

Abstract
Background and Objectives
De novo gain-of-function variants in the CACNA1D gene, encoding the L-type voltage-gated Ca2+ channel CaV1.3, cause a multifaceted syndrome. Patients show variable degrees of autism spectrum disorder, developmental delay, epilepsy, and other neurologic and endocrine abnormalities (primary aldosteronism and/or hyperinsulinemic hypoglycemia). We study here a novel variant [c.3506G>A, NM_000720.4, p.(G1169D)] in 2 children with the same CACNA1D mutation but different disease severity.
Methods
The clinical data of the study patients were collected. After molecular analysis and cloning by site-directed mutagenesis, patch-clamp recordings of transfected tsA201 cells were conducted in whole-cell configuration. The functional effects of wild-type and mutated channels were analyzed.
Results
One child is a severely affected boy with a novel de novo CACNA1D variant with additional clinical symptoms including prenatal-onset tremor, congenital respiratory insufficiency requiring continuous positive airway pressure ventilation, and sensorineural deafness. Despite episodes of hypoglycemia, insulin levels were normal. Aldosterone:renin ratios as a screening parameter for primary aldosteronism were variable. In the second patient, putative mosaicism of the p.(G1169D) variant was associated with a less severe phenotype. Patch-clamp electrophysiology of the p.(G1169D) variant in a heterologous expression system revealed pronounced activity-enhancing gating changes, including a shift of channel activation and inactivation to more hyperpolarized potentials, as well as impaired channel inactivation and deactivation. Despite retained sensitivity to the Ca2+ channel blocker isradipine in vitro, no beneficial effects of isradipine or nifedipine treatment were observed in the index case.
Discussion
Through this report, we expand the knowledge about the disease presentation in patients with CACNA1D variants and show the novel variant's modulatory effects on CaV1.3 gating.
Introduction
CACNA1D encodes the α-1D subunit of the voltage-dependent L-type calcium channel CaV1.3. It is a member of a family of Ca2+ channels that are sensitive to clinically approved Ca2+ channel blockers, such as the dihydropyridines (DHPs) nifedipine and isradipine. Ca2+ influx through CaV1.3 in neurons, cochlear hair cells, sinoatrial node, and endocrine cells is essential for neuronal development, different types of learning and memory, hearing, cardiac pacemaking, and insulin and aldosterone secretion.1,2
In line with this widespread expression, sequence variations in the CACNA1D gene have been linked to several human pathologies.3 Biallelic loss-of-function variants cause autosomal recessive sinoatrial node dysfunction and deafness.4 By contrast, de novo heterozygous CACNA1D mutations have been reported in patients with a neurodevelopmental phenotype of variable severity.2,5 These variants induce characteristic gating changes and enable increased Ca2+ influx through the mutant channel, e.g., by shifting the activation curve to less depolarized potentials.2,6,7 Apart from developmental delay and autism spectrum disorder, neurologic symptoms such as cerebral palsy mimicry, epilepsy, and muscular hypotonia have been reported, and some patients show endocrine abnormalities (hyperinsulinemic hypoglycemia and/or primary aldosteronism).6,8-11 A rather severe clinical manifestation has been described as PASNA (primary aldosteronism, seizures, and neurologic abnormalities) syndrome.5,6 2 recently published knock-in mouse models expressing such gating-modifying CACNA1D variants recapitulate various aspects of the human phenotype.12,13
In this study, we report a severely affected boy with a novel de novo CACNA1D variant extending the previously described phenotype. We also describe a second patient with presumed mosaicism for the same variant and a less severe clinical phenotype. We perform patch-clamp studies of the mutant CaV1.3 channel and demonstrate its modified gating properties.
Methods
Study Patients
Written informed consent to publish this article was obtained from the parents of both cases. The study adhered to the World Health Association Declaration of Helsinki (2013) and was approved by the local ethics committee of the Charité - Universitätsmedizin Berlin, Germany (approval no. EA2/084/18).
Genetic Analysis
Index Patient
Blood samples were collected from the index patient and both parents. DNA extraction and clinical whole-exome sequencing were performed as routine genetic diagnostics at the Praxis für Humangenetik/CeGaT GmbH Tübingen, Germany.
Patient With Presumed Mosaicism
Blood samples were collected from the patient and both parents. Whole-exome sequencing (KAPA HyperExome (Roche), 2x 150 bp paired-end Illumina sequencing, RoDa5 pipeline (VUmc), Alissa Interpret v5 (VUMC), GRCh37/hg19) was performed by the Laboratory of Genome Diagnostics, Department of Human Genetics, Amsterdam UMC, University of Amsterdam. >30× coverage on target was >94%.
Molecular Cloning
Site-directed mutagenesis of the canonical isoform of human CACNA1D in the pCMV6-XL6 plasmid was performed, and coding sequences were validated as previously published.6
Heterologous Expression in tsA201 Cells
CaV1.3 channel complexes were transiently transfected into tsA201 cells (ECACC: 96121229, passage number ≤19) as previously described.8,14 cDNA constructs were as follows: 3 µg of α1 (human, NM_000720, WT, or p.(G1169D) mutant), 2 µg of β3 (rat, NM_012828), 2.5 µg of α2δ1 subunits (rabbit, NM_001082276), and 1.5 µg of EGFP as the transfection marker.8
Electrophysiologic Recordings
Ca2+ currents were recorded 48–72 hours after transfection in the whole-cell patch-clamp configuration using the Axopatch 200A amplifier (Axon Instruments) and analyzed as described.8,14 Bath solution included the following (in mM): 15 CaCl2, 150 choline-Cl, 10 HEPES, and 1 MgCl2 (pH 7.3 with CsOH).8 Pipette solution included the following (in mM): 135 CsCl, 1 MgCl2, 10 HEPES, 10 Cs-EGTA, and 4 ATP-Na2 (pH 7.3 with CsOH).8 All voltages were corrected for the liquid junction potential of −9.3 mV.15
The holding potential was set to −109.3 mV or −139.3 mV. The voltage dependence of Ca2+ current activation (30-ms pulses to various test potentials, Δ 5 mV), steady-state inactivation (ratio of 2 20-ms long pulses to the voltage of maximal activation Vmax, separated by a 5-second conditioning pulse to various test potentials, Δ 10 mV, sweep start-to-start interval 30 seconds), and inactivation kinetics (5-second pulse to Vmax) were determined and analyzed as previously described.8,14 Current densities were calculated by normalizing inward currents to the cell size (pF).8 Window currents were derived by multiplying the mean current density with the normalized steady-state inactivation for the respective potential.8,14 Deactivation was measured after a 30-ms depolarization to the reversal potential, followed by 40-ms long repolarizations to various test potentials (Δ 10 mV). Owing to the slow p.(G1169D) tail current kinetics, no deactivation time constants could be calculated.8 Instead, tail currents at each repolarization potential were normalized to the peak tail current of the given sweep and integrated over the 40 ms of repolarization.8
Pharmacology
Pharmacologic experiments were performed as previously described.8 In brief, cells from 3 independent transfections were depolarized using 100-ms square pulses to Vmax (0.1 Hz, holding potential: −89.3 mV) and constantly perfused with a flow rate of ∼350 μL/mL with bath solution with or without isradipine (Sigma Aldrich, D8418). Stocks were prepared in DMSO, kept at −20°C, and diluted freshly 1:1000 in bath solution every day. After at least 3 constant control sweeps during bath solution perfusion, cumulative inhibition by 30 nM, 100 nM, and 3 µM (for complete inhibition) of isradipine was quantified and corrected for linear current decay (“run-down”) measured in individual control cells (recordings with bath solution only, using the same test tubes that were later used for drug administration).8
Statistics
Statistical significance was computed with unpaired Student t tests, 2-way ANOVA with Šídák multiple comparison post hoc analysis, or multiple Mann-Whitney tests (GraphPad Prism 9 software) as indicated.8 Data are given as mean ± SEM; statistical significance was set at p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
Results
Clinical Findings
The index patient (patient 1) with variant p.(G1169D) was conceived through in vitro fertilization by healthy, nonconsanguineous parents of Turkish descent. There was no family history of neurodevelopmental disease. In the second trimester, the Hadlock method–estimated fetal weight was 631 g (95th centile) and macrocephaly (head circumference of 220.9 mm, 98th centile) was diagnosed. The mother was treated with sertraline for depressive symptoms in late pregnancy until birth. In the 3rd trimester, the mother noted fetal tremor, which was confirmed by ultrasound. The boy was born at term by cesarean section in breech position (Apgar 8/8/8, arterial umbilical cord pH 7.21). He was born large for gestational age (birth weight 4,390 g, >99th centile). He was admitted to an intensive care unit (ICU) because of respiratory insufficiency and required continuous positive airway pressure (CPAP, high-flow nasal cannula during sleep) support for the following 24 months because of severe tremor of the diaphragm, central apnea, and muscular hypotonia. Extensive workup revealed no other cause of the respiratory insufficiency. The patient showed tongue, limb, and facial tremor. A unilateral humeral fracture was discovered immediately after birth and confirmed by sonography and X-ray, without signs of primary bone pathology. Dysmorphic features included a broad forehead, low-set ears, midface hypoplasia, microstomia, adducted hands and feet, and distal leg hypotrophy. He required gastric tube feedings and IV glucose treatment because of poor feeding and recurrent episodes of hypoglycemia.
Transcranial ultrasound and spinal ultrasound in the first week of life were normal. Cranial MRI on the 11th day of life revealed acute small subarachnoid hemorrhages in the left occipital lobe and parafalcine region. Visually evoked potentials revealed severely impaired conductance bilaterally. No brainstem-evoked response audiometry potentials were recordable, and cochlear implants were placed at 12 months of age. Nerve conduction and somatosensory-evoked potential results were normal. The boy had mild pulmonary hypertension, mild right ventricular hypertrophy, a patent foramen ovale, and a QTc time of 437 ms (normal <440 ms). No electrolyte imbalances were present in the first months of life. The neonatal systolic blood pressure was between 5th and 10th centiles. Abdominal ultrasound revealed undescended testicles, but no signs of adrenal hyperplasia or other abnormalities. Elevated l-Dopa levels were detected in CSF. Otherwise, results of routine CSF, blood, and urine analysis including human cytomegalovirus screening, homocysteine, amino acids, carnitine, acylcarnitine, biogenic amines, fT3, fT4, cathepsin D activity, vitamin profile, ionized Ca2+, protein-bound Ca2+, calcidiol chromosomal analysis, and array comparative genomic hybridization were normal.
Tremors were triggered by acoustical and tactile stimuli. However, hyperekplexia was ruled out because muscle hypertonia, startle reaction, and myoclonus were absent. Amplitude-integrated EEG in the first week of life showed no signs of seizures or burst suppression patterns. Because no event-related EEG changes were noted, even during episodes of intensified tremor, epilepsy was largely ruled out as cause of the tremor. Follow-up EEGs showed continuous slowing; however, background activity was almost age-appropriate. In the third month of life, the boy's parents reported increased frequencies of apnea and cyanotic episodes. Antiseizure medication with levetiracetam (30 mg/kg/d) was started but terminated after 8 weeks because of a lack of effect. Tremors, central apnea with oxygen desaturations as low as 70%, dysphagia with breathing-swallowing discoordination, and muscular hypotonia persisted throughout the first 2 years of life. Caffeine treatment initiated postnatally to prevent apnea was sustained until 10 months of age.
A SARS-CoV-2 infection at the age of 19 months was life-threatening and resulted in an interstitial pneumonia that required noninvasive positive pressure ventilation (NIV), followed by 5 weeks of invasive ventilation on the ICU and again NIV. EEGs recorded during the ICU stay revealed an abnormal background activity, burst suppression patterns, and persistent sharp waves predominantly from the left parietal and right frontal region, consistent with a diagnosis of epilepsy and epileptic encephalopathy. Antiseizure medication with levetiracetam (mg/kg/d) and clobazam (mg/kg/d) was started. Oxybutynin (0.07 mg/kg/d) and intermittent catheterization were initiated for the neurogenic bladder. Intraglandular botulinum toxin injections were performed for hypersalivation.
After the boy's breathing parameters improved, he was transferred to a rehabilitation center with an endotracheal cannula. In the first weeks after ICU treatment, he received CPAP support during his sleep for 10 hours daily, which could be terminated during the 5 months of rehabilitation. Parents reported paroxysmal events that followed a fixed sequence: Initially, the boy's eyes were wide open and deviated to the right side, breathing and heart rate increased, and legs and sometimes arms were repeatedly extended for a second. The sequences typically ended with agitated breathing and repeated sneezing and were followed by 30–60 minutes of restlessness. These symptoms were attributed to the genetic epilepsy, and antiseizure medication was maintained.
Plasma levels of aldosterone and renin and aldosterone:renin ratios were normal at ages 7 months and 26 months. However, renin was low despite diuretic therapy (3.54 ng/L, normal range 6.30–149) once at age 24 months, with normal aldosterone (255 ng/L, normal range 52–290), but a high aldosterone:renin ratio (72.0, normal <20) as a potential sign of primary aldosteronism. During the first and second year of life, systolic and diastolic arterial blood pressure remained normal. Although the boy experienced repeated episodes of hypoglycemia throughout his first year of life, plasma insulin, plasma C-peptide, and urinary C-peptide levels were always normal.
At the last follow-up at the age of 46 months, severe global developmental delay was still present, with no visual fixation and no responsive smile to the child's primary caregivers.
Owing to inconclusive endocrinologic results, we diagnosed our patient with incomplete PASNA syndrome.
Patient 2 with the p.(G1169D) variant and MAF of 17%: We identified one additional case with the identical variant in the Leiden Open Variation Database (LOVD, DB-ID CACNA1D_000160). Consultation with the submitters revealed a 10-month-old boy, the first child of healthy, nonconsanguineous parents of Dutch descent. The mother was treated with methyldopa for pregnancy-induced hypertension. The maternal glucose tolerance was normal. Fetal macrosomia was suspected during pregnancy. A cesarean section was performed at 34 weeks of gestational age because of reduced variability on cardiotocography, reduced fetal movements, and reversed flow in the umbilical artery. Apgar scores were 9 and 10 after 1 and 5 minutes, respectively. The birth weight was 3,865 g (SDS +3.22), and the occipital-frontal circumference was 36 cm (SDS +2,38). The boy was admitted to the neonatal ICU at Amsterdam University Medical Center because of congenital hyperinsulinemic hypoglycemia (insulin 224 pmol/L, normal range 12–96 pmol/L). A cardiac ultrasound demonstrated biventricular hypertrophy, with the left ventricle being more affected. Furthermore, persistent pulmonary hypertension and hepatomegaly were diagnosed. During admission, tremors not related to hypoglycemia were initially noted. There were no convulsions. At first, a brain MRI scan suggested impaired maturation of gyri. A follow-up MRI scan indicated further maturation of the brain, but an old intraventricular hemorrhage grade II, and white matter injury due to prematurity. Except for vertical creases behind the ears, no other dysmorphic features were identified. The boy was treated with continuous feeding, IV glucose infusions, and initially diazoxide and hydrochlorothiazide. Because of the lack of effect and development of side effects, including persistent pulmonary hypertension, diazoxide treatment was replaced by lanreotide and octreotide. Plasma levels of aldosterone and renin and the aldosterone:renin ratio were normal at the age of 2 months. The boy was discharged from hospital at the age of 2 months. At 10 months, blood glucose levels were stable with once-monthly injection of lanreotide and tube feeding. Motor development was slightly delayed because of muscular hypotonia affecting the trunk.
Genetic Analysis
Patient 1
Whole-exome sequencing revealed a variant in the CACNA1D gene [c.3506G>A, NM_000720.4, p.(G1169D)] (Figure 1) that was absent in both parents. No other clinically relevant variants that could account for the phenotype were detected. This variant is not present in ClinVar or gnomAD but has been reported once as a variant of unknown significance in the LOVD (mentioned later). It affects an evolutionary highly conserved glycine at the cytoplasmic interface of helix 6 within repeat III in the α1-subunits of the CaV1 and CaV3 family. Variant p.(G1169D) is predicted to be pathogenic by Mutation Taster, fathmm, Mutation Assessor, SIFT, fathmm-MKL coding, LRT, and PROVEAN. Clinically, it was classified as pathogenic and disease causing.
Figure 1. Cav1.3 Structure and Position of Identified De Novo Mutations.

CACNA1D encodes the pore-forming CaV1.3 α1 subunit. Each of its 4 homologous repeats comprises 6 transmembrane helices. Helices S5 and S6 and their connecting loops of each repeat form the Ca2+-conducting ion pore, and helices S1-S4 the voltage-sensing domain. The auxiliary subunits α2δ and β support channel function and membrane targeting. p.(G1169D) found in the patients presented here is highlighted (G1169D) along with previously identified de novo pathogenic CACNA1D germline variants implicated in neurodevelopmental disease.2
Patient 2
De novo variants (dominant model) and hemi/homozygous or compound heterozygous variants (recessive model) in the entire exome were analyzed. The CACNA1D variant [c.3506G > A, NM_000720.4, p.(G1169D)] was detected in 17% of the reads. No additional genetic variants explaining the observed phenotype were identified. This variant could not be identified in parents, suggesting mosaicism because of a postzygotic de novo mutation.
Biophysical and Pharmacologic Characterization of p.(G1169D)-Containing Human CaV1.3 Ca2+ Channels
To functionally characterize the p.(G1169D) variant, we transiently transfected human kidney tsA201 cells with wild-type (WT) or mutated p.(G1169D) CaV1.3 channels and performed whole-cell patch-clamp recordings (Figure 2). The p.(G1169D) variant significantly shifted the voltage dependence of activation and steady-state inactivation by ∼35 mV toward more negative potentials (Figure 2A, eTables 1 and 2). This enabled a significantly increased constant background Ca2+ influx at subthreshold potentials (“window current”, Figure 2B). This leads to an expanded voltage range in which activated channels do not inactivate completely. During a prolonged (5 seconds) depolarization to Vmax, the p.(G1169D) variant displayed significantly slower inactivation kinetics (Figure 2C). Deactivation kinetics (reporting closure of open channels after repolarization) were also significantly slowed by the p.(G1169D) variant (Figure 2, D and E). Together, these biophysical changes strongly suggest that enhanced channel activity underlies the observed phenotype. Because DHPs were chosen as a treatment option for the index patient (mentioned in the next section), we tested whether the p.(G1169D) mutation affects the sensitivity to the DHP isradipine, as previously described for other pathogenic variants.8,12,14 No statistically significant difference in isradipine inhibition was observed between WT and p.(G1169D)-mutated CaV1.3 Ca2+ channels at 2 concentrations tested (30 and 100 nM). Comparable isradipine potency suggests that p.(G1169D)-containing channels retain full sensitivity to DHPs (Figure 2F).
Figure 2. Biophysical and Pharmacologic Characterization of p.(G1169D)-Containing Human CaV1.3 Ca2+ Channels.
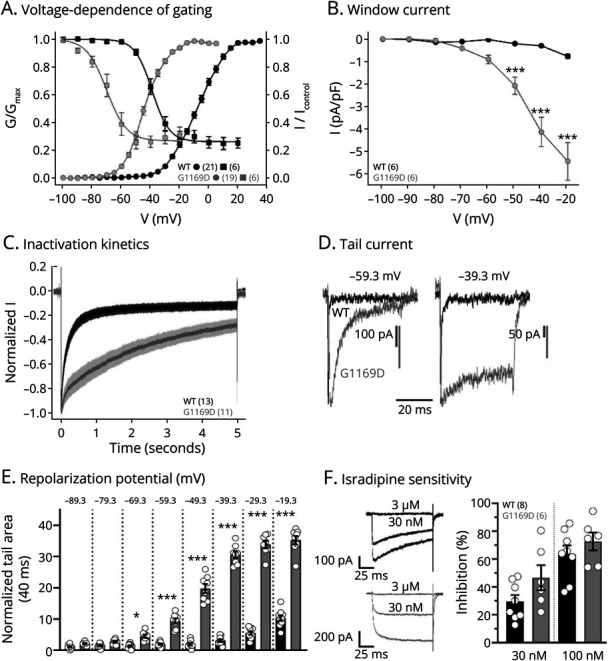
Whole-cell patch-clamp recordings in tsA201 cells of transiently transfected wild-type (WT, black) or p.(G1169D) (grey/G1169D) CaV1.3 Ca2+ channel complexes (coexpressed with β3 and α2δ1, 15 mM Ca2+). Data were measured using a holding potential of −109.3 mV (A–E) and are given as mean ± SEM for the indicated number of recordings. Parameters and statistics are provided in eTables 1–3. (A) The p.(G1169D) variant shifted the voltage dependence of activation (G-V curve, circles) and steady-state inactivation (squares) by ∼35 mV toward more negative potentials. (B) This enabled a significantly increased constant background Ca2+ influx (“window current”) at subthreshold potentials. Two-way ANOVA (interaction, potential, genotype: p < 0.001) with the Šídák multiple comparison post hoc test as indicated. (C) During a prolonged (5 s) depolarization to the voltage of maximal activation (Vmax), the p.(G1169D) variant displayed slower inactivation kinetics. Averaged current traces (mean ± SEM) are shown. (D–E) Deactivation kinetics of the p.(G1169D) variant were significantly slower, as shown by representative traces of tail currents elicited by 40-ms long repolarization from the reversal potential (Vrev) to −59.3 mV or −39.3 mV (D) and quantification of the normalized tail current area for all tested repolarization potentials (details on normalization in Methods). Two-way ANOVA (interaction, repolarization potential, genotype: p < 0.001) with the Šídák multiple comparison post hoc test as indicated. (F) Sensitivity to the L-type Ca2+ channel inhibitor isradipine (30 and 100 nM) was evaluated during 100-ms long depolarizations to the Vmax (holding potential −89.3 mV, 0.1 Hz, 3 independent transfections). Normalized representative current traces for 30 nM and subsequent full block with 3-µM isradipine are shown (left, tail currents were cut). Inhibition was corrected for current run-down (details in Methods) and is presented as means ± SEM (right). No statistically significant difference in isradipine inhibition was observed between WT and p.(G1169D) CaV1.3 Ca2+ channels (two-way ANOVA). This provides class IV evidence. It is an observational study without controls.
Therapy With DHP Ca2+ Channel Inhibitors
Because clinical symptoms and the overall severe phenotype of the index patient (patient 1) fit well within the CACNA1D variant spectrum5 and the observed gating changes of the mutant channel cause enhanced activity, off-label treatment with oral nifedipine was initiated at the age of 4 months. This DHP was selected because of a previous report indicating symptom improvement in a 6-month-old child harboring the L271H CACNA1D variant and the drug availability as oral immediate-release formulation, allowing for weight-adjusted dosing through a nasogastric tube. Treatment was initiated as an inpatient at 0.5 mg/kg/d divided into 2 doses every 12 hours, which was gradually increased to 3 mg/kg/d over 5 days. Treatment was terminated after 2 weeks for severe arterial hypotension and reflex tachycardia. Despite dose-limiting side effects, there was no obvious improvement of the tremor or other clinical parameters. A second treatment attempt with oral nifedipine was initiated at the age of 22 months as an inpatient, administered continuously with a nasogastric tube through a syringe driver with a much lower starting dose of 0.015 mg/kg/d. No cardiovascular side effects were observed. Treatment was discontinued after 2 days because of a transient rise in liver enzymes (alanine aminotransferase and aspartate aminotransferase). Analysis of archived blood samples, however, showed that this rise had already started before nifedipine treatment. Alanine aminotransferase levels reached a 46-fold increase on the fifth day after discontinuation of nifedipine. 2 months later, liver enzyme levels were normal again. Glutamate dehydrogenase and bilirubin levels were never elevated during this episode. The parents opted not to reinitiate nifedipine.
At the age of 38 months, an oral treatment attempt with the DHP isradipine was initiated, with a dosage titration in the pediatric ICU. Initially, oral isradipine in an extended-release formulation (Vascal Uno) suitable for weight-adjusted dosing was administered at a dose of 0.02 mg/kg/d in 2 doses every 12 hours through a nasogastric tube. This corresponds to approximately one-third of the weight-adjusted daily adult dose. The dosage was gradually increased to 0.06 mg/kg/d within 6 days and maintained at this level for 3 months. Three days after reaching the target dose, the patient was discharged home. During the course of the treatment, no reduction in tremors or seizure frequency was observed. Temporarily, the parents noted an increased alertness in their child. Owing to the lack of identifiable effectiveness of the medication, the medication was gradually tapered after 3 months. Because the parents had the impression of an increase in seizure frequency on discontinuation of isradipine, medication was continued at 0.035 mg/kg/d at parental request and terminated after 5 months at the conclusion of the observation period. Overall, a clinically quantifiable reduction in seizure frequency until the 46th month of life could not be confirmed. There was no evidence that tremors or other symptoms improved under isradipine therapy. Long-term treatment was well tolerated, liver enzymes did not rise, and no cardiovascular side effects were observed.
Discussion
Our genetic, clinical, and biophysical findings confirm and extend the disease spectrum reported for de novo missense CACNA1D variants. Our findings allow the classification of the p.(G1169D) variant as pathogenic (strong evidence according to ACMG16) and suggest that treatment with DHP Ca2+ channel blockers nifedipine and isradipine at the doses of maximum 3 mg/kg/d and 0.06 mg/kg/d, respectively, does not significantly ameliorate neurologic symptoms in patients with this variant.
Our index patient (patient 1) was a boy harboring the de novo CACNA1D variant p.(G1169D) with the clinically remarkable presentation of a prenatal-onset severe tremor. Maternal sertraline therapy was initially suspected as a cause, but low sertraline serum levels after birth, persistence of tremor, and the presence of trembling in 2 other cases with de novo CACNA1D mutation and in a CACNA1D knock-in mouse model all support a genetic origin.10,13,14 Patient 1 suffered from seizures and other severe neurologic symptoms. Routine plasma aldosterone and renin measurements were inconclusive regarding the presence of primary aldosteronism, in part also due to interfering medication. Although hypoglycemic episodes were noted, plasma insulin and C-peptide levels and urinary C-peptide levels were normal. It was pointed out that most de novo dominant variants cause no endocrine pathologies, and therefore, hyperinsulinism and hyperaldosteronism are not considered as obligatory criteria for the disease spectrum observed with de novo CACNA1D missense variants.2
The presentation of patients with de novo CACNA1D variants is highly variable, ranging from severe cases described here to less affected cases with autism spectrum disorder but no other neurologic and endocrine symptoms.2,6,14 Mutation-induced gating changes, i.e., the degree to which inactivation is affected or voltage-dependent gating is shifted, differ between variants.2 Whether the variability in clinical presentation is due to these functional differences in the causative CACNA1D variants remains to be determined. Moreover, improvement of hyperinsulinism and hyperaldosteronism with age has been reported.2
There is uncertainty whether the index patient's deafness is attributable to the CACNA1D variant because hearing impairments have so far not been described in association with gain-of-function variants.2,5 Current evidence suggests that deafness is associated with a homozygous loss of channel function.4 However, the possibility that de novo (heterozygous) variants with gating changes as described for p.(G1169D) can also cause deafness (e.g., by causing Ca2+ toxicity and cell loss of cochlear hair cells) cannot be excluded.
Another potential explanation for variable severity of the disease is genetic mosaicism due to postzygotic mutation. The low MAF in patient 2 points to mosaicism. Despite sharing key symptoms (including preterm birth, postnatal tremor, postnatal episodes of hypoglycemia, pulmonary hypertension, and developmental delay), the clinical presentation of patient 2 was milder. Similarly, variability of the clinical symptoms due to genetic mosaicism has been discussed for Timothy syndrome caused by pathogenic CACNA1C (CaV1.2) variants.17
Neuronal CaV1.3 channels are key regulators of cellular excitability, dendritic spine morphology, and excitation-transcription coupling.1,18 However, the exact mechanisms underlying neurologic impairment associated with de novo CACNA1D variants remain to be determined. Increased l-Dopa levels in the CSF of our index case indicate a role of CaV1.3 in dopaminergic signaling. Owing to their relatively hyperpolarized activation threshold, CaV1.3 channels can contribute to the oscillatory activity in mesencephalic dopaminergic neurons and the excitability of striatal medium spiny neurons.1,2,19-22 Abnormal CaV1.3 activity may, therefore, cause a dysfunction within the dopamine midbrain system contributing to some of the symptoms. This is in line with the observed changes in the excitability of these 2 neuron populations within the dopamine midbrain system in a mouse model expressing the pathogenic A769G CACNA1D variant (found in a patient with autism spectrum disorder and intellectual disability).12
The p.(G1169D) variant in our index patient exhibited characteristic gating changes that enable enhanced channel activity (Figure 2). This includes impaired inactivation and a pronounced shift of the voltage dependence of activation to more hyperpolarized potentials (−70 to −50 mV, close to the neuronal resting potential). These gating changes are typical for pathogenic CACNA1D variants associated with neurodevelopmental disease, thereby confirming the pathogenicity of this variant. Notably, the observed gating changes are more pronounced than in other so far characterized variants.2 One other recently described variant (Figure 1), F767S, also exhibits a similar electrophysiologic phenotype14 including a slightly less pronounced hyperpolarizing shift in voltage-dependent activation and inactivation, slowed inactivation, and deactivation kinetics and an increase in the window currents.14 Intriguingly, this variant also displays a motor phenotype referred to as “jittering.”14 In line with the human phenotype, tremors were also observed in a mouse model carrying the I772M mutation.13 Therefore, it is possible that quantitatively larger changes of voltage-dependent channel gating associate with more severe motor disturbances such as jittering or (intrauterine) tremors. p.(G1169D) is located at the cytoplasmic interface of helix 6 within repeat III and is part of a conserved, functional motif (“G/A/G/A ring”23) that forms part of the activation gate and is coupled to the voltage-sensing domain of the channel.23 Cav1.3 variants affecting other residues within this motif, i.e., p.(G403) in domain I and p.(A749) in domain II, have been shown to alter channel function and induce disease: p.(G403D): PASNA6; p.(A749G/T): neurodevelopmental disease phenotype.2,24 Similarly, mutations of corresponding residues of Cav1.2 cause Timothy syndrome [p.(G402S) in domain I,23 p.(A1473) in domain IV,25 residues numbered according to NM_000719.7] or alter channel gating,26 for p.(G1163) in domain III [ = Cav1.3 p.(G1169)] even with a similarly, strong shift in the voltage dependence of activation.26,27
Furthermore, aldosterone-producing adenomas show several somatic mutations near p.(G1169D), highlighting that this region has functional relevance.2 The gating changes observed with p.(G1169D) have implications for the function of neurons and hormone-producing cells, especially for pacemaker neurons that are active around potentials of −50 to −70 mV.28 In pacemaker neurons, Ca2+ conductance is often voltage-critical and time-critical; thus, even small changes can have significant implications for bursting rhythms and overall pacemaker activity.29 The shift of the voltage dependence of gating to more negative potentials (Figure 2A) leads to a large increase in the so-called window current at negative voltages (Figure 2B). They can, therefore, contribute to constant background Ca2+ influx at potentials at which conductivity in WT is much smaller or absent. Impaired inactivation and deactivation further contribute to gain-of-channel function.
Our study provides a detailed clinical assessment of the therapeutic effects of CNS-permeable DHP Ca2+ channel inhibitors on the neurologic phenotype in a patient with CACNA1D. No major effects of nifedipine therapy in the index patient were observed, with the known hypotensive treatment-limiting adverse drug reactions. We also used the highly potent DHP isradipine, given its even better brain permeation in preclinical studies compared with nifedipine and, therefore, potentially better treatment of CNS symptoms in patients with CACNA1D mutations.30 Although 1 therapeutic trial with immediate-release isradipine had to be stopped early in another patient affected by a high-risk CACNA1D variant because of side effects,2 our long-term treatment regimen with an extended-release formulation was well tolerated. However, no clinically meaningful effects on seizure frequency, severity of tremors, or alertness could be observed.
Taken together, our data extend the disease spectrum reported for de novo missense CACNA1D variants and provide evidence that treatment with DHP Ca2+ channel blockers does not ameliorate neurologic symptoms in patients with the newly described p.(G1169D) variant.
Acknowledgment
The authors thank our patient and his family for taking part in this study and the physicians and therapists involved in his care. Parts of this article (content, graphs, and tables) have also been used and published in the MD thesis of the first author, Fabian Dannenberg.
Glossary
- CPAP
continuous positive airway pressure
- DHP
dihydropyridine
- ICU
intensive care unit
- WT
wild-type
Appendix. Authors
| Name | Location | Contribution |
| Fabian Dannenberg | Department of Pediatric Neurology; Center for Chronically Sick Children, Charité-Universitätsmedizin Berlin, Germany | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
| Arpad Von Moers, MD, PD | Department of Pediatrics, DRK Kliniken Berlin Westend, Berlin, Germany | Acquisition of data; analysis or interpretation of data |
| Petra Bittigau, MD, PD | Department of Pediatric Neurology; Center for Chronically Sick Children, Charité-Universitätsmedizin Berlin, Germany | Drafting/revision of the manuscript for content, including medical writing for content |
| Jörn Lange, MD | Department of Neuropediatrics, VAMED Klinik Hohenstücken, Brandenburg an der Havel, Germany | Analysis or interpretation of data |
| Sylvia Wiegand, MD | Department of Neuropediatrics, VAMED Klinik Hohenstücken, Brandenburg an der Havel, Germany | Analysis or interpretation of data |
| Ferenc Török, PhD | Department of Pharmacology and Toxicology, Institute of Pharmacy, Center for Molecular Biosciences Innsbruck, University of Innsbruck, Austria | Major role in the acquisition of data; analysis or interpretation of data |
| Gabriel Stölting, MD | Center of Functional Genomics, Berlin Institute of Health at Charité - Universitätsmedizin Berlin, Germany | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Jörg Striessnig, MD, Prof. | Department of Pharmacology and Toxicology, Institute of Pharmacy, Center for Molecular Biosciences Innsbruck, University of Innsbruck, Austria | Drafting/revision of the manuscript for content, including medical writing for content; study concept or design; analysis or interpretation of data |
| M. Mahdi Motazacker, MD, PhD | Department of Human Genetics, Amsterdam UMC, University of Amsterdam, The Netherlands | Major role in the acquisition of data |
| Marjoleine F. Broekema, MD, PhD | Department of Human Genetics, Amsterdam UMC, University of Amsterdam, The Netherlands | Major role in the acquisition of data |
| Markus Schuelke, MD, Prof. | Department of Pediatric Neurology; Center for Chronically Sick Children, Charité-Universitätsmedizin Berlin, Germany | Analysis or interpretation of data |
| Angela M. Kaindl, MD, Prof. | Department of Pediatric Neurology; Center for Chronically Sick Children; Institute for Cell Biology and Neurobiology, Charité-Universitätsmedizin Berlin, Germany | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Ute I. Scholl, MD, Prof. | Center of Functional Genomics, Berlin Institute of Health at Charité - Universitätsmedizin Berlin; Department of Nephrology and Medical Intensive Care, Charité - Universitätsmedizin Berlin, Germany | Drafting/revision of the manuscript for content, including medical writing for content; analysis or interpretation of data |
| Nadine J. Ortner, PhD | Department of Pharmacology and Toxicology, Institute of Pharmacy, Center for Molecular Biosciences Innsbruck, University of Innsbruck, Austria | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
Study Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, project-ID 431984000, CRC1453 to U.I.S., CRC1315 to A.M.K., FOR3004 to A.M.K.), the Günter Endres Fond via the Einstein Stiftung to AMK, the Stiftung Charité (BIH_PRO_406 to U.I.S.), the University of Innsbruck (Erika-Cremer habilitation fellowship to N.J.O.), the Austrian Science Fund (FWF, P35087 to N.J.O., P35722 to J.S.) and the Marie Skłodowska-Curie Actions COFUND to FT (ARDRE, H2020 No. 847681).
Disclosure
The authors report no relevant disclosures. Go to Neurology.org/NG for full disclosures.
References
- 1.Zamponi GW, Striessnig J, Koschak A, Dolphin AC. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev. 2015;67(4):821-870. doi: 10.1124/pr.114.009654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ortner NJ, Kaserer T, Copeland JN, Striessnig J. De novo CACNA1D Ca2+ channelopathies: clinical phenotypes and molecular mechanism. Pflügers Arch. 2020;472(7):755-773. doi: 10.1007/s00424-020-02418-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Striessnig J. Voltage-gated Ca2+-channel α1-subunit de novo missense mutations: gain or loss of function - implications for potential therapies. Front Synaptic Neurosci. 2021;13:634760. doi: 10.3389/fnsyn.2021.634760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baig SM, Koschak A, Lieb A, et al. Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci. 2011;14(1):77-84. doi: 10.1038/nn.2694 [DOI] [PubMed] [Google Scholar]
- 5.Ortner NJ. CACNA1D-Related Channelopathies: From Hypertension to Autism. Springer Berlin Heidelberg; 2023. [DOI] [PubMed] [Google Scholar]
- 6.Scholl UI, Goh G, Stölting G, et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet. 2013;45(9):1050-1054. doi: 10.1038/ng.2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Azizan EAB, Poulsen H, Tuluc P, et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet. 2013;45(9):1055-1060. doi: 10.1038/ng.2716 [DOI] [PubMed] [Google Scholar]
- 8.Hofer NT, Tuluc P, Ortner NJ, et al. Biophysical classification of a CACNA1D de novo mutation as a high-risk mutation for a severe neurodevelopmental disorder. Mol Autism. 2020;11(1):4. doi: 10.1186/s13229-019-0310-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Semenova NA, Ryzhkova OR, Strokova TV, Taran NN. [The third case report a patient with primary aldosteronism, seizures, and neurologic abnormalities (PASNA) syndrome de novo variant mutations in the CACNA1D gene]. Zh Nevrol Psikhiatr Im S S Korsakova. 2018;118(12):49-52. Tretiĭ sluchaĭ sindroma pervichnogo al'dosteronizma, sudorog i nevrologicheskikh narusheniĭ (PASNA), obuslovlennogo variantom mutatsii de novo v gene CACNA1D. doi: 10.17116/jnevro201811812149 [DOI] [PubMed] [Google Scholar]
- 10.De Mingo Alemany MC, Mifsud Grau L, Moreno Macián F, Ferrer Lorente B, León Cariñena S. A de novo CACNA1D missense mutation in a patient with congenital hyperinsulinism, primary hyperaldosteronism and hypotonia. Channels (Austin). 2020;14(1):175-180. doi: 10.1080/19336950.2020.1761171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flanagan SE, Vairo F, Johnson MB, et al. A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatr Diabetes. 2017;18(4):320-323. doi: 10.1111/pedi.12512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ortner NJ, Sah A, Paradiso E, et al. The human channel gating-modifying A749G CACNA1D (Cav1.3) variant induces a neurodevelopmental syndrome-like phenotype in mice. JCI Insight. 2023;8(20):e162100. doi: 10.1172/jci.insight.162100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stölting G, Dinh HA, Volkert M, et al. Isradipine therapy in Cacna1dIle772Met/+ mice ameliorates primary aldosteronism and neurologic abnormalities. JCI Insight. 2023;8(20):e162468. doi: 10.1172/jci.insight.162468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Török F, Tezcan K, Filippini L, et al. Germline de novo variant F747S extends the phenotypic spectrum of CACNA1D Ca2+ channelopathies. Hum Mol Genet. 2023;32(5):847-859. doi: 10.1093/hmg/ddac248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lieb A, Ortner N, Striessnig J. C-terminal modulatory domain controls coupling of voltage-sensing to pore opening in Cav1.3 L-type Ca2+ channels. Biophysical J. 2014;106(7):1467-1475. doi: 10.1016/j.bpj.2014.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bauer R, Timothy KW, Golden A. Update on the molecular genetics of Timothy syndrome. Front Pediatr. 2021;9:668546. doi: 10.3389/fped.2021.668546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stanika R, Campiglio M, Pinggera A, et al. Splice variants of the CaV1.3 L-type calcium channel regulate dendritic spine morphology. Scientific Rep. 2016;6(1):34528. doi: 10.1038/srep34528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guzman JN, Ilijic E, Yang B, et al. Systemic isradipine treatment diminishes calcium-dependent mitochondrial oxidant stress. J Clin Invest. 2018;128(6):2266-2280. doi: 10.1172/jci95898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shin J, Kovacheva L, Thomas D, et al. Cav1.3 calcium channels are full-range linear amplifiers of firing frequencies in lateral DA SN neurons. Sci Adv. 2022;8(23):eabm4560. doi: 10.1126/sciadv.abm4560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olson PA, Tkatch T, Hernandez-Lopez S, et al. G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J Neurosci. 2005;25(5):1050-1062. doi: 10.1523/JNEUROSCI.3327-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vergara R, Rick C, Hernández-López S, et al. Spontaneous voltage oscillations in striatal projection neurons in a rat corticostriatal slice. J Physiol 2003;553(Pt 1):169-182. doi: 10.1113/JPHYSIOL.2003.050799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hering S, Zangerl-Plessl EM, Beyl S, Hohaus A, Andranovits S, Timin EN. Calcium channel gating. Pflugers Arch. 2018;470(9):1291-1309. doi: 10.1007/s00424-018-2163-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pinggera A, Lieb A, Benedetti B, et al. CACNA1D de novo mutations in autism spectrum disorders activate Cav1.3 L-type calcium channels. Biol Psychiatry. 2015;77(9):816-822. doi: 10.1016/j.biopsych.2014.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gillis J, Burashnikov E, Antzelevitch C, et al. Long QT, syndactyly, joint contractures, stroke and novel CACNA1C mutation: expanding the spectrum of Timothy syndrome. Am J Med Genet A. 2012;158a(1):182-187. doi: 10.1002/ajmg.a.34355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Depil K, Beyl S, Stary-Weinzinger A, Hohaus A, Timin E, Hering S. Timothy mutation disrupts the link between activation and inactivation in Ca(V)1.2 protein. J Biol Chem. 2011;286(36):31557-31564. doi: 10.1074/jbc.M111.255273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beyl S, Depil K, Hohaus A, et al. Physicochemical properties of pore residues predict activation gating of Ca V1.2: a correlation mutation analysis. Pflugers Arch. 2011;461(1):53-63. doi: 10.1007/s00424-010-0885-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guzman JN, Sanchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in Substantia Nigra dopaminergic neurons. J Neurosci. 2009;29(35):11011-11019. doi: 10.1523/jneurosci.2519-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramirez JM, Koch H, Garcia AJ 3rd, Doi A, Zanella S. The role of spiking and bursting pacemakers in the neuronal control of breathing. J Biol Phys. 2011;37(3):241-261. doi: 10.1007/s10867-011-9214-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uchida S, Yamada S, Nagai K, Deguchi Y, Kimura R. Brain pharmacokinetics and in vivo receptor binding of 1,4-dihydropyridine calcium channel antagonists. Life Sci. 1997;61(21):2083-2090. doi: 10.1016/s0024-3205(97)00881-3 [DOI] [PubMed] [Google Scholar]