

Abstract
Data-independent acquisition (DIA) has revolutionized the field of mass spectrometry (MS)-based proteomics over the past few years. DIA stands out for its ability to systematically sample all peptides in a given m/z range, allowing an unbiased acquisition of proteomics data. This greatly mitigates the issue of missing values and significantly enhances quantitative accuracy, precision, and reproducibility compared to many traditional methods. This review focuses on the critical role of DIA analysis software tools, primarily focusing on their capabilities and the challenges they address in proteomic research. Advances in MS technology, such as trapped ion mobility spectrometry, or high field asymmetric waveform ion mobility spectrometry require sophisticated analysis software capable of handling the increased data complexity and exploiting the full potential of DIA. We identify and critically evaluate leading software tools in the DIA landscape, discussing their unique features, and the reliability of their quantitative and qualitative outputs. We present the biological and clinical relevance of DIA-MS and discuss crucial publications that paved the way for in-depth proteomic characterization in patient-derived specimens. Furthermore, we provide a perspective on emerging trends in clinical applications and present upcoming challenges including standardization and certification of MS-based acquisition strategies in molecular diagnostics. While we emphasize the need for continuous development of software tools to keep pace with evolving technologies, we advise researchers against uncritically accepting the results from DIA software tools. Each tool may have its own biases, and some may not be as sensitive or reliable as others. Our overarching recommendation for both researchers and clinicians is to employ multiple DIA analysis tools, utilizing orthogonal analysis approaches to enhance the robustness and reliability of their findings.
Keywords: data-independent acquisition, clinical proteomics, mass spectrometry
Graphical Abstract
Highlights
-
•
Overview of historical developments and key advancements in DIA strategies.
-
•
Summary of key DIA aspects: PTM analysis, statistical analysis, and clinical applicability.
-
•
Comparison of popular DIA software tools and published benchmark studies.
-
•
Perspective on future directions and upcoming challenges of DIA.
In Brief
Data-independent acquisition (DIA)has revolutionized mass spectrometry–based proteomics by yielding unbiased, high-accuracy, and reproducible data. This review evaluates the capabilities and challenges of leading DIA analysis software tools as well as the latest DIA method developments and discusses their critical role in advancing clinical and biological research.
From the detection of posttranslational modifications (PTMs) to the measurement of protein stoichiometry, mass spectrometry (MS)-based proteomics is a powerful tool to answer a plethora of different questions related to proteins (1). Due to their low cost, straightforward sample preparation, and scalability, label-free approaches have been popular in quantitative proteomics for decades. In label-free proteomics experiments, data can either be acquired using data-dependent acquisition (DDA) or data-independent acquisition (DIA).
In DDA, following peptide separation by the liquid chromatography system, peptides are ionized and a survey scan (MS1) is performed. After this survey scan, usually the n top abundant m/z signals are isolated and fragmented and their respective fragment spectra (MS2) are recorded. As relative protein abundances may vary between samples, stochastic sampling often occurs, leading to missing values. Additionally, quantitation is restricted to the area under the curve of MS1 scans or spectral counting. The great advantage of DDA is the low complexity of the resulting fragment spectra and over the years multiple tools have been developed to not only perform standard proteome searches but also searches with large search spaces, such as PTM searches, semispecific, or unspecific searches or even open searches (2, 3, 4).
In DIA, fragment spectra are recorded in a predefined acquisition scheme (5, 6, 7, 8). While MS1 scans can be performed optionally, they do not impact the fragmentation spectra recorded during the data acquisition. As the whole mass range of interest shall be covered (usually between 400 and up to 1200 m/z), wider isolation windows have to be employed as compared to DDA, resulting in higher fragment spectra complexity and necessitating elaborate analysis software and/or the usage of spectral libraries. In recent years, DIA has gained momentum and is slowly replacing DDA methods for many applications, as coverage and quantitative precision of DIA methods often supersede classical DDA methods and circumvent stochastic sampling during measurement (8, 9, 10, 11).
The consistent acquisition scheme in DIA yields fewer missing values across multiple measurements, for example, in cohort studies, which in turn enhances statistical power. Furthermore, as compared to isobaric labeling approaches, where missing values are also negligible, DIA leads to the sample-specific assignment of precursor, peptide, or protein identification confidence, allowing the investigation of extremely heterogeneous samples in one experiment (e.g., knock out) (12).
Historically, the generation and analysis of DIA-type proteomics data have been challenging due to the increased complexity of fragment spectra as compared to DDA. For most DIA analyses, mass spectrometers require a high-resolution mass analyzer in combination with a quadrupole to detect fragment ions at a high speed. TOF and Orbitrap instruments were therefore often used in combination with long gradients. Recently, there have been fascinating developments in the field of MS concerning the ability of mass spectrometers to acquire DIA data. While the general speed of mass spectrometers increased over the past decade for both Orbitrap and TOF instruments, additional inventions such as trapped ion mobility spectrometry (TIMS) and high-field asymmetric waveform ion mobility spectrometry (FAIMS) have substantially improved the range of possible applications of DIA (13, 14). As the speed of mass spectrometers increases, the isolation width of fragmentation windows in DIA approaches the same width as windows employed in classical DDA experiments (e.g., 2 m/z) (15).
As for the analysis of DIA-type data, often laborious library generation had to be performed in the past before DIA data could be analyzed (8, 16). Spectral libraries were often generated employing DDA runs (offline prefractionation) or gas-phase fractionation (GPF). This step can nowadays be circumvented, as numerous strategies have been described to directly search the DIA data, for example, by generating pseudo-MS2 spectra, which can be searched using conventional DDA analysis tools, or by using predicted libraries (17, 18, 19). Such predicted libraries enable unbiased and reproducible data analysis, which is required for individual samples such as in clinical applications. Furthermore, predicted libraries promote projects for which project- and sample-specific libraries are not feasible due to limited sample amounts and measurement time.
In this review, we want to present an overview of current DIA analysis software tools regarding their capabilities, limitations, and reliability. Additionally, we present the latest developments in instrumentation as this greatly influences the analysis software and the potential biological and clinical relevance of the interplay between LC-MS/MS instrumentation, DIA methods, and analysis software. We conclude with some perspectives and emerging trends in the field of DIA-based proteomics.
Development of DIA
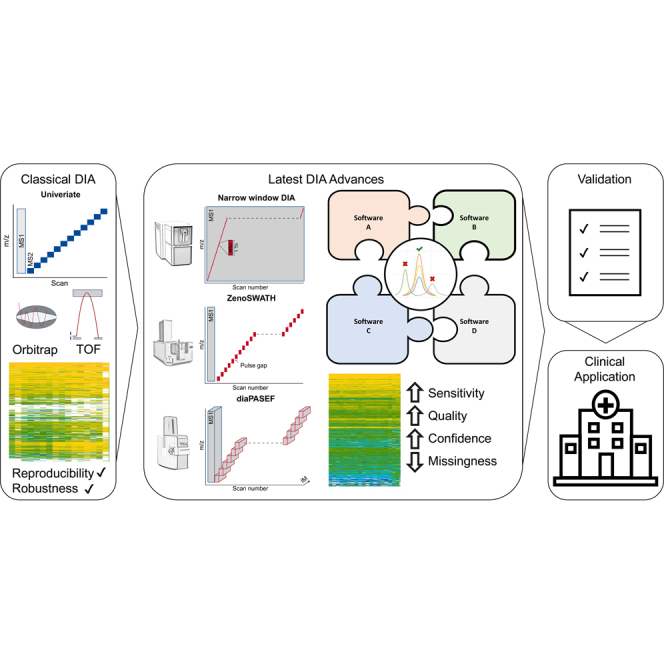
The advancement of DIA methodologies has undergone significant acceleration over the past 2 decades alongside rapid progression in both instrumental technologies and software tools. The advent of DIA-evolution lies in one of the first conceptualizations by Purvine et al. in 2003, which introduced the one-shot collision-induced dissociation of all peptides over the whole mass range as an alternative to serial fragmentation, termed shotgun collision-induced dissociation (5). In this, the whole MS duty cycle is being utilized and was later commercialized by Waters Corporation on QqTOF instruments as MSE. MSE enabled absolute label-free quantification by inducing fragmentation of precursor ions across the full spectrum in a single chromatographic run due to the simultaneous acquisition of both low and high collision energy scans (6). An increase in resolution and sensitivity was then achieved by leveraging the potential of ion mobility-dependent fragmentation in Synapt HDMS and UDMSE instruments (20, 21). Parallely, Geiger and colleagues introduced all-ion fragmentation (AIF) in 2010, which utilized higher-energy collisional dissociation fragmentation (22). AIF was originally designed for Orbitrap detectors, debuting in Thermo's Exactive model, which omitted mass filtering by excluding the quadrupole (21). Later on, wide-window scans, including the iterative isolation of precursors from windows of >10 m/z enabling slicing through the mass range replaced the detection of all ions in one shot (Fig. 1A) (23, 24).
Fig. 1.

Key principles and developments toward state-of-the-art DIA-MS.A, most common isolation window schemes in DIA-MS. B, implementation of DIA-MS over time on different instrument platforms and their usage approximated by citations in Google Scholar. C, schematic overview of state-of-the-art MS instruments with their respective key developments and features highlighted in bold text. The ion flux through the respective ion optics is depicted as a red line with the direction from left to right. D, state-of-the-art DIA-MS strategies based on respective key features of each instrument type. DIA, data-independent acquisition; IM, ion mobility; MS, mass spectrometry.
While simultaneous low and high-energy approaches laid the foundations for MSE and AIF, another paradigm emerged with stepwise isolation fragmentation. In the early proof-of-concept work, Venable et al. introduced the term DIA. This targeted approach involved the sequential isolation and fragmentation of precursor ions in a linear ion trap and allowed the detection of less than 250 proteins (23).
Since then, DIA has evolved to encompass innovative strategies that provide enhanced proteome coverage, reproducibility, increased sensitivity, and quantitative accuracy. An exemplar of this progress is the parallel accumulation serial fragmentation ion chromatography method, introduced in 2009 by Panchaud et al., which systematically pursued a comprehensive interrogation of MS2 scans across a given m/z range, benefiting from parallel ion accumulation (7). However, the scans had to align with the chromatographic peak retention time (RT), necessitating multiple injections of the same sample and thereby demanding a considerable amount of instrument time.
Further improvements in mass spectrometers with faster acquisition time and duty cycle led to more recent developments of new DIA strategies such as sequential window acquisition of all theoretical fragment ion spectra MS (SWATH-MS), introduced in 2012 by Gillet et al., where the DIA method is extended to include a targeted data extraction strategy. SWATH-MS was originally widely used on SCIEX quadrupole-TOF instruments (Fig. 1B). Distinctive to other approaches, it integrates the advantages of both DDA and DIA for the simultaneous detection and quantification of proteins. The library of spectra was pregenerated in advance in DDA mode, while in a distinct DIA run, tandem mass spectra were systematically acquired over marginally overlapping precursor isolation windows, typically set to 25 m/z (8).
Progressively, similar methodologies evolved to a wider spectrum of platforms such as Orbitrap-based instruments—albeit not being designed for DIA approaches initially. Orbitraps were mostly used for DDA, owing to their high resolution and mass accuracy but slower acquisition speed. However, DIA-MS increasingly gained traction for these instruments since recurrent refinement expanded the toolbox. Initially, the acquisition of chimeric spectra due to limited scanning speed in Orbitraps remained challenging. Yet, early adaptations proposed by Amodei et al. circumvented that by overlapping the isolation windows leading to a noticeable improvement in peptide identifications (Fig. 1A) (25). Furthermore, the full potential of DIA-MS on widely available MS instruments just recently became evident (Fig. 1B). DIA-MS resulted in ∼7000 protein identifications by optimizing the precursor isolation schemes based on the actual precursor distribution across the m/z range and the RT from a 60-min gradient on a QExactive Plus (26). Hence, DIA-MS has been demonstrated to be a widely applicable and versatile tool in proteomics. Nevertheless, DIA-MS on Orbitrap instruments was limited by the scanning speed, especially in comparison to typically faster TOF analyzers, which exhibit a higher sensitivity toward noise at lower resolution.
With the emergence of TIMS technology into high-resolution mass spectrometers, the full potential of TOF analyzers for DIA-MS came into play (Fig. 1B) (27, 28). Besides the addition of the collisional cross section dimension for improved peptide separation capacity, TIMS contributes to a decreased noise resulting in cleaner spectra. Thereby, timsTOF instruments paved the way for ultrasensitive, high-scanning speed DIA-MS approaches (29).
Consequently, ion mobility–based separation upstream of precursor fragmentation increasingly gained attention leading to further adaptations like FAIMS. FAIMS is a powerful technique, which can be combined with Thermo’s Orbitrap instruments allowing the filtering for specifically charged ions right after ionization, separating ions according to their mobility under the influence of strong and weak electric fields (11, 30, 31). This additional ion mobility separation dimension at the front end of Orbitrap instruments reduces chemical noise, often resulting in cleaner, high-quality MS and MS/MS spectra (32).
Consequently, the possibility of highly sensitive, high-throughput DIA-MS drove the development of parallel accumulation and serial fragmentation (DIA-PASEF) and its methodological offspring ushering in the concept of 4D proteomics (32, 33). Importantly, all these hardware developments were accompanied by software approaches to analyze the novel-shaped data contributing to the success of these approaches (34, 35).
Data-Independent Acquisition–Parallel Accumulation and Serial Fragmentation
This advancement builds upon classical 3D proteomics, which encompasses the chromatographic RT, m/z values, and ion intensities and introduces a novel fourth separation dimension: ion mobility (Fig. 1, C and D). This augmentation brings significant benefits in terms of detection sensitivity and scanning speed, ultimately increasing both the depth and precision of protein quantification (27, 36). The introduction of TIMS has facilitated the implementation of the PASEF principle (29). In DIA-PASEF, the ions are first sorted and time-focused in the TIMS funnel, followed by a selection based on the respective m/z values through the coordinated motion of the quadrupole, whose movement is synchronized with the ion mobility elution profile. This synergistic approach results in up to a 10-fold increase in sequencing speed, which leads to improved analysis of complex proteomes within significantly shorter gradients. DIA-PASEF also incorporates a novel windows scheme, which can be more efficient in ion utilization compared to classical DIA approaches. This enhanced efficiency leverages the aforementioned correlation between ion mobility and m/z. Specifically, the isolation windows start at high m/z values since larger species elute early in TIMS, gradually progressing to lower m/z, ultimately achieving a 100% duty cycle. Furthermore, tools such as pydiAID published by Skowronek and colleagues democratized the design of optimized DIA-PASEF acquisition schemes on timsTOF instruments leveraging the potential of an automated evidence-driven parameter refinement (37).
Synchronized PASEF, midia-PASEF, and Slice-PASEF
Synchronized PASEF represents a pioneering evolution of DIA-PASEF proposed by Skowronek et al. (38). This methodology introduced a continuous fragmentation spectrum that operates seamlessly within both the m/z and ion mobility dimensions. Its distinctive feature revolves around the diagonal traversal of the quadrupole selection window through the m/z-ion mobility plane, eliminating the presence of artificial borders characteristic of DIA-PASEF, where the quadrupole rapidly transitions between different mass ranges. However, in synchronized PASEF, the peptide precursors are sliced by the quadrupole, thereby segregating the fragment ion signals of the individual precursor into consecutive scans. This poses a challenge for subsequent downstream analysis, as currently there is a deficiency in available tools capable of effective and accurate handling of the data structures generated by synchro scans.
In an attempt to overcome the precursor slicing, Distler et al. proposed an approach termed midia-PASEF, which uses a similar sliding quadrupole movement to effectively cover the ion cloud; overlapping quadrupole windows ensure scanning of all fragment ions in three consecutive midia scans, thereby maximizing information content in DIA-PASEF (39).
Introduced in 2022 by Szyrwiel et al., Slice-PASEF comprises a set of associated acquisition strategies, incorporating diagonal scans of quadrupole selection windows. By keeping the initial mobility window ranges constant while slicing through the m/z range in cycles, all ions within each m/z slice undergo fragmentation. This adaptable method can also be applied to multiframe schemes with overlapping windows in the m/z dimension, maximizing the selectivity of precursor ions and facilitating effective fragmentation (40).
ZENO-Sequential Window Acquisition of All Theoretical Fragment Ion Spectra
In addition to DIA-PASEF, various other specialized approaches have been developed for specific MS instruments, exemplified by techniques such as Zeno-SWATH. Zeno-SWATH, created for the ZenoTOF instrument from SCIEX, exploits a linear ion trap, coined Zeno trap (Fig. 1, C and D). Positioned strategically at the exit of the collision cell, this trap facilitates precise control over ion transmission to the TOF analyzer, resulting in a higher duty cycle of nearly >90%. In Zeno-SWATH DIA, activation of the Zeno trap serves to enhance MS2 sensitivity for each acquired variable window, reaching a sensitivity gain of 4- to 20-fold (41, 42).
Narrow Window DIA-MS
The very recent advances in MS-based proteomics further leveraged the potential of DIA-MS finally proposing a very narrow data-independent selection of precursors for fragmentation covering the whole mass range of interest. Historically, when employing narrow window DIA-MS, only a specific mass range was investigated (11, 43, 44) or samples were injected multiple times. The hardware that facilitated the realization of a very narrow DIA-MS is the Asymmetric Track Lossless (ASTRAL) analyzer introducing the loss-less and fast scanning of fragments with high resolution (Fig. 1, C and D) (45). The ASTRAL analyzer is a long-track TOF-like analyzer with ion optics that enable high accuracy and high sensitivity facilitating an ultrafast scanning of MS2 spectra. In this, the parallelization of high-resolution MS scans in the Orbitrap with the 200 Hz frequency of the ASTRAL analyzer for MS2 scans (46). Subsequently, the application of new DIA modes emerged like narrow-window DIA-MS with isolation windows of ∼2 m/z, allowing for a DDA-like precursor selection in a nondata driven mode, contributing to increased throughput and depth for MS-based proteomics (47). Thus, the instrumentation is capable of moving DIA-MS in a novel direction putatively resulting in a foreseeable convergence of DDA and DIA. Reproducible and robust data acquisition is one of the most pressing challenges in the field of MS-based proteomics. Furthermore, consistent data analysis is pivotal for established and emerging methodologies. Consequently, further improvements in interoperability, comparability, and reproducibility as well as the ever-widening use cases for DIA-MS in clinics permanently challenge state-of-the-art principles.
PTM Analysis Using DIA
State-of-the-art DIA-MS enables the investigation of a variety of PTMs, promoting deep proteome coverage and high reproducibility (48). As fragment patterns of posttranslationally modified peptides typically show higher complexity, bioinformatic advances for PTM identification and localization paved the way for the utilization of DIA-MS (49, 50, 51, 52). DIA-MS has been shown to yield a higher coverage while maintaining quantitative accuracy reaching >20,000 phosphosites with a QExactive HF-X or an Orbitrap Lumos (53, 54). Subsequently, DIA-MS has already been widely applied for phosphoproteomic studies (55, 56), as well as for the investigation of ubiquitination, acetylation, and succinylation (57, 58). Further driven by innovations in sample preparation, the accurate detection of phosphopeptides from low input has enabled the quantification of tens of thousands of phosphosites from limited input amounts such as 10 μg of peptides (59) Technological progress has also enabled the detection of >40,000 phosphopeptides in a single 1-h measurement (60). The refinement of libraries for the phosphoproteomic approach also benefited from machine learning tools assisting the data analysis in turn increasing the depth of coverage (61).
Even in the field of glycoproteomics, known for being challenging due to the generation of glycan and peptide fragments, DIA-MS is widely used as instrumentation and data analysis capabilities advance (62, 63, 64). Still facing limitations in terms of data complexity, the potential of increased coverage and decreased missing values for glycoproteomics by DIA-MS is evident when DDA-refined glycoproteomic libraries together with recent rescoring algorithms come into play (65, 66, 67).
As more and more PTM-scoring and localization tools are being implemented for DIA-MS data as well exemplified by the PTM-Shepherd implementation in the Trans-Proteomic Pipeline, the potential of a robust DIA-MS–driven PTMs analysis is fostered (68). With reliable and robust workflows being implemented on a global scale for PTMs, throughput, and data quality harbor the potential of narrow window open-search analysis for a yet unexplored field of open searches on the whole proteome scale.
Statistical Analysis of DIA Data
To facilitate the statistical analysis of differentially abundant proteins based on DIA data, MSstats, a popular tool for statistically analyzing bottom-up MS-based proteomic experiments, provides direct converters for the output of Skyline, Spectronaut, DIA-Umpire, OpenSWATH, and DIA-NN (69). Furthermore, Fahrner et al. implemented an open-source DIA analysis suite in the web-based Galaxy framework supporting reproducible and transparent analyses (70).
Compared to DDA datasets, datasets generated in the DIA mode feature more detected peptides and proteins, fewer missing values, and higher measurement reproducibility (68, 71, 72), which benefits the statistical power of large-scale proteomics studies by allowing for the integration of data from different instruments and over longer periods (73, 74). The reduction of missing values also broadens the applicability of approaches that can only partially handle the occurrence of missing values. For instance, statistical tests and multiple regression models provide only partial results if there are proteins for which all data points are missing across all replicates of an experimental condition. Hierarchical clustering, as another example, is only applicable if there are sufficient nonmissing data points to calculate a chosen distance measure for all protein pairs. However, although missing values are not as prominent in DIA data as in DDA data, they can still strongly impact what is concluded from the data, for example, by reducing statistical power (75). Finally, compared to DDA datasets, DIA datasets in theory contain less random intensity-independent missing values, which can impact the choice of a possible imputation (76).
While in the case of DDA quantification is conducted based on the signal of only intact peptides (MS1 level), DIA can also use the quantitative information of fragmented peptides (MS2 level). In contrast to SWATH-type DIA, which traditionally uses the MS2 information for quantification, Huang et al. proposed a statistical approach, which treats the quantitative MS1 and MS2 signals as technical replicates from the same biological sample. By combining MS1 and MS2 signals, they showed that the precision of the measurement and, in turn, the statistical power of detecting differentially abundant peptides and proteins could be increased (77). They found this to be especially true for comparisons with, for example, small fold changes and few replicates. The precision of the MS2 level was shown to be consistently higher than that of the MS1 level. Furthermore, as possible coeluting inferences on the MS1 level are independent from those on the MS2 level, combining both levels is also beneficial in that it reduces the negative impact of the interferences in one of the MS levels on downstream statistical analyses (77).
Biological and Clinical Relevance of DIA
A vast majority of diseases have predominantly been studied on the genomic and transcriptomic level, lacking information on proteins as other biologically and functionally relevant molecules (78, 79). The limited correlation between mRNA and respective protein levels as well as the occurrence and stoichiometry of PTMs and protein proteoforms, which can only be addressed by direct investigation of proteins, highlights the urgent need and potential of proteomics (53, 60, 80, 81, 82).
The integration of MS-based proteomics into clinical applications such as molecular diagnostics has gained tremendous interest over the last few years (79, 83, 84, 85, 86). This growing interest in clinical proteomics is mainly driven by a) providing complementary information to genomic and transcriptomic molecular diagnostics and b) detailed investigation of proteins as primary therapeutic targets in personalized treatments and precision medicine. A limited number of immuno-based and targeted MS approaches have already been implemented in molecular diagnostics (85, 87, 88). However, targeted assays require a priori knowledge about target proteins and peptides and yield only information on a limited number of analytes. Additionally, immuno-based protocols tend to be biased and dependent on the availability and specificity of the established antibodies (72, 83, 89). Thus, in clinical proteomics, targeted assays for sensitive and accurate quantification of proteins of interest can be complemented by applying explorative approaches yielding in-depth information on the proteome (90).
DIA-MS provides such an unbiased, explorative, and high-throughput approach that could complement current molecular diagnostics by promoting in-depth molecular and functional characterization of clinical specimens (73, 91, 92, 93). DIA-MS with state-of-the-art LC-MS/MS instrumentation enables large cohort studies for biomarker screening and discovery (94). Furthermore, advancements in sensitivity during sample preparation and measurement with DIA-MS enable proteomic profiling using minute sample amounts (95, 96, 97). Notably, patient-derived material is often limited concerning availability and amount such as needle biopsies that are frequent input material for molecular and pathological diagnostics.
However, the integration of proteomics into molecular diagnostics requires extensive quality control and standardization of the whole workflow, including sample preparation, measurement, data analysis, and reporting. Semiautomated sample preparation in combination with DIA-MS on modern, fast-scanning LC-MS/MS instrumentation enables key aspects of clinical applications, such as robustness, reproducibility, sensitivity, and high throughput (79, 91, 95). The selected software profoundly influences the quantitation of peptides and proteins, as emphasized by the discussed benchmark studies in this review. In the clinical context, particularly for therapy recommendations, prioritizing highly confident and accurately quantified proteins is crucial. Therefore, in clinical proteomics, it is essential to extensively benchmark the chosen software for the specific use case and the clinical question that should be addressed. Without certification and accreditation for diagnostic use of the chosen software, performing orthogonal searches with different software may be advantageous for detecting certain proteins with high confidence, such as biomarkers for diagnosis and therapy recommendations (98, 99, 100).
DIA Analysis Tools
Over the last few years, a plethora of different software tools have been developed for different aspects of DIA analysis. A truly comprehensive review discussing every software option lies beyond the scope of this review and we therefore will focus on the most important software tools. To assess which software tools are important to the field of proteomics, we have aggregated citation and usage statistics for different DIA analysis tools.
These tools include (in alphabetical order) DeepNovo-DIA (101), DIA-NN (102), DIA-Umpire (17), DreamDIA (103), EncyclopeDIA (104), FragPipe (35), Group-DIA (105), MaxDIA (106), MSPLIT-DIA (107), OpenSWATH (108), PECAN (109), PIQED (110), ScaffoldDIA, Skyline (111), Specter (112), Spectronaut (68) and SWATHProphet (113).
We first analyzed the annual citation frequency of the original manuscript of each software tool as indexed by Google Scholar (Fig. 2A). For us, citations represent a general impact on the field of proteomics. However, some software tools may not be developed by academic groups and may therefore lack an original publication. Other software tools may be capable of analyzing different data as well, such as DDA data, and therefore their citation numbers might not be strictly connected to their ability to analyze DIA data. Furthermore, multiple manuscripts for one tool might have been published and different manuscripts may be cited by users. It is also possible that users do not cite the software tool’s original manuscript, although data was analyzed with said tool.
Fig. 2.

Usage metrics of DIA analysis tools between 2010 and 2023.A, annual number of citations of each primary manuscript as indexed by Google Scholar. B, annual co-occurrence of “SoftwareTool” with “proteomics” and either “DIA” or “SWATH” in PMC literature searches (in the case of EncyclopeDIA, “Searle” was additionally used in the co-occurrence search, which probably leads to an underrepresentation). DIA, data-independent acquisition; PMC, PubMed Central; SWATH, sequential window acquisition of all theoretical fragment ion spectra.
We therefore complemented our citation statistics with usage statistics. To approximate usage statistics, we searched PubMed Central (PMC), which allows searching whole manuscripts for the co-occurrence of “[SoftwareTool]” with “proteomics” and either “DIA” or “SWATH” per year for all software tools (Fig. 2B). As only a fraction of all published research is available via PMC, this only serves as a representation of usage statistics. We expect that most DIA analysis tools being examined here are being used more frequently.
Despite the inaccuracy of both methods, we think that the relative impact of all tools on the field of proteomics and their usage can be approximated when combining both metrics.
Judging from the co-occurrence metric, the most frequently used tools currently are as follows: Spectronaut, DIA-NN, Skyline, FragPipe, OpenSWATH, DIA-Umpire, and MaxDIA. It has to be noted that PMC searches yield a very high number of co-occurrences for “EncyclopeDIA”, “DIA,” and either “SWATH” or “DIA,” as PMC searches are case insensitive, and many manuscripts mention “encyclopedia” independent of the software suite EncyclopeDIA. Hence, we refined the search for EncyclopeDIA by including the additional term “Searle.” This probably led to an underrepresentation of “EncyclopeDIA” in the co-occurrence metric. Judging by the citations per year, EncyclopeDIA also has had a major impact on the field of proteomics over the last few years, so it will also be included in this review.
Overview of DIA Software Tools and Their Features
We have summarized the most important features supported by different DIA software tools in Table 1.
Table 1.
Overview of features supported by different DIA software tools
| Feature | SpectropectroNaut 18 |
DIA-NN 1.8.1 |
Open SWATH (part of OpenMS 3.0.0) | MSFragger-DIA 21 | Skyline 23.1 | EncyclopeDIA 2.12 | MaxDIA 2.4 |
|---|---|---|---|---|---|---|---|
| Free for Academia | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Open source | ✗ | ✗ | ✓ | ✗ | ✓ | ✓ | ✗ |
| Windows | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Linux | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Orbitrap | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| TOFa | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| DIA-PASEF | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ | ✓ |
| PTM searches | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| PTM localization scoring | ✓ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ |
| Multiplex (e.g., SILAC, mTRAQ) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Interference removalb | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✗ |
| Visualization of MS data | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ |
| QC metric reporting | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Statistical analysis (diff. abundance)c | ✓ | ✗ | ✗ | ✓ | ✓ | ✗ | ✗ |
| No a priori knowledge DIA search (FASTA > DIA search) | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✗ |
| Library generation/refinement directly from DIA | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ |
| Library generation from DDA | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Combined library generation: DDA, DIA, GPF |
✓ | ✗ | ✗ | ✓ | ✓ | ✓ | ✓ |
| Identification/peak detection with neural networks/machine learning | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Integration of upstream tools for library generation | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Visualization/analysis capabilities of upstream tools’ search results | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ |
| Interactive visualization of data (personal opinion of the authors) | + + | + | + | + + + | + | + |
DDA, data-dependent acquisition; DIA, data-independent acquisition; FAIMS, high-field asymmetric waveform ion mobility spectrometry; GPF, gas-phase fractionation; MS, mass spectrometry; PASEF, parallel accumulation and serial fragmentation; PTM, posttranslational modification; QC, quality control; SWATH, sequential window acquisition of all theoretical fragment ion spectra.
Depending on the specific software version and instrument file format combination, not all combinations might be possible for all software tools and MS vendors.
OpenSWATH and MaxDIA offer background subtraction.
Built-in functionality for differential statistics, to compare protein abundances between different samples/conditions.
Spectronaut
Spectronaut is a commercial software tool specifically aimed at the analysis of DIA data developed by Biognosys. Originally developed for Windows, Spectronaut as of version 18 is “cloud-ready and comes with Linux support.” Spectronaut offers many DIA analysis modes, including multiplexing (e.g., isotopic labeling strategies) and PTM searches with site-confidence estimation (53). Additionally, Spectronaut offers data visualization, quality control metrics, and quality control visualizations as well as post-processing statistical analysis and visualization options. Spectronaut aims to provide a user-friendly and powerful DIA analysis tool also for nonexpert users. To this end, Spectronaut has been focusing a lot of effort on a “DirectDIA” workflow, which does not require any a priori spectral library information and can deal with large search spaces including many PTMs and unspecific searches.
Data-Independent Acquisition–NN
DIA-NN is a free, closed-source software suite developed for the analysis of DIA data with a focus on short gradients, which makes DIA-NN especially useful when sample throughput is a priority. DIA-NN supports TOF, Orbitrap, and DIA-PASEF data. DIA-NN offers multiplex DIA analysis and PTM searches with site-confidence estimation (52). The graphical user interface of DIA-NN focuses on the most commonly used input parameters empowering researchers without bioinformatic background to perform DIA data analysis. Sophisticated and more customized searches can be performed by adding command line settings. DIA-NN itself does not offer any data visualization or post-processing statistical analysis options. However, multiple software suites can handle DIA-NN input for visualization and statistical analysis of results, such as AlphaMap (114). DIA-NN also offers a built-in prediction of spectral libraries, allowing searches without a priori spectral libraries.
MaxDIA
MaxDIA is a software platform for analyzing DIA data developed by the Cox lab, which is integrated into MaxQuant. MaxQuant is a freely available, closed-source software that runs on both Windows and Linux systems and is particularly renowned for its proficiency in analyzing DDA MS data. MaxDIA supports TOF, Orbitrap, and DIA-PASEF data. MaxQuant comes with an elaborate graphical user interface that enables a broad variety of researchers to perform their proteomic data analysis. While MaxQuant itself does not offer statistical analysis of proteomics data directly, the Cox lab has also developed the Perseus computational platform. Perseus enables analysis of nearly all proteomics software tool outputs including DDA, DIA, as well as other omics data (115). At present, MaxDIA is designed to process DIA data exclusively when a preexisting spectral library, created using DDA data, is available, or if the data pertains to one of the most commonly studied organisms for which predicted libraries are accessible in a compatible format. However, future updates, specifically in version 2.5, are expected to introduce native support for analyzing DIA data using only a FASTA file as the initial knowledge base.
OpenSWATH
OpenSWATH is an open-source application, which is fully integrated into the OpenMS software ecosystem. It is vendor-neutral and can be used for the analysis of Orbitrap and TOF data. Within OpenSWATH a vast amount of advanced settings and parameters can be defined allowing for specialized use cases and promoting fully customizable DIA data analysis. OpenSWATH supports modular analysis strategies and is most efficiently applied using the OpenSWATHWorkflow (OSW) executable (116). With module extensions such as Mobi-DIK and diapysef, the OSW software suite enables analysis of all commonly used DIA data types including DIA-PASEF. Noteworthy, OSW among other related software tools can be used on public cloud infrastructures via the Galaxy framework offering an easy-to-use graphical user interface and circumventing the need for private computational resources (70). While OpenSWATH itself does not directly support statistical analysis of proteomics data, it is fully compatible with MSstats (69). OpenSWATH is especially interesting for developers, based on its open-source and modular properties as well as the multitude of analysis parameters.
EncyclopeDIA
EncyclopeDIA is a free and open-source Java application for Windows, Linux, and Mac. EncyclopeDIA allows the user to either build spectral libraries from various sources (DIA, GPF, and DDA) or use prebuilt libraries (such as the Pan-human library or Prosit libraries) (19, 117). Via the Walnut module, DIA data can also be directly searched against a FASTA. The integrated Thesaurus module can be used to search for PTMs and offers localization scoring. EncyclopeDIA itself does not offer statistical analysis of proteomics data. Recently, EncyclopeDIA has also been integrated into both the Galaxy framework and the Skyline ecosystem, facilitating usability and accessibility.
Skyline
Skyline is a free, extremely versatile, open-source Windows client application, which has actively shaped the proteomics data analysis tool landscape for nearly 2 decades. Skyline was originally developed to analyze targeted proteomics data, for example, selected reaction monitoring. It is vendor-neutral and supports a plethora of different proteomics data, including targeted and explorative (DDA and DIA) data, which can all be used to generate spectral libraries for DIA analysis. Skyline can also generate isolation schemes and acquisition methods for proteomics data acquisition. Skyline offers advanced data visualization and post-processing statistical analysis. As of version 23.1.1.318, Skyline has integrated a complete EncyclopeDIA workflow in their pipeline, enabling narrow and wide window DIA searches. Historically speaking, as Skyline was developed for targeted proteomics analyses, a focus of Skyline has always been the interactive visualization of data. Skyline is currently the only tool in our comparison that can import other DIA software tools’ results to either further inspect and validate these results by visualization or refine the analysis altogether. Skyline also offers a built-in MSstats module allowing the users to perform statistical analyses of their proteomics data within the Skyline software.
DIA-Umpire
Originally, DIA-Umpire was developed by the Nesvizhskii lab as an open-source DIA analysis software. Recently, it was integrated into the FragPipe environment and ultimately superseded by the development of MSFragger-DIA.
FragPipe (MSFragger-DIA in Combination with DIA-NN)
FragPipe is a graphical user interface for the analysis of proteomics data that runs on Windows and Linux systems, which incorporates either DIA-Umpire or MSFragger-DIA in combination with DIA-NN for DIA analysis. DIA-Umpire or MSFragger-DIA builds a spectral library from DDA, DIA, or GPF measurements (or a combination thereof), leveraging spectral indexing for fast searches, even with large search spaces such as semispecific searches. The resulting spectral library can then be used to search DIA data using the built-in DIA-NN module (35, 118) It supports data from Orbitrap and TOF data and can analyze DIA-PASEF data. MSFragger-DIA supports PTM analyses as well as multiplex DIA. Currently, DIA-PASEF data cannot be used during the library building stage, which limits the direct analysis of DIA-PASEF data. It is planned to address this limitation in future updates.
Performance of Different Software Tools
In recent years, several systematic studies have been conducted to compare the performance of different DIA analysis tools. We will focus on different metrics that are compared in these studies: proteome coverage, false identifications, accuracy and precision of quantitation, PTMs, and their site localization.
Identification Sensitivity and Specificity
We define the sensitivity of a DIA analysis software as the ability to detect many precursors as this generally indicates that a software tool can I) best deal with high spectral complexity and II) needs a lower number of fragment ions to detect a precursor.
We define specificity as the likelihood that an identification is correct. As each software applies its approach to estimate false discovery rate (FDR), independent benchmark studies use different strategies to compare this metric. Sensitivity and specificity as defined above are interdependent. By relaxing the specificity of reported results, the sensitivity may be increased, that is, while reporting more identified precursors and corresponding proteins, the proportion of incorrect identifications may also increase. As the field of proteomics historically is a numbers game, an independent validation of FDR is warranted. Comparing FDRs between different software suites can be achieved by using entrapment searches (119, 120, 121, 122, 123). In an alternative approach, samples are analyzed with a spectral library that contains entries that have not been present in the samples, thereby allowing a false positive comparison (18).
We deliberately choose to define sensitivity in a manner distinct from that used in receiver operator characteristics analysis. While the sensitivity of a receiver operator characteristics is an invaluable tool to assess the performance of a given software solution within its framework, it should not be used in our view to compare different lists of identifications. In the context of DIA analysis, it is more intuitive to define sensitivity as the ability of a software tool to detect a wide array of peptide precursors.
Quantitation Accuracy and Precision
We define accuracy as the ability of DIA software to estimate the magnitudes of differential abundances of precursors and the corresponding inferred protein abundances across samples without bias. This entails the software’s proficiency in determining the numerical values that accurately reflect the underlying relative changes in protein abundance between different experiment conditions and samples. It is a common practice to display these relative changes in logarithmic fold changes instead of ratios. While accuracy refers to the existence of bias, the term precision relates to the variance of the estimates. These two aspects are intertwined in the so-called bias-variance trade-off; hence variance can be reduced at the expense of introducing bias (124). While accuracy, that is, unbiased estimates might play an important role in many applications, it has been shown that in some settings a ratio overestimation or underestimation (“compression”) might be tolerated, as it is often more important to reduce the variance of estimated expression changes rather than estimating absolute fold changes without bias (125). Especially, in the field of isobaric labeling, this has been investigated and discussed at length (126, 127). We define precision as each software’s reproducibility of quantitation regarding variance over replicates. This concept encompasses the software’s ability to yield consistent numerical values for precursor and protein abundances, even if these absolute values might be systematically skewed or biased. As briefly discussed above, precision is often deemed more important than accuracy, as lower quantitation variance leads to higher statistical power during the detection of differentially abundant proteins.
PTMs and Site Localization
Some DIA analysis software suites provide the option to identify PTMs. A major challenge while analyzing PTMs in proteomics experiments is the reliable localization of said PTM to a single amino acid inside the peptide sequence (2, 128). Currently, all investigated tools provide a PTM site localization probability or score estimation except for Skyline.
Benchmark Studies
While many benchmark studies have been published in the past few years, there is only a limited amount of publications dedicated to comparing different DIA software tools. Other benchmark studies focused, for example, on comparing DDA to DIA (9), DIA to isobaric labeling quantitation (12) or focused on testing the reproducibility of DIA across different laboratories (round robin studies) (129, 130).
Navarro et al. focused on the analysis of SWATH data generated by SCIEX TOF instruments and a three-species sample mix was employed for the assessment of performance (131). An R package was developed to compare the different software tools. The analysis was performed by the software developers to ensure optimal parameter selection. A first round of analyses by the developers was followed by sharing and discussing the benchmark results with all participants, after which a second round of analysis was performed by the participants. Of the software suites, we are focusing on in this review the following software suites are compared in this study: DIA-Umpire, Skyline, Spectronaut, and OpenSWATH. OpenSWATH, Skyline, and Spectronaut identified a comparable number of proteins in this setting, while the number of peptides slightly varied between software suites. Precision was measured as the CV of human identifications on both peptide and protein levels. On the protein level, these data suggest that Spectronaut leads to the highest precision. Accuracy was measured to each other within each species separated into tertiles (0–⅓ intensity, ⅓–⅔ intensity, and ⅔–1 intensity). Based on this report, DIA-Umpire overall had the worst accuracy results, while for the other software options the performance varied, based on the species, whether peptide or protein level was investigated, and which tertile was investigated.
We previously compared different search strategies for DIA data analysis and provided a comprehensive view of all steps of data analysis from spectral library generation to statistical analysis for differentially abundant proteins (18). DIA-NN, OpenSWATH, Skyline, and Spectronaut were compared in our study. We found that different software packages performed vastly differently when using different spectral libraries. DIA-NN, in combination with a GPF-refined library, identified the most precursors and protein groups overall. In general, GPF-refined libraries led to the highest identification rates independent of which software was used. However, the amount of false positives was also higher than other spectral libraries. In general, using Spectronaut and DIA-NN led to fewer false identifications than OpenSWATH and Skyline. For OpenSWATH, this could be reduced by employing an automated alignment strategy called transfer of identification confidence (132). The accuracy was assessed by comparing the theoretical fold change between two spike-in conditions to the experimentally observed fold change. Skyline and OpenSWATH led to the most accurate measurement of fold change, while DIA-NN and Spectronaut consistently (for all used spectral libraries) overestimated the fold change. We evaluated the quantitative precision of all tools by investigating the variance of Escherichia coli quantitations. All tools employing all spectra libraries showed similar precision.
Gotti et al. added universal proteomics standard (UPS1) in different concentrations to an E. coli background to assess the ability of software tools to analyze different Orbitrap DIA methods (133). The four isolation windows that were investigated covering different mass ranges were: narrow (8 m/z for 350–950 m/z), wide (15 m/z for 350–1475 m/z), mixed (8 m/z for 455–711, 15 m/z for 350–455 and 711–1251), and overlapped (8 m/z, 50% overlap for 350–954 m/z). The software tools of interest to this review that were investigated are DIA-NN, OpenSWATH, Skyline, and Spectronaut.
DIA-NN consistently identified most peptides and proteins of the E. coli background in FASTA mode, while Spectronaut identified the most peptides in library mode. Skyline identified the most proteins in library mode but, according to the study, without protein FDR filtering (which has to be performed using additional tools, such as MSstats) (134).
The variance was assessed for both the E. coli peptides and proteins and the UPS1 peptides and proteins.
Gotti et al. also show that with different spike-in concentrations, the DIA analysis software tools quantify varying numbers of UPS1 peptides and proteins. However, as the authors did not include a “zero” spike-in condition, it is not possible to distinguish whether the software tools merely quantified noise signals or very low real UPS1 signals at the lower spike-in concentrations. Therefore, we emphasize the limitation of these results to assess the trade-off between sensitivity and specificity for different software tools. This study is focused on the interplay between different software tools and different DIA methods.
Shahbazy et al. evaluated the capabilities of different DIA software tools to analyze immunopeptidomics DIA data in the form of two monoallelic datasets, of which each provides a distinct diversity of peptide sequences. In the C1R-B57 dataset, DIA-NN and Spectronaut achieved higher peptide coverage, while in the C1R-A02 dataset, DIA-NN and PEAKS achieved higher peptide coverage. Across replicates, DIA-NN showed the highest number of consistently identified and quantified peptides. The specificity of identification was estimated with a hybrid library approach, in which DIA-NN provided lower external FDR rates during external validation in the C1R-B57 dataset, whereas in the C1R-A02 dataset, Spectronaut and DIA-NN showed lower empirical FDR errors. All tools demonstrated reasonable correlations in quantifying precursors of human leukocyte antigen-bound peptides.
In a seminal work, Lou et al. recently investigated the performance of DIA-NN, MaxDIA, Skyline, and Spectronaut (135). The authors generated a two-species dilution series experiment (murine membrane proteins spiked into yeast background) and measured the samples on both an Orbitrap and a timsTOF mass spectrometer. The resulting data was analyzed using a range of different spectral libraries. The identification performance varied as expected with different spectral libraries and the instrument used for the data acquisition. For the Orbitrap data, a universal library generated by FragPipe based on prefractionated DDA runs led to comparable murine protein identifications between DIA-NN, Skyline, and Spectronaut. Using a software-specific DDA library, Spectronaut identified the most murine proteins. Using an in silico–generated library, DIA-NN achieved the best identification performance. For the timsTOF data, DIA-NN and Spectronaut identified the most murine proteins using a universal library. Using a software-specific DDA library, Spectronaut identified the most murine proteins. When using an in silico–generated library, DIA-NN identified the most murine proteins.
Lou et al. also evaluated the false identification percentage employing an entrapment-like strategy using decoy databases comprising different amounts of Arabidopsis sequences. DIA-NN and Spectronaut both showed adequate false discovery rates, with DIA-NN slightly outperforming Spectronaut. Quantitative accuracy was evaluated by comparing the expected fold changes of murine proteins with the reported fold changes. DIA-NN showed the best performance for the Orbitrap and the timsTOF data. Quantitative precision was assessed by calculating protein CVs over replicates, with DIA-NN performing best for both Orbitrap and timsTOF data.
To investigate the performance of the four software tools mentioned above regarding their ability to detect and locate phosphosites, Lou et al. reanalyzed a dataset published by Kitata et al. (136). In this dataset, synthetic human phosphopeptides (with known phosphorylation positions) were spiked into a yeast background and the data were recorded on an Orbitrap system using DIA. The first decoy library, which included isomeric phosphopeptides with different phosphosite positions, revealed that while Skyline identified the most phosphopeptides, it also had the highest FDR, indicating less reliable error rate control. In contrast, DIA-NN showed significantly lower FDRs, especially when adjusting the site confidence score cut-off. The second decoy library, featuring isomeric phosphopeptides with altered site positions, demonstrated that DIA-NN and Spectronaut maintained high sensitivity in detecting synthetic phosphopeptides, with DIA-NN showing a notably lower local site-level FDR. Overall, DIA-NN exhibited more stringent FDR control, albeit at the cost of a lower phosphopeptide identification rate, than Spectronaut, which offered higher sensitivity but with a higher FDR.
Overall, the available benchmark studies present an incomplete and heterogeneous picture regarding performance differences, especially in regard to PTM site localization, which remains an understudied aspect of DIA analyses. DIA-NN and Spectronaut performed best in most cases, although it should be noted that they also represent the software suites most frequently evaluated in those benchmark studies. Since the choice of a software tool has a profound impact on the results and, as emphasized above, especially in the clinical context, clearly defined analysis workflows and guidelines for choosing analysis methods are essential, there is a great need for further benchmark studies aimed at deriving clear guidelines.
Future Directions and Emerging Trends in Clinical Applications
To capture accurate tumor proteomes, macrodissection of tumorous tissue is essential due to adjacent nontumorous areas in patient samples. Techniques like laser capture microdissection enable precise isolation of specific tumor regions and individual cell populations, facilitating focused analysis and representing tumor heterogeneity at a spatial level (137, 138, 139). The advanced sensitivity of DIA-MS state-of-the-art instrumentation allows in-depth proteomic investigation of minute sample amounts, such as from dissected clinical specimens or single-cell experiments (140, 141, 142, 143). Furthermore, the increasing depth of DIA-MS–based proteomics paved the way for comprehensive and integrative investigations such as proteogenomics (144, 145). Proteogenomics focuses on a) corroboration of determined sequence variants (DNA or RNA) on the protein level to address proteomic penetrance of individual mutations and b) determining the impact/correlation of genetic mutations on the global proteome composition and certain signaling pathways of interest (146, 147, 148). Perspectively, short liquid chromatography gradients coupled with fast scanning and sensitive MS instrumentation enable vast numbers of measurements (>300 samples per day) using minimal sample inputs. This has paved the way for fast and deep proteome profiling in the scope of single-cell and large cohort studies, rendering the measurement of thousands of samples feasible within reasonable time frames. Furthermore, targeted measurements of relevant biomarkers with already established methods such as reverse phase protein arrays or MS-based panels (multiple reaction monitoring and parallel reaction monitoring), might be complemented with explorative proteomics using DIA-MS in routine molecular diagnostics. Leveraging vast archives of published proteomic data paves the way for a) a reference-based comparison of DIA-MS measurements from individual samples or patients (as is often the case in molecular diagnostics) and b) the development of novel approaches, such as clinical immunopeptidomics using DIA-MS (149). Comprehensive molecular characterization might promote more sophisticated diagnosis as well as the proposal of individualized and effective treatment options for particularly challenging malignancies and other diseases.
Ultimately, the integration of MS-based proteomics in routine molecular diagnostics will require standardized and certified sample preparation as well as data analysis, which falls beyond the scope of individual benchmarking studies. Hence, certification of analysis strategies for diagnostic use will be an upcoming challenge independent of the data acquisition strategy in the field of MS-based clinical proteomics.
Conclusion
DIA is a versatile method for MS-based proteomics. Developments in LC-MS/MS instrumentation mainly contributed to a higher separation capacity and scan speed together with cleaner fragment spectra (32, 41, 42, 45). Hence, MS-based proteomics is on the verge of full proteome coverage in practicable acquisition times paving the way for a manifold of applications in research and potentially molecular diagnostics. Perspectively, the trend of faster acquisition and narrow isolation windows promotes hybrid approaches of DDA and DIA-like measurements yielding reproducible and in-depth proteome coverage and reliable and sensitive quantification using minute sample input.
In parallel with the hardware developments, there have been innovations in software and analysis such as machine learning–dependent strategies that drastically improved the analysis and interpretation of DIA-MS data. Other exciting software developments are the implementation of artificial intelligence for pattern recognition in spectrum identification and quantification. An ever-increasing spectrum of applications of DIA-MS is possible due to the extensive toolbox of various MS acquisition and data analysis strategies. Each tool comes with considerable strengths and weaknesses but the toolbox in its entirety enables a suitable way for a sample- and data-driven implementation of DIA-MS in proteomics and beyond. When evaluating different DIA software tools, it is crucial to recognize that each tool is designed with specific objectives and functionalities in mind, leading to significant feature variations. We would like to stress that in our opinion the choice of a particular tool is less critical than ensuring the chosen tool aligns with the user's methodology and objectives with tool-specific limitations in mind. The key factor is the user's proficiency with the tool; an expert who thoroughly understands a tool can more effectively assess the strengths and limitations of an analysis than someone using a highly rated tool without in-depth knowledge of it, which was impressively demonstrated by Choi et al. (129).
For newcomers to DIA, we recommend starting with tools that offer visualization capabilities, such as Skyline, MaxDIA, FragPipe, or Spectronaut, as this aids in grasping the fundamentals of DIA and its data analysis more intuitively. Other factors, such as cost, also play a role; for instance, while Spectronaut provides a comprehensive DIA analysis pipeline, its licensing fees may be prohibitive for individual researchers and laboratories. On the other hand, DIA-NN could be an excellent choice for data scientists skilled in visualizing and exploring proteomics data independently.
We believe it is beneficial to become acquainted with multiple DIA data analysis tools. Cross-referencing results from different tools not only validates findings but also accelerates the learning process in identifying the strengths and weaknesses of various tools. Regardless of the chosen DIA analysis workflow, we recommend Skyline as an interactive visualization tool. Its ability to import results from other tools enhances its utility and supports a more comprehensive analysis approach. Shahbazy et al. also advocated for the use of a "two orthogonal tools'' strategy in their immunopeptidomics DIA software benchmarking study (150).
Over the last decade, the amount of identified features from DIA data has drastically increased. While the development of novel software algorithms allows researchers to investigate the proteome at greater depths, not every software version may have the same level of reliability. Therefore, we caution against transferring any attribute of software from one version to another, as the underlying algorithms might have changed, which might heavily influence data properties. However, this also directly impacts our ability to transfer our DIA analysis approaches, which might work well in research settings, to clinically relevant settings. If DIA analyses are not reliable enough, we may not be able to implement DIA in routine diagnostics despite its huge potential to combine the ability to extract fragment signals in a targeted manner and at the same time allow for explorative analyses. For the integration of proteomics into clinical applications, such as molecular diagnostics, reliable and highly confident identification and quantification are, therefore, of utmost importance.
Funding and Additional Information
O. S. acknowledges funding by the Deutsche Forschungsgemeinschaft (DFG, Projects 446,058,856, 466,359,513, 444,936,968, 405,351,425, 431,336,276, 43,198,400 (SFB 1453 “NephGen”), 441,891,347 (SFB 1479 “OncoEscape”), 423,813,989 (GRK 2606 “ProtPath”), 322,977,937 (GRK 2344 “MeInBio”), the ERA PerMed programme (BMBF, 01KU1916, 01KU1915A), the German-Israel Foundation (Grant No. 1444), and the German Consortium for Translational Cancer Research (project Impro-Rec), funding by the MatrixCode research group, FRIAS, and support by the Fördergesellschaft Forschung Tumorbiologie, Freiburg im Breisgau. A. S. acknowledges funding by the University of Basel. This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy (CIBSS-EXC-2189-2100249960-390939984) (C. K. and E. B.).
Conflict of interest
The authors declare no competing interests.
Acknowledgments
Author contributions
K. F., M. F., C. K., A. S., and O. S. conceptualization; K. F., M. F., E. B., A. S., M. M., C. K., A. S., and O. S. writing–original draft; K. F., M. F., E. B., A. S., M. M., C. K., A. S., and O. S. writing–review and editing; E. B., A. S., and M. M. visualization; O. S. resources; O. S. supervision.
Footnotes
Alexander Schmidt and Oliver Schillin shared last authorship.
References
- 1.Aebersold R., Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 2.Yu F., Teo G.C., Kong A.T., Haynes S.E., Avtonomov D.M., Geiszler D.J., et al. Identification of modified peptides using localization-aware open search. Nat. Commun. 2020;11:4065. doi: 10.1038/s41467-020-17921-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang W.H., Halpern B.R., Shilov I.V., Seymour S.L., Keating S.P., Loboda A., et al. Discovering known and unanticipated protein modifications using MS/MS database searching. Anal. Chem. 2005;77:3931–3946. doi: 10.1021/ac0481046. [DOI] [PubMed] [Google Scholar]
- 4.Fahrner M., Kook L., Fröhlich K., Biniossek M.L., Schilling O. A systematic evaluation of semispecific peptide search parameter enables identification of previously Undescribed N-Terminal peptides and Conserved Proteolytic processing in cancer cell lines. Proteomes. 2021;9:26. doi: 10.3390/proteomes9020026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Purvine S., Eppel J.-T., Yi E.C., Goodlett D.R. Shotgun collision-induced dissociation of peptides using a time of flight mass analyzer. Proteomics. 2003;3:847–850. doi: 10.1002/pmic.200300362. [DOI] [PubMed] [Google Scholar]
- 6.Silva J.C., Denny R., Dorschel C.A., Gorenstein M., Kass I.J., Li G.-Z., et al. Quantitative proteomic analysis by accurate mass retention time pairs. Anal. Chem. 2005;77:2187–2200. doi: 10.1021/ac048455k. [DOI] [PubMed] [Google Scholar]
- 7.Panchaud A., Scherl A., Shaffer S.A., von Haller P.D., Kulasekara H.D., Miller S.I., et al. Precursor acquisition independent from ion count: how to dive deeper into the proteomics ocean. Anal. Chem. 2009;81:6481–6488. doi: 10.1021/ac900888s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gillet L.C., Navarro P., Tate S., Röst H., Selevsek N., Reiter L., et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dowell J.A., Wright L.J., Armstrong E.A., Denu J.M. Benchmarking quantitative performance in label-free proteomics. ACS Omega. 2021;6:2494–2504. doi: 10.1021/acsomega.0c04030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muntel J., Gandhi T., Verbeke L., Bernhardt O.M., Treiber T., Bruderer R., et al. Surpassing 10 000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy. Mol. Omics. 2019;15:348–360. doi: 10.1039/c9mo00082h. [DOI] [PubMed] [Google Scholar]
- 11.Kawashima Y., Nagai H., Konno R., Ishikawa M., Nakajima D., Sato H., et al. Single-shot 10K proteome approach: over 10,000 protein identifications by data-independent acquisition-based single-shot proteomics with ion mobility spectrometry. J. Proteome Res. 2022;21:1418–1427. doi: 10.1021/acs.jproteome.2c00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muntel J., Kirkpatrick J., Bruderer R., Huang T., Vitek O., Ori A., et al. Comparison of protein quantification in a complex background by DIA and TMT workflows with fixed instrument time. J. Proteome Res. 2019;18:1340–1351. doi: 10.1021/acs.jproteome.8b00898. [DOI] [PubMed] [Google Scholar]
- 13.Swearingen K.E., Moritz R.L. High-field asymmetric waveform ion mobility spectrometry for mass spectrometry-based proteomics. Expert Rev. Proteomics. 2012;9:505–517. doi: 10.1586/epr.12.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meier F., Beck S., Grassl N., Lubeck M., Park M.A., Raether O., et al. Parallel accumulation-serial fragmentation (PASEF): multiplying sequencing speed and sensitivity by synchronized scans in a trapped ion mobility Device. J. Proteome Res. 2015;14:5378–5387. doi: 10.1021/acs.jproteome.5b00932. [DOI] [PubMed] [Google Scholar]
- 15.Heil L.R., Damoc E., Arrey T.N., Pashkova A., Denisov E., Petzoldt J., et al. Evaluating the performance of the Astral mass analyzer for quantitative proteomics using data independent acquisition. bioRxiv. 2023 doi: 10.1101/2023.06.03.543570. [preprint] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins B.C., Gillet L.C., Rosenberger G., Röst H.L., Vichalkovski A., Gstaiger M., et al. Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14-3-3 system. Nat. Methods. 2013;10:1246–1253. doi: 10.1038/nmeth.2703. [DOI] [PubMed] [Google Scholar]
- 17.Tsou C.-C., Avtonomov D., Larsen B., Tucholska M., Choi H., Gingras A.-C., et al. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods. 2015;12:258–264. doi: 10.1038/nmeth.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fröhlich K., Brombacher E., Fahrner M., Vogele D., Kook L., Pinter N., et al. Benchmarking of analysis strategies for data-independent acquisition proteomics using a large-scale dataset comprising inter-patient heterogeneity. Nat. Commun. 2022;13:2622. doi: 10.1038/s41467-022-30094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gessulat S., Schmidt T., Zolg D.P., Samaras P., Schnatbaum K., Zerweck J., et al. Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods. 2019;16:509–518. doi: 10.1038/s41592-019-0426-7. [DOI] [PubMed] [Google Scholar]
- 20.Distler U., Kuharev J., Navarro P., Levin Y., Schild H., Tenzer S. Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat. Methods. 2014;11:167–170. doi: 10.1038/nmeth.2767. [DOI] [PubMed] [Google Scholar]
- 21.Pringle S.D., Giles K., Wildgoose J.L., Williams J.P., Slade S.E., Thalassinos K., et al. An investigation of the mobility separation of some peptide and protein ions using a new hybrid quadrupole/travelling wave IMS/oa-ToF instrument. Int. J. Mass Spectrom. 2007;261:1–12. [Google Scholar]
- 22.Geiger T., Cox J., Mann M. Proteomics on an Orbitrap benchtop mass spectrometer using all-ion fragmentation. Mol. Cell. Proteomics. 2010;9:2252–2261. doi: 10.1074/mcp.M110.001537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venable J.D., Dong M.-Q., Wohlschlegel J., Dillin A., Yates J.R. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods. 2004;1:39–45. doi: 10.1038/nmeth705. [DOI] [PubMed] [Google Scholar]
- 24.Carvalho P.C., Han X., Xu T., Cociorva D., Carvalho Mda G., Barbosa V.C., et al. XDIA: improving on the label-free data-independent analysis. Bioinformatics. 2010;26:847–848. doi: 10.1093/bioinformatics/btq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amodei D., Egertson J., MacLean B.X., Johnson R., Merrihew G.E., Keller A., et al. Improving precursor selectivity in data-independent acquisition using overlapping windows. J. Am. Soc. Mass Spectrom. 2019;30:669–684. doi: 10.1007/s13361-018-2122-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doellinger J., Blumenscheit C., Schneider A., Lasch P. Increasing proteome depth while maintaining quantitative precision in short-gradient data-independent acquisition proteomics. J. Proteome Res. 2023;22:2131–2140. doi: 10.1021/acs.jproteome.3c00078. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez-Lima F., Kaplan D.A., Suetering J., Park M.A. Gas-phase separation using a trapped ion mobility spectrometer. Int. J. Ion Mobil. Spectrom. 2011;14 doi: 10.1007/s12127-011-0067-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michelmann K., Silveira J.A., Ridgeway M.E., Park M.A. Fundamentals of trapped ion mobility spectrometry. J. Am. Soc. Mass Spectrom. 2015;26:14–24. doi: 10.1007/s13361-014-0999-4. [DOI] [PubMed] [Google Scholar]
- 29.Meier F., Park M.A., Mann M. Trapped ion mobility spectrometry and parallel accumulation-serial fragmentation in proteomics. Mol. Cell. Proteomics. 2021;20 doi: 10.1016/j.mcpro.2021.100138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hale O.J., Illes-Toth E., Mize T.H., Cooper H.J. High-field asymmetric waveform ion mobility spectrometry and native mass spectrometry: analysis of intact protein assemblies and protein complexes. Anal. Chem. 2020;92:6811–6816. doi: 10.1021/acs.analchem.0c00649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.A Shvartsburg A., A Anderson G., D Smith R. Pushing the frontier of high-definition ion mobility spectrometry using FAIMS. Mass Spectrom. 2013;2:S0011. doi: 10.5702/massspectrometry.S0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Faivre D.A., McGann C.D., Merrihew G.E., Schweppe D.K., MacCoss M.J. Comparing peptide identifications by FAIMS versus quadrupole gas-phase fractionation. bioRxiv. 2023 doi: 10.1101/2023.09.01.552989. [preprint] [DOI] [Google Scholar]
- 33.Meier F., Brunner A.-D., Frank M., Ha A., Bludau I., Voytik E., et al. diaPASEF: parallel accumulation-serial fragmentation combined with data-independent acquisition. Nat. Methods. 2020;17:1229–1236. doi: 10.1038/s41592-020-00998-0. [DOI] [PubMed] [Google Scholar]
- 34.Prianichnikov N., Koch H., Koch S., Lubeck M., Heilig R., Brehmer S., et al. MaxQuant software for ion mobility enhanced shotgun proteomics. Mol. Cell. Proteomics. 2020;19:1058–1069. doi: 10.1074/mcp.TIR119.001720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Demichev V., Szyrwiel L., Yu F., Teo G.C., Rosenberger G., Niewienda A., et al. dia-PASEF data analysis using FragPipe and DIA-NN for deep proteomics of low sample amounts. Nat. Commun. 2022;13:3944. doi: 10.1038/s41467-022-31492-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mun D.-G., Budhraja R., Bhat F.A., Zenka R.M., Johnson K.L., Moghekar A., et al. Four-dimensional proteomics analysis of human cerebrospinal fluid with trapped ion mobility spectrometry using PASEF. Proteomics. 2023;23 doi: 10.1002/pmic.202200507. [DOI] [PubMed] [Google Scholar]
- 37.Skowronek P., Thielert M., Voytik E., Tanzer M.C., Hansen F.M., Willems S., et al. Rapid and in-depth coverage of the (Phospho-)Proteome with deep libraries and optimal window design for dia-PASEF. Mol. Cell. Proteomics. 2022;21 doi: 10.1016/j.mcpro.2022.100279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skowronek P., Krohs F., Lubeck M., Wallmann G., Itang E.C.M., Koval P., et al. Synchro-PASEF allows precursor-specific fragment ion extraction and interference removal in data-independent acquisition. Mol. Cell. Proteomics. 2023;22 doi: 10.1016/j.mcpro.2022.100489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Distler U., Łącki M.K., Startek M.P., Teschner D., Brehmer S., Decker J., et al. midiaPASEF maximizes information content in data-independent acquisition proteomics. bioRxiv. 2023 doi: 10.1101/2023.01.30.526204. [preprint] [DOI] [Google Scholar]
- 40.Szyrwiel L., Sinn L., Ralser M., Demichev V. Slice-PASEF: fragmenting all ions for maximum sensitivity in proteomics. bioRxiv. 2022 doi: 10.1101/2022.10.31.514544. [preprint] [DOI] [Google Scholar]
- 41.Wang Z., Mülleder M., Batruch I., Chelur A., Textoris-Taube K., Schwecke T., et al. High-throughput proteomics of nanogram-scale samples with Zeno SWATH MS. Elife. 2022;11 doi: 10.7554/eLife.83947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun W., Lin Y., Huang Y., Chan J., Terrillon S., Rosenbaum A.I., et al. Robust and high-throughput Analytical Flow proteomics analysis of cynomolgus monkey and human matrices with zeno SWATH data-independent acquisition. Mol. Cell. Proteomics. 2023;22 doi: 10.1016/j.mcpro.2023.100562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zmyslia M., Fröhlich K., Dao T., Schmidt A., Jessen-Trefzer C. Deep proteomic investigation of Metabolic adaptation in Mycobacteria under different Growth conditions. Proteomes. 2023;11:39. doi: 10.3390/proteomes11040039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fröhlich K., Furrer R., Schori C., Handschin C., Schmidt A. Robust, precise, and deep proteome profiling using a small mass range and narrow window data-independent-acquisition scheme. J. Proteome Res. 2024;23:1028–1038. doi: 10.1021/acs.jproteome.3c00736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stewart H.I., Grinfeld D., Giannakopulos A., Petzoldt J., Shanley T., Garland M., et al. Parallelized acquisition of orbitrap and Astral analyzers enables high-throughput quantitative analysis. Anal. Chem. 2023;95:15656–15664. doi: 10.1021/acs.analchem.3c02856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heil L.R., Damoc E., Arrey T.N., Pashkova A., Denisov E., Petzoldt J., et al. Evaluating the performance of the Astral mass analyzer for quantitative proteomics using data-independent acquisition. J. Proteome Res. 2023;22:3290–3300. doi: 10.1021/acs.jproteome.3c00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guzman U.H., Martinez Del Val A., Ye Z., Damoc E., Arrey T.N., Pashkova A., et al. Narrow-window DIA: ultra-fast quantitative analysis of comprehensive proteomes with high sequencing depth. bioRxiv. 2023 doi: 10.1101/2023.06.02.543374. [preprint] [DOI] [Google Scholar]
- 48.Yang Y., Qiao L. Data-independent acquisition proteomics methods for analyzing post-translational modifications. Proteomics. 2023;23 doi: 10.1002/pmic.202200046. [DOI] [PubMed] [Google Scholar]
- 49.Sidoli S., Fujiwara R., Kulej K., Garcia B.A. Differential quantification of isobaric phosphopeptides using data-independent acquisition mass spectrometry. Mol. Biosyst. 2016;12:2385–2388. doi: 10.1039/c6mb00385k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martinez-Val A., Bekker-Jensen D.B., Hogrebe A., Olsen J.V. Data processing and analysis for DIA-based phosphoproteomics using Spectronaut. Methods Mol. Biol. 2021;2361:95–107. doi: 10.1007/978-1-0716-1641-3_6. [DOI] [PubMed] [Google Scholar]
- 51.Riley N.M., Coon J.J. Phosphoproteomics in the Age of rapid and deep proteome profiling. Anal. Chem. 2016;88:74–94. doi: 10.1021/acs.analchem.5b04123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steger M., Demichev V., Backman M., Ohmayer U., Ihmor P., Müller S., et al. Time-resolved in vivo ubiquitinome profiling by DIA-MS reveals USP7 targets on a proteome-wide scale. Nat. Commun. 2021;12:5399. doi: 10.1038/s41467-021-25454-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bekker-Jensen D.B., Bernhardt O.M., Hogrebe A., Martinez-Val A., Verbeke L., Gandhi T., et al. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun. 2020;11:787. doi: 10.1038/s41467-020-14609-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wen C., Wu X., Lin G., Yan W., Gan G., Xu X., et al. Evaluation of DDA library-free strategies for phosphoproteomics and Ubiquitinomics data-independent acquisition data. J. Proteome Res. 2023;22:2232–2245. doi: 10.1021/acs.jproteome.2c00735. [DOI] [PubMed] [Google Scholar]
- 55.Gao E., Li W., Wu C., Shao W., Di Y., Liu Y. Data-independent acquisition-based proteome and phosphoproteome profiling across six melanoma cell lines reveals determinants of proteotypes. Mol. Omics. 2021;17:413–425. doi: 10.1039/d0mo00188k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qu L., Liu M., Zheng L., Wang X., Xue H. Data-independent acquisition-based global phosphoproteomics reveal the diverse roles of casein kinase 1 in plant development. Sci. Bull. (Beijing) 2023;68:2077–2093. doi: 10.1016/j.scib.2023.08.017. [DOI] [PubMed] [Google Scholar]
- 57.Hansen F.M., Tanzer M.C., Brüning F., Bludau I., Stafford C., Schulman B.A., et al. Data-independent acquisition method for ubiquitinome analysis reveals regulation of circadian biology. Nat. Commun. 2021;12:254. doi: 10.1038/s41467-020-20509-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wei L., Meyer J.G., Schilling B. Quantification of site-specific protein Lysine acetylation and succinylation stoichiometry using data-independent acquisition mass spectrometry. J. Vis. Exp. 2018 doi: 10.3791/57209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oliinyk D., Will A., Schneidmadel F.R., Humphrey S.J., Meier F. μPhos: a scalable and sensitive platform for functional phosphoproteomics. bioRxiv. 2023 doi: 10.1101/2023.04.04.535617. [preprint] [DOI] [Google Scholar]
- 60.Lancaster N.M., Sinitcyn P., Forny P., Peters-Clarke T.M., Fecher C., Smith A.J., et al. Fast and deep phosphoproteome analysis with the orbitrap Astral mass spectrometer. bioRxiv. 2023 doi: 10.1101/2023.11.21.568149. [preprint] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen M., Zhu P., Wan Q., Ruan X., Wu P., Hao Y., et al. Anal. Chem. 2023;95:7495–7502. doi: 10.1021/acs.analchem.2c05414. [DOI] [PubMed] [Google Scholar]
- 62.Dong M., Lih T.-S.M., Ao M., Hu Y., Chen S.-Y., Eguez R.V., et al. Data-independent acquisition-based mass spectrometry (DIA-MS) for quantitative analysis of intact N-Linked glycopeptides. Anal. Chem. 2021;93:13774–13782. doi: 10.1021/acs.analchem.1c01659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chang D., Klein J., Hackett W.E., Nalehua M.R., Wan X.-F., Zaia J. Improving statistical certainty of Glycosylation Similarity between Influenza A Virus variants using data-independent acquisition mass spectrometry. Mol. Cell. Proteomics. 2022;21 doi: 10.1016/j.mcpro.2022.100412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pan K.-T., Chen C.-C., Urlaub H., Khoo K.-H. Adapting data-independent acquisition for mass spectrometry-based protein site-specific N-Glycosylation analysis. Anal. Chem. 2017;89:4532–4539. doi: 10.1021/acs.analchem.6b04996. [DOI] [PubMed] [Google Scholar]
- 65.Lin C.-H., Krisp C., Packer N.H., Molloy M.P. Development of a data independent acquisition mass spectrometry workflow to enable glycopeptide analysis without predefined glycan compositional knowledge. J. Proteomics. 2018;172:68–75. doi: 10.1016/j.jprot.2017.10.011. [DOI] [PubMed] [Google Scholar]
- 66.Toghi E.S., Shah P., Yang W., Li X., Zhang H. GPQuest: a spectral library Matching algorithm for site-specific assignment of tandem mass spectra to intact N-glycopeptides. Anal. Chem. 2015;87:5181–5188. doi: 10.1021/acs.analchem.5b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ye Z., Vakhrushev S.Y. The role of data-independent acquisition for glycoproteomics. Mol. Cell. Proteomics. 2021;20 doi: 10.1074/mcp.R120.002204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bruderer R., Bernhardt O.M., Gandhi T., Miladinović S.M., Cheng L.-Y., Messner S., et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteomics. 2015;14:1400–1410. doi: 10.1074/mcp.M114.044305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kohler D., Staniak M., Tsai T.-H., Huang T., Shulman N., Bernhardt O.M., et al. MSstats version 4.0: statistical analyses of quantitative mass spectrometry-based proteomic experiments with chromatography-based quantification at scale. J. Proteome Res. 2023;22:1466–1482. doi: 10.1021/acs.jproteome.2c00834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fahrner M., Föll M.C., Grüning B.A., Bernt M., Röst H., Schilling O. Democratizing data-independent acquisition proteomics analysis on public cloud infrastructures via the Galaxy framework. Gigascience. 2022;11 doi: 10.1093/gigascience/giac005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kelstrup C.D., Bekker-Jensen D.B., Arrey T.N., Hogrebe A., Harder A., Olsen J.V. Performance evaluation of the Q Exactive HF-X for shotgun proteomics. J. Proteome Res. 2018;17:727–738. doi: 10.1021/acs.jproteome.7b00602. [DOI] [PubMed] [Google Scholar]
- 72.Ludwig C., Gillet L., Rosenberger G., Amon S., Collins B.C., Aebersold R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol. Syst. Biol. 2018;14 doi: 10.15252/msb.20178126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Poulos R.C., Hains P.G., Shah R., Lucas N., Xavier D., Manda S.S., et al. Strategies to enable large-scale proteomics for reproducible research. Nat. Commun. 2020;11:3793. doi: 10.1038/s41467-020-17641-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Krasny L., Huang P.H. Data-independent acquisition mass spectrometry (DIA-MS) for proteomic applications in oncology. Mol. Omics. 2021;17:29–42. doi: 10.1039/d0mo00072h. [DOI] [PubMed] [Google Scholar]
- 75.Kong W., Hui H.W.H., Peng H., Goh W.W.B. Dealing with missing values in proteomics data. Proteomics. 2022;22 doi: 10.1002/pmic.202200092. [DOI] [PubMed] [Google Scholar]
- 76.Liu M., Dongre A. Proper imputation of missing values in proteomics datasets for differential expression analysis. Brief. Bioinform. 2021;22 doi: 10.1093/bib/bbaa112. [DOI] [PubMed] [Google Scholar]
- 77.Huang T., Bruderer R., Muntel J., Xuan Y., Vitek O., Reiter L. Combining precursor and fragment information for improved detection of differential abundance in data independent acquisition. Mol. Cell. Proteomics. 2020;19:421–430. doi: 10.1074/mcp.RA119.001705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Coscia F., Lengyel E., Duraiswamy J., Ashcroft B., Bassani-Sternberg M., Wierer M., et al. Multi-level proteomics Identifies CT45 as a Chemosensitivity Mediator and Immunotherapy target in Ovarian cancer. Cell. 2018;175:159–170.e16. doi: 10.1016/j.cell.2018.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thiery J., Fahrner M. Integration of proteomics in the molecular tumor board. Proteomics. 2023;24 doi: 10.1002/pmic.202300002. [DOI] [PubMed] [Google Scholar]
- 80.Chen G., Gharib T.G., Huang C.-C., Taylor J.M.G., Misek D.E., Kardia S.L.R., et al. Discordant protein and mRNA expression in lung adenocarcinomas. Mol. Cell. Proteomics. 2002;1:304–313. doi: 10.1074/mcp.m200008-mcp200. [DOI] [PubMed] [Google Scholar]
- 81.Maier T., Güell M., Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009;583:3966–3973. doi: 10.1016/j.febslet.2009.10.036. [DOI] [PubMed] [Google Scholar]
- 82.Koussounadis A., Langdon S.P., Um I.H., Harrison D.J., Smith V.A. Relationship between differentially expressed mRNA and mRNA-protein correlations in a xenograft model system. Sci. Rep. 2015;5 doi: 10.1038/srep10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Phipps W.S., Kilgore M.R., Kennedy J.J., Whiteaker J.R., Hoofnagle A.N., Paulovich A.G. Clinical proteomics for solid organ tissues. Mol. Cell. Proteomics. 2023;22 doi: 10.1016/j.mcpro.2023.100648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Doll S., Kriegmair M.C., Santos A., Wierer M., Coscia F., Neil H.M., et al. Rapid proteomic analysis for solid tumors reveals LSD1 as a drug target in an end-stage cancer patient. Mol. Oncol. 2018;12:1296–1307. doi: 10.1002/1878-0261.12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.DeMarco M.L., Nguyen Q., Fok A., Hsiung G.-Y.R., van der Gugten J.G. An automated clinical mass spectrometric method for identification and quantification of variant and wild-type amyloid-β 1-40 and 1-42 peptides in CSF. Alzheimers Dement. (Amst) 2020;12 doi: 10.1002/dad2.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dasari S., Theis J.D., Vrana J.A., Rech K.L., Dao L.N., Howard M.T., et al. Amyloid typing by mass spectrometry in clinical practice: a comprehensive review of 16,175 samples. Mayo Clin. Proc. 2020;95:1852–1864. doi: 10.1016/j.mayocp.2020.06.029. [DOI] [PubMed] [Google Scholar]
- 87.Whiteaker J.R., Sharma K., Hoffman M.A., Kuhn E., Zhao L., Cocco A.R., et al. Targeted mass spectrometry-based assays enable multiplex quantification of receptor tyrosine kinase, MAP Kinase, and AKT signaling. Cell Rep. Methods. 2021;1 doi: 10.1016/j.crmeth.2021.100015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shi J., Phipps W.S., Owusu B.Y., Henderson C.M., Laha T.J., Becker J.O., et al. A distributable LC-MS/MS method for the measurement of serum thyroglobulin. J. Mass Spectrom. Adv. Clin. Lab. 2022;26:28–33. doi: 10.1016/j.jmsacl.2022.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Carr S.A., Abbatiello S.E., Ackermann B.L., Borchers C., Domon B., Deutsch E.W., et al. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol. Cell. Proteomics. 2014;13:907–917. doi: 10.1074/mcp.M113.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Werner J., Bernhard P., Cosenza-Contreras M., Pinter N., Fahrner M., Pallavi P., et al. Targeted and explorative profiling of kallikrein proteases and global proteome biology of pancreatic ductal adenocarcinoma, chronic pancreatitis, and normal pancreas highlights disease-specific proteome remodelling. Neoplasia. 2023;36 doi: 10.1016/j.neo.2022.100871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Boys E.L., Liu J., Robinson P.J., Reddel R.R. Clinical applications of mass spectrometry-based proteomics in cancer: where are we? Proteomics. 2023;23 doi: 10.1002/pmic.202200238. [DOI] [PubMed] [Google Scholar]
- 92.Kim Y.J., Chambers A.G., Cecchi F., Hembrough T. Targeted data-independent acquisition for mass spectrometric detection of RAS mutations in formalin-fixed, paraffin-embedded tumor biopsies. J. Proteomics. 2018;189:91–96. doi: 10.1016/j.jprot.2018.04.022. [DOI] [PubMed] [Google Scholar]
- 93.Barbieux C., Bonnet des Claustres M., Fahrner M., Petrova E., Tsoi L.C., Gouin O., et al. Netherton syndrome subtypes share IL-17/IL-36 signature with distinct IFN-α and allergic responses. J. Allergy Clin. Immunol. 2022;149:1358–1372. doi: 10.1016/j.jaci.2021.08.024. [DOI] [PubMed] [Google Scholar]
- 94.Messner C.B., Demichev V., Wendisch D., Michalick L., White M., Freiwald A., et al. Ultra-high-throughput clinical proteomics reveals Classifiers of COVID-19 Infection. Cell Syst. 2020;11:11–24.e4. doi: 10.1016/j.cels.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Müller T., Kalxdorf M., Longuespée R., Kazdal D.N., Stenzinger A., Krijgsveld J. Automated sample preparation with SP3 for low-input clinical proteomics. Mol. Syst. Biol. 2020;16 doi: 10.15252/msb.20199111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bekker-Jensen D.B., Martínez-Val A., Steigerwald S., Rüther P., Fort K.L., Arrey T.N., et al. A Compact quadrupole-orbitrap mass spectrometer with FAIMS interface Improves proteome coverage in short LC gradients. Mol. Cell. Proteomics. 2020;19:716–729. doi: 10.1074/mcp.TIR119.001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kuras M., Woldmar N., Kim Y., Hefner M., Malm J., Moldvay J., et al. Proteomic workflows for high-quality quantitative proteome and post-translational modification analysis of clinically relevant samples from formalin-fixed paraffin-embedded archives. J. Proteome Res. 2021;20:1027–1039. doi: 10.1021/acs.jproteome.0c00850. [DOI] [PubMed] [Google Scholar]
- 98.Fu Z., Li S., Han S., Shi C., Zhang Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022;7:93. doi: 10.1038/s41392-022-00947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kennedy J.J., Whiteaker J.R., Kennedy L.C., Bosch D.E., Lerch M.L., Schoenherr R.M., et al. Quantification of human Epidermal Growth factor receptor 2 by Immunopeptide Enrichment and targeted mass spectrometry in formalin-fixed paraffin-embedded and Frozen breast cancer tissues. Clin. Chem. 2021;67:1008–1018. doi: 10.1093/clinchem/hvab047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Doroshow D.B., Bhalla S., Beasley M.B., Sholl L.M., Kerr K.M., Gnjatic S., et al. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2021;18:345–362. doi: 10.1038/s41571-021-00473-5. [DOI] [PubMed] [Google Scholar]
- 101.Tran N.H., Qiao R., Xin L., Chen X., Liu C., Zhang X., et al. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nat. Methods. 2019;16:63–66. doi: 10.1038/s41592-018-0260-3. [DOI] [PubMed] [Google Scholar]
- 102.Demichev V., Messner C.B., Vernardis S.I., Lilley K.S., Ralser M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods. 2020;17:41–44. doi: 10.1038/s41592-019-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gao M., Yang W., Li C., Chang Y., Liu Y., He Q., et al. Deep representation features from DreamDIA improve the analysis of data-independent acquisition proteomics. Commun. Biol. 2021;4:1190. doi: 10.1038/s42003-021-02726-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Searle B.C., Pino L.K., Egertson J.D., Ting Y.S., Lawrence R.T., MacLean B.X., et al. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat. Commun. 2018;9:5128. doi: 10.1038/s41467-018-07454-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li Y., Zhong C.-Q., Xu X., Cai S., Wu X., Zhang Y., et al. Group-DIA: analyzing multiple data-independent acquisition mass spectrometry data files. Nat. Methods. 2015;12:1105–1106. doi: 10.1038/nmeth.3593. [DOI] [PubMed] [Google Scholar]
- 106.Sinitcyn P., Hamzeiy H., Salinas Soto F., Itzhak D., McCarthy F., Wichmann C., et al. MaxDIA enables library-based and library-free data-independent acquisition proteomics. Nat. Biotechnol. 2021;39:1563–1573. doi: 10.1038/s41587-021-00968-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang J., Tucholska M., Knight J.D.R., Lambert J.-P., Tate S., Larsen B., et al. MSPLIT-DIA: sensitive peptide identification for data-independent acquisition. Nat. Methods. 2015;12:1106–1108. doi: 10.1038/nmeth.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Röst H.L., Rosenberger G., Navarro P., Gillet L., Miladinović S.M., Schubert O.T., et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat Biotechnol. 2014;32:219–223. doi: 10.1038/nbt.2841. [DOI] [PubMed] [Google Scholar]
- 109.Ting Y.S., Egertson J.D., Bollinger J.G., Searle B.C., Payne S.H., Noble W.S., et al. PECAN: library-free peptide detection for data-independent acquisition tandem mass spectrometry data. Nat. Methods. 2017;14:903–908. doi: 10.1038/nmeth.4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Meyer J.G., Mukkamalla S., Steen H., Nesvizhskii A.I., Gibson B.W., Schilling B. PIQED: automated identification and quantification of protein modifications from DIA-MS data. Nat. Methods. 2017;14:646–647. doi: 10.1038/nmeth.4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pino L.K., Searle B.C., Bollinger J.G., Nunn B., MacLean B., MacCoss M.J. The Skyline ecosystem: informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 2020;39:229–244. doi: 10.1002/mas.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Peckner R., Myers S.A., Jacome A.S.V., Egertson J.D., Abelin J.G., MacCoss M.J., et al. Specter: linear deconvolution for targeted analysis of data-independent acquisition mass spectrometry proteomics. Nat. Methods. 2018;15:371–378. doi: 10.1038/nmeth.4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Keller A., Bader S.L., Shteynberg D., Hood L., Moritz R.L. Automated validation of results and Removal of fragment ion interferences in targeted analysis of data-independent acquisition mass spectrometry (MS) using SWATHProphet. Mol. Cell. Proteomics. 2015;14:1411–1418. doi: 10.1074/mcp.O114.044917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Voytik E., Bludau I., Willems S., Hansen F.M., Brunner A.-D., Strauss M.T., et al. AlphaMap: an open-source Python package for the visual annotation of proteomics data with sequence-specific knowledge. Bioinformatics. 2022;38:849–852. doi: 10.1093/bioinformatics/btab674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 116.Röst H.L., Aebersold R., Schubert O.T. Automated SWATH data analysis using targeted extraction of ion Chromatograms. Methods Mol. Biol. 2017;1550:289–307. doi: 10.1007/978-1-4939-6747-6_20. [DOI] [PubMed] [Google Scholar]
- 117.Rosenberger G., Koh C.C., Guo T., Röst H.L., Kouvonen P., Collins B.C., et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci. Data. 2014;1 doi: 10.1038/sdata.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yu F., Teo G.C., Kong A.T., Fröhlich K., Li G.X., Demichev V., et al. Analysis of DIA proteomics data using MSFragger-DIA and FragPipe computational platform. Nat. Commun. 2023;14:4154. doi: 10.1038/s41467-023-39869-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.The M., Samaras P., Kuster B., Wilhelm M. Reanalysis of ProteomicsDB using an accurate, sensitive, and scalable false discovery rate estimation approach for protein groups. Mol. Cell. Proteomics. 2022;21 doi: 10.1016/j.mcpro.2022.100437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lazear M.R. Sage: an open-source tool for fast proteomics searching and quantification at scale. J. Proteome Res. 2023;22:3652–3659. doi: 10.1021/acs.jproteome.3c00486. [DOI] [PubMed] [Google Scholar]
- 121.Lin A., Short T., Noble W.S., Keich U. Improving peptide-level mass spectrometry analysis via Double Competition. J. Proteome Res. 2022;21:2412–2420. doi: 10.1021/acs.jproteome.2c00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vaudel M., Burkhart J.M., Breiter D., Zahedi R.P., Sickmann A., Martens L. A complex standard for protein identification, designed by evolution. J. Proteome Res. 2012;11:5065–5071. doi: 10.1021/pr300055q. [DOI] [PubMed] [Google Scholar]
- 123.Granholm V., Navarro J.F., Noble W.S., Käll L. Determining the calibration of confidence estimation procedures for unique peptides in shotgun proteomics. J. Proteomics. 2013;80:123–131. doi: 10.1016/j.jprot.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mahoney D.W., Therneau T.M., Heppelmann C.J., Higgins L., Benson L.M., Zenka R.M., et al. Relative quantification: characterization of bias, variability and fold changes in mass spectrometry data from iTRAQ-labeled peptides. J. Proteome Res. 2011;10:4325–4333. doi: 10.1021/pr2001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bernhard P., Feilen T., Rogg M., Fröhlich K., Cosenza-Contreras M., Hause F., et al. Proteome alterations during clonal isolation of established human pancreatic cancer cell lines. Cell. Mol. Life Sci. 2022;79:561. doi: 10.1007/s00018-022-04584-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Savitski M.M., Mathieson T., Zinn N., Sweetman G., Doce C., Becher I., et al. Measuring and managing ratio compression for accurate iTRAQ/TMT quantification. J. Proteome Res. 2013;12:3586–3598. doi: 10.1021/pr400098r. [DOI] [PubMed] [Google Scholar]
- 127.Ahrné E., Glatter T., Viganò C., von Schubert C., Nigg E.A., Schmidt A. Evaluation and improvement of quantification accuracy in isobaric mass Tag-based protein quantification experiments. J. Proteome Res. 2016;15:2537–2547. doi: 10.1021/acs.jproteome.6b00066. [DOI] [PubMed] [Google Scholar]
- 128.Shteynberg D.D., Deutsch E.W., Campbell D.S., Hoopmann M.R., Kusebauch U., Lee D., et al. PTMProphet: fast and accurate mass modification localization for the Trans-proteomic pipeline. J. Proteome Res. 2019;18:4262–4272. doi: 10.1021/acs.jproteome.9b00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Choi M., Eren-Dogu Z.F., Colangelo C., Cottrell J., Hoopmann M.R., Kapp E.A., et al. ABRF proteome Informatics research group (iPRG) 2015 study: detection of differentially abundant proteins in label-free quantitative LC-MS/MS experiments. J. Proteome Res. 2017;16:945–957. doi: 10.1021/acs.jproteome.6b00881. [DOI] [PubMed] [Google Scholar]
- 130.Kirkpatrick J., Stemmer P.M., Searle B.C., Herring L.E., Martin L., Midha M.K., et al. 2019 association of Biomolecular resource Facilities multi-Laboratory data-independent acquisition proteomics study. J. Biomol. Tech. 2023;34 doi: 10.7171/3fc1f5fe.9b78d780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Navarro P., Kuharev J., Gillet L.C., Bernhardt O.M., MacLean B., Röst H.L., et al. A multicenter study benchmarks software tools for label-free proteome quantification. Nat. Biotechnol. 2016;34:1130–1136. doi: 10.1038/nbt.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Röst H.L., Liu Y., D’Agostino G., Zanella M., Navarro P., Rosenberger G., et al. TRIC: an automated alignment strategy for reproducible protein quantification in targeted proteomics. Nat. Methods. 2016;13:777–783. doi: 10.1038/nmeth.3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gotti C., Roux-Dalvai F., Joly-Beauparlant C., Mangnier L., Leclercq M., Droit A. Extensive and accurate benchmarking of DIA acquisition methods and software tools using a complex proteomic standard. J. Proteome Res. 2021;20:4801–4814. doi: 10.1021/acs.jproteome.1c00490. [DOI] [PubMed] [Google Scholar]
- 134.Choi M., Chang C.-Y., Clough T., Broudy D., Killeen T., MacLean B., et al. MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics. 2014;30:2524–2526. doi: 10.1093/bioinformatics/btu305. [DOI] [PubMed] [Google Scholar]
- 135.Lou R., Cao Y., Li S., Lang X., Li Y., Zhang Y., et al. Benchmarking commonly used software suites and analysis workflows for DIA proteomics and phosphoproteomics. Nat. Commun. 2023;14:94. doi: 10.1038/s41467-022-35740-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kitata R.B., Choong W.-K., Tsai C.-F., Lin P.-Y., Chen B.-S., Chang Y.-C., et al. A data-independent acquisition-based global phosphoproteomics system enables deep profiling. Nat. Commun. 2021;12:2539. doi: 10.1038/s41467-021-22759-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pin E., Stratton S., Belluco C., Liotta L., Nagle R., Hodge K.A., et al. A pilot study exploring the molecular architecture of the tumor microenvironment in human prostate cancer using laser capture microdissection and reverse phase protein microarray. Mol. Oncol. 2016;10:1585–1594. doi: 10.1016/j.molonc.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Longuespée R., Alberts D., Pottier C., Smargiasso N., Mazzucchelli G., Baiwir D., et al. A laser microdissection-based workflow for FFPE tissue microproteomics: important considerations for small sample processing. Methods. 2016;104:154–162. doi: 10.1016/j.ymeth.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 139.Patel V., Hood B.L., Molinolo A.A., Lee N.H., Conrads T.P., Braisted J.C., et al. Proteomic analysis of laser-captured paraffin-embedded tissues: a molecular portrait of head and neck cancer progression. Clin. Cancer Res. 2008;14:1002–1014. doi: 10.1158/1078-0432.CCR-07-1497. [DOI] [PubMed] [Google Scholar]
- 140.Gebreyesus S.T., Siyal A.A., Kitata R.B., Chen E.S.-W., Enkhbayar B., Angata T., et al. Streamlined single-cell proteomics by an integrated microfluidic chip and data-independent acquisition mass spectrometry. Nat. Commun. 2022;13:37. doi: 10.1038/s41467-021-27778-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Petrosius V., Aragon-Fernandez P., Üresin N., Kovacs G., Phlairaharn T., Furtwängler B., et al. Exploration of cell state heterogeneity using single-cell proteomics through sensitivity-tailored data-independent acquisition. Nat. Commun. 2023;14:5910. doi: 10.1038/s41467-023-41602-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Huffman R.G., Leduc A., Wichmann C., Di Gioia M., Borriello F., Specht H., et al. Prioritized mass spectrometry increases the depth, sensitivity and data completeness of single-cell proteomics. Nat. Methods. 2023;20:714–722. doi: 10.1038/s41592-023-01830-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang Y., Lih T.-S.M., Chen L., Xu Y., Kuczler M.D., Cao L., et al. Optimized data-independent acquisition approach for proteomic analysis at single-cell level. Clin. Proteomics. 2022;19:24. doi: 10.1186/s12014-022-09359-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Blank-Landeshammer B., Richard V.R., Mitsa G., Marques M., LeBlanc A., Kollipara L., et al. Proteogenomics of colorectal cancer liver metastases: complementing precision oncology with phenotypic data. Cancers. 2019;11:1907. doi: 10.3390/cancers11121907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lehtiö J., Arslan T., Siavelis I., Pan Y., Socciarelli F., Berkovska O., et al. Proteogenomics of non-small cell lung cancer reveals molecular subtypes associated with specific therapeutic targets and immune evasion mechanisms. Nat. Cancer. 2021;2:1224–1242. doi: 10.1038/s43018-021-00259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ellis M.J., Gillette M., Carr S.A., Paulovich A.G., Smith R.D., Rodland K.K., et al. Connecting genomic alterations to cancer biology with proteomics: the NCI clinical proteomic tumor analysis Consortium. Cancer Discov. 2013;3:1108–1112. doi: 10.1158/2159-8290.CD-13-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nesvizhskii A.I. Proteogenomics: concepts, applications and computational strategies. Nat. Methods. 2014;11:1114–1125. doi: 10.1038/nmeth.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mertins P., Mani D.R., Ruggles K.V., Gillette M.A., Clauser K.R., Wang P., et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pak H., Michaux J., Huber F., Chong C., Stevenson B.J., Müller M., et al. Sensitive immunopeptidomics by leveraging available large-scale multi-HLA spectral libraries, data-independent acquisition, and MS/MS prediction. Mol. Cell. Proteomics. 2021;20 doi: 10.1016/j.mcpro.2021.100080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shahbazy M., Ramarathinam S.H., Illing P.T., Jappe E.C., Faridi P., Croft N.P., et al. Benchmarking bioinformatics pipelines in data-independent acquisition mass spectrometry for immunopeptidomics. Mol. Cell. Proteomics. 2023;22 doi: 10.1016/j.mcpro.2023.100515. [DOI] [PMC free article] [PubMed] [Google Scholar]