

Summary
System-level patient health signals, as captured by treatment-emergent adverse events (TEAEs), might contain correlates of immune checkpoint inhibitor (ICI) therapy response. Using all TEAEs and a novel machine learning modeling approach, we derived a composite signature predictive of, and potentially specific to, the response to the anti-PD-L1 ICI durvalumab in patients with non–small-cell lung cancer (NSCLC). We trained on data from the durvalumab arm and chemotherapy arm in the MYSTIC clinical trial and tested on data from four independent durvalumab-containing NSCLC trials using only the first 60 days’ TEAEs. We directly compared our signature performance against that of three different definitions of immune-related adverse events. Only our signature was predictive and identified longer survivors in patients treated with durvalumab but not in patients treated with chemotherapy or placebo. It also identified durvalumab-treated long survivors with stable disease at their first RECIST evaluation and a set of PD-L1-negative long survivors.
Subject areas: health sciences, natural sciences, immunity, computer science
Graphical abstract
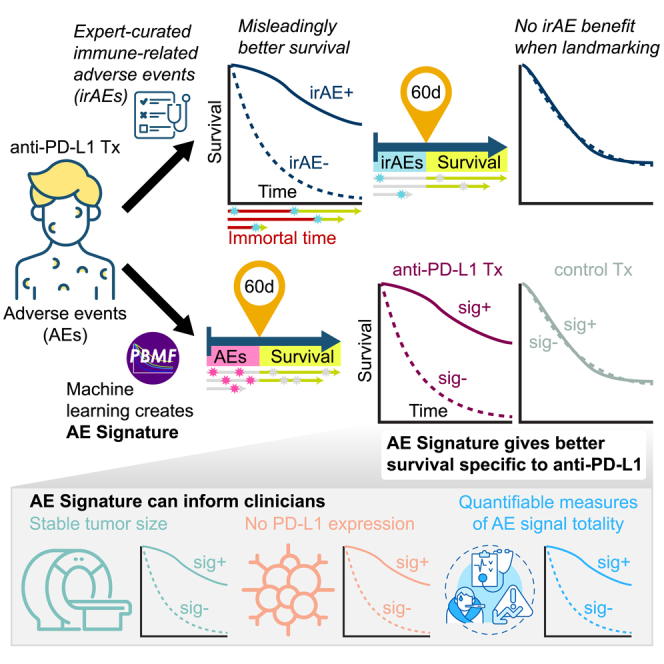
Highlights
-
•
Adverse event signature predictive of long survival for patients on anti-PD-L1 therapy
-
•
Signature identifies long-term survivors with stable disease at first tumor imaging
-
•
Signature identifies long-term survivors in the subset that are PD-L1 negative
Health sciences; Natural sciences; Immunity; Computer science.
Introduction
The advent of immune checkpoint inhibitor (ICI) therapy has led to notable successes in the treatment of a number of cancer types. Nevertheless, we have yet to achieve a complete understanding of the factors contributing to favorable long-term prognosis in patients receiving ICI therapy. This knowledge gap is partly due to the indirect nature of ICIs, which act through the immune system and so are subject to a much broader range of response modifiers than, for example, a tumor cell–targeted therapy. Investigations into ICI response drivers to date have focused primarily on molecular profiling and assessment of the tumor immune microenvironment. In this regard, the expression of programmed cell death 1 ligand (PD-L1) on tumors or in the tumor immune microenvironment has been found to at least partly predict response to anti–PD-(L)1 therapies,1,2 as do molecular features associated with the increased presence of neoantigens2,3,4,5 (e.g., tumor mutational burden or microsatellite instability). However, none of these markers provide optimal predictive value with respect to response to ICI therapy.6 We hypothesized that system-level patient health signals, including those related to immune system function, might also contain correlates of ICI response. Contemporaneous reporting of adverse events (AEs) provides a surrogate for on-treatment patient health in real time. Although on-treatment patient health signals cannot inform decisions regarding the initiation of ICI treatment, they could provide added prognostication value. Further, they could potentially be integrated into future clinical trial designs to help clarify treatment decisions (e.g., switching or intensifying therapy) in patients who are unlikely to benefit.
Previous work has examined a specific subset of TEAEs, immune-related AEs (irAEs), in the context of predicting ICI treatment outcomes, but findings have been equivocal.7,8 Identifying bona fide irAEs is a complex and often subjective process, and irAE reporting practices lack standardization.9,10 Clinicians must weigh imperfect evidence and exclude alternate etiologies. In particular, for open-label clinical studies, clinicians must also guard against preferentially reporting irAEs in the ICI treatment arms. In addition, despite numerous published studies finding that irAEs are associated with ICI efficacy, few correctly account for the critical confounder that longer-surviving individuals have a greater risk of developing an irAE, i.e., survivorship or immortal time bias.11,12,13,14 Finally, few studies have examined whether irAE relation to efficacy is specific to ICIs and not merely prognostic, irrespective of treatment.15,16,17,18
We hypothesized that a combination of AEs, which may include but is not limited to irAEs, can predict whether an individual patient will survive longer on ICI treatment than on chemotherapy or placebo treatment, and in a manner that can be generalized, at least within an indication. Given the aforementioned limitations of prior work, we took an unbiased modeling approach, using all reported TEAEs in a representative late-stage randomized controlled phase 3 clinical trial of ICIs in first-line metastatic non-small-cell lung cancer (NSCLC), the MYSTIC trial (NCT02453282).19 We leveraged our recently developed predictive biomarker modeling framework (PBMF)20 to find a signature that specifically reports on favorable long-term prognosis for anti-PD-L1 therapy (durvalumab) but not chemotherapy. Based on these analyses, we show that patient health signals may indeed contain information related to overall survival (OS) in a manner specific to durvalumab within the examined cohorts. Specifically, we provide proof of concept that a composite signature derived from early TEAEs can provide added prognostication value for ICI therapy beyond that provided by PD-L1 status and that the signature may be assessable before first on-treatment tumor imaging.
Results
Unbiased modeling of adverse events to enrich for patients with non-small-cell lung cancer most likely to benefit from anti-programmed cell death ligand 1 therapy over chemotherapy or placebo
To determine whether system-level patient health signals contained correlates of ICI response, we leveraged the contemporaneous reporting of AEs as a surrogate for on-treatment, real-time patient health. To avoid bias in our analysis, we used all on-treatment AE and OS data from the durvalumab monotherapy and chemotherapy arms of the MYSTIC trial to train a model with our recently developed PBMF (Figure 1A).19,20 To evaluate the model’s performance, we controlled for immortal time bias by performing a landmark analysis with a landmark time of 60 days. Only AEs that occurred before 60 days were considered, and only patients who survived at least 60 days were included. A 60-day landmark was chosen because we wanted a readout proximal to the first tumor imaging assessment.
Figure 1.

Generation and evaluation of a machine learning-derived adverse event signature (AE signature)
(A) Analysis workflow diagram contrasting the use of irAEs versus the AE signature derived with our machine learning framework, the PBMF.
(B) Kaplan-Meier plots for OS, per treatment arm and signature class, for MYSTIC trial data landmarked at 60 days.
(C) Kaplan-Meier plots for OS, per treatment arm and signature class, for PACIFIC trial data landmarked at 60 days.
(D) Kaplan-Meier plots for OS, per treatment arm and signature class, for ARCTIC trial data landmarked at 60 days.
(E) Point estimates and 95% confidence intervals for hazard ratios (Cox proportional hazards regression model) with machine learning (ML) AE signature class as the only covariate, computed per treatment arm, per trial, after AE and OS data were landmarked at 60 days.
(F) Point estimates and 95% confidence intervals for hazard ratios (patient-level meta-analysis; Cox proportional hazards regression model with random effects for trial, indication, and treatment; fixed effects for signature class, age, sex, race, geographical region, tumor stage, and histology), after AE and OS data were landmarked at 60 days (excluding training data), and by signature or one of the three different definitions of an irAE. Chemo, chemotherapy; HR, hazard ratio; sig, ML AE signature.
We verified that our model enriched for longer survivors only for durvalumab treatment in 60-day–landmarked data from the MYSTIC trial (Figures 1B and 1E). We then used our model to compute an AE signature on four durvalumab-treated NSCLC patient cohorts landmarked at 60 days that were not seen by the model during training (Figures 1C–1E; Table S1). Although the trial-level AE signature association with survival varied, we observed a clear trend of longer survival times for patients receiving durvalumab treatment but not those receiving chemotherapy or placebo. This trend was also reflected in a patient-level meta-analysis across all test datasets (durvalumab meta-HR = 0.83, 95% CI = 0.71–0.97; chemotherapy or placebo meta-HR = 1.02, 95% CI = 0.78–1.34; Figure 1F).
Given the extensive literature associating irAEs, a special subset of AEs, with ICI survival outcomes,7,8 we also examined whether having an irAE conferred an OS benefit. Examination of these data without controlling for immortal time bias led to a strong association between having an irAE and surviving longer (Figures 1A and S1A). In contrast, landmarked data showed no association between irAE status and survival (durvalumab trial-reported irAE meta-HR = 1.06, 95% CI = 0.83–1.35; Figures 1F and S1B). An association was found only after landmarking at later times (e.g., 6 months; Figure S1B). A similar lack of association or specificity to ICI treatment was observed with two alternative definitions of irAEs (durvalumab investigator-reported irAE meta-HR = 1.00, 95% CI = 0.78–1.29; durvalumab adverse event of special interest meta-HR = 0.90, 95% CI = 0.77–1.06; chemotherapy or placebo adverse event of special interest meta-HR = 0.92, 95% CI = 0.68–1.25; Figure 1F).
The improved ability of the AE signature over irAE to identify longer survivors on durvalumab was not a result of enriching for a subset of irAE-positive patients, as the numbers of patients with AE sig+ was more than double that of patients with irAE+ (66 sig+ vs. 22 irAE+ for the MYSTIC trial; 159 sig+ vs. 75 irAE+ for the PACIFIC trial). Together, these results suggest that although irAEs are not robustly associated with survival outcomes, unbiased modeling of all on-treatment AEs can be informative, albeit modestly, of survival outcomes specific to durvalumab therapy.
Adverse event signature identifies long surviving patients that are programmed cell death ligand 1 negative or have stable disease at first tumor imaging assessment
The encouraging trend for longer OS specific to durvalumab treatment led us to explore how the signature associates with other established predictors and measures of response. Multiple clinical studies have demonstrated an association of PD-L1 status with outcome after ICI treatment,21 consistent with PD-L1 being a known predictive biomarker for anti-PD-1 and anti-PD-L1 therapy. Interestingly, the AE signature tended to enrich for longer OS in patients with tumors with PD-L1 tumor cell expression <1% (PD-L1 <1%) who were treated with durvalumab (meta-HR = 0.82, 95% CI = 0.58–1.15), but not those treated with chemotherapy or placebo (meta-HR = 1.37, 95% CI = 0.73–2.60; Figures 2A and 2C). In contrast, within the cohort of patients with tumors with PD-L1 tumor cell expression ≥1% (PD-L1 ≥1%), AE signature status appeared to be prognostic for OS (Figures 2B and 2D).
Figure 2.

Evaluation of AE signature in the context of PD-L1 status and tumor response to therapy
(A and B) Kaplan-Meier plots for OS, per treatment arm and sig class, for ARCTIC trial data landmarked at 60 days for (A) PD-L1 <1% and (B) PD-L1 ≥1%.
(C–F) Point estimates and 95% confidence intervals for hazard ratios (Cox proportional hazards regression model) with ML AE signature class as the only covariate, computed per treatment arm, per trial, after AE and OS data were landmarked at 60 days. Boxed in purple is a patient-level meta-analysis (NSCLC only and excluding MYSTIC training data) Cox proportional hazards regression model with random effects for trial, indication, treatment, and fixed effects for signature class, age, sex, race, geographical region, tumor stage, and histology. (C) Patients with <1% PD-L1 tumor cells. (D) Patients with ≥1% PD-L1 tumor cells. (E) Patients with SD at their first RECIST evaluation. (F) Patients with CR or PR at their first RECIST evaluation. Chemo, chemotherapy; HR, hazard ratio; sig, ML AE signature.
Examination of progression-free survival (PFS) in the context of PD-L1 expression revealed that the AE signature did not affect survival outcomes in any PD-L1 subgroup (Figures S2A and S2B). In general, despite its association with longer OS, we observed no clear enrichment of longer PFS with the AE signature (Figure S2C). To understand the difference between OS and PFS within AE signature groups and in relation to PD-L1 status, we examined the disease control rate. We observed a slight trend for improved disease control rate in the PD-L1 <1% patient groups who were AE sig+ compared with AE sig– (Figure S3).
During treatment, patients undergo longitudinal tumor imaging to determine whether lesions have changed over time, according to standard schedules and ad hoc as required, often using RECIST (version 1.1) to assess treatment efficacy.22 However, many patients may have stable disease (SD) per RECIST 1.1, including during early treatment cycles. Therefore, we next asked whether the AE signature could provide added value over tumor imaging alone. Across all cohorts, most patients were assessed as having SD (according to RECIST v1.1) at their first tumor imaging assessment, which was scheduled for 6 or 8 weeks after treatment start, depending on the trial. We examined our 60-day–landmarked AE signatures for their association with OS, segregated by first RECIST evaluation (SD, complete response [CR]/partial response [PR], and progressive disease [PD]). Across each clinical trial and only within the cohorts with SD, we observed a modest association with survival outcomes specific to durvalumab; a patient-level meta-analysis confirmed this association with OS, consistent with that observed in our original 60-day landmark analysis (durvalumab meta-HR = 0.81, 95% CI = 0.65–1.02; chemotherapy or placebo meta-HR = 1.37, 95% CI = 0.94–1.98; Figure 2E). No association between AE signature status and OS was observed in patients with CR or PR (durvalumab meta-HR = 0.84, 95% CI = 0.46–1.54; Figure 2F). Taken together, our data suggest that the AE signature could potentially help inform prognosis for patients with PD-L1 <1% tumors and SD after initial radiographic evaluations.
Efficacy signal of the adverse event signature is driven by highly prevalent and quantifiable events
Having established how the AE signature was related to, but distinct from, known correlates of ICI efficacy or prognosis, we sought to understand the features that made up the signature and how the signature used the features to score and stratify patients. We hypothesized that AE prevalence in the MYSTIC training dataset may contribute significantly to the biomarker. Indeed, when a new model was trained using only AEs with a prevalence of at least ∼1% (n = 132), the model performed similarly to or better than the model trained on all AEs (Figure 3A; Table S2). Further examination of these AEs revealed that most could be quantitatively measured (i.e., “high diagnostic certainty”; 54/132; e.g., lipase increased), followed by those diagnosed by a medical professional (i.e., “medium diagnostic certainty”; 41/132; e.g., colitis), and finally, those often qualitatively reported (i.e., “low diagnostic certainty”; 37/132; e.g., confusion). When further stratified by these three categories of diagnostic certainty (high, medium, and low) via training a model exclusively on features from a given category, patients were most robustly stratified by AEs that had high diagnostic certainty (durvalumab meta-HR = 0.79, 95% CI = 0.67–0.92; chemotherapy or placebo meta-HR = 0.95, 95% CI = 0.72–1.24; Figure 3A; Table S2).
Figure 3.

Examination of what features are important for scoring the AE signature
(A) Point estimates and 95% confidence intervals for hazard ratios (patient-level meta-analysis; Cox proportional hazards regression model with random effects for trial, indication, treatment; fixed effects for signature class, age, sex, race, geographical region, tumor stage, and histology) after AE and OS data were landmarked at 60 days (excluding training data). Meta-analysis was run based on predictions from a model trained with only high-prevalence AEs or a subset of high-prevalence AEs based on their diagnostic certainty.
(B) Top 20 positive and negative gradients, ordered by magnitude; gradients are shown only from high-prevalence AEs.
(C) Representative neural network model from the PBMF ensemble. A sub-network is shown as follows: only the input features with associated |weight| > 0.1| are shown; only the hidden layer neurons with |weight| > 0.1| fanning in and out are shown. Chemo, chemotherapy; HR, hazard ratio; sig, ML AE signature.
Using gradient-based neural network interpretability methods, we next examined the set of AE features that contributed most to model predictions (Figure 3B). By examining the sign of the gradient for each AE, we were able to determine how each event may have contributed to AE signature positivity (positive gradient) or negativity (negative gradient). Many of the highest-magnitude, positive-gradient, highest-prevalence AEs are known irAEs, such as hypothyroidism and adrenal insufficiency; others may potentially be related to underlying irAEs, such as hyperglycemia and its relation to the irAE type 1 diabetes. AEs with negative gradients are those that may be manifestations of an overextended immune system (leukocytosis, delirium, C-reactive protein) or poor health, including poor hepatic function (edema, pleural effusion, increased gamma-glutamyltransferase). The model and AE signature may therefore capture two unique aspects not accounted for by conventional irAE classification: (1) AEs that may be symptoms of or precede an irAE (e.g., hyperglycemia in type 1 diabetes) and (2) an individual’s overall health status.
To further explore this notion, we examined a representative neural network from our model’s ensemble of two hidden-layer neural networks. Specifically, we directly examined the learned weights among the input feature space, the hidden units, and the output prediction layer. Filtering to only the highest-magnitude weights (|weight| > 0.1), i.e., those that most probably influence the output prediction, we found that the network was dominated by a single unit at each hidden layer (Figure 3C). These units made use of a combination of AEs, some of which are known or related to irAEs, to push toward a higher signature score. These units simultaneously used a different set of AEs, some of which appear to be related to overall patient well-being, to drive toward a lower signature score.
Adverse event signature may generalize to immune checkpoint inhibitor combinations
To understand the potential applicability of our signature and modeling approach to ICI therapies beyond anti–PD-L1, we examined the durvalumab + tremelimumab combination arm of the MYSTIC trial. This combination arm was not seen by the model during training, and our signature still enriched for longer survivors within it (HR = 0.76, 95% CI = 0.57–1.0; Figure 4). This result further supports the notion that our signature captures a bona fide signal.
Figure 4.

Evaluation of AE signature on ICI combination therapy not seen by the model during training
Kaplan-Meier plots for OS, per treatment arm (durvalumab + tremelimumab vs. chemotherapy) and signature class, for MYSTIC trial data landmarked at 60 days. sig, ML AE signature.
Discussion
We conducted a retrospective analysis across three late-stage randomized, controlled, prospective clinical trials (MYSTIC [NCT02453282], PACIFIC [NCT02125461], and ARCTIC [NCT02453282]) and two additional early-phase trials (ATLANTIC [NCT02087423] and CD1108 [NCT01693562]) to determine whether system-level patient health signals correlate with ICI response. An AE signature was derived that enriched for longer OS outcomes with durvalumab monotherapy over chemotherapy in patients with late-stage metastatic NSCLC from a single trial, MYSTIC. We examined the generalizability of the signature across test datasets that our model was not trained on: four independent NSCLC studies (two phase 3 randomized controlled trials) for durvalumab monotherapy, as well as a durvalumab + tremelimumab combination arm (from MYSTIC). The results of our work suggest that a set of early (within 60 days) TEAEs could be predictive of improved survival outcomes for ICI therapy only, while not being merely prognostic across all treatments within the same cohorts.
Contrary to many published reports,13,23,24,25 our results demonstrate that irAEs are not likely to be predictive of ICI therapy, at least within time scales likely to be relevant to inform clinical practice or future clinical trials. This is probably due in large part to insufficient appreciation of immortal time bias. We found that a 180-day landmark was required for irAEs to be predictive of durvalumab efficacy, whereas efficacy would likely be known sooner via other means, such as radiological examination. Most published literature has examined irAEs only in the absence of a comparator, non-ICI arm, and thus it is uncertain as to whether irAEs are merely prognostic. In this work, we included analyses from randomized controlled trials and determined that irAEs are either poorly associated with survival outcomes or not specific to ICI therapy, at least within the disease scenarios investigated. In contrast, our unbiased modeling approach searched for signal amongst all TEAEs rather than the subset of signal captured by various irAE definitions. With this approach, we successfully isolated an AE signature that may be predictive, at least for NSCLC. The efficacy signal of the AE signature is modest and may therefore highlight the limits of signal that can reasonably be extracted from AE data.
Although pre-treatment expression of PD-L1 on tumor cells has been validated as a predictor of response to anti-PD-(L)1 ICIs, patients with PD-L1–negative tumors can still respond to such treatments.26 Our AE signature tended to enrich for longer OS in patients with PD-L1 <1% tumors in a manner that was specific to durvalumab therapy, whereas it was mostly prognostic in patients with PD-L1 ≥1% tumors. Further work is required to validate our observation, especially given that only a subset of patients was evaluated for PD-L1 status in our cohorts. Nonetheless, together with recent findings that anti-CTLA-4 combinations with anti–PD-(L)1 therapy may improve survival outcomes in patients with PD-L1 <1% disease,27,28 our results could help inform decisions for patients on whether to receive ICI therapy.
Our AE signature tended to enrich for longer survivors among those with SD at their first RECIST evaluation. Although we cannot rule out that some patients may have been undetected responders, our analysis showed no added value of our AE signature in patients with CR or PR. This result suggests that such patients are not likely to bias results in favor of the AE signature. Further, our examination of how the AE signature enriches for OS but not PFS outcomes highlights the notion that we may be finding a set of long-surviving yet non-responding patients.
Our AE signature relies on the most prevalent, quantifiably measurable AEs within the training data. This is reassuring, as it suggests that commonly observed, mostly objective dimensions of patient health (i.e., those less biased by human interpretation) do contain correlates of ICI response, rather than rare or qualitatively assessed events. From our examination of features contributing to the AE signature score, we see how different AEs may reflect multiple dimensions of patient health to push toward or pull away from sig+ status. This observation highlights how our modeling approach found not only what AEs are related to ICI response, but also how they should be combined. This might explain in part why irAE definitions did not robustly correlate with ICI responses since they only captured AEs along narrower dimensions of patient health.
Future work will be needed to validate whether our modeling approach can be generalized to other ICI therapies, such as anti-PD-1, anti-CTLA-4, combinations, and bispecifics, as well as to other indications. Our finding that the AE signature generalized to the anti-PD-L1 and anti-CTLA-4 combination arm in the MYSTIC trial is therefore promising. Interestingly, although our signature was trained on first-line durvalumab data from the MYSTIC trial, it appeared to generalize to the post-chemoradiation therapy setting of the PACIFIC trial, as well as to the later-line setting of the ARCTIC trial.29 In sum, the findings of our study highlight the potential future utility of studying signatures of patient health to guide ICI treatment decision-making for patients facing uncertainty in prognosis, and the results warrant prospective validation.
Limitations of the study
In the context of patient safety, our list of AEs contributing to the signature score should be interpreted with care. The list only serves to understand the biological signal. The gradients shown here are averages across all datasets and subjects and are not meant to suggest that a given patient will have all the listed AEs, or that a patient should desire to have some AEs over others, or AEs in general. Similarly, the representative model in our ensemble should not be interpreted as a list of AEs that were observed in a single patient or a list that is representative of the types of safety signals for durvalumab.
Different indications may have different safety signals; the AE profile characterized in the present study might be relevant only to specific stages of NSCLC. Our analyses were performed exclusively in patients with locally advanced/advanced-stage NSCLC. There may also be biases or omissions in TEAE reporting, such as those due to the open-label nature of most of the examined trials. Finally, a limitation of our landmarking approach is that patients surviving less than 60 days were not considered.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Software and algorithms | ||
| Lifelines | https://lifelines.readthedocs.io/ | Version 0.26.0 RRID:SCR_024899 |
| coxme: Mixed Effects Cox Models | https://CRAN.R-project.org/package=coxme | Version 2.2–16 https://doi.org/10.32614/CRAN.package.coxme |
| Tensorflow Keras | https://www.tensorflow.org/ | Version 2.6.0 RRID:SCR_016345 |
| Predictive biomarker modeling framework (PBMF) | https://www.medrxiv.org/content/10.1101/2024.01.31.24302104v1 | https://doi.org/10.1101/2024.01.31.24302104 |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Gerald Sun (gerald.sun@astrazeneca.com).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The AstraZeneca group of companies allows researchers to submit a request to access anonymized patient-level clinical data, aggregated clinical or genomics data (when available), and/or anonymized clinical study documents through the Vivli (https://vivli.org) web-based data request platform, in accordance with AstraZeneca’s clinical data access policy: https://www.astrazenecaclinicaltrials.com/our-transparency-commitments.
All original code is available in this paper’s supplemental information.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Clinical trial cohorts
Trial-specific study designs, including age, sex, gender, ancestry, race, ethnicity, and socioeconomic status, can be found in the publications listed in Table S3. We examined data from three randomized controlled phase 3 trials (MYSTIC, NCT02453282; PACIFIC, NCT02125461; ARCTIC, NCT02453282) and two phase 1/2 trials (ATLANTIC, NCT02087423; CD1108, NCT01693562). Total sample numbers may differ from those officially reported in trials because we conducted our analysis in the actual arm assigned, not in the intent-to-treat population (where applicable). All protocols and amendments for all studies were approved by the Institutional Review Boards or Ethics Committees of the participating centers, and all patients provided written informed consent.
Method details
Immune-related adverse event reporting
Trial-specific study designs can be found in the publications listed in Table S3. Trials examined in the present study had irAEs reported in two ways: directly by a given trial investigator (‘investigator-reported irAE’), and via independent centralized adjudication (‘trial-reported irAE’). Only the latter represents the official set of irAEs reported for each trial (e.g., for regulatory authorities). Treatment emergent adverse events can also be manually triaged into putative irAEs using a proprietary list of curated ‘adverse events of special interest’.
Predictive biomarker modeling framework: Model specification and training
The complete formulation of the PBMF is described in Arango et al.20 Briefly, the PBMF utilizes a neural network model ensemble with a custom loss function that seeks to maximize the difference in target outcome between subgroups with different treatments. The PBMF uses as input time-to-event data with censoring, a treatment label, and a feature matrix (n patients by f features). Through model training, the PBMF learns to use a potentially nonlinear combination of input features to output a signature score, whereby signature-positive patients (sig+; defined as a score of >0.5) survive longer for only one treatment (e.g., durvalumab) but not the other (e.g., chemotherapy). The PBMF model for the AE signature was trained using OS and only on data from the MYSTIC clinical trial and from patients treated with durvalumab monotherapy or chemotherapy. The feature matrix consisted exclusively of unique adverse events observed in the training data, where each value in the matrix was 1 or 0, corresponding to whether the AE was observed for the patient at any point after the start of treatment.
Unless otherwise specified, each model (n = 100) in the reported PBMF ensemble for the AE signature used a neural network with two hidden layers, each with 32 units. Hidden layers were initialized with “glorot_uniform” weights and “zeros” for bias and utilized “relu” activation and L1 regularization set to 0.0001. Models were specified, trained, and evaluated with Tensorflow Keras. Models were trained for 2000 epochs with batch gradient descent and the “adam” optimizer with a learning rate of 0.01.
Contrary to what is reported in the original trial design, we used OS data from the MYSTIC trial relative to the treatment start date, not the randomization date. The same was done for all other randomized controlled trials used in the present study. The treatment label used was the actual treatment given, rather than the assigned treatment, and thus we did not perform our analysis in the intent-to-treat population. AEs were also subsetted only to those that occurred after treatment start. For training the PBMF model with MYSTIC data, the AE feature matrix consisted of the 1036 unique AEs reported on-treatment. Patients who did not experience at least one AE were excluded. This yielded a final training matrix of 721 patients for 1036 AE features.
Because different clinical trials may report slightly different sets of AEs, all test data AEs were aligned to the 1036 training data AEs by a “left join.” That is, for a given test dataset, we discarded the AEs that did not overlap with the 1036 training set AEs and only added nonzero feature values for those AEs for which there were data in the test dataset.
Landmark analysis and model evaluation
To control for immortal time bias, we performed landmark analysis.11 For each cohort, we chose the same landmark day (day 60), assessed survival after that landmark day, and, importantly, considered only AEs up to but not beyond the landmark day. Subsetting to only those patients who were still alive at the landmark day would have been insufficient to control for immortal time bias.
Landmarking resulted in much fewer AEs being available to assess but was critical to the validity of the landmark analysis. For this reason, although we evaluated the performance (of the trained PBMF model, or any of the three irAE definitions) only on landmarked data, we fit the PBMF model to all on-treatment AEs, without landmarking. We reasoned that AEs can present either randomly or, in the case of those already implicated as putative irAEs, across a known, large window of time.30 In this case, we wanted our model to have the maximum information to learn a predictive signature against a background of potential noise and prognostic signatures.
First Response Evaluation Criteria in Solid Tumors (RECIST) evaluations were taken as the most recent evaluations between treatment start and halfway between the scheduled time for first and second evaluations. For instance, evaluations were scheduled every 8 weeks for the PACIFIC clinical trial [NCT02125461], and the most recent evaluation occurring before 12 weeks was taken as the first RECIST evaluation. The model was still landmarked to 60 days, irrespective of when the RECIST evaluation occurred. This allowed for a fair comparison across trials.
Model interpretation: Ablation experiments and feature importance
Ablation experiments were carried out to assess how subsets of features affect the score outputted from the model. We trained a new PBMF model with training data subsetted only to those features that passed specified criteria. After training a new PBMF model with training data subsetted only to those features that passed specified criteria, we evaluated the new PBMF model on test datasets that were aligned to the subsetted training data. Test dataset AEs were aligned to the subsetted training AEs by a “left join.”
We manually annotated the AEs with >1% prevalence into one of three classes—low, medium, and high—corresponding to how well they could be objectively and quantifiably measured (Table S2). This assessment was made in collaboration with internal experts specializing in patient safety and AE reporting for durvalumab. The threshold of 1% prevalence (among the MYSTIC trial training data) for the prevalence ablation experiments was determined semi-empirically by balancing the stratification performance degradation with the number of excluded AEs as evaluated on our training data landmarked at 60 days. We chose 1% because it led to almost no change in stratification performance but allowed removal of ∼85% of the AE features. Stratification would still degrade minimally with further filtering, although with diminishing returns on the number of low-prevalence AEs excluded (data not shown).
AE feature importance was derived by computing neural network gradients for each AE feature across all the 60-day landmarked NSCLC durvalumab monotherapy versus chemotherapy/placebo datasets and reported as the average across all samples. Computing these gradients for each model in the ensemble then generated an empirical distribution. To rank AE features by importance, we computed the neural network gradients as
where is the input feature vector of AEs from a given sample and is the output prediction score.
Quantification and statistical analysis
To obtain hazard ratios and 95% confidence intervals that were computed separately for each dataset, we fit a Cox proportional hazards regression model (lifelines Python package, https://lifelines.readthedocs.io/) to the time-to-event data, with only the binarized AE signature group membership (1 or 0) as a feature. For meta-analyses, we fit a mixed-effects Cox proportional hazards regression model (Coxme R package, https://CRAN.R-project.org/package=coxme) with random effects for trial, indication, and treatment and fixed effects for age, sex, race, geographical region, tumor stage, and histology. C-index was computed, per treatment arm, using the AE signature score or the binary class for irAE status (lifelines Python package).
Acknowledgments
We thank Tom Gibbons for data processing guidance, Paul Metcalfe and Abhijit Das Gupta for data analysis guidance, Ioannis Kagiampakis for scientific input, and Deborah Shuman for help editing the article (all from AstraZeneca). This study was funded by AstraZeneca.
Author contributions
GJS contributed to the conception of the study. GJS, EJ, ZL, and GJD contributed to the design of the study. GJS, GAA, and DEB contributed to analysis of the data. DP provided patient safety guidance. GJD and ACC provided clinical guidance. GJS, EJ, and RAS wrote the article. EJ supervised the work.
Declaration of interests
All authors are employees of AstraZeneca and may have stock ownership, interests, and/or options in the company.
Published: August 2, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110634.
Contributor Information
Gerald J. Sun, Email: gerald.sun@astrazeneca.com.
Etai Jacob, Email: etai.jacob@astrazeneca.com.
Supplemental information
C-index values for all trial-level analyses for test data landmarked at 60 days, related to Figure 1.
Most prevalent adverse events in MYSTIC trial training data, by diagnostic certainty, gradient magnitude, and sign, related to Figure 3.
References
- 1.Garon E.B., Rizvi N.A., Hui R., Leighl N., Balmanoukian A.S., Eder J.P., Patnaik A., Aggarwal C., Gubens M., Horn L., et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 2.Topalian S.L., Taube J.M., Anders R.A., Pardoll D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer. 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le D.T., Durham J.N., Smith K.N., Wang H., Bartlett B.R., Aulakh L.K., Lu S., Kemberling H., Wilt C., Luber B.S., et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le D.T., Uram J.N., Wang H., Bartlett B.R., Kemberling H., Eyring A.D., Skora A.D., Luber B.S., Azad N.S., Laheru D., et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rizvi N.A., Hellmann M.D., Snyder A., Kvistborg P., Makarov V., Havel J.J., Lee W., Yuan J., Wong P., Ho T.S., et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chowell D., Yoo S.K., Valero C., Pastore A., Krishna C., Lee M., Hoen D., Shi H., Kelly D.W., Patel N., et al. Improved prediction of immune checkpoint blockade efficacy across multiple cancer types. Nat. Biotechnol. 2022;40:499–506. doi: 10.1038/s41587-021-01070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Das S., Johnson D.B. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer. 2019;7:306. doi: 10.1186/s40425-019-0805-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Postow M.A., Sidlow R., Hellmann M.D. Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 2018;378:158–168. doi: 10.1056/NEJMra1703481. [DOI] [PubMed] [Google Scholar]
- 9.Naidoo J., Murphy C., Atkins M.B., Brahmer J.R., Champiat S., Feltquate D., Krug L.M., Moslehi J., Pietanza M.C., Riemer J., et al. Society for Immunotherapy of Cancer (SITC) consensus definitions for immune checkpoint inhibitor-associated immune-related adverse events (irAEs) terminology. J. Immunother. Cancer. 2023;11 doi: 10.1136/jitc-2022-006398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie T., Zhang Z., Qi C., Lu M., Zhang X., Li J., Shen L., Peng Z. The Inconsistent and Inadequate Reporting Of Immune-Related Adverse Events in PD-1/PD-L1 Inhibitors: A Systematic Review of Randomized Controlled Clinical Trials. Oncologist. 2021;26:e2239–e2246. doi: 10.1002/onco.13940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dafni U. Landmark analysis at the 25-year landmark point. Circ. Cardiovasc. Qual. Outcomes. 2011;4:363–371. doi: 10.1161/CIRCOUTCOMES.110.957951. [DOI] [PubMed] [Google Scholar]
- 12.Yadav K., Lewis R.J. Immortal Time Bias in Observational Studies. JAMA. 2021;325:686–687. doi: 10.1001/jama.2020.9151. [DOI] [PubMed] [Google Scholar]
- 13.Fan Y., Xie W., Huang H., Wang Y., Li G., Geng Y., Hao Y., Zhang Z. Association of immune-related adverse events with efficacy of immune checkpoint inhibitors and overall survival in cancers: a systemic review and meta-analysis. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.633032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou X., Yao Z., Yang H., Liang N., Zhang X., Zhang F. Are immune-related adverse events associated with the efficacy of immune checkpoint inhibitors in patients with cancer? A systematic review and meta-analysis. BMC Med. 2020;18:87. doi: 10.1186/s12916-020-01549-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eggermont A.M.M., Kicinski M., Blank C.U., Mandala M., Long G.V., Atkinson V., Dalle S., Haydon A., Khattak A., Carlino M.S., et al. Association between immune-related adverse events and recurrence-free survival among patients with stage III melanoma randomized to receive pembrolizumab or placebo: a secondary analysis of a randomized clinical trial. JAMA Oncol. 2020;6:519–527. doi: 10.1001/jamaoncol.2019.5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khan Z., Hammer C., Carroll J., Di Nucci F., Acosta S.L., Maiya V., Bhangale T., Hunkapiller J., Mellman I., Albert M.L., et al. Genetic variation associated with thyroid autoimmunity shapes the systemic immune response to PD-1 checkpoint blockade. Nat. Commun. 2021;12:3355. doi: 10.1038/s41467-021-23661-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan Z., Di Nucci F., Kwan A., Hammer C., Mariathasan S., Rouilly V., Carroll J., Fontes M., Ley Acosta S., Guardino E., et al. Polygenic risk for skin autoimmunity impacts immune checkpoint blockade in bladder cancer. Proc. Natl. Acad. Sci. USA. 2020;117:12288–12294. doi: 10.1073/pnas.1922867117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Socinski M.A., Jotte R.M., Cappuzzo F., Nishio M., Mok T.S.K., Reck M., Finley G.G., Kaul M.D., Yu W., Paranthaman N., et al. Association of Immune-Related Adverse Events With Efficacy of Atezolizumab in Patients With Non-Small Cell Lung Cancer: Pooled Analyses of the Phase 3 IMpower130, IMpower132, and IMpower150 Randomized Clinical Trials. JAMA Oncol. 2023;9:527–535. doi: 10.1001/jamaoncol.2022.7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rizvi N.A., Cho B.C., Reinmuth N., Lee K.H., Luft A., Ahn M.-J., van den Heuvel M.M., Cobo M., Vicente D., Smolin A., et al. Durvalumab with or without tremelimumab vs standard chemotherapy in first-line treatment of metastatic non–small-cell lung cancer: the MYSTIC phase 3 randomized clinical trial. JAMA Oncol. 2020;6:661–674. doi: 10.1001/jamaoncol.2020.0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arango G., Bikiel D.E., Sun G.J., Kipkogei E., Smith K.M., Jacob E. AI-based predictive biomarker discovery via contrastive learning retrospectively improves clinical trial outcome. medRxiv. 2024 doi: 10.1101/2024.01.31.24302104. Preprint at. [DOI] [Google Scholar]
- 21.Arfe A., Fell G., Alexander B., Awad M.M., Rodig S.J., Trippa L., Schoenfeld J.D. Meta-Analysis of PD-L1 Expression As a Predictor of Survival After Checkpoint Blockade. JCO Precis Oncol. 2020;4:1196–1206. doi: 10.1200/PO.20.00150. [DOI] [PubMed] [Google Scholar]
- 22.Eisenhauer E.A., Therasse P., Bogaerts J., Schwartz L.H., Sargent D., Ford R., Dancey J., Arbuck S., Gwyther S., Mooney M., et al. New Response Evaluation Criteria in Solid Tumours: revised RECIST guideline (version 1.1) Eur. J. Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 23.Hussaini S., Chehade R., Boldt R.G., Raphael J., Blanchette P., Maleki Vareki S., Fernandes R. Association between immune-related side effects and efficacy and benefit of immune checkpoint inhibitors: a systematic review and meta-analysis. Cancer Treat Rev. 2021;92 doi: 10.1016/j.ctrv.2020.102134. [DOI] [PubMed] [Google Scholar]
- 24.Zhao Z., Wang X., Qu J., Zuo W., Tang Y., Zhu H., Chen X. Immune-related adverse events associated with outcomes in patients with NSCLC treated with anti-PD-1 inhibitors: a systematic review and meta-analysis. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.708195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou C., Li M., Wang Z., An D., Li B. Adverse events of immunotherapy in non-small cell lung cancer: A systematic review and network meta-analysis. Int. Immunopharmacol. 2022;102 doi: 10.1016/j.intimp.2021.108353. [DOI] [PubMed] [Google Scholar]
- 26.Lipson E.J., Forde P.M., Hammers H.J., Emens L.A., Taube J.M., Topalian S.L. Antagonists of PD-1 and PD-L1 in Cancer Treatment. Semin. Oncol. 2015;42:587–600. doi: 10.1053/j.seminoncol.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paz-Ares L., Ciuleanu T.E., Cobo M., Schenker M., Zurawski B., Menezes J., Richardet E., Bennouna J., Felip E., Juan-Vidal O., et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): an international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22:198–211. doi: 10.1016/S1470-2045(20)30641-0. [DOI] [PubMed] [Google Scholar]
- 28.Johnson M.L., Cho B.C., Luft A., Alatorre-Alexander J., Geater S.L., Laktionov K., Kim S.W., Ursol G., Hussein M., Lim F.L., et al. Durvalumab With or Without Tremelimumab in Combination With Chemotherapy as First-Line Therapy for Metastatic Non-Small-Cell Lung Cancer: The Phase III POSEIDON Study. J. Clin. Oncol. 2023;41:1213–1227. doi: 10.1200/JCO.22.00975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Planchard D., Reinmuth N., Orlov S., Fischer J.R., Sugawara S., Mandziuk S., Marquez-Medina D., Novello S., Takeda Y., Soo R., et al. ARCTIC: durvalumab with or without tremelimumab as third-line or later treatment of metastatic non-small-cell lung cancer. Ann. Oncol. 2020;31:609–618. doi: 10.1016/j.annonc.2020.02.006. [DOI] [PubMed] [Google Scholar]
- 30.Tang S.Q., Tang L.L., Mao Y.P., Li W.F., Chen L., Zhang Y., Guo Y., Liu Q., Sun Y., Xu C., Ma J. The Pattern of Time to Onset and Resolution of Immune-Related Adverse Events Caused by Immune Checkpoint Inhibitors in Cancer: A Pooled Analysis of 23 Clinical Trials and 8,436 Patients. Cancer Res. Treat. 2021;53:339–354. doi: 10.4143/crt.2020.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
C-index values for all trial-level analyses for test data landmarked at 60 days, related to Figure 1.
Most prevalent adverse events in MYSTIC trial training data, by diagnostic certainty, gradient magnitude, and sign, related to Figure 3.
Data Availability Statement
The AstraZeneca group of companies allows researchers to submit a request to access anonymized patient-level clinical data, aggregated clinical or genomics data (when available), and/or anonymized clinical study documents through the Vivli (https://vivli.org) web-based data request platform, in accordance with AstraZeneca’s clinical data access policy: https://www.astrazenecaclinicaltrials.com/our-transparency-commitments.
All original code is available in this paper’s supplemental information.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.