 5.26M-taxa SARS-CoV-2 phylogeny by F1ALA, showing the top 50 lineages by the number of assigned taxa, with the Omicron lineage highlighted (branch lengths are not to scale to allow differentiation of the lineages). Annotation information (annotation node, distance to tree root, F1-score, and number of TPs) is shown when a mouse hovers over the nodes displayed in a browser. (d) The collapsed tree of 2277 PANGO lineages from the 5.26M-taxa SARS-CoV-2 phylogeny. Each lineage is represented by its annotation node in the tree. Branch length shows the number of mutations (instead of substitution rate) (McBroome et al. 2021) and Omicron sublineages (BA.1, BA.2, BA.5, and XBB.1.5) are highlighted. (e) Venn diagram showing the number of individual and shared TPs (proportions over all taxa) for the annotations by F1ALA, PastML, and matUtils.
F1ALA was benchmarked against PastML (1.9.34) and matUtils (pUShER; v0.6.2) using the precision and recall metrics. A TP was defined as the lineage label given to a taxon being the same as its closest annotation node, if not, it was a false positive (FP). Then, precision = TP/(TP + FP) (i.e. the fraction of tips correctly classified as a specific lineage out of all tips the model predicted to belong to that lineage), and recall = TP/(total number of labeled taxa) (i.e. the fraction of tips in a linage that the model correctly classified out of all tips in that lineage). In addition, pairwise single nucleotide polymorphism (SNP) distances between sequences within a lineage and between lineages were also used for evaluation, which were calculated by snp-dists v.0.8.2 (https://github.com/tseemann/snp-dists). A lower mean SNP distance within a lineage indicates a better ALA; and a larger mean SNP distance between lineages indicates a better ALA.
Since PastML may generate multiple clusters for a specific lineage, the biggest cluster was chosen to be annotated as a monophyletic group (McLennan 2010). PastML was run under the DOWNPASS model to minimize changes in ancestral states. matUtils was run using the annotate function with “set-overlap = 0.”
Results
Computational performance
The computational performances of F1ALA, PastML, and matUtils (pUShER) were compared on the 100K-, 660K-, and 5.26M-taxa SARS-CoV-2 phylogenies (Table 1). F1ALA annotated 2277 PANGO lineages in the 5.26M-taxa phylogeny using 12 min and 42 s, roughly an order of magnitude faster than the other methods. F1ALA significantly optimized the memory requirement to be 3.6 GB, a reduction of around 88% of that in PastML, which allows ALA of a huge phylogeny to be run on a laptop or general computer.
Ancestral lineage annotation of PANGO lineages
The accuracy of ALA was evaluated by precision and recall (“Materials and Methods” section) (Fig. 2a and b). F1ALA achieved the highest precision with the 100K-taxa and 660K-taxa phylogenies (higher than PastML by 4.5% and 10.0%, respectively). For the 5.26M-taxa phylogeny, F1ALA ranked the second highest with 99.8% precision, less than 0.2% below PastML. For recall, F1ALA had the best performance on the 660K-taxa phylogeny (higher than PastML by 2.8%) and had a difference of 0.02% and 0.1%, respectively, to PastML on the 100K-taxa and 5.26M-taxa phylogenies. matUtils (pUShER) showed the worst performance on all benchmarks (Supplementary Table S1).
F1ALA achieved significantly smaller mean pairwise SNP distance within a lineage and larger distance between lineages than other compared methods in 100K dataset (P-value < 0.01 in paired t-test; Supplementary Table S2). The calculation of SNP distances in 660K and 5.26M datasets cannot be done within 96 h using 32 threads in an AMD EPYC 9654 Processor, due to a large pairwise computation requirement.
F1ALA can generate an html file to allow visualization of the ALA. An example using the 5.26M-taxa phylogeny is presented in Fig. 2c, which by default shows the 50 largest lineages. F1ALA can also output a lineage-collapsed tree (Fig. 2d), where each lineage is represented by its annotation node and the original tree topology is preserved.
On simulated datasets with labeling errors (Fig. 3a and b and Supplementary Table S3), F1ALA achieved high and robust precision and recall values for the different percentages of taxa with lineage labeling errors. The precision of F1ALA is significantly better than PastML in all settings (P-value < 0.05). The accuracy of matUtils (pUShER) dropped significantly when the error rate increased. For masked labels (part of the lineage labels of taxa were masked) (Fig. 3c and d and Supplementary Table S3), F1ALA achieved high precision and recall though those of PastML were significantly better (P-value < 0.05). matUtils (pUShER) performed more stably with masked labels, but still showed significantly lower precision and recall than F1ALA and PastML in all tests. F1ALA performed accurately given both kinds of errors, with over 99.4% precision and recall when error rates were ≤20%. With errors at 50%, precision and recall decreased by less than 1% for labeling errors and 2% with masked labels relative to the results with 5% of taxa having errors.
Figure 3.
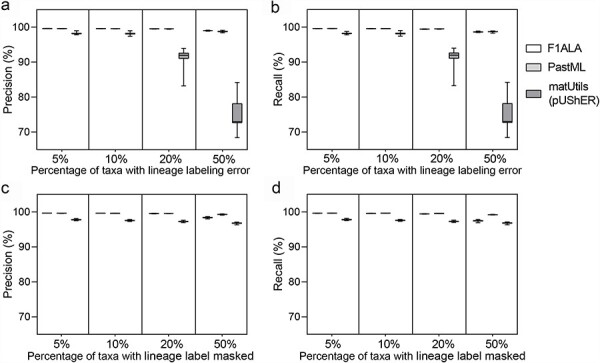
Accuracy of ALA for the datasets with simulated errors. (a) Precision and (b) recall when introducing PANGO lineages labeling errors to 5%, 10%, 20%, and 50% of taxa in the tree (100 replicates). (c) Precision and (d) recall when lineage labels were masked for 5%, 10%, 20%, and 50% of taxa in the tree (100 replicates). Paired t-tests were statistically significant (P-value < 0.05) for all pair-wise comparisons among F1ALA, PastML, and matUtils (pUShER). The whiskers represent the minimum and maximum values while the box shows the lower and upper quartiles with the median crossing the box.
Tree refinement
The proposed tree refinement method was tested on the 100K-taxa dataset with four iterations (Fig. 4). The 660K- and 5.26M-taxa SARS-CoV-2 phylogenies were not tested as calculating the tree log-likelihood is not practical. Using either TIPars (v1.1.0) or UShER (v0.6.2) to update the tree (“Materials and Methods” section), the lineage annotation of the phylogeny was optimized to higher accuracy in precision [7.1% (TIPars), 7.7% (UShER)] and recall (10.8%, 9.6%) (Fig. 4a), larger Gamma20 log-likelihood (0.5%, 0.2%) and smaller tree parsimony score (3.0%, 3.4%) (Fig. 4b).
Figure 4.
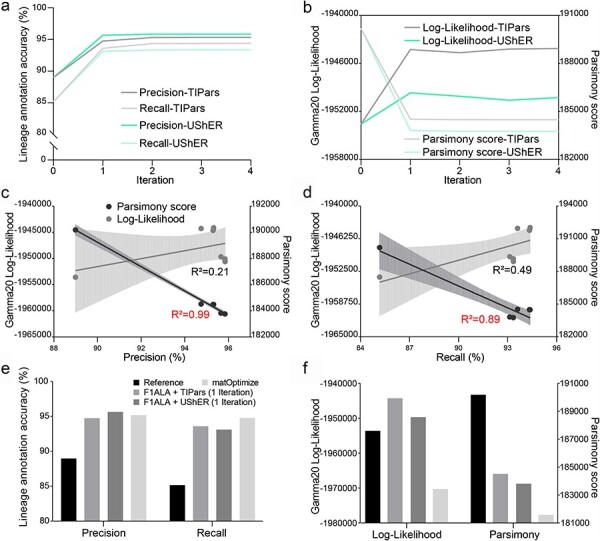
Accuracy of tree refinement. (a) Precision and recall for four iterations of tree refinement. Each iteration contains an ALA by F1ALA and online updating of the tree by TIPars or UShER. (b) Gamma20 log-likelihood and parsimony score for four iterations of tree refinement. Gamma20 log-likelihood was calculated by FastTree2 (reoptimizing the branch lengths with a fixed topology). Tree parsimony score was calculated by UShER. (c) and (d) Linear regression of Gamma20 log-likelihood and parsimony score against precision (c) and recall (d) using trees generated in 0–4 iterations of tree refinement with F1ALA + TIPars and F1ALA + UShER. All regressions, except log-likelihood on precision, are statistically significant (P-value < 0.05). The dashed area shows the 95% confidence interval of the regression. Large R2 values (>0.85) are marked in red. The differences of some points are too small to present in the graphs (overlapping), especially those from the third and fourth iterations of tree refinement. (e) Lineage annotation accuracy after tree refinement. “Reference” is the tree built by IQTREE2. Only one iteration of refinement by “F1ALA+X” (TIPars or UShER) is reported. (f) Gamma20 log-likelihood and parsimony score after tree refinement; other details as in (e).
Linear regression of the Gamma20 log-likelihood and parsimony scores against precision (Fig. 4c) and recall (Fig. 4d) for the original and 4 iterations of tree refinements using TIPars and UShER (data from Fig. 4a and b). Precision and recall explained 99.3% and 89.1% of variance in the tree parsimony score, respectively, showing that their usage as evaluation metrics can reflect the tree parsimony score. matOptimize (v0.6.2) (Ye et al. 2022) is currently applied to optimize the huge SARS-CoV-2 phylogenetic trees in GISAID and Genome Browser, which uses fast subtree pruning and regrafting (SPR) moves. Compared to our proposed method (using F1ALA for ALA and TIPars or UShER for online tree updating; denoted as “F1ALA + TIPars” and “F1ALA + UShER” in Fig. 4e and f), the 100K taxa tree refined by matOptimize achieved the highest recall in ALA (Fig. 4e) and the smallest tree parsimony score, but the lowest tree log-likelihood [even lower than that of without refinement (the reference tree) by 0.9%] (Fig. 4f). “F1ALA + TIPars” improved the tree with the best log-likelihood by 0.5%.
Discussion
ALA, particularly for pathogens affecting public health, has become a more pressing challenge given the extent of sequence data that can be obtained now. This is demonstrated by the need for annotation of PANGO lineages in the huge SARS-CoV-2 phylogenies. We present a novel and practical method, F1ALA, to achieve this, which was demonstrated to be highly efficient, in runtime and memory usage, on an extremely large phylogeny (Table 1) and have high accuracy on empirical and simulated SARS-CoV-2 datasets (Figs 2 and 3).
Lineage assignment can be seen as a multi-class classification problem, where precision and recall are two metrics to measure the quality of model predictions and how well the model did for the actual observations. Notably, a higher precision may come with a lower recall. For example, the model only returns the highly confident prediction such that the precision is high but with a low recall (only a small proportion of instances is reported). F1-score is a trade-off between precision and recall. F1ALA applies F1-score to evaluate the confidence with which ancestral node can be annotated as the clade root of a lineage which allows to emphasize one specific lineage since F1ALA determines annotations of lineages one at a time, even if there are imbalanced classes/lineages, which are real cases in SARS-CoV-2. matUtils is based on a parsimony-based phylogenetic placement (UShER) that places the consensus sequence of each lineage into the tree, where the placed node is the clade root. PastML is a conventional ancestral state reconstruction method that can use either parsimony or maximum likelihood method.
We acknowledged there may be bias toward F1ALA because F1-score, the harmonic mean of the precision and recall, are also used for metrics in ALA performance. To eliminate this potential bias, pairwise SNP distances between sequences within a lineage and between lineages were also used for evaluating ALA performance. The results were consistent with the performance using precision and recall that F1ALA achieved a significantly smaller mean pairwise SNP distance within a lineage and larger distance between lineages (Supplementary Table S2). On the other hand, the regression analysis in Fig. 4c and d shows precision and recall explained 99.3% and 89.1% of variance in the tree parsimony score, respectively, suggesting that their usage as evaluation metrics were practical.
Errors or omissions in the lineage labels assigned to taxa may introduce bias and affect the accuracy of ALA (Fig. 3). F1ALA performed robustly in these cases. PANGO nomenclature labeling errors were introduced and labels were masked to simulate missing data, which are frequent in the real SAR2-CoV-2 sequence data (Shu and McCauley 2017, McBroome et al. 2021, O’Toole et al. 2021). F1ALA and PastML performed well and comparably on these tests but matUtils (pUShER) was worse, particularly for labeling errors (Fig. 3a and b). ALA in matUtils (pUShER) relies on the consensus sequence of each lineage, so labeling errors or omissions lead to an incorrect or inadequately specified consensus sequence that might lead to inaccurate phylogenetic placements (Turakhia et al. 2021).
F1ALA, PastML, and matUtils (pUShER) had higher precision and recall in 5.26M compared to 100K and 660K datasets. A possible reason is the different version of pangolin downloaded for the three datasets according to the timepoint to generate them. PANGO nomenclature system has utilized two inference pipelines for lineage assignment, pangoLEARN (default used in pangolin versions 1 to 3) (O’Toole et al. 2021) and pUShER (default in v4 that was released in April 2022) (O’Toole 2022). pangoLEARN is a machine learning method while pUShER is based on phylogenetic placement. The PANGO lineage labels in 100K and 660K datasets belong to pangolin v2 (downloaded in January 2021) and v3 (downloaded on 6 September 2021), respectively, while those in 5.26M are v4 (downloaded on 19 February 2023). Pangolin v2 and v3 are based on machining learning method for lineage assignment (pangoLEARN) while v4 is based on phylogenetic placement method (pUShER). The ALAs in F1ALA, PastML and matUtils are all based on tree topology rather than machine learning which is expected to be more consistent with pangolin v4 than v2 and v3. A recent study (de Bernardi Schneider et al. 2024) demonstrated only 82.13% and 84.68% concordances between pangoLEARN and pUShER in pangolin v3.1.13 but 97.28% and 97.35% in pangolin v4.0.2 in their two testing datasets that are consistent with our results in Fig. 2. As a double check, we also applied the latest pangolin version v4.3.1 on the 100K and 660K datasets, and both F1ALA and PastML achieved significant higher precision and recall (Supplementary Table S4).
We have proposed a tree refinement method that utilizes the annotations from F1ALA in conjunction with online tree updating software (e.g. TIPars and UShER) to optimize a phylogenetic topology, increasing its log-likelihood and decreasing its parsimony score (Fig. 4a and b). Particularly, the optimized tree using TIPars for tree updating achieved larger Gamma20 log-likelihood than that of UShER [−1 944 123 (TIPars) versus −1 950 256 (UShER)]. However, the tree parsimony score of UShER was smaller [184 487 (TIPars) versus 183 762 (UShER)]. matOptimize, the commonly used method for tree refinement in huge SARS-CoV-2 phylogenies (Ye et al. 2022), improved the tree with the smallest parsimony score compared to our proposed method (F1ALA + TIPars or F1ALA + UShER) but the lowest log-likelihood (even lower than the reference tree) (Fig. 4f). This can be explained by UShER and matOptimize being fully parsimony-based methods that have limited consideration of the tree log-likelihood.
The improvement of tree refinement is mostly observed in the first iteration which suggests a small number of iterations are required (Fig. 4a and b). Updating a tree by TIPars or UShER takes about 21 or 2 s to insert 100 SARS-CoV-2 genomes into a 100K-taxa phylogeny (Ye et al. 2024). These make the proposed tree refinement approach feasible in large trees.
After refinement of the 100K-taxa phylogeny, the precision and recall of ALA was approximately 95% (Fig. 4). Further investigation is needed to determine whether the remaining 5% of inconsistently annotated taxa are positioned incorrectly in the phylogeny due to the tree-building method, an error in ALA or their PANGO lineages being inaccurately labeled.
With the rapid advancement of high-throughput sequencing technology and increasing recognition of the utility of genomic information in studying viruses, a substantial increase in the generation of new genomic sequences for various viruses is expected. When confronted with the huge phylogenetic tree resulting from a vast amount of genomic sequences, our method, F1ALA, is anticipated to be useful in providing efficient and accurate ALA. For example, ALA by F1ALA can be used to infer lineage label for query samples and trace the virus evolution by the visualization of a lineage-collapsed tree (Fig. 2c and d) given a dataset with reference sequences and customized query samples, and the reconstructed phylogenetic tree. The detection of tips with potential mislabeled lineage in the phylogeny for one gene or a segment in a genome, using the lineage labels defined from a phylogeny for another gene or another segment, may provide evidence for reassortment or recombination.
Supplementary Material
Acknowledgements
We gratefully acknowledge the authors from the laboratories responsible for obtaining the specimens and generating and sharing the genetic sequence data used here to GISAID. The acknowledgement table can be found under two EPI_SET-IDs, EPI_SET_20211201vz and EPI_SET_20211206tc.
Contributor Information
Yongtao Ye, State Key Laboratory of Emerging Infectious Diseases, School of Public Health, The University of Hong Kong, Hong Kong SAR, P. R. China; Laboratory of Data Discovery for Health, 19W Hong Kong Science & Technology Parks, Hong Kong SAR, P. R. China.
Marcus H Shum, Laboratory of Data Discovery for Health, 19W Hong Kong Science & Technology Parks, Hong Kong SAR, P. R. China.
Isaac Wu, Laboratory of Data Discovery for Health, 19W Hong Kong Science & Technology Parks, Hong Kong SAR, P. R. China.
Carlos Chau, Laboratory of Data Discovery for Health, 19W Hong Kong Science & Technology Parks, Hong Kong SAR, P. R. China.
Ningqi Zhao, Laboratory of Data Discovery for Health, 19W Hong Kong Science & Technology Parks, Hong Kong SAR, P. R. China.
David K Smith, State Key Laboratory of Emerging Infectious Diseases, School of Public Health, The University of Hong Kong, Hong Kong SAR, P. R. China; Laboratory of Data Discovery for Health, 19W Hong Kong Science & Technology Parks, Hong Kong SAR, P. R. China.
Joseph T Wu, State Key Laboratory of Emerging Infectious Diseases, School of Public Health, The University of Hong Kong, Hong Kong SAR, P. R. China; Laboratory of Data Discovery for Health, 19W Hong Kong Science & Technology Parks, Hong Kong SAR, P. R. China.
Tommy T Lam, State Key Laboratory of Emerging Infectious Diseases, School of Public Health, The University of Hong Kong, Hong Kong SAR, P. R. China; Laboratory of Data Discovery for Health, 19W Hong Kong Science & Technology Parks, Hong Kong SAR, P. R. China; Guangdong-Hongkong Joint Laboratory of Emerging Infectious Diseases, Joint Institute of Virology (Shantou University/The University of Hong Kong), Shantou, Guangdong 515063, P. R. China; EKIH (Gewuzhikang) Pathogen Research Institute, Futian District, Shenzhen City, Guangdong 518045, P. R. China; Centre for Immunology & Infection, 17W Hong Kong Science & Technology Parks, Hong Kong SAR, P. R. China.
Supplementary data
Supplementary data is available at VEVOLU Journal online.
Conflict of interest:
None declared.
Funding
This project is supported by the Theme Based Research Scheme (T11-705/21-N), the Health and Medical Research Fund (COVID1903011-WP1) of the University Grants Commission Hong Kong, the Hong Kong Government’s Innovation and Technology Commission’s InnoHK funding (to D24H).
Data availability
The source codes and benchmark data can be found at https://github.com/id-bioinfo/F1ALA.
References
- Aksamentov I, Roemer C, Hodcroft E. et al. Nextclade: clade assignment, mutation calling and quality control for viral genomes. J Open Source Software 2021;6:3773. doi: 10.21105/joss.03773 [DOI] [Google Scholar]
- de Bernardi Schneider A, Su M, Hinrichs AS. et al. SARS-CoV-2 lineage assignments using phylogenetic placement/UShER are superior to pangoLEARN machine-learning method. Virus Evol 2024;10:vead085. doi: 10.1093/ve/vead085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa SA, Zhukova A, Iwasaki W. et al. A fast likelihood method to reconstruct and visualize ancestral scenarios. Mol Biol Evol 2019;36:2069–85. doi: 10.1093/molbev/msz131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBroome J, Thornlow B, Hinrichs AS. et al. A daily-updated database and tools for comprehensive SARS-CoV-2 mutation-annotated trees. Mol Biol Evol 2021;38:5819–24. doi: 10.1093/molbev/msab264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLennan DA. How to read a phylogenetic tree. Evol: Educ Outreach 2010;3:506–19. doi: 10.1007/s12052-010-0273-6 [DOI] [Google Scholar]
- Minh BQ, Schmidt HA, Chernomor O. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 2020;37:1530–34. doi: 10.1093/molbev/msaa015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole Á. pangolin v4.0. 2022. https://github.com/cov-lineages/pangolin/releases?page=2 5 April 2024, date last accessed.
- O’Toole Á, Scher E, Underwood A. et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol 2021;7:veab064. doi: 10.1093/ve/veab064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers D. Evaluation: from precision, recall and F-factor to ROC, informedness, markedness & correlation. Mach Learn Technol 2008;2:37–63. [Google Scholar]
- Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 2010;5:e9490. doi: 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Holmes EC, O’Toole Á. et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol 2020;5:1403–07. doi: 10.1038/s41564-020-0770-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, McCauley J. GISAID: global initiative on sharing all influenza data - from vision to reality. Eurosurveillance 2017;22:30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turakhia Y, Thornlow B, Hinrichs AS. et al. Ultrafast sample placement on existing tRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic. Nature Genet 2021;53:809–16. doi: 10.1038/s41588-021-00862-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye C, Thornlow B, Hinrichs A. et al. matOptimize: a parallel tree optimization method enables online phylogenetics for SARS-CoV-2. Bioinformatics 2022;38:3734–40. doi: 10.1093/bioinformatics/btac401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Shum MH, Tsui JL. et al. Robust expansion of phylogeny for fast-growing genome sequence data. PLoS Comput Biol 2024;20:e1011871. doi: 10.1371/journal.pcbi.1011871 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source codes and benchmark data can be found at https://github.com/id-bioinfo/F1ALA.