

Abstract
Despite the increasing demand for efficient and sustainable chemical processes, the development of scalable systems using biocatalysis for fine chemical production remains a significant challenge. We have developed a scalable flow system using immobilized enzymes to facilitate flavin-dependent biocatalysis, targeting as a proof-of-concept asymmetric alkene reduction. The system integrates a flavin-dependent Old Yellow Enzyme (OYE) and a soluble hydrogenase to enable H2-driven regeneration of the OYE cofactor FMNH2. Molecular hydrogen was produced by water electrolysis using a proton exchange membrane (PEM) electrolyzer and introduced into the flow system via a designed gas membrane addition module at a high diffusion rate. The flow system shows remarkable stability and reusability, consistently achieving >99% conversion of ketoisophorone to levodione. It also demonstrates versatility and selectivity in reducing various cyclic enones and can be extended to further flavin-based biocatalytic approaches and gas-dependent reactions. This electro-driven continuous flow system, therefore, has significant potential for advancing sustainable processes in fine chemical synthesis.
Subject terms: Biocatalysis, Immobilized enzymes, Oxidoreductases, Flow chemistry
Flavin-based biocatalysis using flavin mononucleotide (FMN) cofactor attracts significant attention for its application in asymmetric alkene reduction and various other reactions, however, the scale-up of flavin-based biocatalysis in flow remains unexplored. Here, the authors develop a closed-loop flow platform for H2-driven regeneration of cofactor FMNH2 and ene-reduction using immobilized Old Yellow Enzyme, achieving >99% conversion of ketoisophorone to levodione.
Introduction
Continuous flow biocatalysis is increasingly recognized as a sustainable manufacturing methodology within the pharmaceutical and fine chemicals sectors1–3. It offers improved control over reactions, minimizes waste generation, and enhances efficient use of energy4. The application of such systems is not limited to conventional chemical synthesis, it can be adapted to biocatalysis, using enzymes as catalysts to enable efficient syntheses under mild conditions5. Biocatalysis in continuous flow has become more feasible due to the advancements in flow chemistry and enzyme immobilization techniques6,7, harnessing the highly stereo- and regioselective catalytic power of enzymes. One of the most useful reactions for generating stereogenic centers involves the asymmetric reduction of activated C=C bonds8,9. This ene-reduction can be catalyzed by an ene reductase from the Old Yellow Enzyme family (OYE)10, which contains a prosthetic flavin mononucleotide (FMN) cofactor. Through trans-hydrogenation, OYE catalyzes the asymmetric reduction of α,β-unsaturated carbonyl compounds11. The commonly recognized process of regenerating enzyme-bound FMNH2 involves the reduced nicotinamide cofactor NAD(P)H, although OYE shows promiscuity with respect to the sources of reductant12–14, including receiving electrons from reduced free-flavins10,15,16. Flavin-based biocatalysis has recently attracted considerable attention not only for its application in C=C double bond reduction, but also for its applicability in epoxidation, hydroxylation and nitro group reduction, due to its low-cost cofactor compared to NAD(P)H17–19. Consequently, alternative regeneration methods such as photochemical and electrochemical approaches have been reported to regenerate the free flavins. These methods, however, are constrained by limitations, such as low conversion rates and stability20–24. In vivo ene-reduction in continuous flow using OYEs has demonstrated high conversion rates25,26. However, in vivo reactions pose challenges in downstream processing, as unwanted materials or contaminants can be co-purified. In- vitro biocatalysis in flow chemistry can significantly simplify the downstream process, and the potential for scaling up flavin-based biocatalysis through the application of flow chemistry remains unexplored4.
Molecular hydrogen (H2) serves as an ideal reductant since it does not produce any waste and can be produced by electrolysis of water using renewable energy. H2 can be reversibly oxidized by the soluble hydrogenase of Cupriavidus necator (SH) to reduce NAD+ to NADH27. This process involves a hydrogenase module, HoxHY, for H2 oxidation and a reductase module, HoxFUI2, for NAD+ reduction. Due to its 100% atomic efficiency28, SH/H2 presents as an excellent system for NADH cofactor regeneration29 compared to existing NAD(P)H-recycling systems, which often suffer from low activity or produce unwanted side products30. The native NAD+-reducing ability of SH can be further extended to NADPH through rational mutagenesis, allowing H2-driven NADPH regeneration31. Additionally, SH exhibits tolerance to ambient O2 condition32, making it a valuable tool for the recycling of NAD(P)H in various biocatalytic reactions for the production of amines and alcohols33–35. The application of hydrogenases has also been explored for regenerating synthetic analogous of NAD(P)H14 and reduced flavin cofactors17–19. Coupling of SH with the thermostable OYE from Thermus scotoductus (TsOYE)36 and styrene monooxygenase allowed the regeneration of FMNH2 for the reduction of C=C double bonds, and reduced flavin adenine dinucleotide (FADH2) for the epoxidation of styrenes, respectively, in small-volume batch reactions17. This work shows that SH is an atom-efficient regeneration system that uses H2 as electron source for regenerating not only NAD(P)H but also flavin cofactors. Compared to Hyd1 from E. coli18, which lacks a reductase module, the SH demonstrates nearly two orders of magnitude higher specific activity (5.8 U mg−1) for FMN reduction17. This indicates a significant contribution of the reductase module of SH to free FMN binding and reduction37,38.
The in- situ production of H2 by water electrolysis effectively minimizes the risks associated with handling large quantities of explosive H2. Photoelectrochemical oxidation of water has been applied to fuel enantioselective reduction of TsOYE as a cathodic reaction39. However, it has a limited efficiency due to low current densities (<1 mA cm−2) when coupling the photocatalysts with the biocatalysts, posing challenges for upscaling the reaction40. The use of electrical energy to drive NADH-dependent biocatalytic processes in a flow system, using H2 as a mediator, has been previously successfully demonstrated. In this process, electricity was used to split water via a simple two-electrode system to produce H2 and O241. The hydrogen gas was then introduced into the flow system by liquid-to-liquid gas transfer, facilitated by gas-permeable Teflon™ AF-2400 tubing41,42. Additionally, a commercial continuous-flow hydrogenation reactor has been applied in flow biocatalysis, designed for optimizing H2 availability which may become a relevant efficiency factor43. In this work, a simple and cost effective method of supplying different gases to the flow system was developed by integrating gas permeable tubing into a closed loop system. This approach allows continuous and efficient delivery of H2 and reduced cofactors without damaging the biocatalysts. For energy-efficient water electrolysis, proton exchange membrane (PEM) electrolyzer provide a suitable method to produce pure H2 with >60% voltage efficiency and >95% faradaic efficiency44. Compared to standard alkaline water electrolyzers, PEM electrolyzers can operate in high current densities, have instant current response, and can be coupled with renewable energy sources45. To introduce H2 produced from the PEM electrolyzer via gas-to-liquid transfer without bubble formation, tube-in-tube methods or the use of gas-permeable tubes in a gas addition module have been selected for application in flow chemistry46,47.
In this study, the main novelty is in development of a closed-loop flow platform for electro-driven flavin-dependent biocatalysis via H2 as a mediator produced by a commercial PEM electrolyzer in combination with a gas addition module. Thermostable TsOYE, as a reduced flavin-dependent model enzyme, was immobilised via coordination bonds and coupled to SH. To enable regeneration of the flavin cofactor, SH was immobilised by affinity binding and applied to the flow system. This system, which facilitates efficient gas-to-liquid H2 transfer, was thoroughly evaluated for biocatalyst immobilization, system stability, reusability, and adaptability with different substrates, establishing its potential for technical-scale flavin-dependent biocatalytic reactions.
Results and discussion
Gas addition module design and H2 supply for H2-driven flow biocatalysis
A closed-loop flow system was developed to serve as a versatile platform for electro-driven enzymatic reactions (Fig. 1). This system contained the H2-dependent SH for the recycling of reduced flavin FMNH2. H2 was supplied from water electrolysis by a commercially available lab-scale PEM electrolyzer with an IrRuOx anode and a mixed PtB/PtC (platinum black, platinum-supported carbon) cathode. The evolved H2 was fed into the flow system through gas-permeable membrane tubing within a constructed metal-encased gas addition module (see SI chapter 3). For H2 transfer from gas-to-liquid phase in the gas addition module, tubes made of polymethylvinylsiloxane (PVMS) with a high gas permeability and polytetrafluoroethylene (PTFE) as inert material were evaluated41,48. PVMS tubes with a membrane thickness of 250 µm and a length of 2 m rolled up in the gas addition module showed an H2 transfer rate of 0.647 µmol min−1 at 1 bar H2 pressure in a single pass through the tubing (see SI chapter 4). Therefore, it was favored for its high gas transfer efficiency. Conversely, PTFE tubing with a lower H2 transfer rate of 0.418 µmol min−1 was used as a non-reactive alternative in cases where chemical interactions with PVMS were observed.
Fig. 1. Platform for electro-driven FMNH2-dependent asymmetric reduction of cyclic enones.
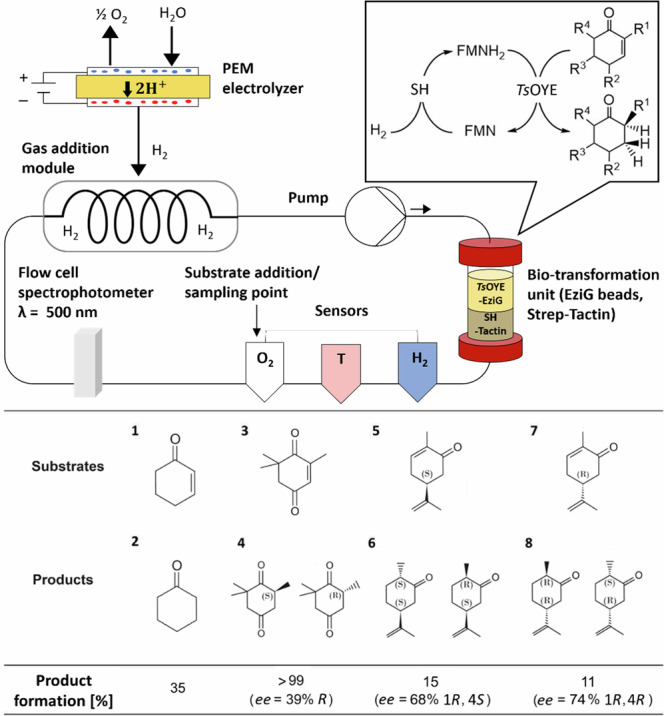
H2 is produced from a PEM electrolyzer using PtB/PtC as hydrogen evolution catalyst. A gas permeable tubing (PVMS or Teflon) transfers the H2 from the gas addition module to the flow system (17 mL, flow rate 2.6 mL min−1). H2 was supplied during biocatalysis to the gas addition module from the PEM electrolyzer (H2 10 mL min−1, 3.4 V, 0.89 A). Clark-type sensors for H2, optical sensors for O2, and temperature sensors and spectrophotometer (FMN) were integrated into the flow system to monitor online the interplay between enzymes and the electrolyzer. Due to overlapping of the substrates and FMNH2 absorbance peak, wavelength of 500 nm was used for FMN detection. The enzymes SH, and TsOYE were immobilized by Strep-Tactin XT 4Flow (down) and EziG beads (up), respectively, and packed into a column within the flow system. Reaction conditions: 17 mL, 50 mM Tris-HCl pH 8 at 20 °C containing SH (5 mg), TsOYE (6.5 mg), FMN (1 mM), catalase (5 mg) substrate 1 and 3 (25 mM), substrate 5 and 7 (5 mM). DMF as a cosolvent was added with the substrate at the ratio of 1:2. Conversions and ee values were determined by GC-FID. The flow system was at room temperature and the column with immobilized biocatalysts in temperature-controlled chamber was set at 30 °C. For upscale reaction, 3 was used at 18.5 mM concentrations.
Closed-loop flow biocatalysis reactions with immobilized enzymes
For facilitated product isolation and allowing reusability of enzymes in the flow setup, immobilization of SH and TsOYE simplifies product isolation and enables the recycling of enzymes within a flow system. However, adsorption of FMN was detected on the material Amberlite FP54TM, previously used in flow setup for SH immobilization49. Therefore, the Strep-Tactin XT 4flow resin was assessed as a potential alternative for application in flow chemistry, aimed at Strep-tagged SH immobilization (SH-Tactin)32,50.
Analogously, TsOYE was equipped with a 6xHis-tag on the N-terminal site and was immobilized on porosity glass metal ion affinity EziG beads from EnginZyme51. Different types of EziG beads varying in hydrophilicity were evaluated for TsOYE immobilization to assess performance (see SI chapter 5). Employing affinity immobilization techniques, His-tagged TsOYE with EziG beads and strep-tagged SH with Strep-Tactin were used in this proof-of-concept study to rationalize the purification and immobilization of both SH and TsOYE into a one-step process. This approach showed good residual activity and high immobilization yields (see SI chapter 2.4), while also simplifying the immobilization process compared to covalent bonding methods.
Subsequently, we investigated the electro-driven asymmetric reduction of cyclic enones using in -situ generated H2 within the enzymatic system. Given that the KM of SH is approximately 680 µM, we used 1 mM FMN to ensure an adequate concentration close to kcat of SH. To avoid reactive oxygen species (ROS) from the reaction of reduced FMNH2 with O2 diffusing into the flow system, which potentially damages metal-dependent enzymes52, catalase was introduced into the circulating system. The immobilized enzymes, SH and TsOYE, were packed into a column which was integrated into the flow reactor. To ensure higher immobilization yield of SH, it was essential for Strep-Tactin resin to be packed and settled for 24 h before immobilization. This led to a defined arrangement of the immobilized biocatalysts in the biotransformation unit where SH-Tactin is positioned beneath the TsOYE-EziG (Figs. 1, S2). Considering the relatively weaker binding affinity between the Strep-tag and Strep-Tactin compared to coordinate bonds, there is potential for SH leaching. This involves the possibility of SH circulating around the flow setup and being reintroduced into the packed bed reactor.
During the reaction, we conducted online monitoring of various parameters, such as H2 evolution and consumption, potential O2 leakage, and FMN reduction (Fig. 2). This was done to monitor each catalytic step and assess whether H2 concentrations and FMNH2 were sufficient to initiate and progress the reaction. In that way, the functionality of each enzyme was comprehensively assessed, ensuring that no parameter within the cascade was limiting. The time at which the reaction ended could be determined by observing the saturation of H2 and the low concentration of oxidised FMN at 19 h and 22.5 h respectively (Fig. 2). It is worth noting that such a monitoring system, as shown in Fig. 1, had previously only been used in-vivo during fermentations and one other in flow chemistry setup41. This novel application in our current study represents a significant advancement in the ability to monitor enzymatic processes and identification of potential bottlenecks.
Fig. 2. On-line monitoring of H2, O2, and FMN during the electro-driven biotransformation.
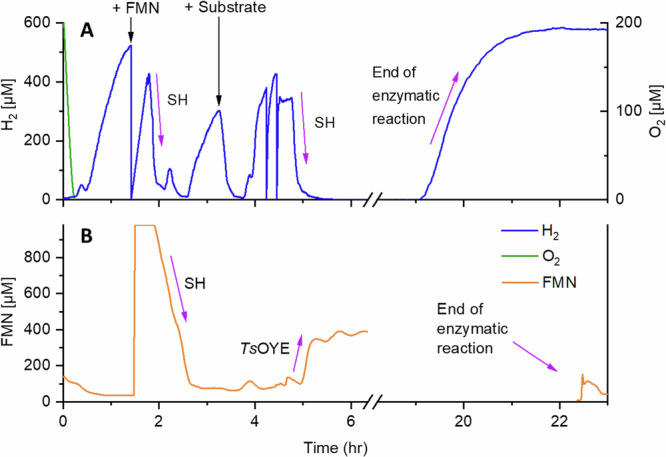
The concentration of H2 (A, blue), O2 (A, green), and oxidized FMN (B, orange) were followed during 24 h of the transformation of 3 to 4 as exemplary biotransformation. H2 was supplied to the system with the rate of 11 mL min−1 by the PEM electrolyzer (3.4 V, 0.89 A) and gas addition module. The electrolysis was performed until H2 concentration reached a plateau and then FMN was added (black arrow). After H2 concentration started to increase again and FMN was fully reduced to FMNH2, substrate was added to ensure reducing conditions for TsOYE activity. Purple arrows indicate the activity of each enzyme: the activity of TsOYE is indicated by oxidizing FMNH2 to FMN, the activity of soluble hydrogenase (SH) is shown in the consumption of H2, and the reduction of FMN to FMNH2. The experiment was performed as described in SI 3.2.
Substrate scope of flow system
A range of substrates was chosen according to the substrate spectrum of TsOYE to assess the adaptability of the flow system in 17 mL volume with various substrates (Fig. 1)16. Cyclohexenone (1) was first selected for its high water solubility and relatively high conversion53. In a 17 mL flow reaction, 25 mM of (1) yielded a 35% conversion to cyclohexanone (2) within 15 h, while no further conversion was detected. This conversion rate is consistent with previous batch experiments17, but this yield is lower compared to other reactions using NADPH53 (Table S4). The lower conversion rate could be due to the propensity of (1) for Michael's addition with water54. This combined with the potential for FMNH2 to generate H2O2 in the presence of O2 traces during substrate injection or sampling32,38,55 could have contributed to side reactions and resulted to low conversions. Our detection of hydroxylated side products via GC-MS in the flow system suggested a potential issue with side reactions (see SI chapter 14.3). Side products were also observed when SH and TsOYE were entrapped in an enzymatic membrane reactor (EMR) with a cutoff membrane of 30 kDa as an alternative immobilization technique (see SI chapter 6)56. (1) yielded a 28% conversion to (2) after 6 days in an EMR. While the utilization of EMR extended the enzymatic activity, it also increased the likelihood of side product formation due to its longer reaction time, rendering it unsuitable for this application.
Subsequently, the reduction of ketoisophorone (3) to levodione (4) was evaluated in the flow system due to the pharmaceutical significance of levodione as a building block. Complete conversion of 25 mM (3) to (4) was achieved 22 h after addition of substrate (>99%, Fig. 1, Figure S6). Despite constantly attaining high conversion rate, the flow reaction produced (4) with a relatively low optical purity, i.e., from ee = 77% R at 2 h after substrate addition to ee = 39% R at 22 h (see SI chapter 7). This decrease of optical purity has been linked to non-enzymatic racemization in water due to keto-enol tautomerization during sample preparations or to prolonged reaction conditions17,57,58, as well as the non-enzymatic reduction of (3) to racemic product (4) by free FMNH221. Additionally, it has been demonstrated that a lower concentration of the enzyme contributes to higher optical purity, which introduces the possibility of reversibility of the reaction by the enzyme57.
(S)- and (R)-carvone (5, 7) were also included to assess the performance of the flow system with chiral centers. The same flow volume, 17 mL, was used with these chiral substrates (5, 7), but at a reduced concentration of 5 mM due to their low solubility in aqueous solution. It is important to note that during the reaction, loss of substrate due to adsorption on PVMS tubing was detected by GC-FID. Hence, PTFE was used as the tubing for H2 transfer for reactions with carvone as substrate. After 16 h substrate addition, (5) resulted in 15% formation of the product (1R, 4S)-dihydrocarvone (6) (ee = 68%), while (7) exhibited an 11% conversion to (1R, 4R)-dihydrocarvone (8) (ee = 74%) (Fig. 1). In flow reaction, substrate conversion was similar to the batch reaction17. However, lower optical purity was observed, which can be also attributed to product racemization due to keto-enol tautomerization59,60.
Reusability of immobilized biocatalysts and upscaling
Reusability of the immobilized enzymes and the scalability of the reaction in flow setup was then investigated. Product (4) was synthesized from (3) with a maintained product formation of >99% throughout seven cycles without replacing the enzymes due to high enzyme loading (Fig. S7), demonstrating reusability of the system (see SI chapter 8). The biocatalysts utilized in these flow reactions achieved a total turnover number (TTN) of 1.01 × 105 for SH-Tactin and 1.69 × 104 for TsOYE-EziG (see SI chapter 9, Table S3). Overall, the consistently high conversion rates (>99%) in multiple cycles, along with the high TTN, especially for sophisticated oxidoreductases, illustrate the remarkable stability of the biocatalysts, indicating that Strep-Tactin XT 4Flow resin and EziG beads are robust immobilization carriers for SH and TsOYE, respectively. These observations corroborate previous findings, where EziG beads served as a stable enzyme carrier for imine reductase (IRED) and IRED-EziG was reused for up to 14 cycles61. The high TTN of SH-Tactin and TsOYE-EziG highly suggests that stable enzymes have the potential to be used in upscaled flow reactions.
We also determined the Faradaic efficiency for the electro-driven reduction of (3) to (4) in the flow system. With an applied voltage of 3.3 V and a current of 500 mA, the Faradaic efficiency of the flow system was calculated to be 0.15% (see SI chapter 12). The limited electron contribution to product formation compared to other work39 stems from the requirement of H2 gas outflow from the gas addition module, which is a drawback for commercially available PEM electrolyzers that have difficulty in managing back pressure if the module remains closed. Additionally, the commercial PEM electrolyzer only facilitated stable electrolysis of water at high voltage (>3 V) to overcome the internal resistance in the cell. To increase Faradaic efficiency or the energy efficiency of the system, zero gap cells can be used where lower amount of electrical energy is used for the production of exact amount of H2 that is needed for the biocatalytic reaction62,63.
Finally, we increased the volume of the flow reaction to 185 mL to demonstrate the scalability of the system. This was done by adding 168 mL segment that increases the volume without any headspace to inhibit O2 introduction to the system. Compound (3) was used as substrate due its high conversion in the flow setup (Fig. 1). Product formation of above 99% was achieved after 77 h (see SI chapter 10), resulting to a theoretical yield of 527 mg (4). The employed procedure ensured a practical product isolation. Simple extraction of the collected dispersion with diethyl ether yielded 89% (471 mg) of (4) in 96% purity. The TTN of mol product per mol SH-Tactin and TsOYE-EziG in the 185 mL upscaled reaction reached up to 3.2 × 105 and 2.6 × 104, respectively, with indications that further upscaling can possibly yield even higher TTN. This approach slightly exceeds the performance of previous biphasic batch reactions for (4) production in TsOYE-TTN as well as by an order of magnitude the TTN achieved by the Hyd1 hydrogenase from E. coli18,64. The high TTN values for SH-Tactin and TsOYE-EziG demonstrate that immobilizing Strep-tagged SH with Strep-Tactin resin and 6xHis-tagged TsOYE with EziG beads are optimal methods for significantly enhancing the robustness of biocatalysts in flavin-dependent biocatalytic reactions, surpassing the performance of previous biphasic batch reactions for (4) production in TsOYE-TTN. Furthermore, the SH TTN during FMN recycling was an order of magnitude lower than during NADH recycling, indicating the physiological electron acceptor improves enzyme stability41.
The specific activity of SH for reducing FMN is 5.8 U × mg−1, with a KM,FMN of 680 μM17. Thus, we decided to use 1 mM FMN in the reaction, which resulted in a slightly lower TTN of mol product per mol FMN in comparison to other studies (see Table S4). It is worth noting that catalase (~5000 U) was added every day to mitigate potential inhibition by ROS, ensuring full product conversion. This highlights the adverse impact of ROS, particularly H2O2, on enzymatic activity. E factor was also quantified to compare the waste generated by product65,66. 17 mL and 185 mL reactions were calculated to E factor of 6.0 and 5.7, respectively (see SI chapter 13), which represents low waste production compared to other FMNH2 regeneration systems19. This approach to electro-driven flavin-based biocatalysis in a flow system demonstrates a promising pathway for scalable and efficient biotransformations.
Conclusion
In this study, we established a closed-loop flow system using immobilized biocatalysts to convert electrical energy into chemical energy for production of fine chemicals. This flow system demonstrates adaptability to a range of substrates, scalability, and serves as an electro-driven system for regenerating flavin cofactors. It can be implemented to diverse biocatalytic processes that are dependent on reduced flavins such as styrene monooxygenase, unspecific peroxygenase-catalyzed hydroxylation via H2O2 from reduced flavin re-oxidation with O2, and nitroreductases10,17–19. Apart from improving the H2-availability for H2-driven biocatalysis with a PEM electrolyzer and highly gas-permeable tubing PVMS, this study introduces Strep-Tactin resin and highlights EziG beads as stable enzyme carriers suitable for flavin-dependent biocatalytic applications in a flow setup. It was highly advantageous that Strep-Tactin resin did not show adsorption of FMN, unlike Amberlite resin, an alternative for immobilizing SH49. Additionally, using coordination bonds between the EziG beads and the 6xHis-tag on the enzyme, high residual activity was maintained while demonstrating remarkable stability, compared to other methods such as covalent irreversible immobilization. However, the use of aqueous solutions led to side reactions, notably in cyclohexenone conversion, with observed low optical purity of the chiral products. The subsequent challenge will be to incorporate immiscible organic solvents to the flow setup to increase the solubility of substrates in water and the address the poor optical purity of chiral products57. TsOYE-EziG can be further investigated regarding its stability in organic solvents within a micro-aqueous environment, following previous studies on TsOYE immobilized via adsorption on Celite67.
It is also crucial to understand the significant differences in enzyme behavior between flow and batch reactions, including effects on activity, stability, and stereoselectivity, in order to further fine-tune conditions and preserve desired enzyme functionalities in flow reactions, especially in large scales. Overall, this scalable platform for electro-driven flow biocatalysis demonstrates high potential for chemical synthesis and can be flexibly adapted to other gas-dependent enzymes by attachment of corresponding gas bottles to the gas addition module. These enzymes include O2-dependent P450 monooxygenase, CH4-/O2-dependent soluble methane monooxygenases, or CO2-dependent formate dehydrogenases for hydroxylation and CO2 fixation reactions, respectively. The integration of advanced monitoring systems, effective biocatalyst immobilization, and careful selection of reaction conditions have provided a robust basis for further investigation and optimization of enzymatic processes in flow systems.
Methods
Plasmid construction, growth conditions, and protein purification
TsOYE (Thermos scotoductus SA-01 ATCC 700910) was recombinantly produced in E. coli BL21 Gold (DE3) cells with the corresponding plasmid in a pET-28a(+) vector, with an N-terminal His-tag (NdeI/EcoRI). For TsOYE purification, 50 mL pre-culture of LB medium containing 50 μg/mL of kanamycin was inoculated with toothpick of a colony from transformed cells with plasmid pET-28a(+)-TsOYE and incubated overnight at 37 °C and 180 rpm. Overexpression was carried out in 2 L of TB medium supplemented with 50 μg/mL of kanamycin. The main cultures were inoculated with 1% of pre-culture and grown at 37 °C and 180 rpm. When an OD600 of ~0.6 was reached (~2 h 30 min), 0.1 mM of IPTG was added. After induction, cultures were incubated overnight at 30 °C and 180 rpm. Cells were harvested by centrifugation (18,692 × g for 30 min at 4 °C). The obtained cell pellets (~27 g wet pellet mass) were washed and suspended in approximately 30 mL MOPS-NaOH buffer (20 mM, pH 7.0), supplemented with a spatula spoon tip of DNase and MgCl2, and one pill of EDTA-free cOmpleteTM protease inhibitor cocktail. Cells were disrupted using Multi Shot Cell Disruption System (1 cycle at 1.36 kbar pressure) and the cell debris was separated from the crude extract by centrifugation (18,692 × g for 30 min at 4 °C). The obtained supernatant was incubated in 50 mL falcon tubes in a water bath at 70 °C for 1 h 30 min. Precipitated proteins were separated by centrifugation at 38,759 × g for 30 min at 4 °C. (See SI chapter 2.2 for TsOYE amino acid and nucleotide sequence respectively).
The production and purification of NAD+-reducing hydrogenase (SH) was performed as described in Lauterbach et al. (2013)32.
Immobilization of biocatalysts
Strep-Tactin® XT 4Flow resin and EziG beads were used to immobilized the enzymes. For SH immobilization, the Strep-Tactin® XT 4Flow resin was first packed inside the C 10/10 column (Cytiva) and let settle for 1 day at 4 °C. Purified SH fused with a Strep-tag was loaded into the column bed at 3.3 mg SH per g resin. For TsOYE immobilization, the carrier used was EziG Amber beads. In a 50 mL centrifuge tube, TsOYE fused with 6×His-tag was loaded to the carrier (14.4 mg g−1 EziG), followed by incubation by overnight at room temperature in a roller shaker (30 rpm). The immobilized TsOYE in EziG was then resuspended and added to the C 10/10 column. Catalase was added on to the top (2,000 U) before closing. The C 10/10 column, with SH-Tactin underneath and TsOYE-EziG on the top, was used as the biotransformation unit and was attached to the flow setup. For further detailed steps see SI chapter 3.1.
Reactions in flow setup
Each experiment in flow setup was done in step-reactions to check the functionality of the gas addition module, SH-Tactin, and TsOYE-EziG. The flow volume (Tris-HCl 50 mM, pH 8, 30 °C) was first saturated with H2 by starting the PEM electrolyzer to 0.89 A to produce rate of 11 mL min−1 H2 gas. The generated H2 was added to the flow volume through the gas-addition module via the gas-permeable tubing (SI chapter 3). Once H2 saturation was observed, 1 mL of H2-saturated stock solution of FMN (concentration depending on the flow volume) was added to the flow volume to prevent O2 from entering the system. After observing reduction of FMN to FMNH2 and checking the functionality of SH, substrate was added (Fig. 2). The consumption of H2 after free FMNH2 was depleted was indicative of TsOYE-EziG and SH-Tactin activities. Upon saturation of H2 was observed, the reaction was stopped and the flow volume was stored for analysis.
For 17 mL flow reactions, flow volume with 1 mM FMN, 2000 U catalase, and substrate (either 25 mM ketoisophorone, 25 mM cyclohexenone, or 5 mM (S)- or (R)-carvone). For the 185 mL upscaled flow volume, the reaction involved a mobile phase with 500 µM FMN, ~5000 U catalase (added every day), and 18.5 mM of substrate ketoisophorone. A segment of 168 mL was added to the existing 17 mL to increases the volume without any headspace. For further detailed information of reaction by substrate see SI chapters 3.2 to 3.5.
Analytics
Compound analysis was carried out on Thermo Scientific Trace GC Ultra instrument with a AS 3000 auto-sampler coupled with flame ionization detector (FID) using helium as carrier gas. Products were confirmed by reference standards. Details of the column and temperature programs used are given in Table S6. All measurements were calibrated with calibration curves. Only conversion rate was quantified with GC-FID. Enantiomeric excess (ee %) of compounds was assessed with Shimadzu GC-2010 equipped with an FID using helium as carrier gas (see SI chapter 14.2). Details of the column and temperature programs used are given in Table S7. Time between experiments and GC measurements was several days which also explained the poor enantiomeric excess obtained (racemization of certain products occurred). Gas chromatography-mass spectrometry was carried out on a Thermo Scientific Trace GC Ultra instrument coupled with a Thermo Scientific ISQ mass selective detector (MSD) and an additional flame ionization detector. Injector temperature: 250 °C. Split mode with a split ratio of 10. Detector temperature: 320 °C. Electron ionization of the analyte with 70 eV acceleration voltage. For further information see SI chapter 14.3. Nuclear magnetic resonance spectra were recorded on a BRUKER Avance Neo 600 spectrometer at 26 °C and were analyzed with the software Mnova 14.2.3 (Mestrelab Research). Chemical shifts δ were reported in parts per million (ppm). The residual solvent signals were used for referencing 1H NMR spectra (7.26 ppm for CDCl3)68,69. A relaxation delay of D1 = 25 s was used for quantitative 1H NMR experiments. For further information see SI chapter 15.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We thank EnginZyme (Stockholm, Sweden) for EziGTM beads. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 955740, from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No. 949910) and from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany´s Excellence Strategy—Cluster of Excellence 2186 “The Fuel Science Center”—ID: 390919832. Further funding was received through a Liebig Fellowship by the Fonds der Chemischen Industrie, grant number Li 210/01 (F.F.M.).
Author contributions
Conceptualization: L.L.; Experiments: G.L., D.C., A.W., F.F.M.; Investigation and analysis: G.L., D.C., A.W., F.M., P.R.F.C., A.D., D.R., C.E.P., L.L.; Writing—original draft: G.L., D.C., P.R.F.C.; Writing—editing and review: G.L., D.C., A.W., F.F.M., P.R.F.C., D.R., D.C.P., L.L.; Supervision and funding: L.L. All authors contributed to the discussion. All authors have given approval to the final version of the manuscript.
Peer review
Peer review information
Communications Chemistry thanks László Poppe and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Data availability
Raw and metadata for this contribution are available at 10.5281/zenodo.10798153.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-024-01288-y.
References
- 1.Meyer, L.-E., Hobisch, M. & Kara, S. Process intensification in continuous flow biocatalysis by up and downstream processing strategies. Curr. Opin. Biotechnol.78, 102835 (2022). 10.1016/j.copbio.2022.102835 [DOI] [PubMed] [Google Scholar]
- 2.Ley, S. V. On being green: can flow chemistry help? Chem. Rec.12, 378–390 (2012). 10.1002/tcr.201100041 [DOI] [PubMed] [Google Scholar]
- 3.Tang, Z., Oku, Y. & Matsuda, T. Application of immobilized enzymes in flow biocatalysis for efficient synthesis. Org. Process Res. Dev. 28, 1308–1326 (2024).
- 4.Britton, J., Majumdar, S. & Weiss, G. A. Continuous flow biocatalysis. Chem. Soc. Rev.47, 5891–5918 (2018). 10.1039/C7CS00906B [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reetz, M. T. Biocatalysis in organic chemistry and biotechnology: past, present, and future. J. Am. Chem. Soc.135, 12480–12496 (2013). 10.1021/ja405051f [DOI] [PubMed] [Google Scholar]
- 6.Romero-Fernández, M. & Paradisi, F. Protein immobilization technology for flow biocatalysis. Curr. Opin. Chem. Biol.55, 1–8 (2020). 10.1016/j.cbpa.2019.11.008 [DOI] [PubMed] [Google Scholar]
- 7.Tamborini, L., Fernandes, P., Paradisi, F. & Molinari, F. Flow bioreactors as complementary tools for biocatalytic process intensification. Trends Biotechnol.36, 73–88 (2018). 10.1016/j.tibtech.2017.09.005 [DOI] [PubMed] [Google Scholar]
- 8.Stuermer, R., Hauer, B., Hall, M. & Faber, K. Asymmetric bioreduction of activated C=C bonds using enoate reductases from the old yellow enzyme family. Curr. Opin. Chem. Biol.11, 203–213 (2007). 10.1016/j.cbpa.2007.02.025 [DOI] [PubMed] [Google Scholar]
- 9.Toogood, H. S. et al. Structure-based insight into the asymmetric bioreduction of the C=C double bond of alpha, beta-unsaturated nitroalkenes by pentaerythritol tetranitrate reductase. Adv. Synth. Catal.350, 2789–2803 (2008). 10.1002/adsc.200800561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toogood, H. S., Gardiner, J. M. & Scrutton, N. S. Biocatalytic reductions and chemical versatility of the old yellow enzyme family of flavoprotein oxidoreductases. ChemCatChem2, 892–914 (2010). 10.1002/cctc.201000094 [DOI] [Google Scholar]
- 11.Stott, K., Saito, K., Thiele, D. J. & Massey, V. Old yellow enzyme. The discovery of multiple isozymes and a family of related proteins. J. Biol. Chem.268, 6097–6106 (1993). 10.1016/S0021-9258(18)53224-5 [DOI] [PubMed] [Google Scholar]
- 12.Toogood, H. S., Knaus, T. & Scrutton, N. S. Alternative hydride sources for ene‐reductases: current trends. ChemCatChem6, 951–954 (2014). 10.1002/cctc.201300911 [DOI] [Google Scholar]
- 13.Knaus, T. et al. Better than nature: nicotinamide biomimetics that outperform natural coenzymes. J. Am. Chem. Soc.138, 1033–1039 (2016). 10.1021/jacs.5b12252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reeve, H. A. et al. A hydrogen-driven biocatalytic approach to recycling synthetic analogues of NAD(P)H. Chem. Commun.58, 10540–10543 (2022). 10.1039/D2CC02411J [DOI] [PubMed] [Google Scholar]
- 15.Zhang, W. & Hollmann, F. Nonconventional regeneration of redox enzymes—a practical approach for organic synthesis? Chem. Commun.54, 7281–7289 (2018). 10.1039/C8CC02219D [DOI] [PubMed] [Google Scholar]
- 16.Bernard, J., van Heerden, E., Arends, I. W. C. E., Opperman, D. J. & Hollmann, F. Chemoenzymatic reduction of conjugated C=C double bonds. ChemCatChem4, 196–199 (2012). 10.1002/cctc.201100312 [DOI] [Google Scholar]
- 17.Al-Shameri, A., Willot, S. J.-P., Paul, C. E., Hollmann, F. & Lauterbach, L. H2 as a fuel for flavin- and H2O2-dependent biocatalytic reactions. Chem. Commun.56, 9667–9670 (2020). 10.1039/D0CC03229H [DOI] [PubMed] [Google Scholar]
- 18.Joseph Srinivasan, S. et al. E. coli nickel-iron hydrogenase 1 catalyses non-native reduction of flavins: demonstration for alkene hydrogenation by old yellow enzyme ene-reductases. Angew. Chem. Int. Ed.60, 13824–13828 (2021). 10.1002/anie.202101186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramirez, M. A. et al. H2-driven reduction of flavin by hydrogenase enables cleaner operation of nitroreductases for nitro-group to amine reductions. Front. Catal.2, 906694 (2022). 10.3389/fctls.2022.906694 [DOI] [Google Scholar]
- 20.Grau, M. M. et al. Photoenzymatic reduction of C=C double bonds. Adv. Synth. Catal.351, 3279–3286 (2009). 10.1002/adsc.200900560 [DOI] [Google Scholar]
- 21.Taglieber, A., Schulz, F., Hollmann, F., Rusek, M. & Reetz, M. T. Light-driven biocatalytic oxidation and reduction reactions: scope and limitations. Chembiochem9, 565–572 (2008). 10.1002/cbic.200700435 [DOI] [PubMed] [Google Scholar]
- 22.Hollmann, F., Hofstetter, K., Habicher, T., Hauer, B. & Schmid, A. Direct electrochemical regeneration of monooxygenase subunits for biocatalytic asymmetric epoxidation. J. Am. Chem. Soc.127, 6540–6541 (2005). 10.1021/ja050997b [DOI] [PubMed] [Google Scholar]
- 23.Mifsud, M. et al. Photobiocatalytic chemistry of oxidoreductases using water as the electron donor. Nat. Commun.5, 3145 (2014). 10.1038/ncomms4145 [DOI] [PubMed] [Google Scholar]
- 24.Ruinatscha, R., Dusny, C., Buehler, K. & Schmid, A. Productive asymmetric styrene epoxidation based on a next generation electroenzymatic methodology. Adv. Synth. Catal.351, 2505–2515 (2009). 10.1002/adsc.200900291 [DOI] [Google Scholar]
- 25.Valotta, A., Malihan-Yap, L., Hinteregger, K., Kourist, R. & Gruber-Woelfler, H. Design and investigation of a photocatalytic setup for efficient biotransformations within recombinant cyanobacteria in continuous flow. ChemSusChem15, e202201468 (2022). 10.1002/cssc.202201468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szczepańska, E. et al. Ene-reductase transformation of massoia lactone to δ-decalactone in a continuous-flow reactor. Sci. Rep.11, 18794 (2021). 10.1038/s41598-021-97585-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lauterbach, L. & Lenz, O. How to make the reducing power of H2 available for in vivo biosyntheses and biotransformations. Curr. Opin. Chem. Biol.49, 91–96 (2019). 10.1016/j.cbpa.2018.11.020 [DOI] [PubMed] [Google Scholar]
- 28.Lauterbach, L., Lenz, O. & Vincent, K. A. H. 2-driven cofactor regeneration with NAD(P)+-reducing hydrogenases. FEBS J.280, 3058–3068 (2013). 10.1111/febs.12245 [DOI] [PubMed] [Google Scholar]
- 29.Ratzka, J., Lauterbach, L., Lenz, O. & Ansorge-Schumacher, M. B. Systematic evaluation of the dihydrogen-oxidising and NAD+-reducing soluble [NiFe]-hydrogenase from Ralstonia eutropha H16 as a cofactor regeneration catalyst. Biocatal. Biotranformation29, 246–252 (2011). 10.3109/10242422.2011.615393 [DOI] [Google Scholar]
- 30.Mordhorst, S. & Andexer, J. N. Round, round we go—strategies for enzymatic cofactor regeneration. Nat. Prod. Rep.37, 1316–1333 (2020). 10.1039/D0NP00004C [DOI] [PubMed] [Google Scholar]
- 31.Preissler, J. et al. Dihydrogen‐driven NADPH recycling in imine reduction and P450‐catalyzed oxidations mediated by an engineered O2 ‐tolerant hydrogenase. ChemCatChem12, 4853–4861 (2020). 10.1002/cctc.202000763 [DOI] [Google Scholar]
- 32.Lauterbach, L. & Lenz, O. Catalytic production of hydrogen peroxide and water by oxygen-tolerant NiFe-hydrogenase during H2 cycling in the presence of O2. J. Am. Chem. Soc.135, 17897–17905 (2013). 10.1021/ja408420d [DOI] [PubMed] [Google Scholar]
- 33.Holzer, A. K. et al. Asymmetric biocatalytic amination of ketones at the expense of NH3 and molecular hydrogen. Org. Lett.17, 2431–2433 (2015). 10.1021/acs.orglett.5b01154 [DOI] [PubMed] [Google Scholar]
- 34.Lonsdale, T. H. et al. H2-driven biotransformation of n-octane to 1-octanol by a recombinant Pseudomonas putida strain co-synthesizing an O2-tolerant hydrogenase and a P450 monooxygenase. Chem. Commun.51, 16173–16175 (2015). 10.1039/C5CC06078H [DOI] [PubMed] [Google Scholar]
- 35.Al-Shameri, A. et al. Synthesis of N-heterocycles from diamines via H2-driven NADPH recycling in the presence of O2. Green. Chem.21, 1396–1400 (2019). 10.1039/C8GC03798A [DOI] [Google Scholar]
- 36.Opperman, D. J. et al. Crystal structure of a thermostable old yellow enzyme from Thermus scotoductus SA-01. Biochem. Biophys. Res. Commun.393, 426–431 (2010). 10.1016/j.bbrc.2010.02.011 [DOI] [PubMed] [Google Scholar]
- 37.Lauterbach, L. et al. The hydrogenase subcomplex of the NAD + ‐reducing [NiFe] hydrogenase from Ralstonia eutropha—insights into catalysis and redox interconversions. Eur. J. Inorg. Chem.2011, 1067–1079 (2011). 10.1002/ejic.201001053 [DOI] [Google Scholar]
- 38.Lauterbach, L., Idris, Z., Vincent, K. A. & Lenz, O. Catalytic properties of the isolated diaphorase fragment of the NAD+-reducing NiFe-hydrogenase from Ralstonia eutropha. PloS one6, e25939 (2011). 10.1371/journal.pone.0025939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Son, E. J. et al. Carbon nanotube–graphitic carbon nitride hybrid films for flavoenzyme‐catalyzed photoelectrochemical cells. Adv. Funct. Mater.28, 1705232 (2018). 10.1002/adfm.201705232 [DOI] [Google Scholar]
- 40.Choi, D. S., Kim, J., Hollmann, F. & Park, C. B. Solar‐assisted eBiorefinery: photoelectrochemical pairing of oxyfunctionalization and hydrogenation reactions. Angew. Chem. Int. Ed. Engl.132, 16020–16024 (2020). 10.1002/ange.202006893 [DOI] [PubMed] [Google Scholar]
- 41.Al-Shameri, A. et al. Powering artificial enzymatic cascades with electrical energy. Angew. Chem. Int. Ed.59, 10929–10933 (2020). 10.1002/anie.202001302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Brien, M., Baxendale, I. R. & Ley, S. V. Flow ozonolysis using a semipermeable Teflon AF-2400 membrane to effect gas-liquid contact. Org. Lett.12, 1596–1598 (2010). 10.1021/ol100322t [DOI] [PubMed] [Google Scholar]
- 43.Poznansky, B., Cleary, S. E., Thompson, L. A., Reeve, H. A. & Vincent, K. A. Boosting the productivity of H2-driven biocatalysis in a commercial hydrogenation flow reactor using H2 from water electrolysis. Front. Chem. Eng. 3, 718257 (2021).
- 44.Lagadec, M. F. & Grimaud, A. Water electrolysers with closed and open electrochemical systems. Nat. Mater.19, 1140–1150 (2020). 10.1038/s41563-020-0788-3 [DOI] [PubMed] [Google Scholar]
- 45.Carmo, M., Fritz, D. L., Mergel, J. & Stolten, D. A comprehensive review on PEM water electrolysis. Int. J. Hydrog. Energy38, 4901–4934 (2013). 10.1016/j.ijhydene.2013.01.151 [DOI] [Google Scholar]
- 46.Mallia, C. J. & Baxendale, I. R. The use of gases in flow synthesis. Org. Process Res. Dev.20, 327–360 (2016). 10.1021/acs.oprd.5b00222 [DOI] [Google Scholar]
- 47.Greiner, L. et al. Membrane aerated hydrogenation: enzymatic and chemical homogeneous catalysis. Adv. Synth. Catal.345, 679–683 (2003). 10.1002/adsc.200303031 [DOI] [Google Scholar]
- 48.Grushevenko, E. A., Borisov, I. L. & Volkov, A. V. High-selectivity polysiloxane membranes for gases and liquids separation (a review). Pet. Chem.61, 959–976 (2021). 10.1134/S0965544121090103 [DOI] [Google Scholar]
- 49.Herr, N., Ratzka, J., Lauterbach, L., Lenz, O. & Ansorge-Schumacher, M. B. Stability enhancement of an O2-tolerant NAD+-reducing [NiFe]-hydrogenase by a combination of immobilisation and chemical modification. J. Mol. Catal. B Enzym.97, 169–174 (2013). 10.1016/j.molcatb.2013.06.009 [DOI] [Google Scholar]
- 50.Cha, J., Lee, J., Jeon, B. W., Kim, Y. H. & Kwon, I. Real flue gas CO2 hydrogenation to formate by an enzymatic reactor using O2- and CO-tolerant hydrogenase and formate dehydrogenase. Front. Bioeng. Biotechnol.11, 1265272 (2023). 10.3389/fbioe.2023.1265272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thompson, M. P. et al. A generic platform for the immobilisation of engineered biocatalysts. Tetrahedron75, 327–334 (2019). 10.1016/j.tet.2018.12.004 [DOI] [Google Scholar]
- 52.Torella, J. P. et al. Efficient solar-to-fuels production from a hybrid microbial-water-splitting catalyst system. Proc. Natl Acad. Sci. Usa.112, 2337–2342 (2015). 10.1073/pnas.1424872112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jongkind, E. P. J. et al. Synthesis of chiral amines via a bi‐enzymatic cascade using an ene‐reductase and amine dehydrogenase. ChemCatChem. 14, e202101576 (2022).
- 54.Resch, V., Seidler, C., Chen, B.-S., Degeling, I. & Hanefeld, U. On the Michael addition of water to α,β‐unsaturated ketones using amino acids. Eur. J. Org. Chem.2013, 7697–7704 (2013). 10.1002/ejoc.201301230 [DOI] [Google Scholar]
- 55.Halliwell, B. & Gutteridge, J. M. C. Free Radicals in Biology and Medicine (Oxford University Press, 2015).
- 56.Kragl, U., Gygax, D., Ghisalba, O. & Wandrey, C. Enzymatic two‐step synthesis of N‐acetyl‐neuraminic acid in the enzyme membrane reactor. Angew. Chem. Int. Ed.30, 827–828 (1991). 10.1002/anie.199108271 [DOI] [Google Scholar]
- 57.Fryszkowska, A. et al. Asymmetric reduction of activated alkenes by pentaerythritol tetranitrate reductase: specificity and control of stereochemical outcome by reaction optimisation. Adv. Synth. Catal.351, 2976–2990 (2009). 10.1002/adsc.200900574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fu, Y., Castiglione, K. & Weuster-Botz, D. Comparative characterization of novel ene-reductases from cyanobacteria. Biotechnol. Bioeng.110, 1293–1301 (2013). 10.1002/bit.24817 [DOI] [PubMed] [Google Scholar]
- 59.Zhang, W. et al. The chemo-catalytic racemization of lactic acid enantiomer derived from biomass via keto-enol tautomerization. Ind. Crop. Prod.193, 116260 (2023). 10.1016/j.indcrop.2023.116260 [DOI] [Google Scholar]
- 60.Russell, G. F. & Hills, J. I. Odor differences between enantiomeric isomers. Science172, 1043–1044 (1971). 10.1126/science.172.3987.1043 [DOI] [PubMed] [Google Scholar]
- 61.Gand, M. et al. A NADH-accepting imine reductase variant: Immobilization and cofactor regeneration by oxidative deamination. J. Biotech.230, 11–18 (2016). 10.1016/j.jbiotec.2016.05.006 [DOI] [PubMed] [Google Scholar]
- 62.Rad, R. et al. A hybrid bioelectrochemical system coupling a zero-gap cell and a methanogenic reactor for carbon dioxide reduction using a wastewater-derived catholyte. Cell Rep. Phys. Sci.4, 101526 (2023). 10.1016/j.xcrp.2023.101526 [DOI] [Google Scholar]
- 63.Baek, G., Rossi, R., Saikaly, P. E. & Logan, B. E. High-rate microbial electrosynthesis using a zero-gap flow cell and vapor-fed anode design. Water Res.219, 118597 (2022). 10.1016/j.watres.2022.118597 [DOI] [PubMed] [Google Scholar]
- 64.Nett, N. et al. A robust and stereocomplementary panel of ene-reductase variants for gram-scale asymmetric hydrogenation. Mol. Catal.502, 111404 (2021). 10.1016/j.mcat.2021.111404 [DOI] [Google Scholar]
- 65.Sheldon, R. A. The E factor 25 years on: the rise of green chemistry and sustainability. Green. Chem.19, 18–43 (2017). 10.1039/C6GC02157C [DOI] [Google Scholar]
- 66.Wu, Y., Paul, C. E. & Hollmann, F. Mirror, mirror on the wall, which is the greenest of them all? A critical comparison of chemo- and biocatalytic oxyfunctionalisation reactions. Green. Carbon1, 227–241 (2023). 10.1016/j.greenca.2023.10.004 [DOI] [Google Scholar]
- 67.Villa, R., Ferrer-Carbonell, C. & Paul, C. E. Biocatalytic reduction of alkenes in micro-aqueous organic solvent catalysed by an immobilised ene reductase. Catal. Sci. Technol.13, 5530–5535 (2023). 10.1039/D3CY00541K [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fulmer, G. R. et al. NMR Chemical shifts of trace impurities: common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics29, 2176–2179 (2010). 10.1021/om100106e [DOI] [Google Scholar]
- 69.Gottlieb, H. E., Kotlyar, V. & Nudelman, A. NMR Chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem.62, 7512–7515 (1997). 10.1021/jo971176v [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and metadata for this contribution are available at 10.5281/zenodo.10798153.