

Abstract
Mitochondria are multifaceted organelles with key roles in anabolic and catabolic metabolism, bioenergetics, cellular signalling and nutrient sensing, and programmed cell death processes. Their diverse functions are enabled by a sophisticated set of protein components encoded by the nuclear and mitochondrial genomes. The extent and complexity of the mitochondrial proteome remained unclear for decades. This began to change 20 years ago when, driven by the emergence of mass spectrometry-based proteomics, the first draft mitochondrial proteomes were established. In the ensuing decades, further technological and computational advances helped to refine these ‘maps’, with current estimates of the core mammalian mitochondrial proteome ranging from 1,000 to 1,500 proteins. The creation of these compendia provided a systemic view of an organelle previously studied primarily in a reductionist fashion and has accelerated both basic scientific discovery and the diagnosis and treatment of human disease. Yet numerous challenges remain in understanding mitochondrial biology and translating this knowledge into the medical context. In this Roadmap, we propose a path forward for refining the mitochondrial protein map to enhance its discovery and therapeutic potential. We discuss how emerging technologies can assist the detection of new mitochondrial proteins, reveal their patterns of expression across diverse tissues and cell types, and provide key information on proteoforms. We highlight the power of an enhanced map for systematically defining the functions of its members. Finally, we examine the utility of an expanded, functionally annotated mitochondrial proteome in a translational setting for aiding both diagnosis of mitochondrial disease and targeting of mitochondria for treatment.
Introduction
Mitochondria are broadly recognizable as the cellular ‘powerhouses’ — a moniker earned through discoveries during the 1940s–1960s that placed these organelles at the centre of aerobic metabolism. Although apt, this familiar nickname implies that mitochondria are simple, fully defined cellular machines. In reality, many fundamental questions about how mitochondria operate persisted in the decades following the discovery of the oxidative phosphorylation (OXPHOS) system. Moreover, new and confounding primary mitochondrial disorders were being discovered during this time, and these organelles were being linked to surprising new cellular processes. To resolve mitochondrial mysteries of the past and illuminate emerging mitochondrial connections to paradigms of the present, the need to revisit the core protein composition of mitochondria became clear. To this end, and motivated by the advent of new mass spectrometry technology, the turn of the century saw multiple efforts to establish comprehensive ‘maps’ of the mitochondrial protein componentry.
The creation of mitochondrial protein maps had two immediate effects on the field of mitochondrial biology. First, they revealed that we had little to no functional information for hundreds of mitochondrial proteins. This fact persists and doubtlessly contributes to our difficulties in defining the genetic underpinnings of mitochondrial disorders and devising therapeutics to rectify mitochondrial dysfunction. However, these maps also provided the very framework to address these challenges: they became the basis for systematic efforts to define functions for orphan proteins and offered a curated set of candidates for investigations seeking to identify missing components of established mitochondrial processes. Indeed, over the past 20 years, mitochondrial protein maps have catalysed numerous biological discoveries and disease diagnoses.
Despite their success, current mammalian mitochondrial maps have limitations that reduce their utility. First, because the original efforts were based largely on whole tissue preparations from a limited set of species, the maps remain coarse-grained. There is limited information available on how the mitochondrial proteome changes across many tissues and cell types, and even within individual cells. Second, these maps focus primarily on resident mitochondrial proteins to the neglect of proteins with multiple localizations or those that ‘moonlight’ in mitochondria. Third, these maps do not discern between proteoforms — the myriad different protein variants based on alternative splicing, protease processing and post-translational modifications (PTMs). Last, many mitochondrial proteins lack robust functional characterization and disease annotations, thereby hindering biological and clinical applications.
The past 20 years have taught us the value of mitochondrial maps, revealed their shortcomings and introduced new technological methods to improve them. Similar to learning a language, whereby an initial set of words empowers further exploration and mastery in a recurring cycle, the quality of the initial maps has nucleated efforts to improve upon them. In this Roadmap, we review the state of current maps, suggest how an enhanced map can be generated and discuss the potential benefits this may have for advancing our understanding of mitochondrial biology and for overcoming the challenges of mitochondrial disease diagnosis and treatment.
The status of contemporary mitochondrial maps
The initial foray into mitochondrial proteomics began with the first isolation of mitochondria in the late 1940s. This landmark event revealed that these organelles are home to central pathways of oxidative metabolism and sparked interest in cataloguing the various proteins required for their function1,2,3. True to the reductionist approaches of the day, this involved aligning protein activities with mitochondria during cellular fractionation. In the early 1950s, these methods helped to assign many central metabolic enzymes to mitochondria, including adenylate cyclase4, glutamate dehydrogenase1, pyruvate decarboxylase5 and many others6. This piecemeal approach was effective at solidifying mitochondria as a metabolic hub; however, a more efficient assessment of mitochondrial protein content would have to await further technological advancements.
The first systematic analysis of mitochondrial proteins would not come for another 50 years. In 1998, empowered by the emergence of mass sprectrometry-based proteomics, 48 mitochondrial proteins were identified from human placenta7. Two years later, ~1,500 unidentified proteins were separated from rat liver mitochondria using 2D gel electrophoresis8. Although these data were based on somewhat impure mitochondria-enriched fractions, they, along with observations from genomic analyses, helped to provide the first estimate of the mitochondrial proteome’s full size. Mass spectrometry technology quickly matured in the ensuing years, leading to rapid growth in the number of identified mitochondrial proteins. In 2003, the first yeast mitochondrial proteome was published that included 750 proteins9. Expanded mammalian mitochondrial proteomes from mouse tissue and human heart were published that same year that included 591 and 615 proteins, respectively10,11.
Further technological improvements augmented these lists12,13,14, but also revealed that mass spectrometry-based approaches lacked the sensitivity required to establish a comprehensive mitochondrial proteome15. To account for this, three large compendia have now been published containing curated lists of mitochondrial proteins assigned through combined experimental and computational evidence. The first, MitoCarta, was published 15 years ago16 (Fig. 1a). The MitoCarta effort consisted of four phases: phase 1, large-scale proteomics analyses of mitochondria from 14 mouse tissues; phase 2, computational integration of the proteomics data with six other publicly available genome-wide data sets; phase 3, high-throughput microscopy analyses; and phase 4, manual literature curation. Phase 1 consisted of both discovery proteomics, which analysed highly purified mitochondria, and subtractive proteomics, which assessed protein enrichment between two stages of mitochondrial purity (crude and pure). The subtractive analysis was highly effective at removing false positive contaminants that were still present in the pure mitochondria sample at low levels. These data were used to assign a probability that each protein detected by mass spectrometry was truly mitochondrial. To do so, training sets of known mitochondrial and non-mitochondrial genes were compiled and used to calculate the method’s sensitivity (based on the proportion of the positive training set that was correctly identified) and its false discovery rate (FDR) (based on identifications from both the positive and the negative training sets)15,16. The training sets were based on genes instead of proteins because the mass spectrometry method typically could not discern specific protein isoforms and because a gene-centric analysis enabled these data to be integrated with other gene-centric analyses in phase 2. Although these proteomics analyses were highly effective, they were ill equipped to identify proteins of very low abundance, that lacked tryptic peptides or that are mitochondrial only in certain tissues or under certain cellular conditions. Therefore, to increase predictive power, phase 2 combined the proteomics data with six additional data sets that provided clues from homology to yeast mitochondrial and Rickettsia prowazekii proteins, gene expression across tissues and during mitochondrial biogenesis, and predicted mitochondrial targeting signals and protein domains. Using the training sets, each method’s data were used to calculate log-likelihood scores via a naive Bayesian classifier. As each method was largely conditionally independent (naive), the log-likelihood scores for each analysis were combined and used to rank every gene in the mouse genome based on the probability that it encodes a mitochondrial protein. At an arbitrary 10% FDR, this integrated analysis predicted that 951 mouse genes encode mitochondrial proteins. In phase 3, 470 proteins lacking prior support for mitochondrial localization were tagged with GFP and analysed by high-throughput microscopy. This method is useful because, by definition, many genes that fall below the 10% FDR cut-off set in phase 2 will indeed encode mitochondrial proteins (Box 1). This microscopy analysis added experimental evidence for 131 genes, including 54 that fell below the 10% FDR in phase 2. Finally, an additional 93 genes were added to the MitoCarta list based on direct evidence from the literature. These 93 genes were part of the positive training set mentioned above; however, their likelihood score fell below the 10% FDR in phase 2. Summing the 951 genes from phases 1 and 2 with the 54 genes from phase 3 and the 93 from phase 4 yielded the final 1,098 genes included in the original MitoCarta compendium.
Fig. 1:
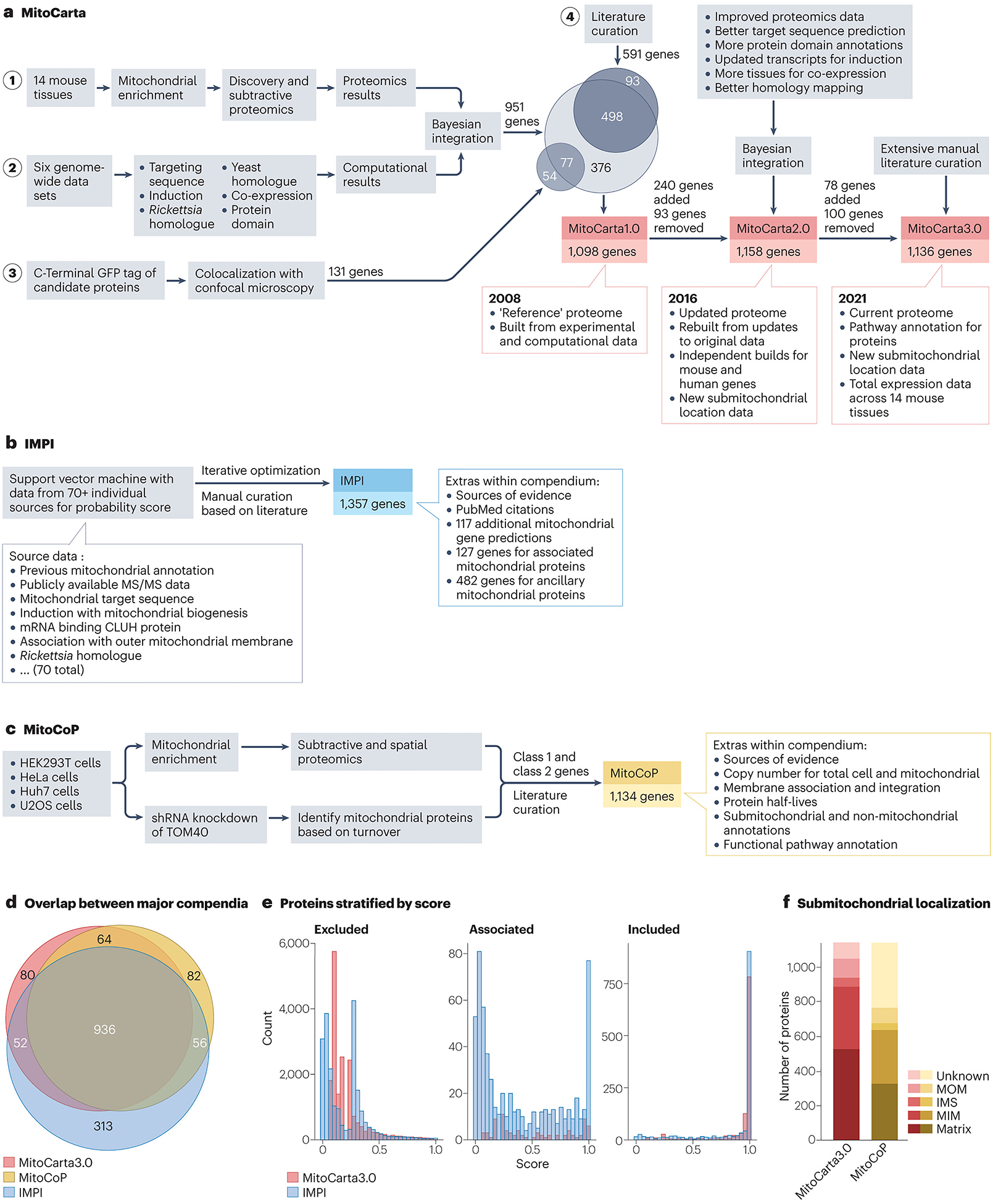
Comparison of the three main mitochondrial protein compendia.
a, The methodology and scoring behind the MitoCarta mitochondrial protein compendia. MitoCarta1.0 began with deep proteomics analyses on mitochondria from 14 mouse tissues (phase 1). The resulting data were then integrated with data from six distinct genome-wide analyses (phase 2) using a naive Bayesian classifier (called Maestro) resulting in a combined weighted score for mitochondrial protein localization. These genome-wide analyses assessed homology to yeast and Rickettsia prowazekii proteins, the presence of mitochondrial targeting signals and protein domains, and RNA co-expression across tissues and induced expression during mitochondrial biogenesis. Weighting for each category was determined using a training set of 591 hand-curated ‘Gold Standard’ mitochondrial genes and 2,519 genes with ‘non-mitochondrial’ annotations in external databases (for example, Gene Ontology (GO)). At a 10% false discovery rate (FDR) — an arbitrary but conventional cut-off — 951 genes were classified as encoding mitochondrial proteins. These genes were integrated with genes validated to encode mitochondrial proteins by GFP microscopy (131 total, 54 new that did not meet the Maestro cut-off) (phase 3) and the Gold Standard mitochondrial genes from literature curation (591 total, 93 that did not meet the Maestro cut-off) (phase 4). MitoCarta2.0 used a similar scoring system to MitoCarta1.0 but began with a training set that contained 60% more genes than the original. The scoring of each individual category was also improved through technological advancements in mass spectrometry search algorithms and updates to protein/homology databases. These improvements in scoring allowed the FDR cut-off to be lowered to 5%, and genes that surpassed this cut-off were combined with the improved training set to form the final list. In total, 240 additional genes were added to MitoCarta2.0 from MitoCarta1.0 and 94 genes were removed. MitoCarta3.0 used the same scores as MitoCarta2.0, and thus changes in membership were determined through manual curation. In total, 78 additional genes were added and 100 were removed, for a new cumulative total of 1,136 genes in the human version of the database. b, Construction of the Integrated Mitochondrial Protein Index (IMPI) began with the manual curation of both mitochondrial and non-mitochondrial genes generated, in part, through the accumulation of data from the MitoMiner4.0 database181 that catalogues evidence of mitochondrial localization. These lists were used as positive and negative training sets, respectively, to train a support vector machine learning model to identify additional mitochondrial proteins. The support vector machine model integrated data from more than 70 different weighted inputs as dimensions for classification. The model then classified genes based on their position within this dimensional space and generated a probability of mitochondrial localization. Genes passing a certain probability threshold were added to the positive training set and the model was iteratively retrained and reapplied to the whole set. The set of potential mitochondrial proteins was then divided manually into three different categories: 1,357 verified mitochondrial proteins with strong localization evidence (for example, from GFP-based confocal imaging); 127 associated proteins with less-definitive localization evidence (for example, from proximity based labelling studies); and 485 ancillary proteins that lack evidence of mitochondrial localization or have strong evidence of non-mitochondrial localization, but that are known to affect mitochondrial function. Finally, the model identified another group of 117 genes that had high (>90%) probability of mitochondrial localization but lacked direct evidence. c, The mitochondrial high-confidence proteome (MitoCoP) compendium was generated primarily based on experimental data using high-resolution mass spectrometry. Subtractive and spatial proteomics were performed on mitochondria enriched from multiple cell lines, and proteins were grouped into distinct classes or clusters based on their enrichment profile. Proteins highly enriched in mitochondrial fractions were placed in the top two classes. In addition, ‘importomics’ was performed using cells deficient in the mitochondrial importer TOM40. Here, all proteins were grouped based on their abundance change between knockdown and wild-type cells. Proteins that fail to be imported into mitochondria are often degraded, and therefore a comparison of proteomes from wild-type and TOM40-knockdown cells can nominate mitochondrial proteins by virtue of their lower abundance in the knockdown cells. The top classes from all the analyses were combined along with genes determined through literature curation to form the 1,134 gene compendium. d, Venn diagram illustrating the overlap in common proteins between MitoCarta3.0, IMPI and MitoCoP. Extensive overlap is expected given the commonalities between approaches. e, Histograms of the likelihood scores within MitoCarta3.0 and IMPI. Scores were generated by each compendium’s respective algorithm and used to establish mitochondrial membership along with manual curation, as described above. A higher score indicates a greater probability of mitochondrial localization. Proteins are stratified into three categories: excluded from each compendium’s mitochondrial list (left), listed as ‘mito-interacting’ in MitoCarta3.0 or ‘ancillary/associated’ in IMPI (middle), and included (right). To allow for comparison between compendia, (1 – FDR) was used for the MitoCarta3.0 score. f, Breakdown of the listed submitochondrial localizations for proteins within MitoCarta3.0 or MitoCoP. For MitoCoP proteins with multiple localizations, partial scores were assigned to each respective localization. MitoCarta3.0 assigns localization values through manual curation whereas MitoCoP uses the GO assignment. IMS, intermembrane space; MIM, mitochondrial inner membrane; MOM, mitochondrial outer membrane; MS/MS, tandem mass spectrometry; shRNA, short hairpin RNA.
Box 1. Mitochondrial proteomes — what’s in a list?
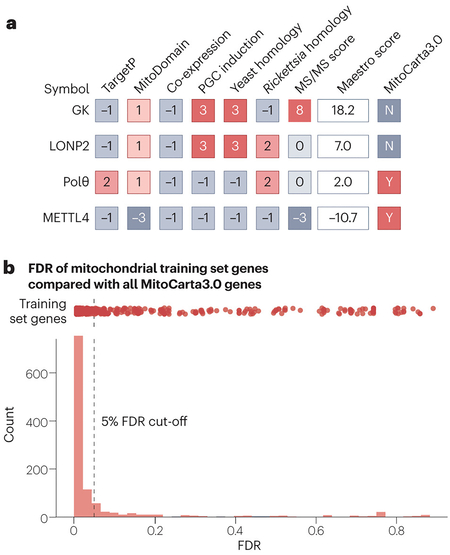
Modern mitochondrial compendia are often mistakenly viewed as rigid lists of mitochondrial and non-mitochondrial proteins. In reality, these resources provide probabilities for mitochondrial localization based on various criteria and then set an arbitrary cut-off to determine which proteins will populate the final ‘list’ (see the figure, part a). Effective use of these resources requires an understanding of how the scoring is performed to enable users to determine which criteria are most relevant to an analysis at hand. For example, in MitoCarta, the protein glucokinase (GK) achieves a high score overall by the Maestro Bayesian classifier, driven largely by its mitochondrial enrichment tandem mass spectrometry (MS/MS) score. However, focused experiments suggest that this MS/MS score is likely due to GK existing in a glycolytic complex at the outer mitochondrial membrane182. Therefore, it was subsequently removed from MitoCarta3.0 and placed in the ‘mito-interacting’ group. This speaks to how the ‘mitochondrial’ classification, which may differ between resources, can be ambiguous. Approximately one quarter of all proteins removed from MitoCarta3.0 fall into this ‘mito-interacting’ category. Although no longer on the official core list, these proteins remain highly relevant to mitochondrial biology. Similarly, the protein LONP2 was also removed from MitoCarta3.0. Closer examination of LONP2, a peroxisomal protein, revealed that its original scores were likely inflated by its high homology to the mitochondrial LONP.
By definition, the differences in scores between proteins just above and below set cut-offs are minor. As noted by the authors of these compendia, such cut-offs can be either raised or lowered to fit experimental goals. It may be of use for large genetic screens to set lower cut-offs to cast a wider net for capturing potentially interesting hit genes for further investigation. The DNA polymerase θ (Polθ) is a good example of a protein with a borderline score. Despite scoring within the top 7% of all proteins for mitochondrial localization, it did not meet the strict cut-off criteria for MitoCarta2.0. Subsequently, however, Polθ was shown to play an important role in mitochondrial DNA (mtDNA) replication183 and was added to MitoCarta3.0. Finally, it is important to consider that although the scoring for these algorithms has been painstakingly refined, it is not perfect. The fact that most, but not all, of the genes used to train the Maestro algorithm do pass the false discovery rate (FDR) cut-off of 5% set in MitoCarta2.0 (see the figure part b, red dots) serves as a reminder that bona fide mitochondrial proteins may be found further down the list. Such proteins, once validated as mitochondrial via direct experimental means, can still benefit from additional information available within the MitoCarta data set (for example, information on tissue distribution or co-regulated genes). Such is the case of the methyltransferase METTL4, which was added to MitoCarta3.0 after its mitochondrial localization was validated experimentally184 despite scoring poorly in all criteria (see the figure, part a).
MitoCarta has since served as the mammalian ‘reference mitochondrial proteome’ in much the same way as the sequencing efforts at the turn of the century provided the ‘reference human genome’17,18. In both cases, subsequent efforts have refined these reference resources and enhanced them in important ways. MitoCarta2.0 was published 8 years ago based on updated versions of all seven original MitoCarta data sets19. For example, the MitoCarta2.0 analysis leveraged updated mass spectrometry search algorithms that identified 10% more peptides from the original MitoCarta data set; improved gene transcript models and homology mapping based on newer NCBI Reference Sequence updates that enabled more sensitive and specific identification of yeast mitochondrial and Rickettsia homologues, induction of genes during mitochondrial proliferation and mitochondrial targeting sequences; broader tissue expression profiles for co-expression analyses; and substantially updated databases of protein domains. MitoCarta2.0 also added new information on submitochondrial protein localization from recent literature and proximity labelling approaches. Notably, unlike the original MitoCarta study, MitoCarta2.0 performed each analysis separately for all human and mouse genes. For features that were specific to mouse data (for example, the mass spectrometry, co-expression and induction analyses), human orthologues were mapped using protein–protein BLAST. MitoCarta3.0 was published 3 years ago20 following a monumental manual literature curation effort that further refined the MitoCarta list and provided rich information on biochemical pathway membership and submitochondrial localization for most of its members. This current version of MitoCarta includes 1,140 mouse genes (1,136 human genes) and the proteins they encode.
The other two modern mammalian compendia are the Integrated Mitochondrial Protein Index (IMPI) and the mitochondrial high-confidence proteome (MitoCoP)21. IMPI is a purely computational approach that began with lists of established mitochondrial and non-mitochondrial proteins curated from various literature sources22 (Fig. 1b). IMPI then applied iterative support vector machine learning modelling to predict whether a protein is mitochondrial based on these positive and negative training sets. The final support vector machine model combined more than 56 different metrics for scoring, including experimental data from MitoCarta and the Human Protein Atlas (HPA)23, resulting in probability scores that justified a list of 1,357 ‘verified’ human mitochondrial proteins and an additional 117 proteins with high (90%) assigned probability. Beyond these, IMPI also provides a curated list of auxiliary, non-mitochondrial proteins that may have functions related to mitochondrial biology. Unlike MitoCarta or IMPI, MitoCoP does not include computational predictions and, instead, relies on experimental evidence from subtractive proteomics, spatial proteomics and importomics (Fig. 1c). It pairs these data with literature curation, including from MitoCarta, to arrive at a list of 1,134 mitochondrial proteins. MitoCoP is unique in that it leverages its extensive mass spectrometry analyses to provide exact copy numbers of mitochondrial proteins. Although not covered further here, excellent recent efforts have established detailed mitochondrial protein maps for Saccharomyces cerevisiae24,25,26.
Collectively, these resources highlight how far mitochondrial proteomics has come in the past 20 years. Each compendium arrives at a similar total number of mitochondrial proteins and there are nearly 1,000 common members between them (Fig. 1d), suggesting that we are likely approaching a complete mammalian mitochondrial proteome. The distribution of likelihood scores (used to define cut-offs for mitochondrial proteome membership) within the entire IMPI and MitoCarta also reflect the commonalities between data sets (Fig. 1e). Yet these undertakings simultaneously demonstrate the difficulty in establishing a fully comprehensive proteome and reveal how little we know about what many mitochondrial proteins do. For example, there are 80, 313 and 82 unique proteins listed as being mitochondrial in the MitoCarta3.0, IMPI and MitoCoP resources, respectively (Fig. 1d), and thus uncertainty about their mitochondrial status remains. There are also differences in the submitochondrial annotations between data sets (Fig. 1f). MitoCarta3.0 lists 100 proteins lacking any pathway affiliation, and large swaths of mitochondrial proteins across these compendia have few to no associated functional studies cited in the literature27. Each compendium is constructed from a limited set of tissues/cells, and there is no information on the distinct protein isoforms (for example, splice variants) that may exhibit distinct functionality. Moving forward, addressing these and related challenges will lead to a more robust mitochondrial proteome map to guide biological and clinical investigations.
The road to an enhanced proteome
The goal of any mitochondrial protein map is to advance our biological understanding of this organelle and elucidate how its dysfunction underlies human disease. The discovery of new mitochondrial proteins will further expand our understanding of what mitochondria can do. Defining the various isoforms of and modifications to these proteins will add an essential level of detail for assessing how mitochondrial processes are regulated and disrupted in disease. This enhanced map will both highlight where our understanding of mitochondrial function falls short and provide the means for filling these knowledge gaps. Finally, a map replete with functional annotations will empower more efficient and accurate disease diagnoses and catalyse the development of new therapeutic strategies. By addressing each of these aspects, we can create a protein map that is not only comprehensive but also a powerful tool for biomedical research and personalized medicine.
Extending mitochondrial protein membership
Future mitochondrial maps will include proteins that have thus far evaded our detection or failed to meet confidence cut-offs. Although the protein membership of current maps approaches the theoretical maximum, individual investigations are revealing that other mitochondrial proteins await discovery. In particular, we predict that new mitochondrial proteins will come primarily from three sources: specific tissue/cell type-enriched proteins; proteins with multiple subcellular localizations; and small open reading frame (smORF) proteins (Fig. 2). Similar to the advent of new mass spectrometry technologies that catalysed the first systematic mitochondrial proteomics efforts, new technologies are offering the opportunity to identify proteins from these categories.
Fig. 2:
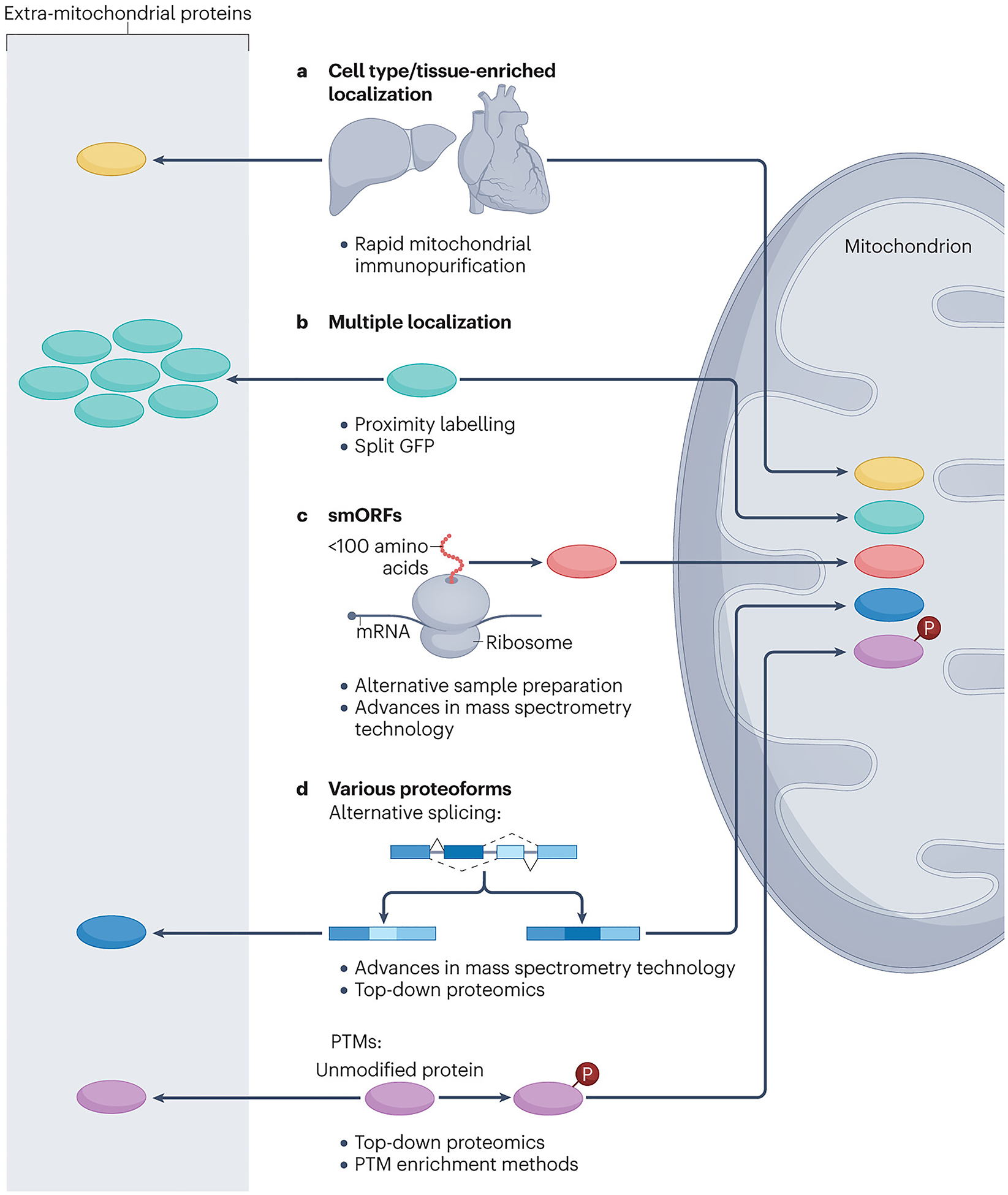
Potential sources of new mitochondrial proteins.
New mitochondrial proteins can derive from different sources. a, Protein expression is differential across different tissues and cell types; therefore, the expression of mitochondrial proteins is variable too. To capture mitochondrial proteins in specific tissues, epitope-tagged outer mitochondrial membrane proteins can be utilized as handles for immunocapture. b, Proteins with multiple localizations (including mitochondrial localization) cannot be found by means of subtractive proteomics if the protein of interest is low in abundance in the mitochondrial fraction. These proteins can be identified through proximity labelling, which allows specific tagging of proteins that reside in different mitochondrial subcompartments. c, Small open reading frame (smORF) proteins can be identified as mitochondrial proteome members through the use of alternative digestion strategies and by mass spectrometry search strategies that include peptides corresponding to predicted smORFs to address the unique features of these proteins. d, Identifying the variety of proteoforms can be facilitated by adjusting mass spectrometry search strategies to reflect the different peptide compositions of protein isoforms and by enrichment techniques for post-translational modifications (PTMs), for example, phosphorylated peptides (Box 2).
The original mammalian mitochondrial maps were based on mitochondrial purifications from large tissues. This was necessary to obtain sufficient amounts of mitochondria at high purity; yet this means that these maps are based on a limited number of cell types, and that resolution of diverse cell types within tissues was lost. However, recent technological innovations are allowing for the swift processing of tissues28,29 and rapid isolation of mitochondria from individual cell types in mice30,31. Coupled with more sensitive and quantitative mass spectrometry methods, these technologies are enabling mitochondrial proteomics analyses from scratch, using a much broader array of sources. This is likely to reveal new mitochondrial proteins, as it has long been known that mitochondria from different tissues participate in specialized functions (for example, steroidogenesis in adrenal mitochondria and ketogenesis primarily in the liver). Moreover, recent studies have revealed discrete mitochondrial populations within the same cell, each of which may harbour bespoke mitochondrial proteomes32,33.
A second likely source of new mitochondrial proteins are those with more than one cellular location. The initial map-making efforts aimed primarily at identifying ‘resident’ mitochondrial proteins — those whose primary or sole cellular residence is within the organelle. Proteins that have a bona fide mitochondrial localization, but that primarily reside elsewhere in the cell, would likely have scored poorly on metrics assessing enrichment (for example, via subtractive proteomics) or mitochondria targeting sequences. Notable examples include DNA glycosylases, whose translocation to mitochondria is driven by alternative splicing and differential transcriptional start sites that unveil mitochondrial targeting sequences34, or apoptosis-related and signalling-related proteins that translocate to mitochondria after being phosphorylated35,36. Moreover, these proteins are difficult to validate via imaging as their overlap with mitochondrial markers is obscured by signals from other cellular locations. Proximity labelling and crosslinking methods are helping to validate proteins that interact with different mitochondrial compartments37,38. One common proximity labelling method, termed BioID, involves linking a mutant biotin ligase enzyme to a ‘bait’ protein of interest39. This ligase promiscuously biotinylates other proteins within its vicinity, allowing these proteins to be isolated and identified. By using a set of baits known to localize to a common compartment, a local protein interaction network can be established that can help to identify additional proteins within that compartment. For example, by using 100 diverse baits from various mitochondrial subcompartments in a BioID approach40, almost 1,500 proteins were identified of which only 35% overlapped with MitoCarta3.0 (ref.41). In a similar approach, termed TurboID, the bait proteins are attached to an engineered biotin ligase with faster kinetics42. Using this technique, >100 proteins at endoplasmic reticulum (ER)–mitochondria contact sites were identified43. Additionally, a creative new split-GFP approach (Box 2) is enabling the detection of dual-localized proteins upon their entry into mitochondria. In this method, a portion of GFP is first encoded in the mitochondrial DNA (mtDNA) and is expressed in the mitochondrial matrix. Proteins are then tagged with the complementary piece of GFP on their carboxy termini. Those proteins that are imported into mitochondria are united with the second GFP component and emit a clear mitochondrial signal. This method has been applied in yeast, where it was used to confirm the mitochondrial relocation of cytosolic tRNA synthases and to discover non-conventional mitochondrial targeting signals for dually localized proteins44. Collectively, these technologies promise to reveal diverse cellular proteins that have function in mitochondria, endowing them with new functionalities or regulating existing pathways and processes.
Box 2. Overview of promising strategies for further mapping of mitochondrial proteins.
Numerous methodologies exist to enhance the future mitochondrial protein map, ranging from refined mitochondrial purification techniques to advanced mass spectrometry methods. Here, we present an overview of some of the most promising approaches to expand the information of the future mitochondrial map.
The mitochondrially localized epitope tag (MITO-Tag) enables rapid and cell type-specific immuno-isolation of mitochondria from various tissues30. Although this technique has been mainly used for the study of mitochondria-specific metabolites185, it has the potential to advance proteomics analyses by enabling the collection of highly pure, rapidly collected mitochondria that may reduce time-sensitive alterations of the proteome.
Chemical crosslinking uses small molecules to covalently tether proteins (or other molecules) that reside in close proximity. Following protease digestion, mass spectrometry-based analyses can identify the interacting proteins and the crosslinked sites. New photoactivatable and cleavable crosslinkers, as well as novel methods that combine blue native PAGE with in-gel crosslinking, are rapidly advancing our understanding of protein interactions115,116,117. The application of these techniques can enhance our ability to resolve mitochondrial proteins and their interactions and shed new light on their possible functions.
Proximity labelling involves expressing a protein of interest (for example, with known mitochondrial localization) that is fused to a promiscuous labelling enzyme, such as biotin ligase or ascorbate peroxidase37,39. Delivery of the promiscuous enzyme’s substrate leads to the covalent tagging of nearby proteins, enabling their subsequent isolation by affinity purification and identification via mass spectrometry. This technique allows for subcompartmental resolution of the mitochondrial proteome and for detection of transient interactions; however, the latter means that non-specific labelling can occur.
In complexome profiling, proteins are extracted and separated by techniques such as size exclusion chromatography or blue native PAGE. Proteins within each separated band or fraction are then identified by mass spectrometry. This gentle technique preserves protein complexes whose membership can be inferred by identifying sets of co-migrating proteins118.
The Split-GFP method is a technique used to study protein–protein interactions (PPIs) and protein localization within cells. It involves dividing GFP into two fragments, where the first fragment is fused to a protein with known mitochondrial localization or expressed from mitochondrial DNA (mtDNA)44, and the second to a protein of interest. The fragments can reconstitute a functional GFP molecule when the fused proteins interact or are in close proximity. This innovative method can detect transient interactions, which may also introduce a risk of detecting false positives.
New mass spectrometry sample processing methods and optimized workflows can decrease the sample processing time28, enrich for small open reading frame (smORF) encoded polypeptides186 and allow for parallel preparation of lipids, metabolites and proteins for mass spectrometry analysis29. These advances increase sensitivity, reduce sample variability and enable the analysis of larger sample sizes and more diverse arrays of tissues and cell types. In addition, peptide generation prior to mass spectrometry-based proteomics can be performed with non-trypsin enzymes, including chymotrypsin, LysC, LysN, AspN, GluC and ArgC21,187. These alternative digestion methods provide depth and flexibility, allowing for increased coverage of the mitochondrial proteome and targeting of specific peptide regions that may not be digested by trypsin.
Prior to mass spectrometry-based proteomics, samples can be enriched for specific post-translational modifications (PTMs) to increase their detection. Techniques that are often employed include immunoaffinity188 and chemical affinity enrichment189. These methods have the potential to greatly expand the number of known mitochondrial proteoforms.
Innovations in mass spectrometry hardware, including ion mobility technology and novel types of mass analysers, have increased the ability both to detect more low-abundance peptides and to do so more rapidly. These technologies thereby increase both the number of proteins identified and sample throughput. Furthermore, advances in computing and machine learning-driven search algorithms are rapidly advancing proteomics data analysis. These updated and more extensive libraries will aid the detection of new mitochondrial proteoforms.
Top-down proteomics differs from traditional bottom-up proteomics in that it involves performing mass spectrometry on intact proteins rather than on peptide fragments generated through enzymatic digestion59,60. Measuring intact proteins enables the detection of proteoforms that could be missed by other methods, such the co-occurrence of distal PTMs that would track with distinct peptides in a bottom-up approach.
A third category of new mitochondrial proteins are those encoded by smORFs. The current proteome maps rely heavily on mass spectrometry detection, which in turn relies on well-annotated genomes. That is, standard mass spectrometry search algorithms are based on the theoretical tryptic peptides that would result from the digestion of proteins encoded by annotated genes. Peptides identified by mass spectrometry that find no match in these search lists are typically discarded. smORFs, which are fewer than 100 amino acids long, were considered too small to be included in the initial ORF predictions of the human genome project. In the relatively small mitochondrial genome, eight smORFs have been functionally characterized, but there may be tenfold more based on the hundreds of small (<30 nucleotides) transcripts expressed in the organelle45. These smORFs are proving to possess key functions. The 24 amino acid Humanin, encoded with the 16S rRNA, is involved in cell signalling and is critical for protection against neurotoxicity46,47. The smORF MOTS-c has wide-ranging effects on metabolism, even being proposed to act as an exercise mimetic48. Although the true number of smORFs encoded by the mtDNA is limited by its small size, the much larger nuclear genome theoretically contains millions of possible smORFs49. The vast majority of these likely will not encode for any expressed protein, and even fewer of those expressed may have meaningful functions related to mitochondria; however, even a minute fraction of this number would increase the size of the known mitochondrial proteome significantly. Already, nuclear-encoded smORFs have been discovered that affect respiratory complex assembly50, the mitochondrial uncoupling response51, mitochondrial translation52 and respiratory efficiency53.
Finally, although perhaps a semantic argument, future mitochondrial maps may wish to include proteins that do not physically localize to mitochondria but still affect their function. Many non-mitochondrial proteins can modulate mitochondrial function through routes including biogenesis, turnover, gene expression and metabolite abundance. The significance of non-mitochondrial proteins as the source of some mitochondrial diseases underscores their importance54, but identifying them will demand significant systematic efforts. Genomic screens targeted at discovering genes critical for respiratory growth have been informative in this regard55, but thus far have been limited to a singular aspect of mitochondrial function. Notably, the IMPI compendium already includes an extensive list of 485 such ancillary proteins22. As this list expands and is refined, it will be an invaluable resource for elucidating mitochondrial biology.
Expanding to proteoforms
Original mitochondrial maps such as MitoCarta are gene-centric (that is, their entries are based on genes, not proteins). This was necessary because their construction relied on genomic information for their predictions (for example, co-expression, induction of gene expression during mitochondrial biogenesis and gene ancestry). Although effective for establishing a core mitochondrial proteome, gene-centric compendia lack information on proteoforms — the vast array of individual protein species that can arise from a single gene56. Current estimates suggest that, on average, ~100 proteoforms exist per gene57, composed of proteins resulting from changes in genetic variation, PTMs or alternative splicing58. These proteoforms can be difficult to detect using traditional bottom-up mass spectrometry-based proteomics techniques, which contributes to their lack of characterization. Top-down proteomics approaches (Box 2) that analyse intact proteins rather than their peptide fragments hold great promise in this regard and have already proven successful in identifying mitochondrial proteoforms59,60. Distinct proteoforms for a single gene can exhibit markedly distinct activities and cellular locations; therefore, it is imperative that future mitochondrial maps include proteoform-centric information (Fig. 2).
A common contributor to proteome diversity is alternative splicing. The proteomic analyses that undergirded contemporary mitochondrial maps lacked the depth required to distinguish most splice isoforms with certainty. This is unsurprising because the unambiguous identification of a specific splice isoform typically requires capturing a tryptic peptide that spans the exon–exon junction. However, as for the DNA glycosylases noted above, focused investigations have revealed the importance of distinct splice isoforms in mitochondria. For example, the dynamin-related GTPase OPA1, which is critical for facilitating proper mitochondrial fusion, has eight known isoforms in humans that exhibit tissue-specific expression patterns61,62. These isoforms undergo different levels of post-translational processing, which helps to fine-tune mitochondrial fusion to the needs of the cell63. These differences can also affect other functions influenced by OPA1, including cristae organization, cytochrome c compartmentalization and calcium homeostasis61,64. Beyond OPA1, a muscle-specific splice variant of the mitochondrial protein MICU1 was shown to greatly alter the efficiency of mitochondrial calcium transport65, and splice variants of the proteins BNIP3 and MCL1 can change the functions of these proteins from pro-apoptotic to pro-cell survival or vice versa based on different conditions66,67. A recent report introduced a method for the global detection of variants and isoforms by deep proteome sequencing68. Through a combination of methodological advancements and the use of six proteases for protein digestion, the authors achieved a median protein sequence coverage of 80% across more than 17,000 protein groups. Application of this approach to isolated mitochondria from diverse sources would add extensive proteoform information to a new mitochondrial map.
PTMs are a second, and often underappreciated, source of mitochondrial proteoform diversity. These modifications come in many flavours, several of which have been reviewed in depth elsewhere69,70,71,72. The functional relevance of some mitochondrial PTMs has been known for decades. For example, reversible phosphorylation was first established as a regulatory mechanism for the pyruvate dehydrogenase complex in the 1960s (refs.73,74). However, save for select enzymes, such as the branched-chain ketoacid dehydrogenase complex, few mitochondrial phosphoproteins were added in the ensuing decades69,75. Here, once again, the advent of new mass spectrometry technologies enabled a shift from studying a single protein or phosphorylation event to surveying these modifications at a global scale76,77,78. Using MitoCarta as a reference, it was found that approximately 91% of mitochondrial proteins have at least one experimentally determined phosphorylation site, yet only 4.5% of known mitochondrial phospho-sites have any functional annotation69. The few investigated phosphorylation events have proven to possess significant functional relevance69,79. Additionally, deletion of the conserved mitochondrial protein phosphatase PPTC7 has dire consequences for organellar function and organismal health80,81. These studies reinforce the importance of cataloguing and characterizing mitochondrial protein PTMs as part of a future map.
Beyond phosphorylation, mitochondrial PTMs derived from metabolic substrates and products of cellular respiration, including acyl groups and reactive oxygen or nitrogen species, are prominent. Acetylation is the most well-known acylation event and more than 60% of all mitochondrial proteins have at least one known acetylation site — a list that includes enzymes from many metabolic pathways and processes, including the TCA cycle, OXPHOS and fatty acid oxidation71,82. However, other alternative acylations, such as succinylation or malonylation, have also been implicated in metabolic regulation83,84. The current understanding of mitochondrial protein acylation comes predominantly from studies of the Sirtuin protein family, particularly SIRT3, SIRT4 and SIRT5. Each of these proteins resides in the mitochondrial matrix and can respond to cellular energy status by deacylating their targets (for example, the SIRT3-mediated deacetylation of long-chain acyl-CoA dehydrogenase85, pyruvate dehydrogenase86 and acetyl-CoA synthetase 2 (ref.87)). Advances in mass spectrometry and acetylation enrichment strategies (Box 2) have allowed for global studies under various conditions, such as heart failure88 or changes in nutrient abundance89.
Interestingly, half of all nitrosylated proteins identified in the heart, and approximately a quarter in the brain, kidney, liver, lung and thymus, are localized to mitochondria90. Mitochondrial protein nitrosylation is critical for repair against cardiac ischaemia–reperfusion injuries91, neurodegeneration92 and immune cell function93. Despite this importance, little is known about how this modification affects protein function outside a handful of examples, such as cytochrome c oxidase (COX)94, very long-chain acyl-CoA dehydrogenase95 and branched-chain amino acid aminotransferase90,95. Recently, in a study on macrophage activation, nitric oxide was shown to covalently modify the lipoic arms of the pyruvate dehydrogenase and oxoglutarate dehydrogenase complexes, thereby blocking their catalytic activity93. Additionally, there exists a wealth of other mitochondrial protein PTMs that remain largely understudied. Mitochondrial proteins with diverse roles in many metabolic pathways are O-GlcNacylated96, which may explain why the modification has been linked to metabolic diseases such as type 2 diabetes mellitus97. Finally, little to nothing is known about how some less prominent PTMs, such as ADP-ribosylation, affect mitochondrial function, although the increase in this modification has been associated with decreased respiration98. In total, these other PTMs remain a fertile area of study whose impact on mitochondrial protein function is likely just beginning to be realized.
Having a complete mitochondrial protein map will also be helpful for filling existing knowledge gaps regarding enzymes that catalyse these modifications, or for suggesting which other PTMs might occur within the organelle. For example, there are few known kinases in the present iterations of the mitochondrial proteome maps, and those with established mitochondrial localization are thought to possess a limited substrate clientele. Experimentally, it has been observed that ‘non-mitochondrial’ enzymes such as protein kinase A, protein kinase C and SRC kinases localize to mitochondria under specific conditions79,99; however, the conditions and mechanisms for this translocation are still poorly understood. Similarly, there are no known protein lysine acetyltransferases or nitric oxide synthases in current versions of the mitochondrial proteome, although inducible nitric oxide synthase can reside at the outer mitochondrial membrane under certain conditions100 and the histone acetyltransferase MOF has been shown to regulate OXPHOS gene expression by binding mtDNA101. Although it is known that some PTMs occur non-enzymatically, a thorough examination of potential regulatory enzymes in mitochondria — including via the methods noted above — is critical to understanding the PTM influence on mitochondrial processes.
Defining protein function
The future mitochondrial map will continue to evolve from a mere parts list into a ‘wiring diagram’ of protein functions, interactions and disease annotations. The original MitoCarta catalogue included nearly 300 proteins lacking any annotation in the Gene Ontology (GO) database, demonstrating the enormity of this task. However, the construction of this catalogue immediately empowered new analyses to assign protein function. For example, a phylogenetic profiling analysis of the full MitoCarta list associated 19 new proteins with respiratory complex I on the basis of their shared evolutionary history16, many of which are now established complex I ‘assembly factors’ known to harbour causative mutations in human disease.
The systematic analyses enabled by mitochondrial maps take two general forms, top-down and bottom-up approaches, which can also be integrated in a complementary and synergistic manner (Fig. 3). First, traditional top-down approaches seek to fill a defined knowledge gap. Various mitochondrial activities have been known for decades but lack associated protein machinery. Here, mitochondrial maps accelerate discovery by providing a prioritized list of proteins that may fulfil these functions. This approach catalysed the discovery of transporters for calcium102, pyruvate103,104 and NAD105. Mitochondrial maps can also assist in first defining where such knowledge gaps exist, similar to approaches based on metabolite data106. One exemplary study of this type is the recent discovery of the putative mitochondrial glutathione importer SLC25A39 (ref.107). Glutathione is an essential mitochondrial molecule, yet mitochondrial maps indicate that the known machinery to produce this molecule is not present within the organelle, suggesting the existence of an undefined transporter. Mitochondrial maps contain 53 members of the SLC25A carrier family. Proteomic analysis on isolated mitochondria from glutathione-deprived cells revealed SLC25A39 as the main upregulated protein, and thus the prime candidate. A related study also identified SLC25A39 as the likely glutathione transporter via a gene-by-gene-by-environment interaction CRISPR screen of all 53 SLC25 carrier family members108 (Fig. 3a). Further experiments from both studies confirmed the requirement of this protein for glutathione import. Top-down analyses can likewise be motivated by unresolved primary mitochondrial diseases. Here, the knowledge gap is the identity of the protein harbouring causative mutations (that is, what is causing the disease). Extensive clinical and biochemical phenotyping of patients often results in the suspicion of a primary mitochondrial disease, of which there are many that remain without an identified underlying molecular cause. These knowledge gaps can be exploited by investigating a common biochemical phenotype observed in primary mitochondrial diseases, such as isolated complex I deficiency (OMIM: 252010). Indeed, targeted inspection of sequencing data with regard to the 65 genes encoding complex I subunits or assembly factors led to the functional annotation of NDUFA6 as an accessory subunit of complex I109.
Fig. 3:
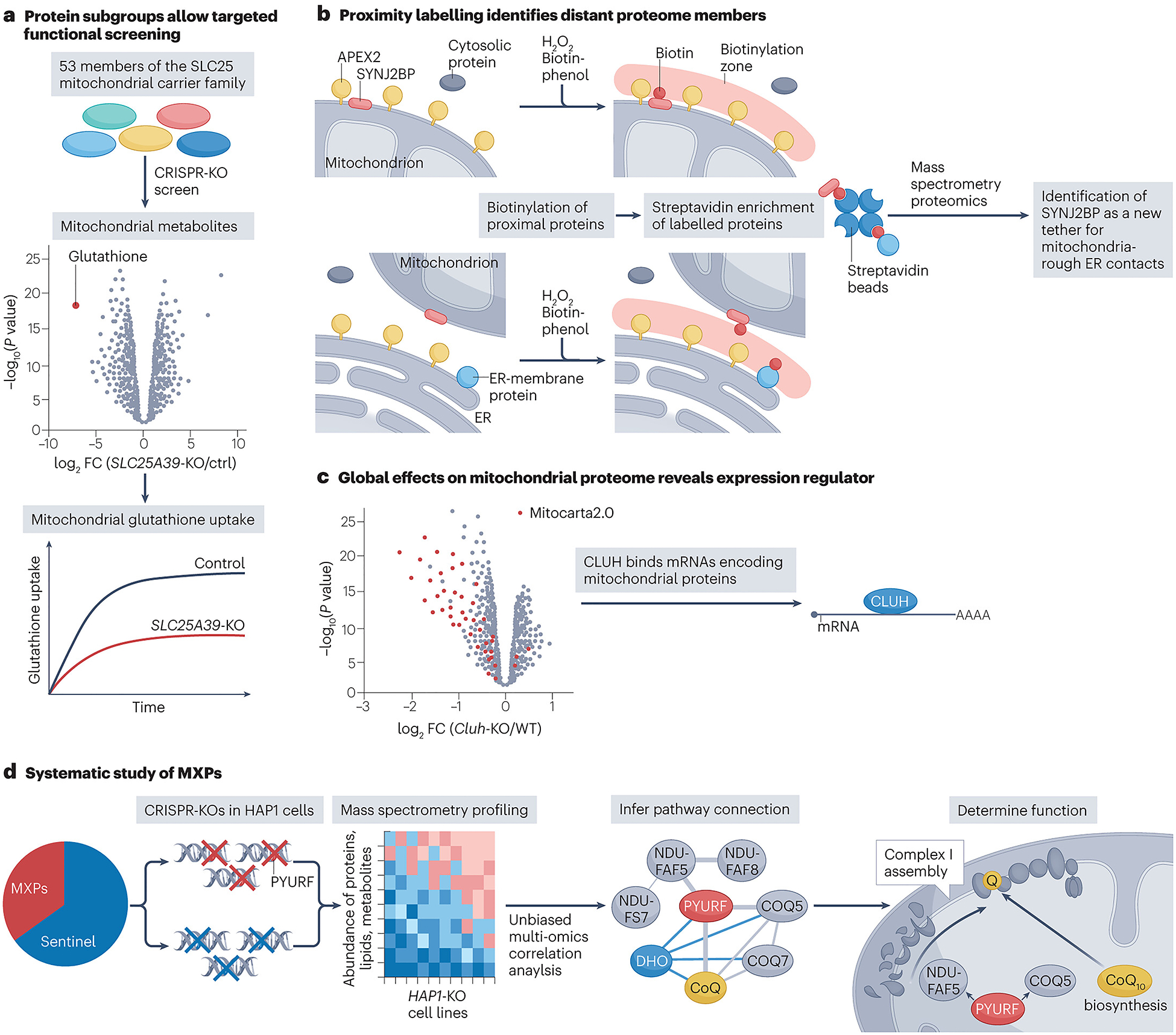
Examples of innovative uses of mitochondrial protein maps.
a, The role of SLC25A39 as a putative mitochondrial glutathione transporter was first suggested through proteomics analyses of isolated mitochondria from glutathione-starved cells107. This function was further validated by a unique gene-by-gene-by-environment CRISPR screen108. The screen used the mitochondrial proteome to design a guide library containing all SLC25 family members known to localize to the organelle, thus narrowing the search space for discovery. CRISPR-mediated deletion of SLC25A39 was subsequently shown to reduce mitochondrial glutathione levels and uptake. b, Knowledge of mitochondrial localization for one protein can lead to the identification of many others through proximity labelling. This is how the protein SYNJ2BP was first identified as an outer mitochondrial membrane protein. This labelling was accomplished by fusing APEX2 to the targeting domain of the established outer mitochondrial membrane protein MAVS. Expression of the APEX–MAVS fusion protein resulted in the biotinylation of other outer mitochondrial membrane proteins within close proximity, which were enriched and identified using proteomics. SYNJ2BP was one of these enriched proteins and subsequent experimentation showed the protein was critical for regulating the formation of mitochondria-rough endoplasmic reticulum (ER) contact sites114. c, Filtering large data sets for mitochondrial genes can associate non-mitochondrial proteins with the organelle. For example, the mRNAs bound by the cytosolic protein CLUH were shown to be strongly enriched for transcripts encoding mitochondrial proteins. Further analysis of proteomics data from the liver-specific Cluh-knockout mouse filtered for mitochondrial proteins also showed a global decrease in mitochondrial proteins. These data firmly established CLUH as an mRNA-binding protein that can regulate mitochondrial protein levels119,120 d, Comparing the multiomic profiles of cells with mitochondrial perturbations has been used multiple times to reveal novel mitochondrial biology. Most recently, proteomics, lipidomics and metabolomics analysis was performed on >200 haploid cell lines containing CRISPR–Cas9-generated gene deletions in both uncharacterized (MXPs) or characterized (sentinel) mitochondrial genes. MXP function can be inferred based on its correlation with other molecules in the data set. Using this type of analysis, the MXP PYURF was linked to coenzyme Q10 (CoQ10) biosynthesis and complex I assembly through its correlation with CoQ10 and complex I assembly factors, respectively. Further investigations demonstrated that this protein, now named NDUFAFQ, acts as a chaperone for requisite methyltransferases in each of these functionally related pathways127. FC, fold change; KO, knockout.
The second variety of systematic analyses enabled by mitochondrial maps are bottom-up approaches that seek to ascribe functions to uncharacterized proteins without focusing on an established knowledge gap. Generally, these studies begin with one or more proteins that lack connections to an established pathway or that are associated with a pathway for unclear reasons. Among these, efforts to infer protein function by virtue of protein–protein interactions (PPIs) are the most common. PPI studies take on many forms and have been boosted by the rapid development of new technologies. An affinity-enrichment mass spectrometry approach that assessed mitochondrial PPIs in differing cell types and media conditions identified a new complex I assembly factor that is disrupted in Leigh syndrome110,111. Rapid short-term labelling of proteins using proximity labelling techniques such as BioID40 or APEX labelling112 have not only been used to identify new members of the mitochondrial proteome as described above but have also been critical for identifying new protein functions. A large-scale BioID investigation of 192 proteins in human cells linked multiple uncharacterized proteins, including C18orf32, to the outer mitochondrial membrane and mitochondrial fission. The authors were subsequently able to show that overexpression or knockdown of C18orf32 resulted in mitochondrial fragmentation or hyperfusion, respectively113. APEX labelling of the outer mitochondrial membrane helped to reveal that the protein SYNJ2BP has a role in stabilizing mitochondria–ER contact sites through its binding with RRBP1 (ref.114) (Fig. 3b). Similarly, advanced crosslinking mass spectrometry techniques have also provided a productive avenue for assigning novel protein function. Crosslinking mass spectrometry was used to discover that the mitochondrial protein AIFM1 forms a complex with subunits of COX115, and the authors speculate that this may explain the OXPHOS defects seen in patients with AIFM1 deficiency. In yeast, crosslinking mass spectrometry helped to assign a novel function in COX assembly to the previously uncharacterized Min8 (refs.116,117). Furthermore, complexome profiling (Box 2) has emerged as a gentle and effective means to identify PPIs. By using this technique to characterize the native mitochondrial complexome, TMEM126B was identified as a complex I assembly factor118. Notably, these methods are also effective at establishing the sub-organelle location of mitochondrial proteins, thereby offering important initial clues when unambiguous new functions cannot yet be established. Finally, mitochondrial protein maps enable the study and functional association of non-mitochondrial proteins with the organelle. An exemplary bottom-up approach uncovered CLUH as a cytosolic mRNA-binding protein that regulates parts of the mitochondrial proteome by directly binding to mRNAs that encode mitochondrial proteins119,120 (Fig. 3c).
Recently, our group has taken a complementary bottom-up approach to assigning mitochondrial protein function based on deep multiomic profiling. Major advancements in mass spectrometry have greatly accelerated the pace and scale at which proteins, lipids and metabolites can be measured29,121,122,123. We hypothesized that deep profiling of these interdependent molecular classes could tease out novel properties of uncharacterized proteins. Our initial use of this strategy in S. cerevisiae revealed an RNA-binding protein124 and a mitochondrial protease125 that regulate the production and maturation of coenzyme Q (CoQ)-related proteins, and a long-missing enzyme that generates the CoQ head group precursor126. In a recent effort127, we advanced this methodology and applied it to a series of >200 mitochondria-related gene knockouts in a common haploid cell line (HAP1). Through this ‘MITOMICS’ screen, we discovered that the orphan protein PYURF (now NDUFAFQ) coordinates CoQ biosynthesis with the assembly of complex I by chaperoning distinct methyltransferases required for these interrelated pathways (Fig. 3d). Furthermore, we revealed that mutations in PYURF underlie a previously unresolved multisystemic mitochondrial disorder and provided functional predictions for many other orphan mitochondrial proteins. Future work could expand upon the MITOMICS effort by adding new layers of information, targeting different genes or conducting new analyses in different cell lines and under contrasting environmental conditions.
The creation of a mitochondrial map replete with accurate functional annotations will require diverse approaches from many researchers. Beyond what has been discussed above, these have included activity-based protein profiling128, evolutionary analyses129, structural biology130 and myriad focused studies on individual proteins. These efforts, when properly incorporated into the map, will have a bootstrapping effect — the more protein functions that are established, the more effective the mitochondrial map will become at fuelling further systematic investigations. These studies will be further enhanced by computational resources such as STRING131, BioGRID132 or GeneMANIA133 that integrate data from many sources. Of course, it would be short-sighted to declare any given protein as fully characterized. Indeed, multiple examples of new functions for well-established mitochondrial proteins have emerged in recent years, such as the unexpected regulation of fatty acid oxidation by the anti-apoptotic BCL-2 family protein MCL1 through its interaction with very long-chain acyl-CoA dehydrogenase134, or involvement of the mitochondrial fusion proteins mitofusin 1 and 2 in regulating lipid metabolism135. Moreover, as discussed in the previous subsection, the function of mitochondrial proteins may be altered by post-translational processes that are just coming to the fore.
Translational applications
Although generating comprehensive and functionally annotated maps of the mitochondrial proteome will advance the understanding of key processes that underlie mitochondrial health and deficiency, an important further goal is to put these maps to use for applications in diagnosis of and therapy for mitochondrial diseases. In this section, we discuss how integration of additional information on the mitochondrial proteome has the potential to be used for improved diagnosis of, and targeted therapy development for, mitochondrial diseases.
Enabling mitochondrial disease diagnosis
A mitochondrial protein map can help to improve the insufficient diagnostic rates of mitochondrial disease, namely by establishing novel links between dysfunctional mitochondrial proteins and a corresponding mitochondrial disease, and by limiting the search space for the causative variant(s) within the genome through prioritization of selected gene groups that are based on functional, pathway or interaction data. In the case of primary mitochondrial diseases, the diagnostic process aims at identifying the cause of a disease phenotype at the molecular level, that is, monogenic pathogenic variants that lead to a dysfunctional gene product. Within the ~1,500 different inborn errors of metabolism136, there are currently ~400 primary mitochondrial diseases with an assigned disease gene137,138, making this group the most common inherited metabolic disorder, with a prevalence of 1:2,000–1:5,000 (ref.139). However, based on the current estimate of 1,000–1,500 core mitochondrial proteins, it is assumed that the list of clinically apparent primary mitochondrial diseases will continue to grow through establishing new gene–disease links.
One of the key factors contributing to suboptimal diagnostic rates for primary mitochondrial diseases is the absence of a comprehensive catalogue of disease annotations in current mitochondrial protein maps. Real-world diagnostic studies show primary mitochondrial disease diagnosis rates between 30% and 60% depending on the composition of the examined cohort and on the diagnostic method employed (whole genome or exome sequencing)140,141,142,143,144,145. Complementary RNA sequencing was shown to boost diagnostic rates to some extent146 and the clinical implementation of new transcriptomic tools147,148 is becoming a reality149,150. However, the unsatisfactory diagnosis rate of primary mitochondrial diseases151 persists, partly due to important limitations of current mitochondrial protein maps. For example, by comparing the mitochondrial proteome with information from online databases (UniProt and OMIM) and literature152, MitoCoP estimates that currently ~40% of the mitochondrial proteome is linked to human diseases21. In addition, in a recent study that diagnosed primary mitochondrial diseases, only 44% involved MitoCarta3.0 proteins153. These numbers indicate both an incomplete catalogue of map members as well as the incomplete link of present map members with a mitochondrial disease — two challenges that we propose should be addressed in the further development of the mitochondrial protein map.
However, to facilitate the usability of the mitochondrial protein map as an efficient diagnostic tool, mere annotation of diseases is not sufficient. In fact, the diagnostic process of a primary mitochondrial disease is usually initiated by the seemingly suggestive combination of phenotypic features. These traits include clinical signs and symptoms as well as biochemical and imaging properties obtained through initial testing. Currently, mitochondrial protein maps are unable to further guide the diagnostic process at this stage because they are lacking standardized phenotype annotations. An exemplary diagnostic study demonstrated how the primary mitochondrial disease phenotype of biochemical complex I deficiency can prioritize genes to be investigated, informed by the mitochondrial protein map154 (Fig. 4). The future mitochondrial map would therefore benefit from including phenotypic trait information based on established phenotype catalogues, such as the Human Phenotype Ontology155. This phenotype compendium is detailed and easily searchable and could be used as the basis for phenotype curation of the future map. Inclusion of disease phenotype annotations per compendium member would allow for streamlined diagnostic pipelines through systematic generation of select gene lists for diagnostic prioritization.
Fig. 4:
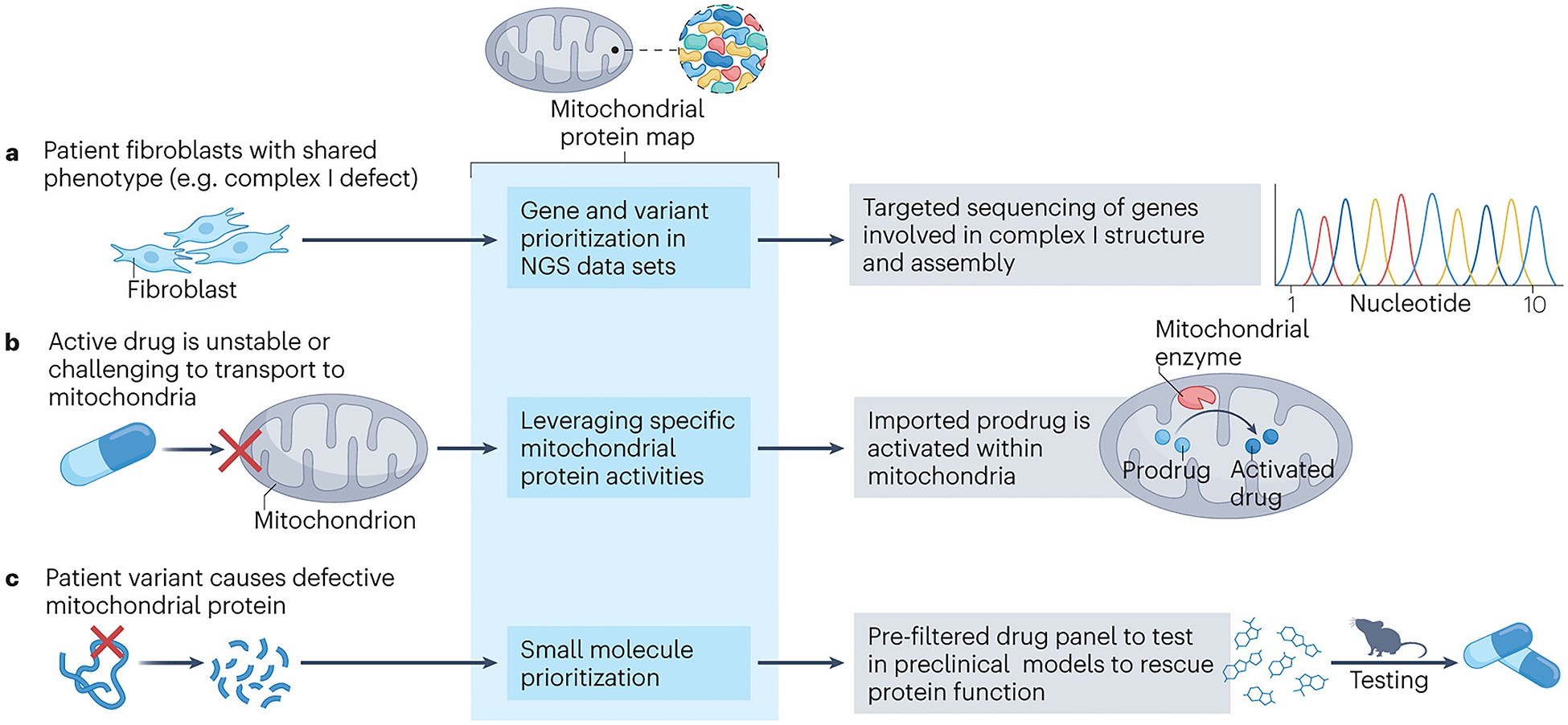
Translational utility of the mitochondrial protein map.
The mitochondrial protein map can prove useful in multiple translational scenarios of which three examples are depicted. a, The diagnostic challenge posed by primary mitochondrial diseases can be effectively addressed through an elegant gene prioritization approach facilitated by the mitochondrial protein map. By comparing phenotypic traits in diagnostic samples with protein function information in the mitochondrial protein map, a specific group of genes can be triaged for diagnostic sequencing. For instance, when faced with patient samples displaying biochemical complex I deficiency, employing a prioritized variant search within the genes associated with complex I function and assembly, as annotated in the mitochondrial protein map, could expedite genetic diagnosis. b, Drug targeting of mitochondrial proteins is challenging due to insufficient transport of specific substances to their intended mitochondrial targets or premature drug degradation. However, this challenge can be addressed by devising prodrugs that exhibit both stability and the ability to traverse mitochondrial membranes. Once these prodrugs reach the mitochondrial matrix, they can undergo processing, such as cleavage, which activates them and enables them to exert their desired effect on the target mitochondrial protein. The rational design of prodrug processing within mitochondria can be facilitated by the comprehensive information provided by the mitochondrial protein map, which offers detailed insights into the resident mitochondrial enzymes that can be harnessed for effective prodrug processing. c, Primary mitochondrial diseases arise from variants that directly impact the optimal functionality of mitochondrial proteins. In the specific case shown here, the variant leads to a disruption in protein stability, presenting a potential avenue for intervention through protein stabilization by small molecules. To optimize this approach, we recommend that the future mitochondrial protein map incorporates a comprehensive library of small molecules, characterized for their specific interactions with mitochondrial proteins. Leveraging this information, small molecules can be prioritized for testing purposes, focusing on those with promising potential to exert beneficial effects on the deficient protein. NGS, next-generation sequencing.
Even with complete disease and phenotypic annotations, arriving at a molecular diagnosis might still be challenging. In some of these cases, accurate assessment of protein abundance as part of the mitochondrial protein map21 could help to guide the diagnostic process. This valuable information allows the comparison of diagnostic proteomics data sets with ‘standard’ protein abundances catalogued in the mitochondrial protein map, potentially indicating abnormal levels of multiple or single proteins. Such findings can again guide the analysis of genetic data sets by narrowing the search space to a group of target genes. This seems especially valuable in the context of variant types that might be missed on exome analysis, including genomic rearrangements, copy-number variants or deep intronic splicing variants156,157. Further, knowledge on protein abundance aids the validation of variants of the missense type that can have various consequences on the protein level158, or of stop codon variants that could potentially escape nonsense-mediated mRNA decay146. Similarly, if the disease protein is implicated in protein complex formation, the pathogenic variant might lead to abundance changes of its interaction partner(s) that can indirectly lead to the correct diagnosis110,159. Such diagnostic interactome studies are only feasible with an enhanced reference mitochondrial interactome map at hand.
Currently, mitochondrial disease diagnostics is moving towards an integration of different information layers from various omics planes to arrive at the best possible diagnostic rate137,159,160,161. In this future framework, the mitochondrial protein map will play a central role in establishing firm links between disease genes and phenotypes and in stratifying genes sets to be prioritized for analysis. This process makes apparent the dynamic nature of the mitochondrial protein map as well as its self-reinforcing properties: it is a discovery tool as well as a compendium of knowledge that, in turn, again enables novel discoveries.
Facilitating rational drug design
Finally, we propose several roles that the mitochondrial protein map could play in the development of mitochondrial disease therapeutics (Fig. 4). There is an unmet need for primary mitochondrial disease treatments in the clinic, illustrated by only two primary mitochondrial disease-related drugs on the market (omaveloxolone, approved for treatment of Friedreich’s ataxia162; and idebenone, approved for treatment of Leber hereditary optic neuropathy163). There is hope that a more refined mitochondrial protein map will facilitate the development of precise medicines by enabling targeted drug design aimed at specific proteins and metabolic pathways. A better characterized mitochondrial proteome, including comprehension of pathomechanisms at the molecular level, would serve as the foundation for rationalized drug development. Examples of these concepts can be seen for the CoQ pathway, where compounds have been identified that specifically target COQ8A164 or that bypass a defective step of CoQ head group modification165,166,167,168. In the context of specific targeting, it could become crucial to recognize that some druggable proteins have secondary ‘moonlighting’ functions within mitochondria, and that it is the mitochondrial form that requires targeting. Moreover, expanding the future mitochondrial protein map to a complete catalogue of proteoforms will prove useful as PTMs have been directly or indirectly implicated in the pathophysiology of inborn errors of metabolism and can be targeted for therapy169,170.
Furthermore, the availability of mitochondrial proteome information at the single-cell or organellar level could lead to the development of therapies aimed at specific subpopulations of healthy or defective cells or mitochondria. This approach is similar to that of transcription activator-like effector nuclease (TALEN)-based and zinc-finger nuclease (ZFN)-based tools, which reduce the number of mutated mtDNA genomes to promote the propagation of healthy mtDNA, thereby shifting heteroplasmy levels171,172,173. Conversely, information on the exact composition of the mitochondrial proteome might allow the selection of healthy mitochondria to be used for ‘mitochondrial transplantation’174 — an approach that has potential utility in the treatment of Alzheimer disease175 or Parkinson disease176. Further harnessing our understanding of the varying proteome compositions across different spatial regions can enable the design of prodrugs that are activated by specific enzymes within their designated mitochondrial subcompartments177,178. This approach would enhance the specificity of drug action, thereby improving overall efficacy.
As mitochondrial protein maps are expanding beyond yeast and metazoans to pathogenic organisms, differences between these maps can be revealed and serve as the basis for the development of specific antipathogenic drugs. One such example includes the PlasmoMitoCarta179 — a replica of the MitoCarta proteome analysis for Plasmodium falciparum — which can be investigated to identify drug targets to treat malaria.
On a more forward-looking note, the future mitochondrial protein map would benefit from a systematic catalogue of small molecules along with a characterization of how they interact with each member of the map, similar to the Cancer Therapeutics Response Portal that lists the effects of 354 compounds on 242 different cancer cell lines180. Such a complementary compendium would allow one to quickly identify inhibitors, activators or chaperone compounds that can be tested in preclinical treatment studies of mitochondrial diseases.
Conclusions and perspectives
Refining mitochondrial protein maps and using them to discover new biology is a reciprocal process: current maps provide a foundation for scientific discoveries that, in turn, lead to improved maps and stronger foundations for new analyses (Fig. 5). This underscores the necessity of leveraging new technologies and strategies to compose and characterize the mitochondrial proteome map as completely as possible. Yet it is important to consider which immediate efforts may be most impactful. Given the quality and high overlap in protein membership of the current compendia, systematic efforts to identify further mitochondrial proteins, although important, will likely have diminishing returns. We suggest that further efforts to define mitochondrial protein function and their regulation by PTMs will lead to the largest leaps in our understanding of mitochondrial biology and best position the field to explore new therapeutic avenues.
Fig. 5:
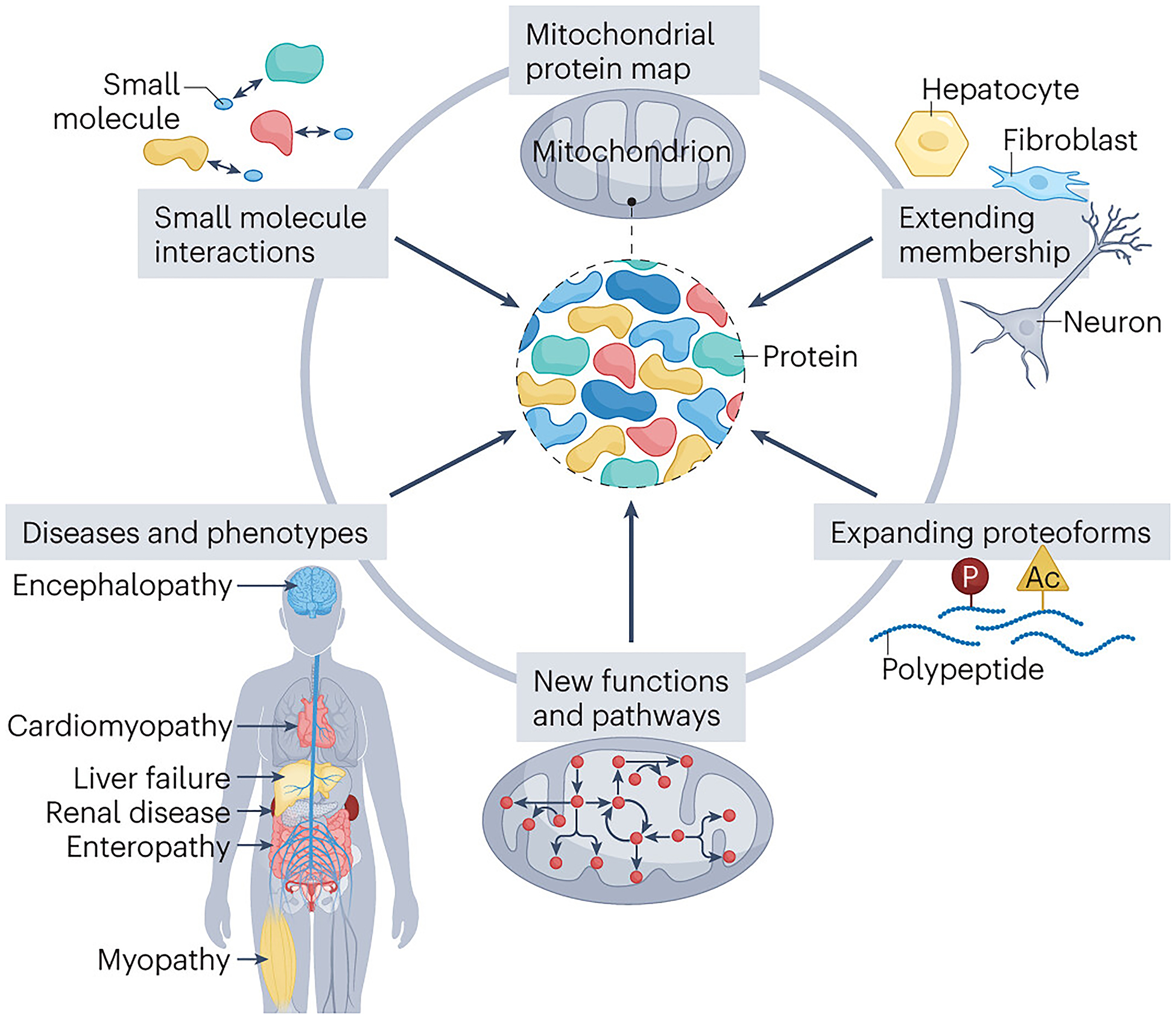
The future mitochondrial protein map.
The future mitochondrial protein map is set to be enhanced with extensive data on its members, including their diverse proteoforms, functions and integration into pathways. Moreover, map members will be linked to both diseases and phenotypes, as well as drug interactions, thus propelling innovative biological discoveries that can be applied in the medical context to improve the diagnosis and treatment of mitochondrial disorders.
Updating and refining the mitochondrial proteome map in all the ways we describe here is an enormous undertaking that will require the cooperation of many groups. We suggest that the time might be right for establishing a consortium charged with establishing and managing a new consensus map. Similar efforts, such as the Saccharomyces Genome Database, have become powerful, multifaceted, central resources that provide a clear point of entry for those entering their respective fields. A comparable effort for the field of mitochondrial biology would provide a ‘living resource’ that could be constantly updated and improved. Whatever form it eventually takes, the future mitochondrial map promises to help reveal new mitochondrial biology and advance the nascent field of mitochondrial therapeutic medicine.
Acknowledgements
The authors thank R. Guerra for critical feedback on this manuscript. This work was funded by National Institutes of Health (NIH) grants R35GM131795 and R01DK098672 and funds from the BJC Investigator Program (to D.J.P.), and fellowships from the European Molecular Biology Organization (ALTF 263-2022) and the Swiss National Science Foundation (P500PB_211038) (to P.F.).
Glossary
- Activity-based protein profiling
A proteomic method that uses enzyme-specific chemical probes to elucidate protein–small molecule interactions. Probes can be fluorescent species, biotin, alkynes or other small molecules that bind to their target proteins and enable enrichment for downstream mass spectrometry analyses.
- Affinity-enrichment mass spectrometry
A technique that couples affinity purification of a protein or protein complex with mass spectrometry analysis to identify protein–protein interactions (PPIs).
- Crosslinking mass spectrometry
A common technique for identifying protein–protein interactions (PPIs). Small molecules with reactive head groups interact with proteins in close proximity and ‘crosslink’ them together. The crosslinked proteins, and the sites of crosslinking, can then be identified using liquid chromatography–mass spectrometry.
- Discovery proteomics
An open-ended proteomics analysis that aims to identify as many proteins in a sample as possible without directly targeting any particular protein species. Often used as the first step in defining a particular proteome.
- False discovery rate
(FDR). A value that estimates the proportion of false positives among all positive findings in a statistical analysis where FDR = false positives / (false positives + true positives). This value can be cross-validated by withholding a portion of a training set and measuring the rate at which the algorithm can correctly assign values.
- Importomics
A method used in the mitochondrial high-confidence proteome (MitoCoP) study to identify proteins that are typically imported into mitochondria by quantifying their degradation following disruption of the mitochondrial import machinery.
- Inborn errors of metabolism
A group of monogenic diseases caused by pathogenic variants in genes that code for proteins involved in metabolism.
- Iterative support vector machine learning
Iterative support vector machine is a supervised machine learning algorithm that uses a set of training data to build a model that can then be used to classify new data. The algorithm seeks to define a boundary within dimensional space that is used to separate and define classes. This boundary is defined by maximizing the distance between it and the closest individual data points (support vectors) within the training set and is then used by the support vector machine model to make further classification decisions.
- Mass spectrometry
An analytical method capable of identifying and quantifying diverse chemical species, including proteins and peptides, by virtue of their mass and related properties.
- Monogenic pathogenic variants
Genetic lesions that can cause or increase the risk of developing an inherited disease.
- Naive Bayesian classifier
Naive Bayes is a probabilistic machine learning algorithm that uses Bayes’ theorem to classify data points. Bayes theorem defines the probability of an outcome or class (for example, ‘mitochondrial’) being true based on prior knowledge of information related to the outcome. The naive Bayes model will assume that the predictors of the model are independent of one another and assign weights to each predictor.
- Nonsense-mediated mRNA decay
A cellular process that degrades mRNAs containing premature stop codons, thereby helping to prevent the production of truncated proteins.
- Primary mitochondrial diseases
A group of genetic disorders that are caused by pathogenic mutations in genes that code for proteins that affect mitochondrial function.
- Proximity labelling approaches
A class of techniques used to analyse macromolecular complexes, protein interaction networks and subcellular protein localization. These methods employ a promiscuous labelling enzyme that is targeted to a specific cellular location through its genetic fusion to a protein of interest. Addition of a small molecule substrate then enables the promiscuous enzyme to covalently tag other proteins within its vicinity, allowing those proteins to be enriched and identified. Commonly used proximity labelling methods include APEX2 (based on an engineered ascorbate peroxidase) and BioID/TurboID (based on a mutant biotin ligase).
- Small open reading frame
(smORF). A compact DNA sequence with fewer than 100 codons that can be translated into one or more proteins.
- Spatial proteomics
In the Mitochondrial high Confidence Proteome (MitoCoP) manuscript, spatial proteomics is defined as a technique whereby tissues or subcellular fractions are isolated and then analysed using liquid chromatography–mass spectrometry proteomics. These data can then be used to establish sub-tissue or subcellular protein mapping.
- Subtractive proteomics
A technique that helps to assign a protein’s subcellular localization by quantifying its enrichment during purification (for example, quantifying the enrichment of a protein from crude to pure mitochondria).
- Tandem mass spectrometry
(MS/MS). A technique that relies on multiple mass analysers assembled in a single mass spectrometer. Following ionization and separation by the first mass analyser, often a quadrupole, ions are fragmented and then analysed in the second mass analyser. This allows for the detection of unique molecules with the same parent (unfragmented) mass.
- Top-down and bottom-up approaches
A top-down approach aims to uncover missing elements in a well-defined process (known unknowns), whereas a bottom-up approach aims to assign functions to poorly understood genes or proteins without a predefined notion of what those functions might be (unknown unknowns).
- Whole genome or exome sequencing
Techniques used to identify causal genetic variants in patients by sequencing the entire genome or only its protein-coding (exome) portion.
References
- 1.Hogeboom G, Schneider WC & Pallade GE Cytochemical studies of mammalian tissues; isolation of intact mitochondria from rat liver; some biochemical properties of mitochondria and submicroscopic particulate material. J. Biol. Chem 172, 619–635 (1948). [PubMed] [Google Scholar]
- 2.Lehninger AL Esterification of inorganic phosphate coupled to electron transport between dihydrodiphosphopyridine nucleotide and oxygen. II. J. Biol. Chem 178, 625–644 (1949). [PubMed] [Google Scholar]
- 3.Kennedy EP & Lehninger AL Oxidation of fatty acids and tricarboxylic acid cycle intermediates by isolated rat liver mitochondria. J. Biol. Chem 179, 957–972 (1949). [PubMed] [Google Scholar]
- 4.Barkulis SS & Lehninger AL Myokinase and the adenine nucleotide specificity in oxidative phosphorylations. J. Biol. Chem 190, 339–344 (1951). [PubMed] [Google Scholar]
- 5.Lardy HA & Adler J Synthesis of succinate from propionate and bicarbonate by soluble enzymes from liver mitochondria. J. Biol. Chem 219, 933–942 (1956). [PubMed] [Google Scholar]
- 6.Ernster L & Schatz G Mitochondria: a historical review. J. Cell Biol 91, 227s–255s (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rabilloud T et al. Two-dimensional electrophoresis of human placental mitochondria and protein identification by mass spectrometry: toward a human mitochondrial proteome. Electrophoresis 19, 1006–1014 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Lopez MF et al. High-throughput profiling of the mitochondrial proteome using affinity fractionation and automation. Electrophoresis 21, 3427–3440 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Sickmann A et al. The proteome of Saccharomyces cerevisiae mitochondria. Proc. Natl Acad. Sci. USA 100, 13207–13212 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mootha VK et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 115, 629–640 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Taylor SW et al. Characterization of the human heart mitochondrial proteome. Nat. Biotechnol 21, 281–286 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Gaucher SP et al. Expanded coverage of the human heart mitochondrial proteome using multidimensional liquid chromatography coupled with tandem mass spectrometry. J. Proteome Res 3, 495–505 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Foster LJ et al. A mammalian organelle map by protein correlation profiling. Cell 125, 187–199 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Reinders J, Zahedi RP, Pfanner N, Meisinger C & Sickmann A Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics. J. Proteome Res 5, 1543–1554 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Calvo S et al. Systematic identification of human mitochondrial disease genes through integrative genomics. Nat. Genet 38, 576–582 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Pagliarini DJ et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Venter JC et al. The sequence of the human genome. Science 291, 1304–1351 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Consortium IHGS et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Calvo SE, Clauser KR & Mootha VK MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 44, D1251–D1257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rath S et al. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49, gkaa1011 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgenstern M et al. Quantitative high-confidence human mitochondrial proteome and its dynamics in cellular context. Cell Metab. 33, 2464–2483.e18 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith AG et al. A curated collection of human mitochondrial proteins — the Integrated Mitochondrial Protein Index (IMPI). Preprint at SSRN Electron. J 10.2139/ssrn.4042282 (2022). [DOI] [Google Scholar]
- 23.Uhlén M et al. Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Schulte U et al. Mitochondrial complexome reveals quality-control pathways of protein import. Nature 614, 153–159 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morgenstern M et al. Definition of a high-confidence mitochondrial proteome at quantitative scale. Cell Rep. 19, 2836–2852 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vögtle F-N et al. Landscape of submitochondrial protein distribution. Nat. Commun 8, 290 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sung AY, Floyd BJ & Pagliarini DJ Systems biochemistry approaches to defining mitochondrial protein function. Cell Metab. 31, 669–678 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughes CS et al. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc 14, 68–85 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Muehlbauer LK et al. Rapid multi-omics sample preparation for mass spectrometry. Anal. Chem 95, 659–667 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bayraktar EC et al. MITO-tag mice enable rapid isolation and multimodal profiling of mitochondria from specific cell types in vivo. Proc. Natl Acad. Sci. USA 116, 303–312 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fecher C et al. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat. Neurosci 22, 1731–1742 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Benador IY et al. Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion. Cell Metab. 27, 869–885.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Najt CP et al. Organelle interactions compartmentalize hepatic fatty acid trafficking and metabolism. Cell Rep. 42, 112435 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saki M & Prakash A DNA damage related crosstalk between the nucleus and mitochondria. Free Radic. Bio Med 107, 216–227 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu J et al. Phosphorylation-dependent mitochondrial translocation of MAP4 is an early step in hypoxia-induced apoptosis in cardiomyocytes. Cell Death Dis. 5, e1424 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X et al. Mitochondria-translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol. Cell 61, 705–719 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rhee H-W et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339, 1328–1331 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hung V et al. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell 55, 332–341 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roux KJ, Kim DI, Raida M & Burke B A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol 196, 801–810 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu X, Salokas K, Weldatsadik RG, Gawriyski L & Varjosalo M Combined proximity labeling and affinity purification−mass spectrometry workflow for mapping and visualizing protein interaction networks. Nat. Protoc 15, 3182–3211 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Antonicka H et al. A high-density human mitochondrial proximity interaction network. Cell Metab. 32, 479–497.e9 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Branon TC et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol 36, 880–887 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho KF et al. Split-TurboID enables contact-dependent proximity labeling in cells. Proc. Natl Acad. Sci. USA 117, 12143–12154 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bader G et al. Assigning mitochondrial localization of dual localized proteins using a yeast bi-genomic mitochondrial-split-GFP. eLife 9, e56649 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mercer TR et al. The human mitochondrial transcriptome. Cell 146, 645–658 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hashimoto Y et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Aβ. Proc. Natl Acad. Sci. USA 98, 6336–6341 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yen K et al. Humanin prevents age-related cognitive decline in mice and is associated with improved cognitive age in humans. Sci. Rep 8, 14212 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee C et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 21, 443–454 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olexiouk V, Van Criekinge W & Menschaert G An update on sORFs.org: a repository of small ORFs identified by ribosome profiling. Nucleic Acids Res. 46, gkx1130 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang C et al. Mitochondrial microproteins link metabolic cues to respiratory chain biogenesis. Cell Rep. 40, 111204 (2022). [DOI] [PubMed] [Google Scholar]
- 51.Chu Q et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat. Commun 10, 4883 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rathore A et al. MIEF1 microprotein regulates mitochondrial translation. Biochemistry 57, 5564–5575 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stein CS et al. Mitoregulin: a lncRNA-encoded microprotein that supports mitochondrial supercomplexes and respiratory efficiency. Cell Rep. 23, 3710–3720.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baker MJ, Crameri JJ, Thorburn DR, Frazier AE & Stojanovski D Mitochondrial biology and dysfunction in secondary mitochondrial disease. Open Biol. 12, 220274 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arroyo JD et al. A genome-wide CRISPR death screen identifies genes essential for oxidative phosphorylation. Cell Metab. 24, 875–885 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith LM et al. Proteoform: a single term describing protein complexity. Nat. Methods 10, 186–187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aebersold R et al. How many human proteoforms are there? Nat. Chem. Biol 14, 206–214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith LM et al. The Human Proteoform Project: defining the human proteome. Sci. Adv 7, eabk0734 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schaffer LV et al. Identification and quantification of murine mitochondrial proteoforms using an integrated top-down and intact-mass strategy. J. Proteome Res 17, 3526–3536 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Catherman AD et al. Large-scale top-down proteomics of the human proteome: membrane proteins, mitochondria, and senescence. Mol. Cell. Proteom 12, 3465–3473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Song Z, Chen H, Fiket M, Alexander C & Chan DC OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol 178, 749–755 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anand R et al. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol 204, 919–929 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dotto VD, Fogazza M, Carelli V, Rugolo M & Zanna C Eight human OPA1 isoforms, long and short: what are they for? Biochim. Biophys. Acta 1859, 263–269 (2018). [DOI] [PubMed] [Google Scholar]
- 64.Dotto VD et al. OPA1 isoforms in the hierarchical organization of mitochondrial functions. Cell Rep. 19, 2557–2571 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Reane DV et al. A MICU1 splice variant confers high sensitivity to the mitochondrial Ca2+ uptake machinery of skeletal muscle. Mol. Cell 64, 760–773 (2016). [DOI] [PubMed] [Google Scholar]
- 66.Gang H et al. PDK2-mediated alternative splicing switches Bnip3 from cell death to cell survival. J. Cell Biol 210, 1101–1115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morciano G et al. Mcl-1 involvement in mitochondrial dynamics is associated with apoptotic cell death. Mol. Biol. Cell 27, 20–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sinitcyn P et al. Global detection of human variants and isoforms by deep proteome sequencing. Nat. Biotechnol 10.1038/s41587-023-01714-x (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Niemi NM & Pagliarini DJ The extensive and functionally uncharacterized mitochondrial phosphoproteome. J. Biol. Chem 297, 100880 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hosp F et al. Lysine acetylation in mitochondria: from inventory to function. Mitochondrion 33, 58–71 (2017). [DOI] [PubMed] [Google Scholar]
- 71.Anderson KA & Hirschey MD Mitochondrial protein acetylation regulates metabolism. Essays Biochem. 52, 23–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Piantadosi CA Regulation of mitochondrial processes by protein S-nitrosylation. Biochim. Biophys. Acta 1820, 712–721 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wieland O & Siess E Interconversion of phospho- and dephospho- forms of pig heart pyruvate dehydrogenase. Proc. Natl Acad. Sci. USA 65, 947–954 (1970). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Linn TC, Pettit FH & Reed LJ α-Keto acid dehydrogenase complexes, X. Regulation of the activity of the pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc. Natl Acad. Sci. USA 62, 234–241 (1969). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Paxton R & Harris RA Isolation of rabbit liver branched chain α-ketoacid dehydrogenase and regulation by phosphorylation. J. Biol. Chem 257, 14433–14439 (1982). [PubMed] [Google Scholar]
- 76.Grimsrud PA et al. A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab. 16, 672–683 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hansen FM et al. Mitochondrial phosphoproteomes are functionally specialized across tissues. Preprint at bioRxiv 10.1101/2022.03.23.485457 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bak S, León IR, Jensen ON & Højlund K Tissue specific phosphorylation of mitochondrial proteins isolated from rat liver, heart muscle, and skeletal muscle. J. Proteome Res 12, 4327–4339 (2013). [DOI] [PubMed] [Google Scholar]
- 79.Kruse R & Højlund K Mitochondrial phosphoproteomics of mammalian tissues. Mitochondrion 33, 45–57 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Niemi NM et al. Pptc7 is an essential phosphatase for promoting mammalian mitochondrial metabolism and biogenesis. Nat. Commun 10, 3197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Niemi NM et al. Pptc7 maintains mitochondrial protein content by suppressing receptor-mediated mitophagy. Preprint at bioRxiv 10.1101/2023.02.28.530351 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim SC et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618 (2006). [DOI] [PubMed] [Google Scholar]
- 83.Park J et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 50, 919–930 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Colak G et al. Proteomic and biochemical studies of lysine malonylation suggest its malonic aciduria-associated regulatory role in mitochondrial function and fatty acid oxidation [S]. Mol. Cell Proteom 14, 3056–3071 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bharathi SS et al. Sirtuin 3 (SIRT3) protein regulates long-chain acyl-CoA dehydrogenase by deacetylating conserved lysines near the active site. J. Biol. Chem 288, 33837–33847 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fan J et al. Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol. Cell 53, 534–548 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hallows WC, Lee S & Denu JM Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl Acad. Sci. USA 103, 10230–10235 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Horton JL et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 1, e84897 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hebert AS et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 49, 186–199 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Doulias P-T, Tenopoulou M, Greene JL, Raju K & Ischiropoulos H Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci. Signal 6, rs1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Prime TA et al. A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia–reperfusion injury. Proc. Natl Acad. Sci. USA 106, 10764–10769 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gu Z, Nakamura T & Lipton SA Redox reactions induced by nitrosative stress mediate protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Mol. Neurobiol 41, 55–72 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Seim GL et al. Nitric oxide-driven modifications of lipoic arm inhibit α-ketoacid dehydrogenases. Nat. Chem. Biol 19, 265–274 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gibson QH & Greenwood C Kinetic evidence for a short lived intermediate in the oxidation of cytochrome c oxidase by molecular oxygen. J. Biol. Chem 240, 957–958 (1965). [PubMed] [Google Scholar]
- 95.Coles SJ et al. S-Nitrosoglutathione inactivation of the mitochondrial and cytosolic BCAT proteins: S-nitrosation and S-thiolation. Biochemistry 48, 645–656 (2009). [DOI] [PubMed] [Google Scholar]
- 96.Cao W et al. Discovery and confirmation of O-GlcNAcylated proteins in rat liver mitochondria by combination of mass spectrometry and immunological methods. PLoS ONE 8, e76399 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hu Y et al. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J. Biol. Chem 284, 547–555 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Andrabi SA et al. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl Acad. Sci. USA 111, 10209–10214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pagliarini DJ & Dixon JE Mitochondrial modulation: reversible phosphorylation takes center stage? Trends Biochem. Sci 31, 26–34 (2006). [DOI] [PubMed] [Google Scholar]
- 100.Gao S et al. Docking of endothelial nitric oxide synthase (eNOS) to the mitochondrial outer membrane a pentabasic amino acid sequence in the autoinhibitory domain of enos targets a proteinase K-cleavable peptide on the cytoplasmic face of mitochondria. J. Biol. Chem 279, 15968–15974 (2004). [DOI] [PubMed] [Google Scholar]
- 101.Chatterjee A et al. MOF acetyl transferase regulates transcription and respiration in mitochondria. Cell 167, 722–738.e23 (2016). [DOI] [PubMed] [Google Scholar]
- 102.Perocchi F et al. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467, 291–296 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Herzig S et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science 337, 93–96 (2012). [DOI] [PubMed] [Google Scholar]
- 104.Bricker DK et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337, 96–100 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Luongo TS et al. SLC25A51 is a mammalian mitochondrial NAD+ transporter. Nature 588, 174–179 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Siddharth T & Lewis N Predicting pathways for old and new metabolites through clustering. Preprint at arXiv 10.48550/arxiv.2211.15720 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang Y et al. SLC25A39 is necessary for mitochondrial glutathione import in mammalian cells. Nature 599, 136–140 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shi X et al. Combinatorial GxGxE CRISPR screen identifies SLC25A39 in mitochondrial glutathione transport linking iron homeostasis to OXPHOS. Nat. Commun 13, 2483 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Alston CL et al. Bi-allelic mutations in NDUFA6 establish its role in early-onset isolated mitochondrial complex I deficiency. Am. J. Hum. Genet 103, 592–601 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Floyd BJ et al. Mitochondrial protein interaction mapping identifies regulators of respiratory chain function. Mol. Cell 63, 621–632 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Alston CL et al. Pathogenic bi-allelic mutations in NDUFAF8 cause Leigh syndrome with an isolated complex I deficiency. Am. J. Hum. Genet 106, 92–101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lam SS et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51–54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Go CD et al. A proximity-dependent biotinylation map of a human cell. Nature 595, 120–124 (2021). [DOI] [PubMed] [Google Scholar]
- 114.Hung V et al. Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. eLife 6, e24463 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hevler JF et al. Selective cross-linking of coinciding protein assemblies by in-gel cross-linking mass spectrometry. EMBO J. 40, e106174 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liu F, Lössl P, Rabbitts BM, Balaban RS & Heck AJR The interactome of intact mitochondria by cross-linking mass spectrometry provides evidence for coexisting respiratory supercomplexes. Mol. Cell Proteom 17, 216–232 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Linden A et al. A cross-linking mass spectrometry approach defines protein interactions in yeast mitochondria. Mol. Cell Proteom 19, 1161–1178 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Heide H et al. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab. 16, 538–549 (2012). [DOI] [PubMed] [Google Scholar]
- 119.Gao J et al. CLUH regulates mitochondrial biogenesis by binding mRNAs of nuclear-encoded mitochondrial proteins. J. Cell Biol 207, 213–223 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schatton D et al. CLUH regulates mitochondrial metabolism by controlling translation and decay of target mRNAs. J. Cell Biol 216, 675–693 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Meyer JG, Niemi NM, Pagliarini DJ & Coon JJ Quantitative shotgun proteome analysis by direct infusion. Nat. Methods 17, 1222–1228 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.He Y et al. Multi-omic single-shot technology for integrated proteome and lipidome analysis. Anal. Chem 93, 4217–4222 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schäfer JA, Sutandy FXR & Münch C Omics-based approaches for the systematic profiling of mitochondrial biology. Mol. Cell 83, 911–926 (2023). [DOI] [PubMed] [Google Scholar]
- 124.Lapointe CP et al. Multi-omics reveal specific targets of the RNA-binding protein Puf3p and its orchestration of mitochondrial biogenesis. Cell Syst. 6, 125–135.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Veling MT et al. Multi-omic mitoprotease profiling defines a role for Oct1p in coenzyme Q production. Mol. Cell 68, 970–977.e11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Stefely JA et al. Mitochondrial protein functions elucidated by multi-omic mass spectrometry profiling. Nat. Biotechnol 34, 1191–1197 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rensvold JW et al. Defining mitochondrial protein functions through deep multiomic profiling. Nature 606, 382–388 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kathayat RS et al. Active and dynamic mitochondrial S-depalmitoylation revealed by targeted fluorescent probes. Nat. Commun 9, 334 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Li Y, Calvo SE, Gutman R, Liu JS & Mootha VK Expansion of biological pathways based on evolutionary inference. Cell 158, 213–225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mühleip A et al. Structural basis of mitochondrial membrane bending by the I–II–III2–IV2 supercomplex. Nature 615, 934–938 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Szklarczyk D et al. The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49, gkaa1074 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Oughtred R et al. The BioGRID database: a comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 30, 187–200 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Franz M et al. GeneMANIA update 2018. Nucleic Acids Res. 46, W60–W64 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Escudero S et al. Dynamic regulation of long-chain fatty acid oxidation by a noncanonical interaction between the MCL-1 BH3 helix and VLCAD. Mol. Cell 69, 729–743.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chung K-P et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat. Commun 10, 3390 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ferreira CR et al. An international classification of inherited metabolic disorders (ICIMD). J. Inherit. Metab. Dis 44, 164–177 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rahman J & Rahman S Mitochondrial medicine in the omics era. Lancet 391, 2560–2574 (2018). [DOI] [PubMed] [Google Scholar]
- 138.Schlieben LD & Prokisch H The dimensions of primary mitochondrial disorders. Front. Cell Dev. Biol 8, 600079 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gorman GS et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol 77, 753–759 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Forny P et al. Diagnosing mitochondrial disorders remains challenging in the omics era. Neurol. Genet 7, e597 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wortmann SB, Koolen DA, Smeitink JA, Heuvel L & Rodenburg RJ Whole exome sequencing of suspected mitochondrial patients in clinical practice. J. Inherit. Metab. Dis 38, 437–443 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Taylor RW et al. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 312, 68–77 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pronicka E et al. New perspective in diagnostics of mitochondrial disorders: two years’ experience with whole-exome sequencing at a national paediatric centre. J. Transl. Med 14, 174 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Theunissen TEJ et al. Whole exome sequencing is the preferred strategy to identify the genetic defect in patients with a probable or possible mitochondrial cause. Front. Genet 9, 400 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Schon KR et al. Use of whole genome sequencing to determine genetic basis of suspected mitochondrial disorders: cohort study. BMJ 375, e066288 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kremer LS et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun 8, 15824 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mertes C et al. Detection of aberrant splicing events in RNA-seq data using FRASER. Nat. Commun 12, 529 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Brechtmann F et al. OUTRIDER: a statistical method for detecting aberrantly expressed genes in rna sequencing data. Am. J. Hum. Genet 103, 907–917 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yépez VA et al. Clinical implementation of RNA sequencing for Mendelian disease diagnostics. Genome Med. 14, 38 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Smirnov D, Schlieben LD, Peymani F, Berutti R & Prokisch H Guidelines for clinical interpretation of variant pathogenicity using RNA phenotypes. Hum. Mutat 43, 1056–1070 (2022). [DOI] [PubMed] [Google Scholar]
- 151.Parikh S et al. Diagnosis of ‘possible’ mitochondrial disease: an existential crisis. J. Med. Genet 56, 123 (2019). [DOI] [PubMed] [Google Scholar]
- 152.Frazier AE, Thorburn DR & Compton AG Mitochondrial energy generation disorders: genes, mechanisms, and clues to pathology. J. Biol. Chem 294, 5386–5395 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tsang MHY et al. Delineation of molecular findings by whole-exome sequencing for suspected cases of paediatric-onset mitochondrial diseases in the southern Chinese population. Hum. Genomics 14, 28 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Calvo SE et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat. Genet 42, 851–858 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Köhler S et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 49, gkaa1043 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Simon MT et al. Novel mutations in the mitochondrial complex I assembly gene NDUFAF5 reveal heterogeneous phenotypes. Mol. Genet. Metab 126, 53–63 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Shaikh SS, Nahorski MS, Rai H & Woods CG Before progressing from “exomes” to “genomes” … don’t forget splicing variants. Eur. J. Hum. Genet 26, 1–4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sahni N et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell 161, 647–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Helman G et al. Multiomic analysis elucidates complex I deficiency caused by a deep intronic variant in NDUFB10. Hum. Mutat 42, 19–24 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wortmann SB et al. How to proceed after “negative” exome: a review on genetic diagnostics, limitations, challenges, and emerging new multiomics techniques. J. Inherit. Metab. Dis 45, 663–681 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Stenton SL, Kremer LS, Kopajtich R, Ludwig C & Prokisch H The diagnosis of inborn errors of metabolism by an integrative “multi-omics” approach: a perspective encompassing genomics, transcriptomics, and proteomics. J. Inherit. Metab. Dis 43, 25–35 (2020). [DOI] [PubMed] [Google Scholar]
- 162.Lynch DR et al. Efficacy of omaveloxolone in Friedreich’s ataxia: delayed-start analysis of the MOXIe extension. Mov. Disord 38, 313–320 (2023). [DOI] [PubMed] [Google Scholar]
- 163.Carelli V et al. Idebenone treatment in Leber’s hereditary optic neuropathy. Brain 134, e188 (2011). [DOI] [PubMed] [Google Scholar]
- 164.Murray NH et al. Small-molecule inhibition of the archetypal UbiB protein COQ8. Nat. Chem. Biol 19, 230–238 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pesini A, Hidalgo-Gutierrez A & Quinzii CM Mechanisms and therapeutic effects of benzoquinone ring analogs in primary CoQ deficiencies. Antioxidants 11, 665 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Pierrel F Impact of chemical analogs of 4-hydroxybenzoic acid on coenzyme Q biosynthesis: from inhibition to bypass of coenzyme Q deficiency. Front. Physiol 8, 436 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Freyer C et al. Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4-dihydroxybensoic acid. J. Med. Genet 52, 779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Guerra RM & Pagliarini DJ Coenzyme Q biochemistry and biosynthesis. Trends Biochem. Sci 48, 463–476 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Baker PR et al. Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain 137, 366–379 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Head PE et al. Aberrant methylmalonylation underlies methylmalonic acidemia and is attenuated by an engineered sirtuin. Sci. Transl. Med 14, eabn4772 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yahata N, Matsumoto Y, Omi M, Yamamoto N & Hata R TALEN-mediated shift of mitochondrial DNA heteroplasmy in MELAS-iPSCs with m.13513G>A mutation. Sci. Rep 7, 15557 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hashimoto M et al. MitoTALEN: a general approach to reduce mutant mtDNA loads and restore oxidative phosphorylation function in mitochondrial diseases. Mol. Ther 23, 1592–1599 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Gammage PA, Rorbach J, Vincent AI, Rebar EJ & Minczuk M Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol. Med 6, 458–466 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Liu Z, Sun Y, Qi Z, Cao L & Ding S Mitochondrial transfer/transplantation: an emerging therapeutic approach for multiple diseases. Cell Biosci. 12, 66 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Nitzan K et al. Mitochondrial transfer ameliorates cognitive deficits, neuronal loss, and gliosis in Alzheimer’s disease mice. J. Alzheimers Dis 72, 587–604 (2019). [DOI] [PubMed] [Google Scholar]
- 176.Chang J-C et al. Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: restoration of mitochondria functions and attenuation of 6-hydroxydopamine-induced neurotoxicity. Transl. Res 170, 40–56.e3 (2016). [DOI] [PubMed] [Google Scholar]
- 177.Ripcke J, Zarse K, Ristow M & Birringer M Small-molecule targeting of the mitochondrial compartment with an endogenously cleaved reversible tag. Chembiochem 10, 1689–1696 (2009). [DOI] [PubMed] [Google Scholar]
- 178.Steenberge LH, Sung AY, Fan J & Pagliarini DJ Coenyzme Q4 is a functional substitute for coenyzme Q10 and can be targeted to the mitochondria. Preprint at bioRxiv 10.1101/2023.07.20.549963 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Esveld S Lvan et al. A prioritized and validated resource of mitochondrial proteins in plasmodium identifies unique biology. mSphere 6, e00614–e00621 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Basu A et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154, 1151–1161 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Smith AC & Robinson AJ MitoMiner v4.0: an updated database of mitochondrial localization evidence, phenotypes and diseases. Nucleic Acids Res. 47, gky1072 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Brandina I et al. Enolase takes part in a macromolecular complex associated to mitochondria in yeast. Biochim. Biophys. Acta 1757, 1217–1228 (2006). [DOI] [PubMed] [Google Scholar]
- 183.Wisnovsky S, Sack T, Pagliarini DJ, Laposa RR & Kelley SO DNA polymerase θ increases mutational rates in mitochondrial DNA. ACS Chem. Biol 13, 900–908 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hao Z et al. N6-deoxyadenosine methylation in mammalian mitochondrial DNA. Mol. Cell 78, 382–395.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chen WW, Freinkman E & Sabatini DM Rapid immunopurification of mitochondria for metabolite profiling and absolute quantification of matrix metabolites. Nat. Protoc 12, 2215–2231 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Fijalkowski I, Peeters MKR & Damme PV Small protein enrichment improves proteomics detection of sORF encoded polypeptides. Front. Genet 12, 713400 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Giansanti P, Tsiatsiani L, Low TY & Heck AJR Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat. Protoc 11, 993–1006 (2016). [DOI] [PubMed] [Google Scholar]
- 188.Swaney DL & Villén J Enrichment of modified peptides via immunoaffinity precipitation with modification-specific antibodies. Cold Spring Harb. Protoc 2016, pdb.prot088013 (2016). [DOI] [PubMed] [Google Scholar]
- 189.Brandi J, Noberini R, Bonaldi T & Cecconi D Advances in enrichment methods for mass spectrometry-based proteomics analysis of post-translational modifications. J. Chromatogr. A 1678, 463352 (2022). [DOI] [PubMed] [Google Scholar]