

Abstract
Temporal development of neural electrophysiology follows genetic programming, similar to cellular maturation and organization during development. The emergent properties of this electrophysiological development, namely neural oscillations, can be used to characterize brain development. Recently, we utilized the innate programming encoded in the human genome to generate functionally mature cortical organoids. In brief, stem cells are suspended in culture via continuous shaking and naturally aggregate into embryoid bodies before being exposed to media formulations for neural induction, differentiation and maturation. The specific culture format, media composition and duration of exposure to these media distinguish organoid protocols and determine whether a protocol is guided or unguided toward specific neural fate. The ‘semi-guided’ protocol presented here has shorter induction and differentiation steps with less-specific patterning molecules than most guided protocols but maintains the use of neurotrophic factors such as brain-derived growth factor and neurotrophin-3, unlike unguided approaches. This approach yields the cell type diversity of unguided approaches while maintaining reproducibility for disease modeling. Importantly, we characterized the electrophysiology of these organoids and found that they recapitulate the maturation of neural oscillations observed in the developing human brain, a feature not shown with other approaches. This protocol represents the potential first steps toward bridging molecular and cellular biology to human cognition, and it has already been used to discover underlying features of human brain development, evolution and neurological conditions. Experienced cell culture technicians can expect the protocol to take 1 month, with extended maturation, electrophysiology recording, and adeno-associated virus transduction procedure options.
Introduction
Human induced pluripotent stem cells (iPSCs) have been used to generate neural tissue to study human brain development and characterize or treat neural disease1,2. Initially, two-dimensional (2D) approaches yielded valuable information regarding human specific features of neural growth and maturation. The development of three-dimensional (3D)-directed differentiation approaches have provided spatially organized, more physiologically relevant neural cultures with increased cellular diversity, maturation and neural circuit complexity, allowing investigation of higher-order features of the human brain3,4.
Development of the protocol
Neural differentiation protocols from iPSCs involve inhibition of BMP and NODAL signaling, resulting in induction of neuroectoderm fate commitment, before further neuronal differentiation and maturation5. ‘Guided’ directed differentiation or ‘unguided’ spontaneous differentiation methods result in the generation of organoids with different cellular composition and maturation. The tuning of brain region-specific differentiation factors and variable lengths of neuronal differentiation and maturation steps allows either more guided differentiation toward a specific brain region or the generation of broad, less regionally defined neural tissue. For example, more guided differentiation protocols may involve longer differentiation steps, the use of additional growth factors, or the use of WNT, BMP or TGFß inhibitors to induce a restricted dorsal forebrain organoid identity. Unguided protocols rely less on the addition of specific patterning molecules and usually have shorter differentiation steps, resulting in organoids with greater cellular heterogeneity and variation in cell maturity and regional identity6. Here, we describe an intermediate option: a guided or ‘semi-guided’ differentiation protocol that, like other guided methods, utilizes growth factors fibroblast growth factor 2 (FGF2) and epidermal growth factor (EGF) to facilitate proliferation and differentiation, and brain-derived growth factor (BDNF) and neurotrophin-3 (NT-3) to facilitate differentiation and maturation, while restricting the use of insulin supplementation and addition of more specific patterning molecules to maintain the benefits of cellular heterogeneity observed in unguided protocols (Fig. 1a and Extended Data Fig. 1). This protocol retains some of the challenges of variability seen in unguided, self-patterned protocols, but it also retains and leverages the self-organizing capacity of iPSCs to produce broad cortical cellular diversity, including functional glutamatergic and GABAergic neurons. Importantly, while different protocols result in organoids with variable cell composition and maturity, as we will now discuss, the method described here results in organoids with cellular composition and functional electrophysiology comparable to the human fetal cortex7,8 (Fig. 2e,f, Extended Data Figs. 1 and 2 and Table 1).
Fig. 1 |. Overview of generation of cortical organoids and resulting features.
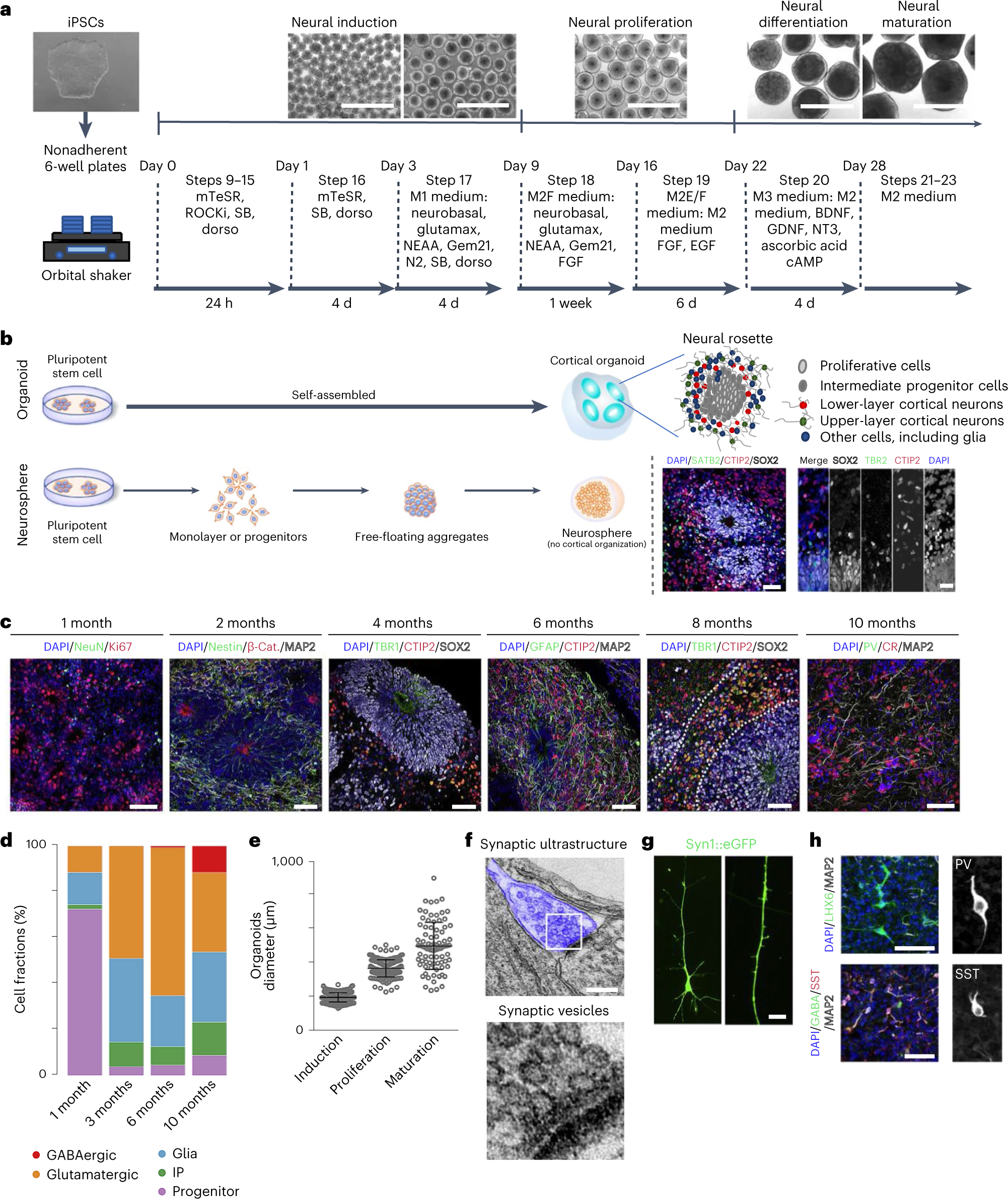
a, A schematic of the protocol used to generate cortical organoids. Scale bars, 1 cm. b, A schematic comparison of brain organoids versus neurosphere, with graphics of neural rosettes and representative immunostainings showing proliferative cells (SOX2), intermediate progenitor cells (TBR2) and lower layer (CTIP2) and upper layer (SATB2) cortical neurons. Scale bars, 50 μm. c, Representative immunostainings that show nuclei (DAPI), β-catenin ((β-cat) stabilized in inner lumen of rosette), proliferating neural progenitor cells (NPCs) (Ki67 and Nestin), neurons (NeuN), lower cortical layer neurons (TBR1 and CTIP2), upper cortical layer neurons (SATB2), interneurons (CR) and glial cells (GFAP), over time. Scale bars, 50 μm. d, The proportion of progenitor cells, intermediate progenitor (IP) cells, glia, and glutamatergic and GABAergic neurons from scRNAseq (~12,000 cells per library prep) at individual timepoints represented as a bar plot. e, The expected organoid size over the course of development, ~200 μm during induction, 375 μm during proliferation and 500 μm during maturation. Induction is around day 5, proliferation is around day 18, maturation is around day 28. f, Electron microscopy of synaptic ultrastructure (blue), staining from a 4-month-old organoid. Scale bar, 200 nm. g, A representative image of a pyramidal neuron and dendritic structures, observed in 4-month-old neurons using a SYN:EGFP reporter. Scale bar, 5 μm. h, Immunostaining of 10-month cortical organoids for the GABAergic neuronal markers parvalbumin (PV) and somatostatin (SST). Scale bars, 50 μm. Panels b–h adapted with permission from ref. 7, Elsevier.
Fig. 2 |. Electrophysiology overview and characterization.
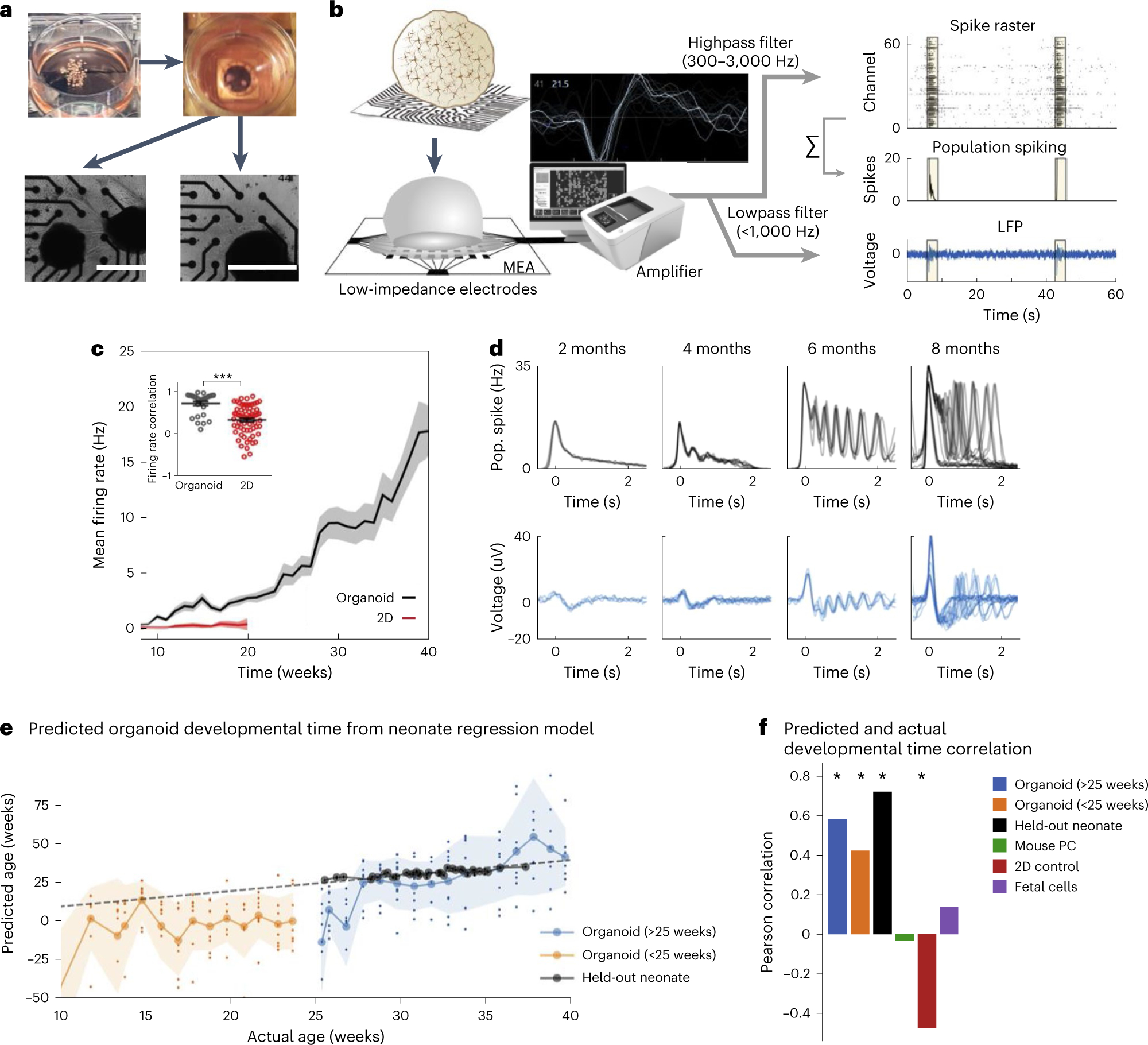
a, Pictures depicting plating of organoids on an MEA plate. Scale bars, 1 mm. b, A schematic for the electrophysiological signal processing pipeline in organoids. Representative waveform shape of neuronal trace/spike. Raw MEA data are analyzed as population spiking and LFP separately. Yellow highlights indicate synchronous network events. c, Compared with 2D neurons, cortical organoids show elevated and continuously increasing mean firing rate (n = 8 for organoid cultures and n = 12 for 2D neurons ± s.d.). Inset: correlation of the firing rate vector over 12 weeks of differentiation (from 8 to 20) between pairs of cultures showing reduced variability among organoid replicates ± s.d. d, A time series of LFP and population (pop.) spiking during network events in cortical organoid development. Each overlaid trace represents a single event during the same recording session. e, Developmental time as predicted by the model (y axis, age in weeks) follows actual weeks that the organoids (orange and blue) were in culture (x axis), as well as the true age of heldout preterm neonate data points in black. The dashed line represents unity and signifies a perfect prediction. Large circles on solid lines and shaded regions denote mean ± s.d. of prediction, respectively, and dots indicate prediction per sample (n = 8 for organoids at all timepoints). f, Pearson’s correlation coefficient between actual and predicted developmental time for organoid and control datasets, including held-out preterm neonate data, mouse primary culture (PC), 2D iPSC culture, and human fetal brain culture. Positive correlations indicate the model’s ability to capture developmental trajectory. Panels b–f adapted with permission from ref. 7, Elsevier.
Table 1 |.
Comparison of main organoid generation approaches
| Reference | Format | Variability | Applications | Considerations/uniqueness |
|---|---|---|---|---|
| Eiraku et al.5 | Guided | Low | Genetic disease, drug dosing, drug screening | Low variability is attractive for drug screening; development and metabolomics may be influenced by presence of insulin in some protocols; may not have spontaneous inhibitory neuron development49 |
| Lancaster et al.19 | Unguided | High | Lissencephalies, genetic disease, whole brain development | Spontaneous development of multiple brain regions; high cell diversity, high organoid to organoid variability. Newer adaptations can produce neural oscillations21 |
| Trujillo et al.7 | Guided or ‘semi-guided’ | Medium (Extended Data Fig. 2) | Genetic disease, metabolic disease, population level spontaneous electrical activity, drug dosing/screening | Spontaneous emergence of physiologically relevant oscillatory activity from functional excitatory–inhibitory neuron development; expected tradeoff of efficiency/variability due to semi-guided nature, long-term developmental scale. Can be used only to model cortical-born interneurons, not ganglionic eminence-born interneurons or migration of interneurons to the cortex. Resultant organoids follow the neurodevelopment of the human brain |
Cellular composition
Derivation of cortical organoids with this approach yields robust neural rosettes, with spatially relevant organization of proliferative cells, intermediate progenitor cells, glia and lower- and upper-layer cortical neurons7,9,10 (Fig. 1b,c and Extended Data Fig. 3). Over the course of organoid maturation, neurons display characteristic pyramidal morphology, mature synaptic ultrastructure and functional populations of inhibitory interneurons emerge, enabling complex neural circuitry (Fig. 1d,f–h). This transcriptional, cellular and organizational evolution of cortical features are known to follow fixed developmental trajectories defined by genetics. In this protocol, organoids are successfully guided toward a cortical fate while still allowing some innate genetic programming to drive development. Of note, while inhibitory interneurons have been shown to develop in the human cortex before mid-gestation11,12, most interneurons develop in the ganglionic eminence, which is not usually modeled in cortical organoids. Thus, the presence and functionality of cortical organoid interneurons are debated. While single-cell transcriptomics performed in other cortical organoid protocols indicate interneuron populations6,8,13,14, in many of these cases it is not clear whether these neurons are releasing GABA or have functional inhibitory properties. In our protocol, we first detect interneurons from single-cell RNA sequencing (scRNAseq) data at 6 months and show GABA staining colocalized with somatostatin-positive interneurons at later timepoints7 (Fig. 1d,h). While GABA staining has been shown in guided subdomain-specific spheroids14,15, this has not been shown in guided cortical organoids6,13,16–18. Of note, calretinin-positive19, GAD67-positive20 and GABA-positive21 interneurons have been shown in unguided organoid protocols, but only recently have neural oscillations been observed in an unguided organoid protocol21. The presence of defined GABAergic neurons is what allows complex neural oscillations, as we confirmed with selective inhibition of GABA receptors7, which has not been shown in tandem in other single circuit (i.e., nonassembloid) cortical organoid protocols6,13,14,16,18,21.
Recently, Tanaka et al. performed a comparative analysis of single-cell transcriptomes from different brain organoid protocols and the human fetal brain8. This paper independently summarizes the key differences between our protocol and others (Extended Data Fig. 1). Notably, pseudotime analysis revealed distinct differentiation trajectories that organoids undergo during development, with respective occupancies of each of these trajectories being similar between our protocol and the fetal brain (Extended Data Fig. 1b–d). As a result, our protocol had a more similar cellular profile to the fetal brain and, importantly, had the lowest enrichment for disease-related processes of the protocols studied (Extended Data Fig. 1e,f). This recapitulation of developmental trajectory, cell type diversity and lack of disease processes with respect to the fetal cortex is important for modeling the complex neural oscillations observed in the fetal cortex, which has not been shown before with high fidelity in single-circuit organoids before our publication7.
Emergence of complex neural oscillations
As neuronal circuits form and mature, they begin to exhibit rhythmic patterns of functional spatiotemporal activity across neurons, resulting in complex neural oscillations. This emergent electrophysiological property can be directly measured via electroencephalography (EEG), and individual features of these oscillations can be resolved, including traveling waves, depolarizing potentials, oscillation time scales and nested oscillations. The spatiotemporal emergence and evolution of these features has been utilized to characterize behavior and disease state in various organisms, both in vivo and in vitro22–25. We characterized these features in organoids generated with this protocol across development (over 10 months) into electrically mature organoids7. From this, we found that neurons from organoids not only have higher firing rates than those from 2D neuron cultures but also exhibit complex neural oscillations that mimic those observed in EEGs from preterm neonates (Fig. 2e). These findings indicate that these organoids have the potential to model electrophysiological features of human brain development; we hypothesize that this is due to conserved genetic features between organoid and in vivo brain development. Thus, the method described here provides a next-generation model for the early human brain and has the potential to be used to discover novel features of neurodevelopmental and degenerative diseases26.
Comparison with other methods
We note that assembly of multiple subdomain-specific spheroids (assembloids) from fully guided protocols can also result in complex neural oscillations. Samarasinghe et al.27 recently generated cerebral cortex–ganglionic eminence assembloids that develop local field potential (LFP) oscillations earlier than this protocol (3 and 6 months, respectively). However, this assembloid method requires the addition of kainate to induce neural oscillations. Furthermore, this approach results in a different cellular profile with respect to the broad cortex (e.g., overrepresentation of interneurons). This may not be a disadvantage depending on the question being studied (e.g., brain region specific or interneuron migration studies), but these assembloids have not been shown to model the cellular diversity, maturation or spontaneous emergence of neural oscillations observed in the fetal cortex. As assembloid approaches require the successful merging of organoids generated via two different brain organoid protocols, they have additional challenges such as additional technical skill complexity, reagent costs, operator time and points of failure.
As discussed above, unguided single-circuit cortical organoid protocols can also generate inhibitory neurons. Recently, Sharf et al.21, using a protocol adapted from the unguided Lancaster protocol19, showed GABA-positive inhibitory neurons, as well as phase-coupled neural oscillations. However, this approach focused on organoid slices and added the use of BDNF, glial cell line-derived neurotrophic factor (GDNF), cAMP and ascorbic acid, which are used in our protocol but not in the Lancaster protocol7,19. Furthermore, organoids generated using this approach have not been directly compared with the fetal brain in terms of cell composition or electrophysiology and require additional culturing steps, such as Matrigel embedding. We anticipate that other protocols could also be adapted using lessons from Trujillo et al.7 to generate spontaneous neural oscillations.
Advantages and limitations
While other methods to generate cortical organoids pose alternative benefits—unguided organoids can recapitulate multiple brain regions in the same organoid, while fully guided organoids are highly regional-specific, reproducible and have been merged into assembloids to study interbrain region connectivity—the protocol presented here represents the only single circuit cortical organoid model with in vivo-like cell heterogeneity and emergence of defined inhibitory neurons, resulting in spontaneous neural oscillations that model the developing fetal cortex7,8,13,28. Neural oscillations, interneuronalopathies and oligodendrocyte differentiation occur slowly in our protocol, as it follows human developmental timescales, but can be achieved more quickly in other protocols not aimed at modeling human developmental timescales. For example, inhibitory–excitatory neuron dynamics and neural oscillations can be modeled in 3 months using subdomain assembloids (compared with 6 months with this protocol). Furthermore, integration of functional vasculature into organoids remains an ongoing challenge in our laboratory and others.
Applications
This protocol is best suited for researchers aiming to (1) study disease or development in an experimentally tractable and broadly human cortical paradigm, (2) balance the reproducibility offered by more guided approaches with the efficacy of less guided approaches or (3) study physiologically relevant developmental neuroelectrophysiology in a single circuit. In support of this, we have successfully used this protocol to model autism and other neurodevelopmental disorders10, the effects of fetal alcohol exposure and neurodevelopment29, drug screening to mitigate the effects of Zika virus infection on the fetal brain30, neurodevelopmental differences between humans and Neanderthals9, learning mechanisms in an organoid–robot integrated interface31, impacts of antidepressants on neurodevelopment32 and various kinds of genetic disease33,34. This protocol has already produced peer-reviewed advances in understanding development, disease and drug testing in neurobiology, and has the potential to elucidate a better understanding of how the developing neocortex is affected in studies ranging from rare neuropathologies to space flight35.
Experimental design
Procedure 1: organoid generation
The master stock or cell bank for iPSCs used for generating organoids should be assessed for pluripotency via successful differentiation into three germ layers and karyotyped to ensure genomic integrity, as suggested by the Standards for Human Stem Cell Use in Research by the International Society for Stem Cell Research36. Newly reprogrammed iPSCs should be passaged at least ten times before use for directed differentiation and freshly thawed iPSCs should be passaged at least twice before use. The iPSCs and organoids should be regularly checked for mycoplasma contamination. Additionally, a power analysis should be conducted before beginning experiments to ensure sufficient replicates. During the first generation of organoids for every new iPSC line, it is important to empirically determine and characterize the number of organoids that spontaneously form per well, their growth rate and the optimal ‘split ratio’ and timing for redistributing growing organoids to additional wells. When culturing control and disease-relevant iPSC lines in comparison to each other, it is important to use a consistent empirically determined split ratio to avoid perceived ‘fish bowl’ effects. These effects may arise from organoids being exposed to different gradients of growth factors as a result of being in a well with substantially more or less organoids than the previous generation at the same timepoint. Neuralization of iPSCs can vary based on genotype; therefore, it is important to assess the neuralization capacity and efficiency of each new iPSC line before beginning and during experimentation (Extended Data Fig. 3). Furthermore, at 1 month after neural induction, organoids should be 250–1,000 μm in diameter (Fig. 1e) and contain neural rosettes (Fig. 1b and Extended Data Fig. 3). Organoids without rosettes should be discarded. The average organoid size will vary between cell lines; average size should be assessed, and notably smaller and/or larger organoids should be discarded at the operator’s discretion.
Variability between organoid generations is inevitable due to the nature of the differentiation approach. We define a replicate as an independent organoid batch generation, starting from the iPSC stage. We note expression of upper- and lower-layer cortical neurons, intermediate neurons and progenitors, and glial cells across our publications7,9,10,29,30. The approximate size of the populations of these cells was validated by immunohistochemistry in our 2019 manuscript7, and we show molecular and functional reproducibility (Extended Data Fig. 2). As the number of controls and replicates needed will depend on the variability associated with the starting iPSC lines, the effect size of the phenotype being studied, the rarity of the disease being studied and the number of consenting donors, we give these considerations rather than specific numerical cutoffs. iPSC lines are known in the field to be variable not only between patients of the same genotype, but also between clones from the same reprogramming event. In disease modeling with iPSCs, it is generally preferred for the control cell lines to be derived from an unaffected family member, usually a parent, so as to have a similar genetic background to the affected patient line. Isogenic cell lines can allow the exploration of a genetic mutation with less of the cell line variability associated with patient-derived iPSCs. Due to the cost-prohibitive nature of scRNAseq, fewer replicates have traditionally been required (or indeed, sometimes only an n of 1), but increasing the number of cells profiled is recommended to better capture the diverse population of cell types in mature organoids. We recommend validating important scRNAseq findings with immunohistochemistry, RT–PCR and other orthogonal assays. We also recommend corroborating phenotypic expression assays with functional measurements. Lastly, operators should have tissue culture experience practicing good sterile techniques and have experience working with iPSCs or human embryonic stem cells before organoid generation.
Procedures 2–4: orthogonal tools for the functional characterization of cortical organoids
Procedure 2: microelectrode array (MEA). Our previously reported functional data7 was recorded using the Axion Biosystems Maestro Pro system (Fig. 2b), which is a user-friendly system that enables biologists to obtain populational-level functional data from plated organoids in a noninvasive way. This allows electrical activity tracking in live organoids over time, without necessarily requiring an electrophysiologist or bioinformatician. For power users and for more advanced data processing and complex analysis, the raw data are amenable to exportation for further processing in collaboration with experienced bioinformaticians. As we have found that the Axion platform is amenable for our project groups that need daily or weekly recordings and it is user friendly enough to accommodate scientists and trainees of varying background in electrophysiology (including no previous experience), we have herein described our protocol for plating onto MEA plates for the use of the Axion Biosystems.
However, other platforms and methods for obtaining electrical functional data from organoids exist and can be explored based on the needs of the project group. The Axion commercial system may be cost prohibitive for project groups that are just beginning to incorporate microelectrode functional experiments or those interested in one-off experiments. A shared core facility purchase of an Axion platform, which also has presets for tracking electrical activity of cardiomyocytes and the growth of cancer cells, may be more financially viable. Other platforms, including Multi Channel Systems and Intan Technologies, also offer traditional low-density electrode arrays like the Axion platform. All three systems offer multiwell plate formats, with a few electrodes per well (usually 16–64), making them well suited for examining overall network activity and basic neuronal functionality. Furthermore, Multi Channel Systems MEA, the Intan Technologies system and OpenEphys software or the Neuropixel technology systems also allow simultaneous recordings and stimulation of organoid’s activity, as well as closed-loop experiments. However, these more modular systems require more knowledge about hardware assembly and software use compared with the Axion Biosystems platform where the user friendliness tradeoff is that experiments cannot currently be customized for closed loop stimulation.
For all of these systems, the MEA plates are made of transparent or glass well bottoms, which allows observation of culture changes over time with a typical inverted microscope. While these multiwell formats, regardless of the platform, provide a higher-throughput format for characterization, assays and drug screening, they often lack enough electrodes to cover the entire well, and therefore require careful placement of organoids at the time of plating and well-optimized culture conditions that promote the outgrowth of neuronal cells to cover the electrode array. Newly emerging MEA technologies may be possible in the future, with high-density MEA platforms, such as those being developed by MaxWell Biosystems and 3D Brain, containing hundreds to thousands of electrodes per well. These systems will offer higher spatial resolution and allow examination of intricate neuronal network interactions, with the tradeoff of being low throughput. A shared limitation of these multiwell MEA formats is that organoids need to be plated, and long-term culture of organoids in this adherent plated format might change the cytoarchitecture of the organoids.
Procedure 3: calcium imaging. One alternative to using an MEA system to assess electrical activity is synthetic calcium indicator staining and imaging. Of note, optical imaging poses certain advantages over direct recording of electrical activity in many applications. Ca2+ imaging enables multiple neurons to be simultaneously observed in a network with accurate spatial resolution, and firing rate estimates from optical reporters align better with those of intracellular recordings. As a result, calcium indicators can reveal more accurate spatiotemporal activity patterns in networks and within neurons37. Finally, calcium imaging assays are more accessible, cheaper and simpler to implement than MEA and other direct recording methods. Of note, cortical organoids can be analyzed on separate MEA and calcium imaging plates or concurrently on the same MEA substrate for more detailed analysis of network and cell dynamics. However, while calcium imaging and MEA recording can be done on the same MEA plate, these recordings cannot be taken at the same time with the Axion Biosystems Maestro Pro. More modular MEA systems, such as the previously mentioned Multi Channel Systems MEA, the Intan Technologies system with OpenEphys software or the Neuropixel technology, will allow simultaneous high-resolution microscopy of calcium imaging and electrical recording on the same substrate, providing an even more comprehensive overview on the network’s dynamics.
The Axion MEA system and calcium imaging using synthetic dyes are thus useful for measuring global and single-cell electrical activity from neurons that have migrated from the organoid surface onto the plate. However, electrical activity from neurons inside the organoid is not assessed. Complementary metal oxide semiconductor (CMOS) shank probes, typically used for in vivo experiments, could potentially be used for intra-organoid measurements, but are currently technically challenging and not well optimized for organoids.
Procedure 4: AAV transduction of cortical organoids. We have previously demonstrated successful lentiviral transduction of cortical organoids with optogenetic probes for intra-organoid neuron stimulation38. Furthermore, adeno-associated viruses (AAVs) have been used previously to transduce organoids. Of interest, AAV7m8 has been recently used to transduce both 2D human neurons39–41 and retinal organoids42,43. We have adapted these protocols to improve transduction approaches for use in cortical organoids, as AAVs can present advantages over lentivirus transduction. Here, we provide directions for AAV transduction of cortical organoids, and further apply this tool to integrate stable optical electrical reporters in neurons within the organoid. Unlike lentiviral vectors, the genetic material delivered by AAVs do not integrate in the host genome, circumventing mutagenesis and oncogenesis events in the host cell. However, the transgenes transduced by AAV vectors can remain as an episome in the host cell, being expressed for years, while being replication defective. Furthermore, AAV vectors can be selected to target specific cell types and tissues, making them an attractive candidate for gene therapies and an effective and less toxic vehicle for gene transfer compared with lentiviral vectors.
First, we established a proper dose to achieve successful transgene expression in our cortical organoids using an AAV7m8 vector coding a GFP reporter (AAV7m8–GFP) and a noncoding sequence (AAV7m8–null) as a negative control in 96- and 6-well plates (Fig. 3d–f). Then, once an appropriate vector dose was established, 45-day-old cortical organoids were transduced with an AAV7m8–GCaMP vector in 6-well plates. Expression of GCaMP was seen throughout the organoid and persisted over time, as best shown in 2-month-old organoids (Fig. 3g,h). GCaMP-expressing cells also showed calcium activity (Supplementary Video 1) demonstrating the potential for targeted and long-term recording of calcium activity in cortical organoids. Genetically encoded calcium ion (Ca2+) indicators enable genetically targeted expression of optical reporters in specific cell types or subcellular locations leading to selective sampling of neuronal subsets37. Moreover, genetically encoded sensors can be stably expressed to study how neuronal dynamics evolve over time44.
Fig. 3 |. Calcium imaging overview and AAV7m8–GCaMP transduction characterization in cortical organoids.
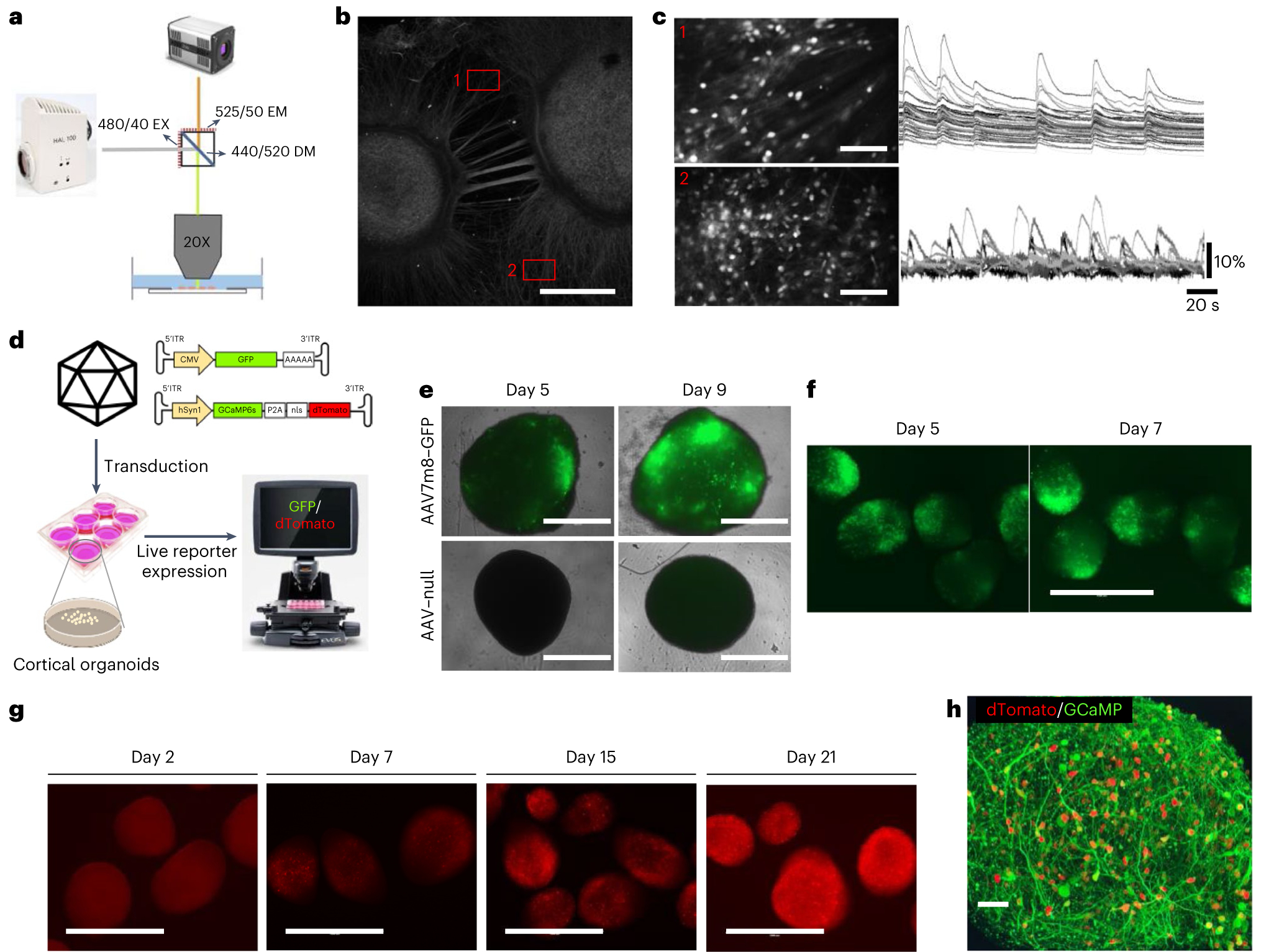
a, The optical setup for one-photon calcium imaging in organoids plated onto imaging plates, with example excitation (EX), emission (EM) and dichroic mirror (DM) specifications. b, A representative image of MAP2-stained cortical organoids and their interconnecting networks on imaging plates. Scale bar, 1,000 μm. c, Calcium traces recorded in two representative FOVs showing highly synchronized calcium activity alternating to individual spiking. Scale bars, 250 μm. d, Experimental design for the transduction of cortical organoids with AAV7m8 and example GFP and GCaMP inverted terminal repeat (ITR) sequence vectors. CMV, cytomegalovirus. e, Representative images of isolated cortical organoids transduced with AAV7m8–GFP and AAV–null at 1 × 1010 vg per well in a 96-well plate on days 5 and 9 after transduction, respectively. Scale bars, 1,000 μm. Replicates: AAV–null = 2, AAV7m8–GFP = 4. f, Representative images of cortical organoids transduced with AAV7m8–GFP at 1 × 1010 vg per well in a 6-well plate on days 5 and 9 after transduction, respectively. Scale bar, 1,000 μm. g, Representative images of cortical organoids showing neuronal expression of GCaMP sensor on days 2, 7, 15 and 21 after transduction, respectively. Scale bars, 1,000 μm. h, Representative confocal image of a 2-month-old organoid expressing GCaMP sensor (green) with physically separate nuclear localized dTomato fluorophore (red). Scale bar, 50 μm.
Materials
Biological materials
-
Human iPSCs, derived de novo by reprogramming consented patient fibroblasts as described in our prior publications7,9,10,29–35
 The experiments using iPSCs in this study were approved by the University of California San Diego Institutional Review Boards and Institutional Stem Cell Research Oversight Committees guidelines and regulations (no. 141223ZF). iPSC lines derived from control individuals used to generate the results presented in this protocol have been previously characterized elsewhere7. Institutional and federal regulations must be followed when using hiPSCs, including institutional review board approval and informed consent.
The experiments using iPSCs in this study were approved by the University of California San Diego Institutional Review Boards and Institutional Stem Cell Research Oversight Committees guidelines and regulations (no. 141223ZF). iPSC lines derived from control individuals used to generate the results presented in this protocol have been previously characterized elsewhere7. Institutional and federal regulations must be followed when using hiPSCs, including institutional review board approval and informed consent.
 The cell lines used in your research should be regularly checked to ensure they are authentic and that cell line misidentification or cross-contamination has not occurred. Additionally, regularly check all cell cultures for mycoplasma using PCR testing.
The cell lines used in your research should be regularly checked to ensure they are authentic and that cell line misidentification or cross-contamination has not occurred. Additionally, regularly check all cell cultures for mycoplasma using PCR testing.
Reagents
Media and supplements
mTeSR 1 kit (StemCell Technologies, cat. no. 85850)
Neurobasal medium (Life Technologies, cat. no. 21103049)
Dulbecco’s modified Eagle medium (DMEM)/F12 with HEPES and phenol red (Thermo Scientific, cat. no. 11330057)
ROCK Inhibitor Y-27632 dihydrocloride (Fisher Scientific, cat. no. 125410)
Stemolecule SB-431542 (SB) (Stemgent, cat. no. 04–0010-10)
Dorsomorphin dihydrochloride (dorso), >98%) (Fisher Scientific, cat. no. 309310)
GlutaMAX Supplement (Life Technologies, cat. no. 35050061)
MEM Nonessential amino acids solution (NEAA; 100×)) (Gibco, cat. no. 11140–050)
Gem21 NeuroPlex (Gemini, cat. no. 400160 010ML; alternatively, Neurocult SM1 (StemCell Technologies, cat. no. 05711) or B27 (Life Technologies, cat. no. 17504044) can be used in place of Gem21
N2 NeuroPlex (Gemini, cat. no. 400163 005ML)
Human BDNF (PeproTech, cat. no. 450–02)
Human GDNF (PeproTech, cat. no 450–10)
Recombinant Human NT-3 (PeproTech, cat. no. 450–03)
l-Ascorbic acid (Sigma-Aldrich, cat. no. A4403–100MG)
N6,2′-O-Dibutyryladenosine 3′,5′-cyclic monophosphate sodium salt (dibutyryl-cAMP),
96% (HPLC), powder) (Sigma-Aldrich, cat. no. D0627–5X1G)
Human FGF (Life Technologies Corporation, cat. no. PHG0263)
Human EGF (Peprotech, cat. no. AF-100–15)
Cell culture reagents
Matrigel Basement Membrane Matrix, growth factor reduced (BD Biosciences, cat. no. 354234)
Dulbecco’s phosphate-buffered saline (DPBS) without CaMg (Corning, cat. no. MT21–031CV)
StemPro Accutase (Life Technologies, cat. no. A1110501)
-
Alcohol, ethyl (ethanol) 100%, 200 proof (Koptec anhydrous 200 proof ethyl alcohol, cat. no. V1005M)
 Ethyl alcohol is flammable and should be stored in a flame protective cabinet.
Ethyl alcohol is flammable and should be stored in a flame protective cabinet. Anti-adherence rinsing solution (StemCell Technologies, cat. no. 07010)
MEA
Laminin mouse protein (Thermo Fisher Scientific, cat. no. 23017015)
Poly-l-ornithine hydrobromide (PLO) (Sigma-Aldrich, cat. no. P3655)
BrainPhys neuronal medium (StemCell Technologies, cat. no. 05790)
NeuroCult SM1 neuronal supplement (StemCell Technologies, cat. no. 05711)
Calcium imaging
PLO (Sigma-Aldrich, cat. no. P3655)
Laminin mouse protein (ThermoFisher Scientific, cat. no. 23017015)
Oregon Green 488 BAPTA-1, AM (Invitrogen, cat. no. O6807)
Pluronic F-127, 20% solution in dimethyl sulfoxide (DMSO) (Invitrogen, cat. no. P3000MP)
BrainPhys imaging optimized medium (StemCell Technologies, cat. no. 05796)
NeuroCult SM1 neuronal supplement (StemCell Technologies, cat. no. 05711)
AAV transduction only
 AAV vectors were produced by the Viral Vector Production Unit and R.B. at the Universitat Autonoma de Barcelona (Spain) with established protocols45.
AAV vectors were produced by the Viral Vector Production Unit and R.B. at the Universitat Autonoma de Barcelona (Spain) with established protocols45.
AAV7m8 vectors encoding GCaMP (AAV7m8–GCaMP) from AAV-hSyn1-GCaMP6s-P2A-nls-dTomato construct (Addgene plasmid no. 51084)
AAV7m8 vectors coding the GFP reporter under the control of the cytomegalovirus promoter (AAV7m8–GFP)
Negative control vectors under the control of the cytomegalovirus promoter (AAV7m8–null)
Bleach (Clorox, mfr. no. 863-CLOX30966; Thomas Scientific, cat. no. 1201R80)
Equipment
General equipment
6 cm tissue culture dishes (Corning, cat. no. 430166)
AggreWell800 (STEMCELL Technologies, cat. no. 34815)
6-well plates (Corning Costar, cat. no. 07–200-83)
Cell strainer, 40 μm (Fisher Scientific, cat. no. 87711)
15 mL, 50 mL centrifuge tubes (BioPioneer, cat. nos. CNT-50, CNT-15)
Portable Pipet-Aid XP pipette controller (Drummondsci, cat. no. 4–000-101)
5 mL, 10 mL, 25 mL and 50 mL serological pipette (Corning Costar, cat. nos. CLS4487, CLS4488, CLS4489, CLS4490)
Pipette tips (Neptune, cat. nos. 89140–932 (CS), 89168–754 (CS), 89140–936 (CS))
Platform shaker, 1.9 cm orbit (Innova 2000 Benchtop Orbital Shaker, Eppendorf, cat. no. M1190–000)
Incubator (Sanyo CO2 incubator, cat. no. MCO-18AIC)
Water bath (Fisher Scientific 2320 isotemp digital control water bath, cat. no. 15–462-20Q)
Benchtop microscope (ZEISS Primovert, item no. 491206–0001-000)
Biosafety cabinet microscope (Invitrogen EVOS FL digital inverted fluorescence, cat. no. 12–563-460)
Cell counter (Bio-Rad TC20 automated cell counter, cat. no. 1450102)
Centrifuge (Eppendorf centrifuge 5702 series, cat. no. 05–413-327)
MEA equipment
MEA 6-well plates (CytoView, cat. no. M384-tMEA-6B)
Maestro Pro multiwell microelectrode array (MEA) system (Axion Biosystems)
Calcium Imaging equipment
MatTek dishes, glass bottom (MatTek, cat. no. P35G-1.5–14-C)
Software
AxIS Software Spontaneous Neural Configuration (Axion Biosystems)
MATLAB
Reagent setup
Reconstitution and storage of growth factor and chemical stock solutions
Dorso: resuspend 10 mg dorso in 20 mL of cell culture grade ultrapure distilled water using a sterile 22 μm filter to obtain a 1 mM stock solution
SB-431542: resuspend 10 mg SB-431542 in 2.6 mL DMSO to obtain a 10 mM stock solution
EGF and FGF growth factors: resuspend in sterile 0.1% (vol/vol) BSA/PBS to 100 μg/mL stock concentration
GDNF, BDNF, NT-3 growth factors: resuspend in sterile 0.1% (vol/vol) BSA/PBS to 20 μg/mL stock concentration
Ascorbic acid, cell culture grade: resuspend 100 mg of cell culture grade ascorbic acid in 2.84 mL of sterile cell culture grade ultrapure distilled water to obtain a 200 mM stock solution
Y-27632 ROCK inhibitor: resuspend 100 mg of cell culture grade Y-27632 ROCK inhibitor in 6.1 mL of sterile cell culture grade ultrapure distilled water to obtain a 5 mM stock solution
cAMP: resuspend one 1 g bottle of cAMP in 9.8 mL sterile cell culture grade ultrapure distilled water to create a 200 mM stock (10.2 mL final volume) and aliquot into clean microcentrifuge tubes using a syringe with attached sterile 22 μm filter
PLO: resuspend PLO solution in 50 mL sterile cell culture grade ultrapure distilled water to make a stock solution (10 mg/mL) for use with calcium imaging and MEA plating experiments Aliquoted stock solutions are prepared in a biosafety cabinet and stored at either −20 °C (for 2–3 months) or −80 °C (for 3–4 months). Once thawed, aliquots can be kept at 4 °C for up to 1 week. Ascorbic acid and SB-431542 are light sensitive and thus should be protected from light. If the SB-431542 aliquot does not freeze at 4 °C, discard it as the hygroscopic nature of DMSO has affected the concentration. For sterilization, filter through a 0.22 μm filter unit. The specific molecular weight may vary from batch to batch, so verify calculations for each batch.
Preparing Matrigel aliquots
Thaw a stock bottle of Matrigel on ice overnight at 4 °C.
Prechill a box of p1000 pipette tips and a new sterile bag of microfuge tubes overnight at −20 °C.
Prepare an ice bucket by filling with fresh ice, then thoroughly disinfecting with 70% alcohol before placing inside a biosafety cabinet. Place the thawed stock bottle of Matrigel and chilled microfuge tubes on ice in the ice bucket.
Using the prechilled p1000 pipette tips, make Matrigel aliquots by transferring 300 μL of undiluted stock Matrigel into each microfuge tube and immediately placing the microfuge tubes on ice.
Store Matrigel aliquots at −20 °C for until the date of expiration as stated on the manufacturer’s Certificate of Analysis.
Preparing Matrigel-coated 6 cm plates
Thaw a Matrigel aliquot by transferring a 300 μL aliquot of Matrigel from the freezer onto ice for 30 min. The aliquot should become completely liquid when thawed on ice.
-
Transfer the Matrigel aliquot into the cell culture hood and dilute it in an appropriate volume of cold DMEM to create a 0.1 mg protein per mL solution. The dilution volume depends on the protein content of the stock bottle of Matrigel. See the manufacturer’s Certificate of Analysis for protein content.
 Matrigel should be handled quickly when not on ice, as it quickly coagulates at room temperature (20–25 °C). Discard if coagulation or visible precipitates occurs.
Matrigel should be handled quickly when not on ice, as it quickly coagulates at room temperature (20–25 °C). Discard if coagulation or visible precipitates occurs. -
Add 2.5 mL of Matrigel/DMEM solution to each 6 cm plate and place in an incubator. Unused Matrigel/DMEM diluted solution can be stored at 4 °C for up to 2 weeks. Matrigel-coated plates should be left in the incubator for at least 1 h before use and preferably should be used after overnight incubation. Matrigel plates can be stored in an incubator for up to 3 d.
 If using precoated Matrigel plates, ensure that the plate is not dried out. Aspirate the Matrigel solution from the plate directly before transferring cells/media to the coated plate.
If using precoated Matrigel plates, ensure that the plate is not dried out. Aspirate the Matrigel solution from the plate directly before transferring cells/media to the coated plate.
General media and reagents guidance
The composition of media and reagents are indicated below. When ready for use, aliquot the required volume of medium into 50 mL sterile tubes and prewarm in a water bath at 37 °C or allow to come to room temperature. Avoid multiple cycles of refrigeration and warming.
 Add growth factors and small molecules before use and only after prewarming aliquoted media.
Add growth factors and small molecules before use and only after prewarming aliquoted media.
- mTeSR 1 medium (for use in maintenance of iPSCs). Prepare the medium as presented in the table below. This medium can be stored for 2 weeks at 4 °C.
Composition Volume (500 mL total) Final concentration mTeSR 1 basal medium 400 mL – mTeSR 1 5× supplement 100 mL 20% (vol/vol) - mTeSR 1 neural induction medium (for use for days 0–2). Prepare the medium as presented in the table below. Only use Y-27632 ROCK inhibitor on day 0. This medium can be stored for 2 weeks at 4 °C. Change medium daily.
Composition Volume (500 mL total) Final concentration Comments mTeSR 1 basal medium 400 mL – – mTeSR 1 5× supplement 100 mL 20% (vol/vol) – Dorso – 1 μM (1: 1,000) Add just before use SB-431542 – 10 μM (1: 1,000) Add just before use Y-27632 (ROCK inhibitor) – 5 μM (1: 1,000) Add just before use. Only use on day 0 - Medium 1 (M1) neural induction medium (use for days 3–8). Prepare medium as presented in the table below. This medium can be stored for 2 weeks at 4 °C. Change medium every other day.
Composition Volume (525 mL total) Final concentration Comments Neurobasal medium 500 mL – M1 medium GlutaMAX 5 mL of 100× 1× NEAA 5 mL of 100× 1% (vol/vol) Gem21 1 bottle (10 mL) 2% (vol/vol) N2 1 bottle (5 mL) 1% (vol/vol) Dorso – 1 μM (1: 1,000 of 1 mM stock) Add just before use SB-431542 – 10 μM (1: 1,000 of 10 mM stock) Add just before use - Medium 2 F (M2F) neural proliferation A medium (use for days 9–15). Prepare medium as presented in the table below. This medium can be stored for 2 weeks at 4 °C.
Composition Volume (520 mL total) Final concentration Comments Neurobasal medium 500 mL – M2 medium GlutaMAX 5 mL of 100× 1× NEAA 5 mL of 100× 1% (vol/vol) Gem21 1 bottle (10 mL) 2% (vol/vol) FGF – 20 ng/mL (1: 5,000 of 100 μg/mL stock) Add just before use - Medium 2 EF (M2EF) neural proliferation B medium (use for days 16–21). Prepare medium as presented in the table below. This medium can be stored for 2 weeks at 4 °C.
Composition Volume (520 mL total) Final concentration Comments Neurobasal medium 500 mL – M2 medium GlutaMAX 5 mL of 100× 1× NEAA 5 mL of 100× 1% (vol/vol) Gem21 1 bottle (10 mL) 2% (vol/vol) FGF – 20 ng/mL (1: 5,000 of 100 μg/mL stock) Add just before use EGF – 20 ng/mL (1: 5,000 of 100 μg/mL stock) Add just before use - Medium 3 (M3) neural ‘speed up’ maturation medium (use for days 22–27). Prepare medium as presented in the table below. This medium can be stored for 2 weeks at 4 °C.
Composition Volume (520 mL total) Final concentration Comments Neurobasal medium 500 mL – M2 medium GlutaMAX 5 mL of 100× 1× NEAA 5 mL of 100× 1% (vol/vol) Gem21 1 bottle (10 mL) 2% (vol/vol) BDNF – 10 ng/mL (1: 2,000 of 20 μg/mL stock) Add just before use GDNF – 10 ng/mL (1: 2,000 of 20 μg/mL stock) Add just before use NT-3 – 10 ng/mL (1: 2,000 of 20 μg/mL stock) Add just before use Ascorbic acid – 200 μM (1: 1,000 of 200 mM stock) Add just before use Dibutyryl-cAMP – 1 mM (1: 200 of 200 mM stock Add just before use - Medium 2 (M2) extended neuronal maturation medium (use for days 28 to endpoint). Prepare medium as presented in the table below. This medium can be stored for 2 weeks at 4 °C.
Composition Volume (520 mL total) Final concentration Comments Neurobasal medium 500 mL – M2 medium GlutaMAX 5 mL of 100× 1× NEAA 5 mL of 100× 1% (vol/vol) Gem21 1 bottle (10 mL) 2% (vol/vol) - Complete BrainPhys medium (for MEA and culture before calcium imaging). Prepare medium as presented in the table below. This medium can be stored for 2 weeks at 4 °C.
Composition Volume (510 mL total) Final concentration Comments BrainPhys neuronal medium 500 mL – – NeuroCult SM1 Neuronal Supplement 1 bottle (10 mL) 2% (vol/vol) – - Complete imaging BrainPhys medium (for calcium imaging). Prepare medium as presented in the table below. This medium can be stored for 2 weeks at 4 °C.
Composition Volume (510 mL total) Final concentration Comments BrainPhys Imaging Optimized medium 500 mL – – NeuroCult SM1 Neuronal Supplement 1 bottle (10 mL) 2% (vol/vol) –
Procedure 1: organoid generation
 Before beginning cell culture, thoroughly clean all cell culture hoods and incubators. Incubator water levels should be regularly checked to ensure proper humidity. Additionally, regularly check all cell cultures for mycoplasma using PCR testing. The timeline for the procedure can be found in Fig. 1a.
Before beginning cell culture, thoroughly clean all cell culture hoods and incubators. Incubator water levels should be regularly checked to ensure proper humidity. Additionally, regularly check all cell cultures for mycoplasma using PCR testing. The timeline for the procedure can be found in Fig. 1a.
Maintenance and passaging of iPSCs
- TIMING 10–15 min per plate, passaging
-
1Culture iPSCs in mTeSR 1 medium on Matrigel-coated 6 cm plates. Plates should be passaged before they reach 100% confluency. Growth rate, and therefore seeding density and passaging frequency, is cell line specific and therefore may need to be optimized for each cell line. Please see considerations in the ‘Experimental design’ section.
-
2Passage plates by first aspirating spent medium then adding 3 mL of fresh prewarmed mTeSR 1 to the plate to be passaged. Place the plate under an EVOS microscope inside a sterile cell culture hood, and use a pipette tip (usually a p1000 tip inserted into a p200 nonfilter tip) to cut or ‘pick’ small pieces from healthy iPSC colonies (~1 mm2). After picking ~300 colony pieces, aspirate the Matrigel from a Matrigel-coated 6 cm plate and transfer the pieces and the 3 mL of fresh medium from the picked ‘parent’ plate to the Matrigel-coated 6 cm plate using a 5 mL serological pipette. This method allows positive selection of iPSCs exhibiting normal morphology and avoids passaging cells that might have begun to differentiate in culture.
 Do not pick iPSCs that exhibit differentiation, death, overgrown density or other unusual phenotypes.
Do not pick iPSCs that exhibit differentiation, death, overgrown density or other unusual phenotypes. -
3Transfer the newly passaged plate into the incubator and move the plate in a ‘figure-8’ motion three to four times to ensure iPSCs are evenly dispersed before closing the incubator. Most healthy picked iPSC colony fragments will then attach to the plate and form new colonies.
-
4Change medium every day using 4 mL of fresh mTeSR 1 medium. Increasing medium to 4.5 mL can be used at later medium changes as needed if the medium quickly turns yellow (usually as the plate approaches confluency).
-
5Monitor cells daily, and when they reach ~85% confluence, proceed to embryoid body (EB) generation (Step 6). Alternatively, continue to grow and passage cells, repeating Steps 1–5 until the desired number of 6 cm plates of iPSCs are generated. Approximately one 6 cm plate of iPSCs will generate one well of a 6-well plate of EBs.
 Remove any differentiation from the plate by pipette, vacuum or scraping/picking differentiating colonies before removing the medium.
Remove any differentiation from the plate by pipette, vacuum or scraping/picking differentiating colonies before removing the medium.
-
1
Days 0–2: dissociation of iPSCs
TIMING 30 min, generation of 3D EBs
-
TIMING 30 min, and beginning of neural induction
 It is important to start the organoid protocol with high-quality iPSCs, characterized by pluripotent cultures with no spontaneous differentiation, in log phase growth and fairly uniform growth state across the culture. Suboptimal iPSC cultures may fail to spontaneous aggregate, and suboptimal handling at day 0 during dissociation and plating may result in aggregates of uneven starting size. If the use of high-quality iPSCs and efficient handling at day 0 continues to produce problems with spontaneous aggregation (such as inconsistent aggregate size) or if using a particularly fragile disease-relevant iPSC line, forced aggregation may be attempted to improve starting aggregate quality and size (Supplementary methods ‘Forced aggregation with AggreWell’).
It is important to start the organoid protocol with high-quality iPSCs, characterized by pluripotent cultures with no spontaneous differentiation, in log phase growth and fairly uniform growth state across the culture. Suboptimal iPSC cultures may fail to spontaneous aggregate, and suboptimal handling at day 0 during dissociation and plating may result in aggregates of uneven starting size. If the use of high-quality iPSCs and efficient handling at day 0 continues to produce problems with spontaneous aggregation (such as inconsistent aggregate size) or if using a particularly fragile disease-relevant iPSC line, forced aggregation may be attempted to improve starting aggregate quality and size (Supplementary methods ‘Forced aggregation with AggreWell’).
 For all supplements throughout differentiation, add supplements directly to the warmed media before changing media. Frozen supplement aliquots, once thawed, can be stored at 4 °C for up to a week.
For all supplements throughout differentiation, add supplements directly to the warmed media before changing media. Frozen supplement aliquots, once thawed, can be stored at 4 °C for up to a week.
-
6Warm mTeSR 1, PBS and Accutase in a 37 °C water bath. Supplement prewarmed mTeSR 1 with 5 μM Y-27632 ROCK inhibitor, 10 μM SB and 1 μM dorso (mTeSR 1 neural induction medium) when ready to be used.
-
7Aspirate the medium from the iPSC plate and wash with PBS.
-
8Gently dissociate iPSCs by removing PBS and adding 2 mL of warm Accutase:PBS (1:1) for 10–20 min, checking for cell detachment every 5 min. Once most of the colonies are mostly detaching, add 4 mL of PBS to the plate and pipette gently across the plate to remove colonies.
-
9Using a serological pipette, collect the cell suspension and pass the suspension through a 40 μm cell strainer placed on top of a 50 mL conical tube to remove undissociated clumps.
-
10Centrifuge the conical tube for 3 min at 100g, 20–27 °C.
-
11Remove supernatant and resuspend in 3–4 mL of mTeSR 1 neural induction medium. If processing multiple plates for dissociation and replating, resuspend cells in an appropriate higher volume of medium (e.g., for three 6 cm plates, resuspend in 5 mL medium).
-
12Take a cell count using Trypan Blue exclusion. Do not proceed if cell viability is below 80%. Ideally, cell viability should be greater than 90% to start the organoid protocol. Plate ~4 million cells in 4 mL of medium into each well of a low-attachment 6-well plate.
 Optimal starting seeding density may vary between iPSC lines, particularly in certain disease genotype iPSC lines. Determine optimal starting seeding density empirically, usually between 3 and 5 million cells per well of a low-attachment 6-well plate. Place the 6-well plate in an incubator (5% CO2, 37 °C) on an orbital shaker at 95 rpm (orbit 1.9 cm/0.75 inches).
Optimal starting seeding density may vary between iPSC lines, particularly in certain disease genotype iPSC lines. Determine optimal starting seeding density empirically, usually between 3 and 5 million cells per well of a low-attachment 6-well plate. Place the 6-well plate in an incubator (5% CO2, 37 °C) on an orbital shaker at 95 rpm (orbit 1.9 cm/0.75 inches). -
13After 24 h, change the medium (mTeSR 1 supplemented with SB and dorso without Y-27632 ROCK inhibitor) by gently swirling the plate in a circular motion until the EBs congregate at the center of the well. Aspirate medium using an aspirating pipette with a filter-free tip to control the aspiration rate, and aspirate in a circular motion along the edges of the well. Perform a 3 mL media change every day.
 For the entirety of the differentiation, the EBs/organoids should be taken out of the incubator only when necessary and returned to the incubator directly after changing media. If the organoids occupy half the diameter of the well or more, then split the EBs/organoids into two new wells.
For the entirety of the differentiation, the EBs/organoids should be taken out of the incubator only when necessary and returned to the incubator directly after changing media. If the organoids occupy half the diameter of the well or more, then split the EBs/organoids into two new wells.

-
6
Days 3–8: neural induction
- TIMING 6 d
-
14Perform media changes every other day with 3–4 mL of prewarmed M1 medium and supplemented with 1 μM dorso and 10 μM SB-431542 (M1 neural induction medium).
-
14
Days 9–15: neural proliferation A
- TIMING 7 d
-
15Perform media changes every day with 3–4 mL of prewarmed M2 medium and supplemented with 20 ng/mL FGF (M2F neural proliferation A medium). Once EBs are large enough, change medium by tilting the plate, letting EBs sink to the bottom of the well and aspirate the medium. This is preferred to remove any slowly sinking debris.
-
15
Days 16–21: neural proliferation B
- TIMING 6 d
-
16Perform media changes every other day with 3–4 mL of prewarmed M2 medium supplemented with 20 ng/mL FGF and 20 ng/mL EGF (M2EF neural proliferation B medium). There should be noticeable organoid growth during this time.

-
16
Days 22–27: neural differentiation
- TIMING 6 d
-
17Perform media changes every other day with 3–4 mL of prewarmed M2 medium supplemented with 10 ng/mL BDNF, 10 ng/mL GDNF, 10 ng/mL NT-3, 200 μM ascorbic acid and 1mM dibutyryl-cAMP (M3 neural ‘speed up’ maturation medium).
-
17
Day 28 onward: extended neural maturation, maintenance and long-term culture
- TIMING up to 18 months
-
18From day 28 onward, switch to M2 medium alone and change medium every 3–4 d. If the medium turns yellow quickly, then perform medium changes every other day.

-
19Monitor organoids for size and morphology at every media change. Organoids should be 250–1,000 μm in diameter (Fig. 1e) and contain neural rosettes (Fig. 1b and Extended Data Fig. 3). Organoids without rosettes should be discarded. The average organoid size will vary between cell lines and the average size should be assessed, and considerably smaller and/or larger organoids should be discarded at the operator’s discretion. If considerable variability is present, consider switching to AggreWell forced aggregation for EBs (Supplementary Methods).
-
20From day 56 onward, use M2 medium and perform half media changes every 3–7 d, depending on the consumption rate of the organoids. If media are consumed quickly (turn yellow quickly), perform half media changes more often or split wells 1:2.
-
18
Procedure 2: MEA
Preparing MEA plates
- TIMING 2 d
-
1Two days before organoid plating, coat MEA plates with PLO solution at 1:100 dilution in ultrapure distilled water (100 μg/mL) and incubate overnight in a 37 °C 5% CO2 humidified incubator. Use 1.0 mL per well of coating solution for 6-well plates.
-
2After overnight incubation, remove PLO solution and rinse wells thoroughly with ultrapure distilled water or dPBS, at least twice, to remove excess poly-l-ornithine.
-
3One day before organoid plating, coat MEA plates with laminin mouse protein solution at 1:100 dilution in PBS (10 μg/mL) and incubate overnight in a 37 °C 5% CO2 humidified incubator. Use 1.0 mL per well of coating solution for 6-well plates.
-
1
Plating organoids
- TIMING <30 min per plate
-
4Choose organoids for MEA plating that best represent the batch. Additionally, plating 1–2 month organoids works best, when they are ~200–400 μm in radius. Visible rosettes should be apparent either as viewed on EVOS under phase contrast or after confirmation of a batch sample by cryosectioning and immunostaining as detailed in Trujillo et al.7 (Fig. 2a,b and Extended Data Fig. 2).
 Avoid plating organoids without rosettes as they may not have neuralized properly and may be more akin to neurospheres than to cortical organoids in spontaneous electrical activity. Two-month-old organoids or older may not have visible rosettes, so confirm presence of rosettes earlier in development.
Avoid plating organoids without rosettes as they may not have neuralized properly and may be more akin to neurospheres than to cortical organoids in spontaneous electrical activity. Two-month-old organoids or older may not have visible rosettes, so confirm presence of rosettes earlier in development. -
5Aspirate laminin coating immediately before adding organoids. Do not rinse.
-
6Plate organoids in neural maturation culture medium. Place two to three organoids per well in an ample amount of fresh medium per well: 2 mL medium for a 6-well plate.
 Less than two and greater than five organoids per well has not demonstrably improved spontaneous electrical activity emergence or capture in MEA plates.
Less than two and greater than five organoids per well has not demonstrably improved spontaneous electrical activity emergence or capture in MEA plates.
 If plating organoids for comparison between cell lines or treatment conditions, plate the same number of organoids per well for every condition.
If plating organoids for comparison between cell lines or treatment conditions, plate the same number of organoids per well for every condition. -
7For very large (>1 mm) organoids only, create a greater attachment surface on the organoids by gentle trituration. Using a cut p1000 tip, transfer large organoids for plating into a 15 mL conical and then pipette up and down slowly so a small amount of cells on the organoid surface loosen.
 Pipette up and down only two or three times to keep organoids intact.
Pipette up and down only two or three times to keep organoids intact. -
8Swirl the plate so that fluid movement brings the organoids into the center, such that they sit on or directly adjacent to the electrode array.
 Organoids do not need to sit directly in the middle of the electrical array, but do need to be at least adjacent to an electrode.
Organoids do not need to sit directly in the middle of the electrical array, but do need to be at least adjacent to an electrode. -
9Carefully place MEA plates onto an isolated shelf in a 37 °C 5% CO2 humidified incubator. Allow organoids to attach by not moving the plate for 1 week. No media changes are performed during attachment week.

-
4
Changing media
-
TIMING once per week
 Use a 5 mL or 10 mL serological pipette for media changes, on the slowest PipetAid setting while applying only half pressure for dispense. Minimize movement as much as possible when changing the media.
Use a 5 mL or 10 mL serological pipette for media changes, on the slowest PipetAid setting while applying only half pressure for dispense. Minimize movement as much as possible when changing the media.
-
10Replace medium with M2 neuronal maturation medium and culture organoids on MEA plates until organoids are 2 months old (total culture age) changing media once or twice per week depending on medium consumption. Then, proceed to Step 11 to begin half media changes in complete BrainPhys medium.
-
11Perform half media changes with complete BrainPhys medium (BrainPhys supplemented with SM1) once per week, or as needed based on the consumption rate of the culture. Do not change media less than once per week.
-
10
Measuring electrical activity
- TIMING 20 min per plate
-
12Perform media changes until ready to measure electrical activity. Organoids begin to exhibit detectable spontaneous electrical activity at 2.5–3 months after the start of the organoid protocol.
-
13After changing media, wait 24 h before recording.
 When preparing for recording and measuring spontaneous activity, be sure to keep the time between the last media change and the time of measurement consistent.
When preparing for recording and measuring spontaneous activity, be sure to keep the time between the last media change and the time of measurement consistent. -
14Allow the Axion Biosystems Maestro Pro to start up and warm up for ~10 min.
-
15Transport plates from the incubator to the Maestro Pro, keeping them warm and sterile.
-
16Place the MEA plate in the machine and allow the plate to equilibrate for 3–5 min in the machine.
-
17Set the Maestro Pro to measure neural spikes if seeking spiking or bursting electrical data, or measure LFPs if seeking data for emerging neural oscillations analysis. Set recording to capture Axis Raw and Axis Spike data.
-
18Measure equilibrated MEA plates by recording for between 3 and 10 min, depending on the needs of the experiment. Be aware that 1 min of recording generates ~1 Gb of data.
 Backup data regularly.
Backup data regularly.

-
12
Quantifying electrical activity
 Spontaneous electrical activity development can be compared among genetic mutations, cell lines and treatment conditions over time in development, etc.
Spontaneous electrical activity development can be compared among genetic mutations, cell lines and treatment conditions over time in development, etc.
-
19
Examine waveform shapes while recording and analyzing, and consider excluding wells that demonstrate nonbiologically relevant waveform recordings, such as constitutively active or constitutively silent electrodes. A representative neuronal waveform, with amplitude below baseline, followed by rapid spike/peak above baseline, can be seen in Fig. 2b.
-
20
Quantify the number of spikes and bursts, and review raster plots, using Axion’s AxIS analysis software. Data can be analyzed with one way ANOVA or two-way ANOVA depending on the parameters being analyzed.
Detecting emerging neural oscillations
-
21
Detect emerging neural oscillations in LFP data using custom MEA analysis code, available at https://github.com/voytekresearch/OscillatoryOrganoids/blob/master/organoid_EEG_age_regression.ipynb. In short, spikes can be detected with AxIS software using an adaptive threshold crossing set to 5.5 times the standard deviation of the estimated noise for each electrode (channel). Classify electrodes that detect at least five spikes/min as active electrodes. Classify bursts in the data recorded from each individual electrode using an interspike interval (ISI) threshold requiring a minimum number of five spikes with a maximum ISI of 100 ms. A minimum of ten spikes under the same ISI with a minimum of 25% active electrodes are required for network bursts in the well. Calculate the synchrony index using a cross-correlogram synchrony window of 20 ms.
Procedure 3: calcium imaging
Preparing calcium imaging plates
- TIMING 2 d
-
1Two days before organoid plating, coat glass-bottom MatTek imaging plates with PLO solution at 1:100 dilution in water (100 μg/mL) and incubate overnight in a 37 °C and 5% CO2 humidified incubator.
 To maximize cell density and avoid waste of cells, coat only the coverslip by adding 200 μL PLO solution to the center of the imaging plate. Move slowly to the incubator to avoid overflowing.
To maximize cell density and avoid waste of cells, coat only the coverslip by adding 200 μL PLO solution to the center of the imaging plate. Move slowly to the incubator to avoid overflowing. -
2After overnight incubation, remove the PLO solution and rinse coated wells thoroughly with ultrapure distilled water or dPBS, at least twice, to remove excess poly-l-ornithine.
-
3One day before organoid plating, coat calcium imaging plates with laminin mouse protein solution at 1:100 dilution in PBS (10 μg/mL) and incubate overnight in a 37 °C and 5% CO2 humidified incubator.
 To maximize cell density and avoid waste of cells, coat only the coverslip by adding 200 μL laminin solution to the center of the imaging plate. Move slowly to the incubator to avoid overflowing.
To maximize cell density and avoid waste of cells, coat only the coverslip by adding 200 μL laminin solution to the center of the imaging plate. Move slowly to the incubator to avoid overflowing.
-
1
Plating organoids
- TIMING <30 min per plate
-
4Choose organoids for plating that best represent the batch (refer to Step 4 of Procedure 2: MEA: plating organoids).
-
5Aspirate laminin coating immediately before adding organoids. Do not rinse.
-
6Add 2 mL warm M2 medium to the plate.
-
7Plate organoids onto the coverslip in the center of the plate. Place two to three organoids per plate to keep consistent with your MEA experiments.
-
8Use a wide-O or cut p1000 tip to place the organoids onto your plate.
-
9Carefully place imaging plates onto an isolated shelf in a 37 °C and 5% CO2 humidified incubator. Allow organoids to attach by not moving the plate for 1 week. No media changes are performed during attachment week.
-
10Perform half media changes after the attachment week. Culture organoids in M2 medium until organoids are 2 months old (total culture age), changing media once or twice per week depending on media consumption based on media color change. After 2 months, perform half media changes with complete BrainPhys medium once per week, or as needed based on the consumption rate of the culture. Do not change media less than once per week.
 To avoid dislodging organoids, use a 5 mL or 10 mL serological pipette for media changes, on the slowest PipetAid setting while applying only half pressure for dispense. Minimize movement as much as possible when changing the media.
To avoid dislodging organoids, use a 5 mL or 10 mL serological pipette for media changes, on the slowest PipetAid setting while applying only half pressure for dispense. Minimize movement as much as possible when changing the media.
-
4
Record calcium activity
- TIMING 1 h per plate
-
11On the recording day, thaw a 50 μg vial of calcium indicator Oregon Green 488 BAPTA-1, AM ester (OGB1) at room temperature.
-
12Dissolve OGB1 in 4 μL of 20% Pluronic F-127 in DMSO.
-
13Dissolve the OGB1 solution in 8 mL of prewarmed complete imaging BrainPhys medium (BrainPhys imaging optimized medium + SM1) to yield a final concentration of 5 μM OGB1. Keep the OGB1 solution in the dark or cover the tube with aluminum foil. OGB1 solution must be prepared fresh every time.
-
14Aspirate medium from previously coated imaging plates.
-
15Wash imaging plates once with PBS and aspirate all the solution very gently to avoid cell detachment.
-
16Add 300 μL OGB1 solution (Step 12) to coated coverslips at the center of the imaging plate.
-
17Incubate for 40 min to 1 h at 37 °C and 5% CO2. Incubating plates must be kept in the dark or covered with aluminum foil.
-
18Gently wash imaging plates with imaging medium.
-
19Add 2 mL imaging medium to imaging plates.
-
20Load individual imaging plates on an epifluorescence microscope and record calcium activity at 20× magnification and a frame rate of 20–50 Hz. Note: the higher the acquisition frequency, the higher the ability to deconvolve calcium waves into burst and spikes events.

-
21Record activity for 3–5 min from at least eight to ten fields of view (FOVs) per imaging plate. Note: recordings can last longer; however, when using microscopes that are not equipped with a recording incubation chamber, do not record for longer than 1–1 h 30 min to avoid sample deterioration at room temperature.
-
22Analyze calcium movies:
-
11
Filter images in time and space to improve the signal-to-noise ratio
Identify regions of interest corresponding to individual neurons and extract corresponding single-neuron calcium time courses. This can be performed manually by drawing the region of interest around each neuron or it can be done in an automated manner as detailed in Puppo et al.46, which was adapted from alternative protocols47. We have tested alternative protocols for image analysis, including the CaImAN algorithm from Giovannucci et al.48, which led to similar results
Compute time courses as percent change relative to the baseline (ΔF/F0)
Procedure 4: AAV transduction of cortical organoids
 This does not represent a fully optimized protocol, but rather an adaption from previous protocols that has been adapted to work with our organoids. Further optimization of AAV serotype or dose may be needed.
This does not represent a fully optimized protocol, but rather an adaption from previous protocols that has been adapted to work with our organoids. Further optimization of AAV serotype or dose may be needed.
Media collection
- TIMING 15 min per 6-well plate
-
1One week before the transduction, collect some conditioned M2 medium from the wells with the 45-day-old organoids to be infected. Perform a half media change and transfer the collected spent media into a conical tube, centrifuge, and store conditioned media at 4 °C until use. All the media will be replaced at time of transduction.
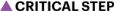 Do not change media later than 2–3 d before transduction.
Do not change media later than 2–3 d before transduction.
-
1
Organoid selection and separation
- TIMING 10 min per well
-
2Select the organoids to be transduced based on their health level (morphology, media consumption, presence of neural rosettes) and batch similarities. Isolate healthy organoids by transferring 20 organoids per well in a 6-well plate. To transfer the organoids, use a wide-O or cut p1000 tip to avoid organoid damage. Keep in the incubator while preparing the AAV mix.
-
2
Preparation of AAV mix
- TIMING 15–30 min
-
3On the day of the infection, thaw the AAV from the −80 °C in regular ice (4 °C) and keep on ice until use.
-
4Mix the AAV with fresh M2 medium and DPBS as follows:
-
3
Determine the number of conditions to test. Make sure to include the appropriate controls (AAV7m8–GFP, AAV7m8–null, nontransduced organoids)
Calculate the total volume. Use a total volume of 2.5 mL per condition (well). The total volume of AAV mix per tested condition will be 2.5 mL multiplied by the number of wells per condition
Calculate the volume of virus based on its titer. The final dose should be 1 × 1010 viral genomes (vg) per well
Use DPBS to dilute the AAV in a total volume of 10 μL per well, according to the titer and the established dose
Add 2,490 μL of M2 medium per well to bring up the AAV mix to its final volume
Example: if the AAv7m8–GCaMP titer is 8.43 × 1012 vg/mL, add 1.19 μL virus, 8.81 μL DPBS and 2,490 μL fresh M2 medium per well. For the control, if the AAV7m8–GFP titer is 6.9 × 1012 vg/mL, add 1.45 μL virus, 8.55 μL DPBS and 2,490 μL fresh M2 per well. Nontransduced organoid media mix would be prepared with 2,490 μL fresh M2 medium and 10 μL DPBS per well.
Transduction
- TIMING 20 min
- 5
-
6Add 2.5 mL of AAV mix to the well.
-
7Incubate at 37 °C and 5% CO2 for at least 5–6 h on an orbital shaker.
-
8After 5–6 h, add 500 μL of conditioned medium per well.
-
9Incubate at 37 °C and 5% CO2 for 2 d.
-
10After 2 d examine organoids under brightfield before changing media:
If organoids do not look healthy (cell death apparent), replace all media with ½ fresh M2 + ½ conditioned M2
- If the organoids look healthy replace only half media with ½ fresh M2 + ½ conditioned M2
-
11Incubate at 37 °C and 5% CO2 and change media every 3–4 d until use.

-
11
Troubleshooting
Troubleshooting advice can be found in Table 2.
Table 2 |.
Troubleshooting table
| Step | Problem | Possible reason | Possible solution |
|---|---|---|---|
| All steps | Contamination | Lapse in sterile handling | Regularly test for mycoplasma |
| Check water sources for contamination and regularly clean water baths and incubator water pans | |||
| Perform regular cleaning, maintenance, and certification of biosafety cabinets and incubators | |||
| Ensure all users are following sterile technique (use 70% vol/vol ethanol spray before moving anything into the hood, regularly change/clean laboratory coats, be careful to not talk while facing an open incubator, proper gloving and degloving technique) | |||
| Procedure 1 | |||
| All steps | EBs/organoids stick or fuse together | Too many EBs/organoids per well | Aspirate joined EBs/organoids. It is usually not possible to separate them once fused, and the mass may cause additional organoids to fuse to it if not promptly removed |
| If using 4 mL of media per well, experiment with decreasing to 3 mL | |||
| If there are too many EBs/organoids and spent media changes color too quickly, split EBs/organoids into more wells (1:2 split is most common) | |||
| Verify that the orbital shaker remains at 95 rpm. Adjacent EBs/organoids can fuse if not under continuous agitation | |||
| Cell death | Dying cells will leak DNA, which is sticky and may cause originally healthy neighboring cells/ organoids to stick together. Be sure to remove poor-quality organoids or dying cells during media changes. The tilt method of media changing (Step 15) allows healthy dense organoids to settle faster than the lighter dying organoids or cell debris, which can then be quickly aspirated away | ||
| Media becomes yellow quickly | Too many EBs/organoids per well | Split EBs/organoids into more wells (1:2 split is most common) | |
| Contamination | Check for mycoplasma contamination | ||
| CO2 sensor needs calibration | Check that the incubator is actually running at 5% CO2—it is possible for sensors to be off, and an incubator running at a slightly higher CO2 level can result in more acidified spent media | ||
| 13 (day 1) | EBs do not form | Contamination or differentiation of iPSCs | Perform mycoplasma testing regularly |
| Perform karyotyping to ensure iPSCs are karyotypically normal. Acquiring mutations that confer proliferative advantages or cause cell population drift in favor of pluripotency can both affect the differentiation potential and efficiency of iPSCs | |||
| Check iPSCs for differentiation and other abnormalities before use for organoid generation | |||
| Be selective when picking iPSCs, picking only iPSCs with good morphology. If there are not enough good iPSCs then continue picking/passaging until sufficient healthy colonies are obtained | |||
| Not enough iPSCs seeded | Be selective when picking iPSCs, picking only iPSCs with good morphology. If there are not enough iPSCs then continue picking/passaging until sufficient healthy colonies are obtained | ||
| If issues persist, try picking 600 fragments when passaging iPSCs before generating organoids. This will result in iPSCs becoming confluent quickly, but will also result in a more spread out/homogeneous plate with less large colonies (which are more likely to have dense areas of iPSCs with slower growth rate) | |||
| Consider adding Y-27632 ROCK inhibitor to iPSC media 1 h before dissociation at 10 μM | |||
| For sensitive iPSC lines that have genotype specific proliferation issues, EBs can be generated using forced aggregation in an AggreWell-800 plate instead of with spontaneous aggregation on the orbital shaker (Supplementary Methods) | |||
| iPSCs were too confluent in plate at time of collection for protocol (growth inhibited) | Perform dissociation when colonies are 80–90% confluent (when they are in log phase growth) | ||
| 16 (day 16 onward) | EBs/organoids disintegrate or are too small | Contamination | Perform mycoplasma testing regularly |
| Check organoid cultures at every media change for signs of contamination visually (turbid media, unusual media color, yeast or fungal growth) and under inverted microscopy at higher magnification (bacterial growth that has directional motion) | |||
| Started protocol with not enough iPSCs or iPSCs were too confluent in plate | Start over and adjust seeding density at next plating | ||
| Use iPSCs in log phase growth. Do not use colonies that are too thin (too sparse) or too overgrown (growth inhibited) | |||
| Being too aggressive with EB/organoid plates (excessive pipetting, splitting organoids too much) | Transfer nondisintegrating EBs/organoids to new wells and ensure sufficient number of organoids per well | ||
| Split only when organoid mass exceeds half the diameter of the well. Splitting too often does not leave enough organoids to properly condition the media | |||
| Perform splitting using a cut p1000 pipette tip or a 5 mL serological pipette, with gentle slow pipetting to not deform the organoids | |||
| Reagent batch issue | Gem21, B27 and SM1 are interchangeable as the media supplement. Experiment with what works best for your iPSC line. This reagent is known to have potential lot-to-lot variability | ||
| iPSC cell line specific issue | For sensitive iPSC lines that have genotype specific proliferation issues, EBs can be generated using forced aggregation in an AggreWell-800 plate instead of with spontaneous aggregation on the orbital shaker (Supplementary Methods) | ||
| For difficult-to-neuralize iPSC lines, try using a different clonal line | |||
| For difficult-to-neuralize iPSC lines, consider extending Neural Induction (Procedure 1, Step 14). Step 14 can be extended from 6 d up to 12 d | |||
| 18 (days 28–30) | Rosettes not observed (Extended Data Fig. 3) | Internal rosettes are difficult to see | Validate lack of rosettes with immunohistochemistry, as rosettes may be internalized. If using brightfield, adjust microscope settings to see if the image resolves to show structures within the organoid |
| Inefficient neural induction, specification, or patterning | Try to have enough organoids per well to condition the media (turn the media yellow) when it comes time to change media. This is important for maintaining the interorganoid secretion of growth factors that help the organoids effectively neuralize. This will change depending on which step of the protocol you are on, and you may not observe full media conditioning on daily media change steps | ||
| For iPSC lines or genetic mutations that repeatedly are difficult to neuralize, consider extending the neural induction step from 6 d to up to 12 d, to ensure sufficient dual SMAD inhibition and the initiation of neural differentiation | |||
| See previous solution for EBs not forming | |||
| Reagent batch issue | Check efficacy of supplements, make sure supplements are not left in refrigerator for more than a week, minimize time supplements spend outside of the refrigerator or are exposed to direct light | ||
| Incubator issue | Check CO2 and water/humidity levels in the incubator. Slightly higher CO2 levels that acidify the media quicker or a too dry incubator will adversely affect organoid differentiation and rosette formation | ||
| Procedure 2 | |||
| 9 (MEA) | Poor organoid attachment onto MEA plate | Problem with coating | When changing media, add 1:1,000 laminin to the media to try to help with reattachment |
| In subsequent plating, Increase the concentration of laminin to 1:50 or 1:25 to help attachment | |||
| In subsequent plating, add a preconditioning step to increase the MEA surface hydrophilicity: treat the surface with freshly prepared 0.07% Terg-a-zyme solution and/or 70% ethanol at room temperature for 1 h; then add fresh culturing media to each well and incubate for 2 d | |||
| Organoids are too big | See Procedure 2, Step 7 and experiment with the amount of gentle trituration needed without breaking up the organoid | ||
| If organoids are too big because later stage/older organoids are being plated, try plating closing to the 1-month-old timepoint to get better attachment, and allow them to mature in plate until desired assay timepoint | |||
| If organoids are too big because they are created with AggreWell using forced aggregation, experiment with seeding fewer cells to create smaller starting organoids | |||
| Organoids are unhealthy | Plate only rosetted organoids that do not exhibit cell death. In the 6-well plate that these organoids are being selected from, there should not be a large amount of cell debris, or neighboring organoids that are breaking apart in the well | ||
| 18 (MEA) | Organoids show low activity | Unhealthy or not well-developed organoids | Plate only rosetted organoids that do not exhibit cell death. Using organoids with well-developed and visible rosettes is important |
| Assess dead cells in the media. After initial plating there should be low cell death | |||
| Due to the higher volume of media relative to the amount of organoids per well of an MEA plate, conditioning of the media may not produce a readily visible change in the phenol red indicator in the spent media. In addition to no or very low cell death and debris observed, operators can look for cell detachment as a negative indicator and neuronal cell outgrowth to cover the treated cell culture surface as a positive indicator of continued cell health. In long-term culture, healthy organoids should demonstrate steady or increasing measurable spontaneous electrical activity | |||
| Poor organoid attachment onto MEA plate | Check the organoids’ position. If the organoids and their network are not in contact with any electrodes, the plate will not record any activity. Improve organoid attachment over the electrode by adding the primary and secondary coating (5–10 μL solution) only onto the active area (electrode array) of the well | ||
| See additional solutions for poor organoid attachment above | |||
| Other factors | If antibiotics were added, remove antibiotics from the culture media. Penicillin/streptomycin can affect neuronal development and activity | ||
| If organoids demonstrate zero activity at 3 months old, start with a new batch | |||
| Procedure 3 | |||
| 20 (Calcium imaging) | Low fluorescence signal | Unhealthy cells | Check the level of health of your cells. There should not be cell debris, or cells that shrink when dye loaded (dying cells are small, round and absorb more dye, becoming abnormally bright) |
| Poorly labeled cells | Prepare your dye solution fresh every time. Warm only enough dye solution for the imaging experiment. Do not re-use previously heated dye solution | ||
| Make sure your dye has not expired | |||
| Check the concentration of your dye solution and try to increase it | |||
| Increase the incubation time | |||
| Incorrect imaging settings | Make sure your optical setup is correct: check the filter settings and make sure they fit the fluorescent dye spectral properties (Oregon Green 488 BAPTA-1 is excited at 494 nm and imaged at 523 nm); check the power of your light source | ||
| Make sure you do not bleach your dye by exposing the cells to extended and/or too intense light stimulation | |||
| Procedure 4 | |||
| 11 (AAV transduction) | Low expression | Unhealthy cells | Check the level of health of your organoids (e.g., is there higher than normal cell shedding, are the organoids consuming media, etc.) |
| Organoid age | AAV transduction in early stage organoids (<4 weeks) can lead to poor expression levels (AAV does not integrate into the cell DNA); wait for older and more developed organoids where the number of mature neurons is higher (5 weeks and older) | ||
| Low transgene expression | Check that vector promoter is appropriate to experimental purpose, e.g., cell specific, constitutive or ubiquitous promoters | ||
| Check the AAV serotype is appropriate to your target cell type | |||
| Increase dose and/or allow more time after transduction before analyzing | |||
| Store AAV appropriately (see next solution) | |||
| Poor AAV storage | Avoid repeated freeze–thaw by storing the AAV in single-use aliquots | ||
| Store the AAV at −80 °C | |||
| Keep the AAV on ice all the time once thawed | |||
Timing
Procedure 1: organoid generation
Reagent setup, preparing Matrigel plates: 1 h to overnight
Steps 1–5, Maintenance and passaging of iPSCs: passage timing, 10–15 min per plate; 2 passages from frozen cells, ~2 weeks
Steps 6–10, Dissociation of iPSCs: 30 min
Steps 10–12, Generation of 3D EBs: 30 min
Step 13, Beginning of neural induction: 3 d
Step 14, Neural induction: 6 d
Step 15, Neural proliferation A: 7 d
Step 16, Neural proliferation B: 6 d
Step 17, Neural differentiation: 6 d
Steps 18–20, Extended neural maturation, maintenance and long-term culture: up to 18 months
Procedure 2: MEA
Steps 1–3, Preparing MEA plates: 2 d
Steps 4–9, Plating organoids on MEA plates: <30 min per plate
Steps 10–11, Changing media on MEA plates: once per week
Steps 12–18, Measuring electrical activity: 30 min per plate
Steps 19–20, Quantifying electrical activity: 1 h per plate
Step 21, Detecting emerging neural oscillations: variable depending on parameters chosen
Procedure 3: calcium imaging
Steps 1–3, Preparing calcium imaging plates: 2 d
Steps 4–10, Plating organoids on calcium imaging plates: <30 min per plate
Steps 11–22, Record calcium activity: 1 h per plate, plus analysis time
Procedure 4: AAV transduction of cortical organoids
Step 1, Media collection: 15 min per 6-well plate
Step 2, Organoid selection and separation: 10 min per well
Steps 3–4, Preparation of AAV mix: 15–30 min
Steps 5–11: Transduction: 20 min
Anticipated results
Cell composition
One 6 cm plate of iPSCs usually yields at least 500 organoids ~0.5–1 mm in diameter 1 month after EB aggregation and neural induction. Organoids should exhibit a relatively spherical morphology without pronounced protrusions or appendages. Organoids 3–5 weeks old should exhibit ‘splotchy’, circular discolorations under confocal microscopy, indicative of neural rosettes (Fig. 1a,c and Extended Data Fig. 3). Older organoids should appear mostly dark. If imaged via immunohistochemistry, organoids should exhibit neural rosettes consisting of a central proliferative region expressing Ki67 and nestin, lower (TBR1 and CTIP2) and upper (CUX1 and SATB2) cortical layer neurons, glial cells (GFAP) and GABAergic (CR) neurons over time (Fig. 1b,c).
Electrical activity
Measurable spontaneous neural spikes should be observable in organoids plated to MEA plates, starting when the organoids are 2.5–3 months old (Fig. 2c,d), and are being cultured in complete BrainPhys medium. Over time, spontaneous neural spikes will coordinate into measurable spontaneous electrical bursts. Spiking and bursting activity will emerge and increase as the organoids increase in age (Fig. 2d). Aberrations in spiking and bursting emergence, frequency, pattern and development over time in certain disease genotype iPSC-derived organoids, relative to appropriate control iPSC-derived organoids, may be observable and measurable. The effects of small-molecule, pharmaceutical compound or toxin exposure may modulate electrical activity and may be measured through MEA recordings pre- and posttreatment. The Axion Biosystems Maestro Pro enables electrical recordings at two filters: neural high-frequency broadband (100–400 Hz) and LFP lowpass/bandpass (0–4 Hz) (Fig. 2b). Recordings using the neural filter allow the capture and quantification of neural spikes and bursts. Recordings using both the neural and LFP filters allow the capture and quantification of emerging oscillatory waves in organoids at later timepoints, using the custom MEA analysis code in Procedure 2: Step 21. LFPs arise at ~6 months, concomitant with development of functional inhibitory interneurons and release of GABA at 6 months (Fig. 2d). This indicates an increased complexity in the communication between neurons, demonstrating enhanced maturation relative to 2D models. To explore the development of these LFPs, EEG features (time of oscillations, time between oscillations, voltage of oscillations, etc.) from preterm neonates was used in a custom unbiased machine learning approach to develop a regularized regression model to predict a developmental ‘age’ of neonates from their EEG features. This model was then applied to the analogous MEA LFP metrics to predict the developmental age of organoids and other neural models. From this, aged organoids (>25 weeks old) exhibit a machine learning-predicted age that closely matches the age of neonate brains during neurodevelopment (Fig. 2e). Notably, this high degree of correlation between predicted age and actual neonate age is recapitulated only in this human organoid model, not in mouse, 2D or other 3D models (Fig. 2f).
Organoids can be plated on Axion Biosystems MEA plates and glass-bottom imaging plates to perform in-parallel analysis of network and single-cell-level activity. After adhesion and growth, organoids form a dense network of neurites and mature neurons, which can be easily stained with commonly used synthetic calcium indicators and imaged with traditional epifluorescence microscopy, as we have previously demonstrated46 (Fig. 3a–c, Procedure 3: calcium imaging). Interestingly, Ca2+ imaging using a synthetic dye show that neurons that have migrated from the organoid and interconnect adjacent organoids reveal much more complex Ca2+ patterns compared with 2D cultures of dissociated human neurons (Supplementary Videos 2 and 3). Single-cell calcium spiking events in neurons from organoids show intense flashes of calcium activity extended to the entire FOV, showing a level of synchrony not observed in 2D-dissociated neurons (Fig. 3c and Supplementary Video 3). In agreement with MEA results, this demonstrates the spontaneous formation of complex network configurations, which give rise to highly synchronized activity in interconnected organoids.
Extended Data
Extended Data Fig. 1 |. Third-party comparison of different brain organoid protocols and the fetal human brain.
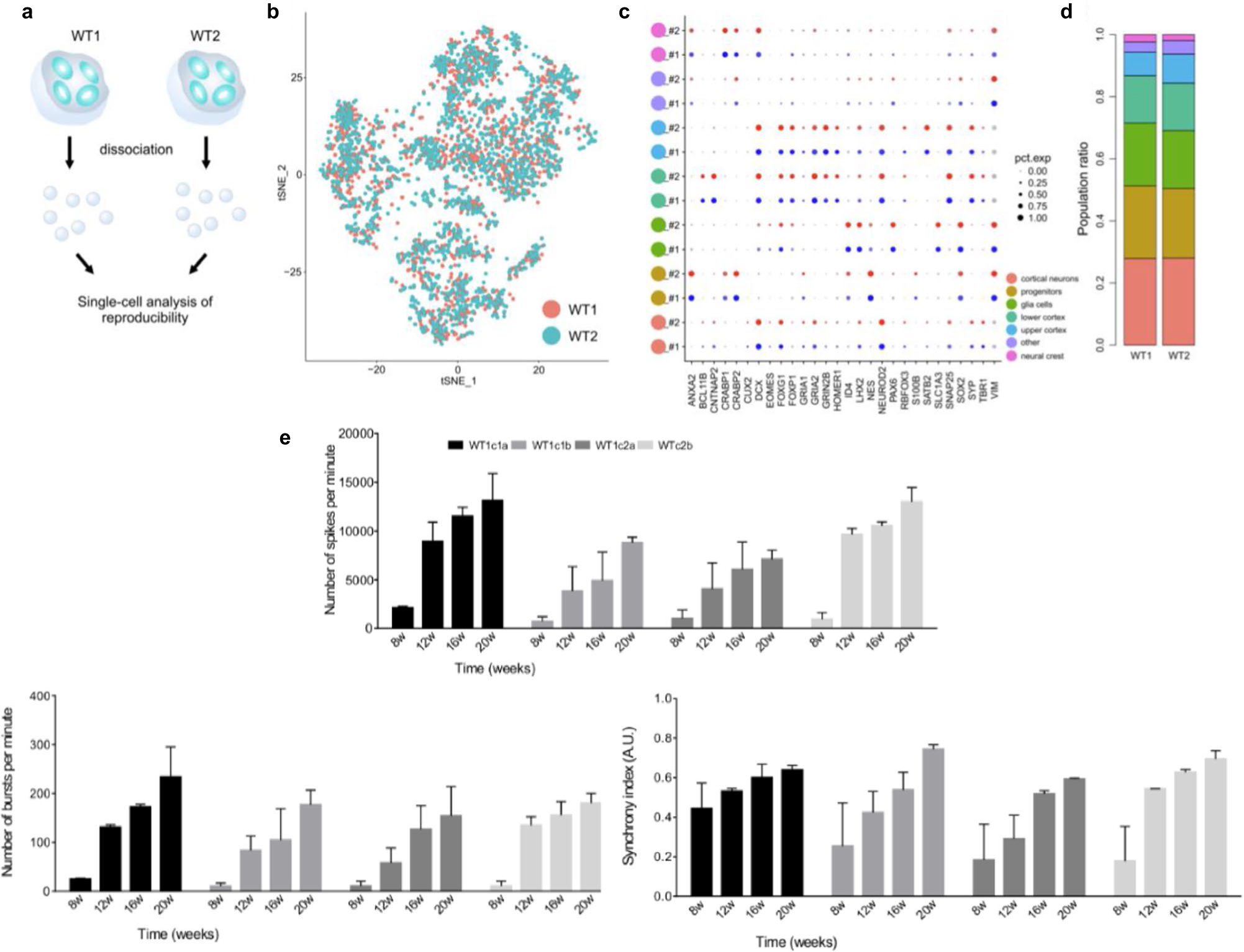
(a) Schematic View of the Culture System Generating human cortical organoids (hCOs). Guided protocols originated from Eiraku et al.5 while non-guided protocols are from Lancaster et al.19. Timeline of neural induction, differentiation, and maturation step is shown across protocols. Note that, while we have use the term ‘semi-guided’ here to describe our protocol and distinguish it from other Eiraku et al.5 -derived directed protocols, all panels in this figure are adapted from ref. 8, which only utilized the terms ‘guided’ and ‘unguided’ and thus correctly classified our protocol7 as guided. (b–d) Shared-nearest-neighbors (SNN) graph visualization for differentiation trajectory. (b) Differentiation directions (arrows) were determined by pseudotime. (c) Estimated trajectory backbone from the SNN graph. (d) Comparison of differentiation trajectory among different protocols. (e) The presence of cell types in each organoid protocol and human fetal brain. Cell types with >0.25% of cells are denoted with a plus sign. F, Fiddes et al.17; V, Velasco et al.6; B, Birey et al.14; M, Madhavan et al.18; T, Trujillo et al.7; X, Xiang et al.15; Q, Quadrato et al.20; G, Giandomenico et al.50. (f) Enrichment of disease-related genes in each organoid protocol. The red boxes indicate data generated from the protocol presented here. Figure adapted with permission from ref. 8, Elsevier.
Extended Data Fig. 2 |. Molecular and functional reproducibility of cortical organoids.
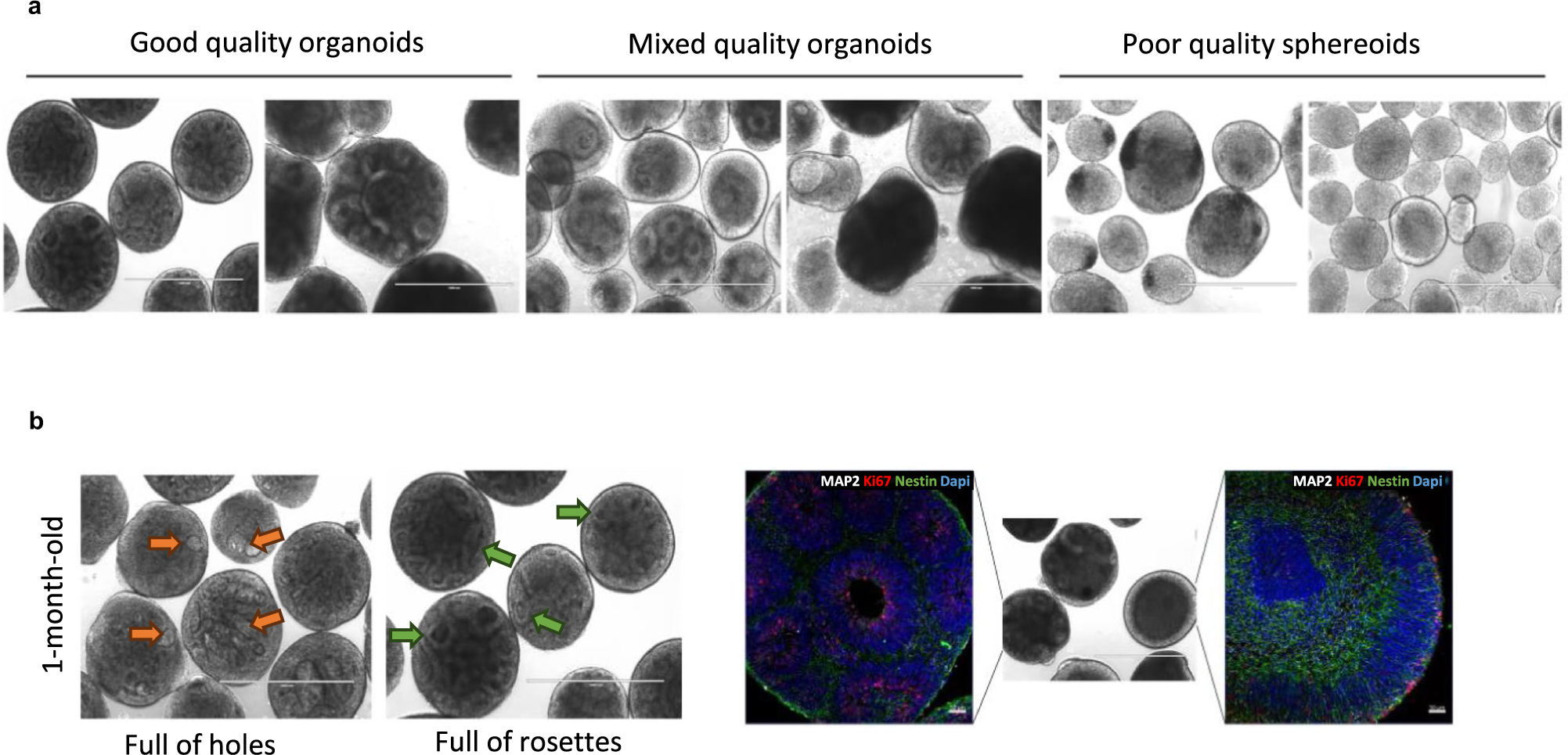
(a) Schematic showing the single-cell approach performed to assess the reproducibility of organoid generation using different iPSC lines (WT1 and WT2). (b) tSNE plot of single-cell mRNA sequencing data from two 6-month-old organoids. (c) Expression of gene markers for various cell types both batches. (d) Population ratio of each cluster by replicate. (e) Consistent and reproducible development of electrical activity in organoids over time across four cell lines, bars represent mean ± s.e.m (n = 8, independent experiments performed in duplicates using two clones of an iPSC line).
Extended Data Fig. 3 |. Selection of organoids for plating.
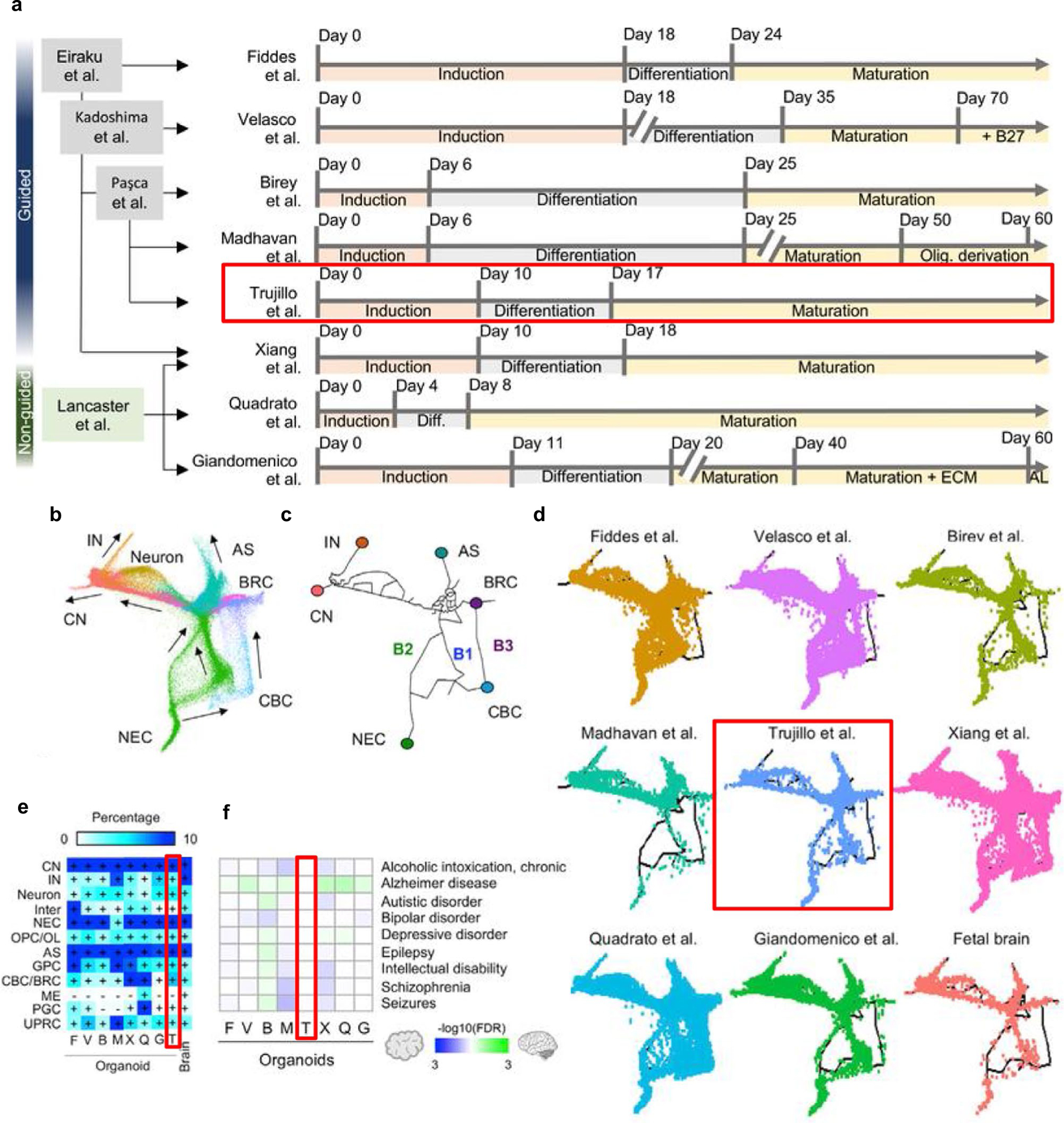
(a) Good-quality 1-month-old organoids with visible spatial arrangement of neural rosette structures. Scale bar, 1,000 μm. Mixed quality organoids, where there is a mixed population of fully differentiated organoids with visible rosette structures and incomplete differentiated organoids—select rosetted organoids only for downstream assays and discard incompletely differentiated organoids. Poor-quality spheroids that did not efficiently neuralize and differentiate into rosetted organoids—discard and start over. (b) Distinguish proper spatial organization and structural development in organoids; orange arrows indicate holes and green arrows indicate rosettes. Scale bar, 1,000 μm. (c) Use of immunohistochemistry to distinguish between good-quality organoids with spatial neural rosette arrangement (smaller circles within the organoid) and incompletely differentiated organoids that contain neurons but lack neural rosette structures and spatial organization. Immunostainings showing nuclei (DAPI), neuron microtubules (MAP2), and proliferating NPCs (Ki67 and Nestin). Scale bar, 50 μm.
Supplementary Material
Key points.
This protocol describes the generation of cortical organoids with complex neural oscillations using a ‘semi-guided’ approach, as well as their functional characterization through microelectrode array measurements, calcium imaging and adeno-associated virus transduction.
The ‘semi-guided’ nature of this approach allows an intermediate between the cellular heterogeneity of unguided approaches and the predictability of guided approaches, which we speculate underlies the emergence of complex oscillations.
Acknowledgements
The authors thank I.-H. Park and colleagues for approving the inclusion of published data (Supplementary Fig. 3) adapted from their figure list in Tanaka et al.8. Additionally, we acknowledge the Muotri Lab for their discussion on best organoid culture practices. This work was supported by US National Institutes of Health grants (R01MH100175, R01NS105969, MH123828, R01NS123642, R01MH127077, R01ES033636, R21MH128827, R01AG078959, R01DA056908, R01HD107788, R01HG012351, R21HD109616, R01MH107367), a California Institute for Regenerative Medicine grant (DISC2-13515) and a grant from the Department of Defense (W81XWH2110306).
Footnotes
Code availability
Code for MEA processing can be found at https://github.com/voytekresearch/OscillatoryOrganoids/blob/master/organoid_EEG_age_regression.ipynb.
Competing interests
A.R.M. is a cofounder of and has an equity interest in TISMOO, a company dedicated to genetic analysis and brain organoid modeling focusing on therapeutic applications customized for autism spectrum disorder and other neurological disorders with genetic origins. The terms of this arrangement have been reviewed and approved by the University of California San Diego in accordance with its conflict-of-interest policies. A.R.M. is an inventor of several patents related to human functional brain organogenesis, including the protocol described here.
Extended data is available for this paper at https://doi.org/10.1038/s41596-024-00994-0.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41596-024-00994-0.
Data availability
Further information for Figs. 1d,e and 2c–f can be found in Trujillo et al.7. Sequencing from Trujillo et al. for Fig. 1 can be found at National Center for Biotechnology Information Gene Expression Omnibus: GSE130238. Further information for Extended Data Fig. 3d–f can be found in Tanaka et al.8.
References
- 1.Scuderi S, Altobelli G, Cimini V, Coppola G & Vaccarino F Cell-to-cell adhesion and neurogenesis in human cortical development: a study comparing 2D monolayers with 3D organoid cultures. Stem Cell Rep. 16, 264–280 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centeno E, Cimarosti H & Bithell A 2D versus 3D human induced pluripotent stem cell-derived cultures for neurodegenerative disease modelling. Mol. Neurodegener 13, 27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adlakha Y Human 3D brain organoids: steering the demolecularization of brain and neurological diseases. Cell Death Discov. 9, 221 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grenier K, Kao J & Diamandis P Three-dimensional modeling of human neurodegeneration: brain organoids coming of age. Mol. Psychiatry 25, 254–274 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Eiraku M et al. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 3, 519–532 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Velasco S et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 570, 523–527 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trujillo C et al. Complex oscillatory waves emerging from cortical organoids model early human brain network development. Cell. Stem Cell 25, 558–569.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka Y, Cakir B, Xiang Y, Sullivan G & Park I Synthetic analyses of single-cell transcriptome from multiple brain organoids and fetal brain. Cell Rep. 30, 1682–1689 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trujillo C et al. Reintroduction of the archaic variant of NOVA1 in cortical organoids alters neurodevelopment. Science 371, eaax2537 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papes F et al. Transcription factor 4 loss-of-function is associated with deficits in progenitor proliferation and cortical neuron content. Nat. Commun 13, 2387 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allison T et al. Defining the nature of human pluripotent stem cell-derived interneurons via single-cell analysis. Stem Cell Rep. 16, 2548–2564 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delgado R et al. Individual human cortical progenitors can produce excitatory and inhibitory neurons. Nature 601, 397–403 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zourray C, Kurian M, Barral S & Lignani G Electrophysiological properties of human cortical organoids: current state of the art and future directions. Front. Mol. Neurosci 15, 839366 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birey F et al. Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiang Y et al. Fusion of regionally-specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell 21, 383–398.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pasca A et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 12, 671–678 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fiddes I et al. Human-specific NOTCH2NL genes affect notch signaling and cortical neurogenesis. Cell 173, 1356–1369.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madhavan M et al. Induction of myelinating oligodendrocytes in human cortical spheroids. Nat. Methods 15, 700–706 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lancaster M et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quadrato G et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 545, 48–53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharf T et al. Functional neuronal circuitry and oscillatory dynamics in human brain organoids. Nat. Commun 13, 4403 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uhlhaas P, Roux F, Rodriguez E, Rotarska-Jagiela A & Singer W Neural synchrony and the development of cortical networks. Trends Cogn. Sci 14, 72–80 (2010). [DOI] [PubMed] [Google Scholar]
- 23.de Hemptinne C et al. Therapeutic deep brain stimulation reduces cortical phase-amplitude coupling in Parkinson’s disease. Nat. Neurosci 18, 779–786 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uhlhaas PJ & Singer W Abnormal neural oscillations and synchrony in schizophrenia. Nat. Rev. Neurosci 11, 100–113 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Khan S et al. Local and long-range functional connectivity is reduced in concert in autism spectrum disorders. Proc. Natl Acad. Sci. USA 110, 3107–3112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dixon T & Muotri A Advancing preclinical models of psychiatric disorders with human brain organoid cultures. Mol. Psychiatry 28, 83–95 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Samarasinghe R et al. Identification of neural oscillations and epileptiform changes in human brain organoids. Nat. Neurosci 24, 1488–1500 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lavazza A ‘Consciousnessoids’: clues and insights from human cerebral organoids for the study of consciousness. Neurosci. Conscious 7, niab029 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams J et al. Impact of alcohol exposure on neural development and network formation in human cortical organoids. Mol. Psychiatry 28, 1571–1584 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mesci P et al. Modeling neuro–immune interactions during Zika virus infection. Hum. Mol. Genet 27, 41–52 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schley L Meet the scientists connecting lab-grown ‘mini brains’ to robots. Discover Magazine https://www.discovermagazine.com/mind/meet-the-scientists-connecting-lab-grown-mini-brains-to-robots (2019). [Google Scholar]
- 32.Marinho L et al. The impact of antidepressants on human neurodevelopment: brain organoids as experimental tools. Semin. Cell Dev. Biol 144, 67–76 (2023). [DOI] [PubMed] [Google Scholar]
- 33.Trujillo C et al. Pharmacological reversal of synaptic and network pathology in human MECP2-KO neurons and cortical organoids. EMBO Mol. Med 13, e12523 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adams JW, Cugola FR & Muotri AR Brain organoids as tools for modeling human neurodevelopmental disorders. Physiology 34, 365–375 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coelho L & Muotri A Cortical brain organoid as a model to study microgravity exposure. Artif. Organs 47, 5–7 (2023). [DOI] [PubMed] [Google Scholar]
- 36.Standards document. International Society for Stem Cell Research https://www.isscr.org/standards-document (2023).
- 37.Lin M & Schnitzer M Genetically encoded indicators of neuronal activity. Nat. Neurosci 19, 1142–1153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Avansini S et al. Junctional instability in neuroepithelium and network hyperexcitability in a focal cortical dysplasia human model. Brain 145, 1962–1977 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dalkara D et al. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci. Transl. Med 5, 189ra76 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Duong T et al. Comparative AAV–eGFP transgene expression using vector serotypes 1–9, 7m8, and 8b in human pluripotent stem cells, RPEs, and human and rat cortical neurons. Stem Cells Int. 2019, 7281912 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu D et al. Overexpressing NeuroD1 reprograms Müller cells into various types of retinal neurons. Neural Regen. Res 18, 1124–1131 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garita-Hernandez M et al. AAV-mediated gene delivery to 3D retinal organoids derived from human induced pluripotent stem cells. Int. J. Mol. Sci 21, 994 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McClements M et al. Tropism of AAV vectors in photoreceptor-like cells of human iPSC-derived retinal organoids. Transl. Vis. Sci. Technol 11, 3 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zarowny L et al. Bright and high-performance genetically encoded Ca2+ indicator based on mneongreen fluorescent protein. ACS Sens. 5, 1959–1968 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Piedra J et al. Development of a rapid, robust, and universal picogreen-based method to titer adeno-associated vectors. Hum. Gene Ther. Methods 26, 35–42 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Puppo F et al. All-optical electrophysiology in hiPSC-derived neurons with synthetic voltage sensors. Front. Cell Neurosci 15, 671549 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mukamel E, Nimmerjahn A & Schnitzer M Automated analysis of cellular signals from large-scale calcium imaging data. Neuron 63, 747–760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giovannucci A et al. CaImAn an open source tool for scalable calcium imaging data analysis. eLife 8, e3817343 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gordon A et al. Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat. Neurosci 24, 331–342 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giandomenico S et al. Cerebral organoids at the air-liquid interface generate diverse nerve tracts with functional output. Nat. Neurosci 22, 669–679 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Key references
- Trujillo C et al. Cell Stem Cell. 25, 558–569.e7 (2019): 10.1016/j.stem.2019.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo C et al. Science 371, eaax2537 (2021): 10.1126/science.aax2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papes F et al. Nat. Commun 13, 2387 (2022): 10.1038/s41467-022-29942-w [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Further information for Figs. 1d,e and 2c–f can be found in Trujillo et al.7. Sequencing from Trujillo et al. for Fig. 1 can be found at National Center for Biotechnology Information Gene Expression Omnibus: GSE130238. Further information for Extended Data Fig. 3d–f can be found in Tanaka et al.8.