

Abstract
Nicotine is the primary addictive component of tobacco products. Through its actions on the heart and autonomic nervous system, nicotine exposure is associated with electrophysiological changes and increased arrhythmia susceptibility. To assess the underlying mechanisms, we treated rabbits with transdermal nicotine (NIC, 21 mg/day) or control (CT) patches for 28 days before performing dual optical mapping of transmembrane potential (RH237) and intracellular Ca2+ (Rhod-2 AM) in isolated hearts with intact sympathetic innervation. Sympathetic nerve stimulation (SNS) was performed at the first to third thoracic vertebrae, and β-adrenergic responsiveness was additionally evaluated following norepinephrine (NE) perfusion. Baseline ex vivo heart rate (HR) and SNS stimulation threshold were higher in NIC versus CT (P = 0.004 and P = 0.003, respectively). Action potential duration alternans emerged at longer pacing cycle lengths (PCL) in NIC versus CT at baseline (P = 0.002) and during SNS (P = 0.0003), with similar results obtained for Ca2+ transient alternans. SNS shortened the PCL at which alternans emerged in CT but not in NIC hearts. NIC-exposed hearts tended to have slower and reduced HR responses to NE perfusion, but ventricular responses to NE were comparable between groups. Although fibrosis was unaltered, NIC hearts had lower sympathetic nerve density (P = 0.03) but no difference in NE content versus CT. These results suggest both sympathetic hypoinnervation of the myocardium and regional differences in β-adrenergic responsiveness with NIC. This autonomic remodeling may contribute to the increased risk of arrhythmias associated with nicotine exposure, which may be further exacerbated with long-term use.
NEW & NOTEWORTHY Here, we show that chronic nicotine exposure was associated with increased heart rate, increased susceptibility to alternans, and reduced sympathetic electrophysiological responses in the intact rabbit heart. We suggest that this was due to sympathetic hypoinnervation of the myocardium and diminished β-adrenergic responsiveness of the sinoatrial node following nicotine treatment. Though these differences did not result in increased arrhythmia propensity in our study, we hypothesize that prolonged nicotine exposure may exacerbate this proarrhythmic remodeling.
Keywords: action potential, arrhythmia, calcium, nicotine, sympathetic
INTRODUCTION
Cigarette smoking is the most preventable cause of cardiovascular morbidity and mortality in the United States (1–3). Although smoking promotes the progression of coronary artery disease and increases the risk of myocardial infarction (MI), heart failure, ischemia-induced arrhythmias, and sudden cardiac death (4–6), short-term secondhand smoke (SHS) exposure alone has been associated with increased incidence of atrial fibrillation and ventricular tachycardia/fibrillation (7). Although the mechanisms for this increased vulnerability to arrhythmias have yet to be fully explored, we have previously shown that the susceptibility to calcium (Ca2+) and action potential duration (APD) alternans was increased with SHS exposure (8). At present, however, the contributions of individual cigarette smoke components to the increased emergence of alternans and arrhythmias are unknown. Whereas cigarette usage has declined, sales of electronic (e)-cigarettes increased by 46.6% between 2020 and 2022 (9). Though often considered less harmful than traditional cigarettes, e-cigarettes also contain numerous chemicals including nicotine (NIC), tobacco-specific nitrosamines, aldehydes, and flavors (10–13). The amount of nicotine present in e-cigarettes is widely variable but, in some cases, has been reported to be higher than an entire pack of conventional cigarettes (14, 15).
Nicotine directly modulates autonomic control of the heart through its binding to nicotinic acetylcholine receptors (nAChRs) found within the brain, adrenal medulla, and postganglionic neurons of the parasympathetic and sympathetic cardiac nerves (16). In studies using labeled norepinephrine (NE), rats given nicotine for 10 days had a higher NE but lower acetylcholine outflow in the hippocampus (16), indicating that the overall net effect from the central nervous system is sympathetically driven. Similarly, acute exposure to e-cigarettes containing nicotine has been linked to reduced heart rate variability (HRV) (17), which is typically associated with increased sympathetic activity and risk of sudden cardiac death (18, 19). Thus, it is possible that long-term nicotine use results in autonomic dysregulation that may play a role in promoting ventricular arrhythmias.
In addition to its actions on the autonomic nervous system, nicotine also directly affects the myocardium, likely through its binding to nonneuronal nAChRs found on ventricular cardiac myocytes (20). Acute nicotine administration has been shown to directly block the membrane stabilizing current Ik1 in isolated myocytes (21, 22), which we have previously demonstrated leads to the generation of ectopic beats in the intact heart (23). Exposure to nicotine is also associated with increased myocardial fibrosis (24–26), which may play a role in creating a heterogeneous substrate that can foster ectopic activity and/or reentrant arrhythmias (27). In line with these findings, in vivo administration of nicotine was reported to result in atrial and ventricular arrhythmias in a canine model of MI (28, 29), with meta-analysis studies also identifying increased risk of rhythm disorders in patients on nicotine replacement therapies (30, 31). As such, at the level of the heart, nicotine exposure is associated with myocardial remodeling alongside alterations in autonomic regulation that appear to be synergistically proarrhythmic. However, despite the importance of these findings, the electrophysiological mechanisms underlying increased arrhythmia susceptibility due to nicotine exposure are unclear. Therefore, the goal of this study was to investigate the effect of chronic nicotine exposure on cardiac electrophysiology, Ca2+ handling, structure, and sympathetic responsiveness in the intact heart to elucidate potential mechanisms of increased arrhythmia risk.
MATERIALS AND METHODS
Ethical Approval
All procedures were approved by the Animal Care and Use Committee of the University of California, Davis and were conducted according to the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (NIH Publication No. 85-23, Revised 2011).
Nicotine Exposure
Male (n = 10) and female (n = 15) New Zealand White rabbits (4–8 mo old, 3.0–3.5 kg) were obtained from Charles River and singly housed with ad libitum access to food and water. Rabbits were randomly assigned to control (CT) or nicotine (NIC) groups with exposure carried out in four similarly sized paired cohorts. Exposure to nicotine occurred via transdermal patches (Nicoderm CQ, 21 mg/day) placed on the ears, with surgical tape used for control rabbits. Transdermal patches and surgical tape were changed daily Monday to Friday with a total exposure duration of 28 days.
Measurement of Serum Cotinine Levels
To confirm the delivery of nicotine, levels of the circulating nicotine metabolite cotinine were assessed using blood drawn from the marginal ear vein of NIC and CT rabbits on days 0, 14, and 28 of exposure. Blood samples were allowed to clot overnight at 2–8°C before centrifugation at 1,000 g (3,000 rpm) for 15 min. Serum was then removed and stored at −80°C. To assess serum cotinine levels, samples were sent overnight on dry ice to the Tobacco and Carcinogen Biomarkers Core at the University of California San Francisco for analysis by liquid chromatography-tandem mass spectrometry (LCMSM) as previously described (32).
Innervated Whole Heart Langendorff Perfusion
At 28 days of exposure, rabbits were euthanized via intravenous injection of pentobarbital sodium (100 mg/kg) following administration of 1,000 IU heparin (Fresenius Kabi USA). Hearts were surgically extracted with the thoracic spinal column (T1–T12) attached to ensure intact sympathetic innervation as in previous studies (Fig. 1) (33, 34). Following extraction, the isolated preparation was flushed with ice-cold cardioplegia, containing (in mmol/L) 110 NaCl, 1.2 CaCl2, 16 KCl, 16 MgCl2, and 10 NaHCO3 via the descending aorta. The preparation was then retrograde perfused and submerged in Tyrode’s solution at 37 ± 0.5°C, consisting of (in mmol/L) 128.2 NaCl, 1.3 CaCl2, 4.7 KCl, 1.05 MgCl2, 1.19 NaH2PO4, 20 NaHCO3, and 11,1 glucose. The excitation-contraction uncoupler (blebbistatin, 10–20 µM, R&D Systems, Minneapolis, MN) (35) and a skeletal muscle paralytic (vecuronium bromide, 6 µM, Cayman Chemical, Ann Arbor, MI) were added to the circulating perfusate to eliminate motion during optical recordings. Two needle electrodes were positioned in the thoracic cavity and a ground was placed in the bottom of the perfusion dish to obtain a continuous ECG recording. Perfusion pressure was maintained at 60–80 mmHg by adjusting perfusion flow rate (80–100 mL/min).
Figure 1.

Innervated rabbit heart preparation.
Optical Mapping
Dual optical mapping was performed as previously described (Fig. 1) (33, 36–38). Briefly, preparations were loaded with Ca2+ (Rhod-2 AM, 50 µL of 1 mg/mL in DMSO + 10% pluronic acid, Biotium, Hayward, CA) and voltage (Vm)-sensitive dyes (RH237, 50 µL of 1 mg/mL in DMSO, Biotium) through the coronary perfusion. A catheter electrode was inserted up the spinal canal to T1–T3 to stimulate the preganglionic sympathetic nerves. A pacing electrode was placed on the center of the left ventricle for epicardial ventricular pacing. The anterior epicardial surface was illuminated with LED light sources at 530-nm bandpass filtered from 511 to 551 nm (LEX-2, SciMedia, Costa Mesa, CA) and focused directly on the surface of the heart. Emitted fluorescence was captured using a THT macroscope (SciMedia) and split by a dichroic mirror set at 630 nm (Omega, Brattleboro, VT). RH237 signals were long-pass filtered at 700 nm, whereas Rhod-2 AM signals were bandpass filtered from 574 to 606 nm. Signals were recorded with two CMOS cameras (MiCam Ultima-L, SciMedia) at a sampling rate of 1 kHz and a 100 × 100 pixel resolution, with a field of view of 3.1 × 3.1 cm and a spatial resolution of 0.31 mm/pixel.
Experimental Protocol
Ex vivo heart rate (HR) was monitored before the addition of blebbistatin and throughout experiments via continuous ECG recording in LabChart (ADInstruments, Colorado Springs, CO). After the addition of blebbistatin, sympathetic nerve stimulation (SNS) thresholds were determined as previously described (36). Specifically, sympathetic nerve fibers were initially stimulated at 0.5 Hz, 7 V for 5 s. Stimulation frequency was then increased in 0.5-Hz increments at a constant voltage of 7 V until a >15% increase in HR was observed. SNS thresholds were then noted and hearts were subsequently stimulated at 5 Hz higher than their threshold value. Ca2+, Vm, and HR recordings were made during sinus rhythm at baseline and with 13 s of SNS. Continuous ventricular pacing was then performed via the epicardial left ventricle (LV) at decreasing pacing cycle lengths (PCLs) from 300 ms until hearts lost capture to assess the emergence of alternans. Finally, hearts were perfused with NE (500 nM) to evaluate β-adrenergic responsiveness.
Optical Mapping Data Analysis
Optical data were analyzed using both Optiq (Cairn Research, UK) and ElectroMap (39) software as previously described (33, 40, 41). Briefly, APD (APD80) and Ca2+ transient duration (CaTD80) were calculated as 80% repolarization minus activation time (with activation time calculated at 50% of peak amplitude). Mean APD80 and CaTD80 for each heart were calculated from the entire mapping field of view. CaTD and APD alternans thresholds were determined using spectral methods as previously described (42, 43) with the threshold defined as the longest PCL at which significant alternans emerged (minimum spectral magnitude ≥ 2).
Because of SNS electrical artifacts on the ECG, data pertaining to changes in HR during SNS were analyzed from optical recordings using ElectroMap. All other HR measurements were obtained from continuous ECG recordings and were analyzed using LabChart (ADInstruments, Colorado Springs, CO). Ex vivo baseline HR was measured 1 min before the addition of blebbistatin and averaged over 1 min. For HR changes in response to NE, HR was measured 1 min before the addition of NE (averaged over 10 s) and 6 min after the addition of NE. To quantify incidence and severity of arrhythmia, ECG recordings were scored according to previous studies (36, 44, 45). Briefly, 0 is no ectopic beats, 1 is premature ventricular contraction (PVC), 2 is bigeminy or salvos (2–4 consecutive beats), 3 is ventricular tachycardia (>5 consecutive, monomorphic beats), and 4 is ventricular fibrillation (>5 consecutive, polymorphic beats). Arrhythmia scores were determined as the most severe arrhythmia observed during the entire experimental protocol.
Tissue Harvesting
After completion of optical mapping experiments, a 2-mm-thick short-axis section was cut from the middle of the LV and sliced into wedges. For immunohistochemical and histological staining, LV tissue wedges were fixed in 4% paraformaldehyde for 1.5 h, washed with phosphate-buffered saline (PBS), and placed in 30% sucrose in PBS overnight. Tissue was then embedded in an optimum cutting temperature (OCT) medium, flash frozen, and stored at −80°C. For high-performance liquid chromatography (HPLC), LV and right atrial (RA) tissue were flash-frozen and stored at −80°C.
Assessment of Fibrosis
To assess fibrosis, tissue sectioning and Masson’s trichrome staining were performed by Acepix Biosciences (Hayward, CA). Tissue was sectioned into 10-µm-thick slices at a step depth of 50–100 µm, thaw mounted on positively charged slides, stained, and stored at −80°C. Imaging was performed on an upright Nikon Eclipse Ni microscope at ×20 magnification with white light excitation. Fibrosis area was color thresholded (blue) and divided by the total tissue area. All images were analyzed by two blinded users and the generated values were averaged.
Assessment of Sympathetic Nerve Density
To assess sympathetic nerve density, tyrosine hydroxylase (TH) labeling was performed as in previous studies (36, 41, 46). Briefly, slides (n = 6 slices/group) were rehydrated with PBS and incubated in sodium borohydride (10 mg/mL) to reduce background autofluorescence. Slides were then blocked with 2% bovine serum albumin (BSA, Sigma) and 0.3% Triton X-100 (Sigma) in PBS (BSA:PBS-T) for 1 h before washing with PBS and incubation with primary anti-TH rabbit polyclonal antibody (EMD Millipore) at 1:300 BSA:PBS-T overnight at room temperature. Slides were subsequently rinsed in PBS and incubated in Alexa Fluor 488 conjugated goat anti-rabbit secondary antibody (1:500; Invitrogen) for 1.5 h at room temperature. After incubation, slides were rinsed with PBS and then briefly dipped in MilliQ water before incubation in a 10 mM copper sulfate/50 mM ammonium acetate solution to reduce autofluorescence (3 × 10 min). Finally, slides were briefly redipped in MilliQ water and a 1:1 glycerol/PBS solution was added to mount a coverslip. Imaging was performed on an upright Nikon Eclipse Ni microscope at ×10 magnification with a FITC filter (Ex/Em: 495/519 nm). Four images per tissue slice were taken and thresholded in ImageJ to determine TH+ sympathetic nerve fiber area and total tissue area. Percent fiber density was defined as the ratio of TH+ fiber area to total tissue area. All images were analyzed by two blinded users and values were averaged between users.
Tissue Norepinephrine Content
NE content was measured via HPLC as previously described (36, 47). Frozen tissue was homogenized at 25°C in perchloric acid (PCA, 0.1 M) containing the internal standard dihydroxybenzylamine (DHBA, 0.25 µM) to correct for NE sample recovery. Catecholamines were purified from a 100-µL aliquot of homogenate using alumina extraction. NE was desorbed from alumina by using PCA (150 µL, 0.1 M) and then measured by C18 reversed-phase HPLC with electrochemical detection (ESA, Coulchem III) and a detection limit of ∼0.05 pmol and >60% recovery from alumina extraction.
Statistics
Data are expressed as means ± SD for n animals. Due to the technical challenges of sequential in vivo blood collection and some ex vivo experimental procedures, sample sizes and sex distribution are unequal for some parameters. Likewise, in some cases, the ex vivo innervated heart preparations did not capture at a particular pacing frequency, which impacted the sample size for some rate-matched parameters. n are specified in each figure legend and data from both sexes are pooled, with O and X symbols indicating female and male sex, respectively (48–50). Statistical analysis was performed using GraphPad Prism 9. Normality was tested using the Shapiro–Wilk test, and significance was assessed using t test, two-way repeated-measures ANOVA, two-way ANOVA with mixed-effects analysis, or Mann–Whitney test as appropriate and specified in the figure legend. Statistical significance was attained when the P value < 0.05.
RESULTS
Baseline Measurements following Nicotine Administration
To confirm nicotine delivery, cotinine was measured in serum blood samples. As expected, CT rabbits had no detectable cotinine at all time points (Fig. 2A). In NIC rabbits, there was no detectable cotinine at day 0, but 455.3 ± 374.0 and 694.6 ± 376.4 ng/mL of cotinine at 2 and 4 wk of exposure, respectively, which is comparable with cotinine levels in humans that smoke or vape (14, 51). Although CT and NIC rabbit body weights were comparable at the start of the study (3.1 ± 0.3 vs. 3.1 ± 0.2 kg, P = 0.34), in accordance with nicotine being an appetite suppressant, body weight was lower in NIC versus CT from 2 to 4 wk of exposure (body weight at 4 wk = 3.3 ± 0.1 kg in CT vs. 2.9 ± 0.3 kg, P = 0.0087). To determine the impact of nicotine on ex vivo HR; baseline HR measurements were obtained from the innervated perfused heart preparations before the addition of blebbistatin and dyes. Baseline HR was 23 ± 32.7% higher in NIC hearts compared with CT (Fig. 2B, 206.3 ± 24.5 vs. 166.6 ± 38.8 beats/min, P = 0.004). SNS stimulation thresholds were also higher in NIC hearts (Fig. 2C, 7.3 ± 5.3 vs. 2.1 ± 1.0 Hz, P = 0.003).
Figure 2.

Cotinine concentration, heart rate, and sympathetic nerve stimulation threshold in control and nicotine-exposed hearts. A: mean cotinine concentration at 0, 2, and 4 wk of nicotine exposure. n = 6–10/group. B: ex vivo baseline heart rate. Measurements taken before the addition of dyes and blebbistatin. C: stimulation frequency threshold for sympathetic nerve stimulation. Spinal cord was stimulated at T1–T2 with constant voltage and frequency was increased at 0.5-Hz increments until a >15% increase in heart rate was observed. n = 8–11/group. Data are represented as means ± SD. *P < 0.05 and **P < 0.01 by two-way ANOVA with mixed-effects analysis (A) or by Mann–Whitney test (B and C). X, males; O, females.
Electrophysiological Responses to SNS
To determine the effect of SNS on electrophysiological parameters, HR, APD80, and CaTD80, were measured during sinus rhythm immediately before and at 13 s of SNS. SNS resulted in comparable increases in HR in both CT and NIC hearts (Fig. 3B, 41.1 ± 45.52 vs. 29.2 ± 32.66% increase, P = 0.51). Similarly, SNS shortened APD80 to a similar degree in both CT and NIC hearts (Fig. 3, C and D). Interestingly, SNS reduced CaTD80 compared with baseline values in CT but not in NIC hearts (Fig. 3D, P = 0.018); however, the percent change in CaTD80 with SNS was not different between groups (Fig. 3E, P = 0.411).
Figure 3.

Effect of sympathetic nerve stimulation (SNS) on control and nicotine-exposed hearts. Heart rate (A and B), action potential duration (APD80, C and D), and calcium transient duration (CaTD80, E and F) changes with SNS during sinus rhythm. Baseline data assessed just before SNS. n = 8–11/group. Data are represented as means ± SD. *P < 0.05 by two-way ANOVA with repeated measures (A, C, and E), Mann–Whitney test (B), or two-tailed, unpaired t test (D and F). X, males; O, females.
APD and CaTD Alternans
To evaluate alternans susceptibility, hearts were paced at progressively shorter PCLs. As sympathetic stimulation typically shortens the PCL at which alternans emerge because of accelerated Ca2+ handling (33, 42, 52), this was first performed without and subsequently with SNS. In both the absence and presence of SNS, APD alternans emerged at longer PCLs in NIC compared with CT hearts (Fig. 4A, 238.8 ± 15.5 vs. 197.5 ± 22.5 ms, P = 0.0022 and 230.0 ± 22.7 vs. 180.0 ± 28.3 ms, P = 0.0003, respectively). Although SNS was associated with alternans emerging at shorter PCLs compared with baseline in CT (Fig. 4A, P = 0.0194), no difference in APD alternans threshold was observed with SNS in NIC hearts (Fig. 4A, P = 0.2896). Similar results were obtained for CaTD alternans (Fig. 4B), whereby alternans emerged at shorter PCLs in CT animals with SNS versus baseline (P = 0.0013), and the threshold PCL for alternans in the presence of SNS was longer and thus occurred at slower rates in NIC versus CT (P = 0.003).
Figure 4.
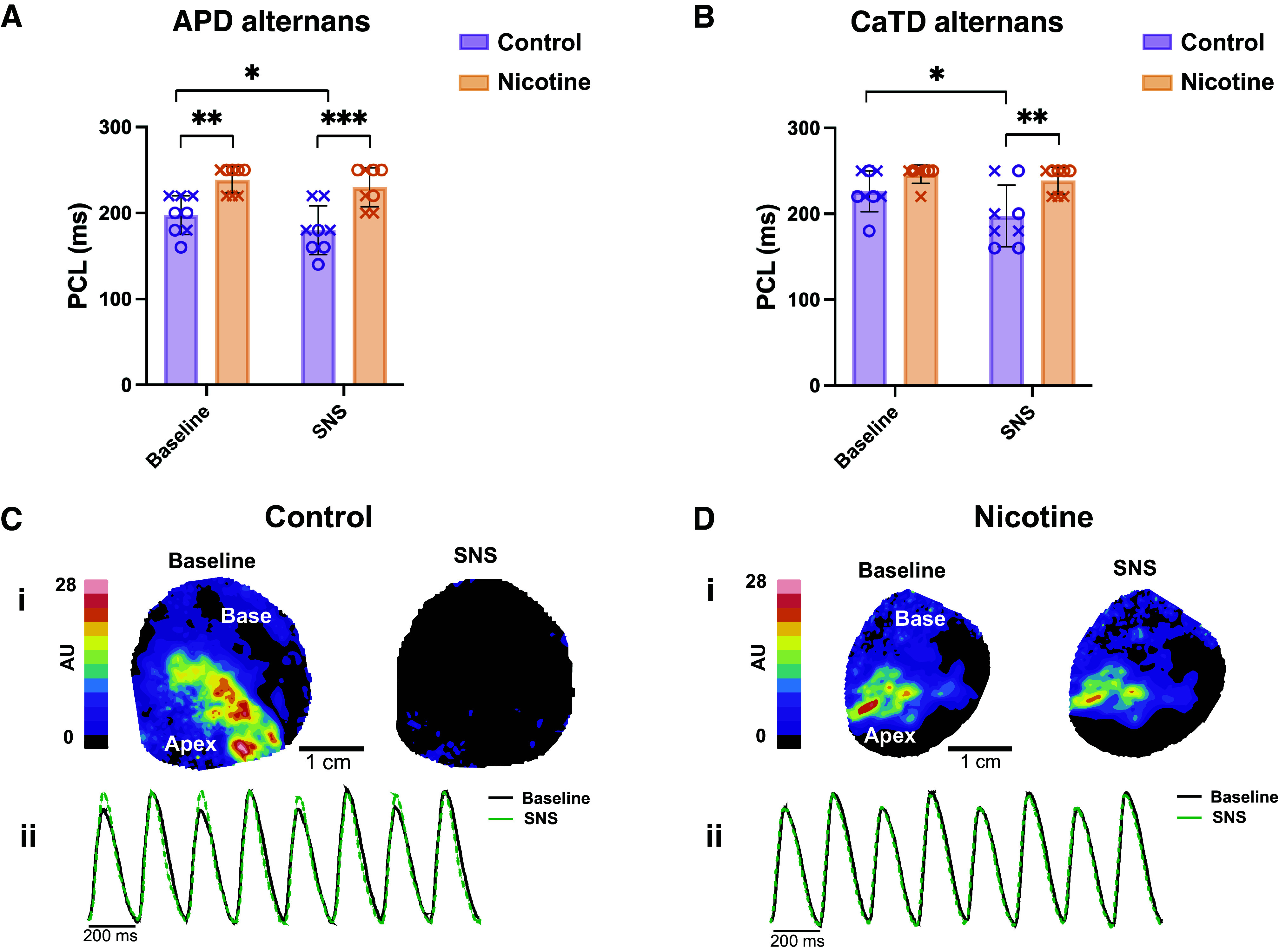
Alternans threshold and magnitude in control and nicotine-exposed hearts. A: pacing cycle length (PCL) at which action potential duration (APD) alternans emerged at baseline and with sympathetic nerve stimulation (SNS). B: PCL at which Ca2+ alternans emerged. C and D: representative contour maps (i) and corresponding Ca2+ traces (ii) demonstrating Ca2+ alternans magnitude at baseline and with SNS at PCL = 200 ms (C: control; D: nicotine). Ca2+ trace averaged from entire field of view to demonstrate mean alternans magnitude. n = 8/group. Data are represented as means ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001 by two-way ANOVA with repeated measures. X, males; O, females.
β-Adrenergic Responsiveness
Electrophysiological differences in response to SNS may be due to altered nerve density, function (e.g., NE release), β-adrenergic responsiveness of cardiomyocytes, or a combination of these factors. To specifically assess β-adrenergic responses of the heart, 500 nM NE was added to the perfusate. HR before the addition of NE (time 0, t0) and up to 6 min after the addition of NE is shown in Fig. 5A. To account for differences in baseline HR, the percent change in HR over 6 min of NE perfusion was also calculated (Fig. 5B). Both groups had faster HRs at 2 and 3 min after NE compared with t0 (Fig. 5C), but only CT hearts showed significant elevation at 1 min (P = 0.0005), suggesting a potentially slower HR response to NE in NIC hearts. CT hearts tended to have a larger relative change in HR from initial values compared with NIC (Fig. 5D, CT: 68.2 ± 41.0 vs. NIC: 34.1 ± 29.8%, P = 0.0591). Maximal HRs after the addition of NE were not different between groups (Fig. 5E, CT: 216.4 ± 51.4 vs. NIC: 205.2 ± 45.5 beats/min, P = 0.86). Although NIC hearts tended to show lower HR responses to NE (reflective of sinoatrial [SA] nodal responses), ventricular responses to NE were comparable between groups (Fig. 5, F–I). APD80 decreased with NE by 18.6 ± 14.9% in CT (P = 0.0062) and 10.6 ± 11.1% in NIC (P = 0.0269), with CaTD80 decreased by 23.5 ± 12.3% in CT (P = 0.0011) and 15.8 ± 13.2% in NIC (P = 0.0027). No difference in APD80 or CaTD80 responses to NE was observed between groups (P = 0.2078 and P = 0.2371, respectively).
Figure 5.

Responses to norepinephrine (NE) in control and nicotine-exposed hearts. A and B: heart rate changes over time immediately before and after the addition of 500 nM NE to the perfusate. C: heart rate measurements after the addition of NE at 1 min (t1), 2 min (t2), and 3 min (t3). D: percent change from baseline to maximum heart rate with NE. E: maximum heart rate after NE added. F–I: action potential duration (APD; F and G) and calcium transient duration (CaTD; H and I) changes in response to NE in sinus rhythm. n = 7–11/group, except A–C where n = 4–11 depending on time point. Data are represented as means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by two-way ANOVA with mixed-effects analysis (A–C, F, and H) Mann–Whitney test (D), or t test (E, G, and I). X, males; O, females.
Arrhythmia Susceptibility
Arrhythmia susceptibility was measured from pseudo-ECG recordings obtained from needle electrodes inserted into the posterior thoracic cavity (hence atypical ECG morphology, Fig. 1). PVCs and bigeminy/salvos were observed in both CT and NIC hearts with no difference in arrhythmia score determined between groups (Fig. 6, CT: 0.88 ± 0.83 vs. NIC: 1.5 ± 0.71, P = 0.1354).
Figure 6.

Arrhythmia susceptibility in control and nicotine-exposed hearts. A: pseudo-ECG recording demonstrating a premature ventricular contraction (PVC) during sinus rhythm. Recordings were measured from the posterior thoracic cavity in the bath. B: average arrhythmia scores. n = 8–10/group. Data are represented as means ± SD. Significance tested by Mann–Whitney test. X, males; O, females.
Fibrosis, Sympathetic Nerve Density, and Norepinephrine Content
Chronic nicotine administration has been previously associated with increased myocardial fibrosis (24–26). Therefore, fibrotic density in CT and NIC hearts was evaluated from Masson’s trichrome images. However, no difference in fibrotic density was found (Fig. 7, A and B, 21.78 ± 3.68 vs. 19.30 ± 3.68%, P = 0.53). To assess whether changes in sympathetic nerve density were responsible for altered SNS responses, the percent TH+ fiber density was quantified from LV tissue. CT hearts had a higher percent area of TH+ fibers compared with NIC hearts (Fig. 7, C and D, 10.18 ± 2.45 vs. 7.55 ± 0.91%, P = 0.0339). NE content was also measured in the LV and RA in CT and NIC hearts (Fig. 7E). In line with greater sympathetic nerve density in the atrium versus ventricle (53), we observed increased NE content in the RA versus LV (P = 0.0037, two-way ANOVA main effect). Pairwise, we found increased NE content in the RA versus LV of NIC hearts (P = 0.0194), and a tendency for this in CT (P = 0.1264). Interestingly, despite the lower ventricular nerve density, we saw no difference in NE content in the NIC LV compared with CT (29.2 ± 9.5 vs. 25.6 ± 8.4 pmol/mg, P = 0.7431), though a tendency for increased NE was observed in the RA (44.6 ± 12.1 vs. 34.8 ± 9.3 pmol/mg, P = 0.1970).
Figure 7.
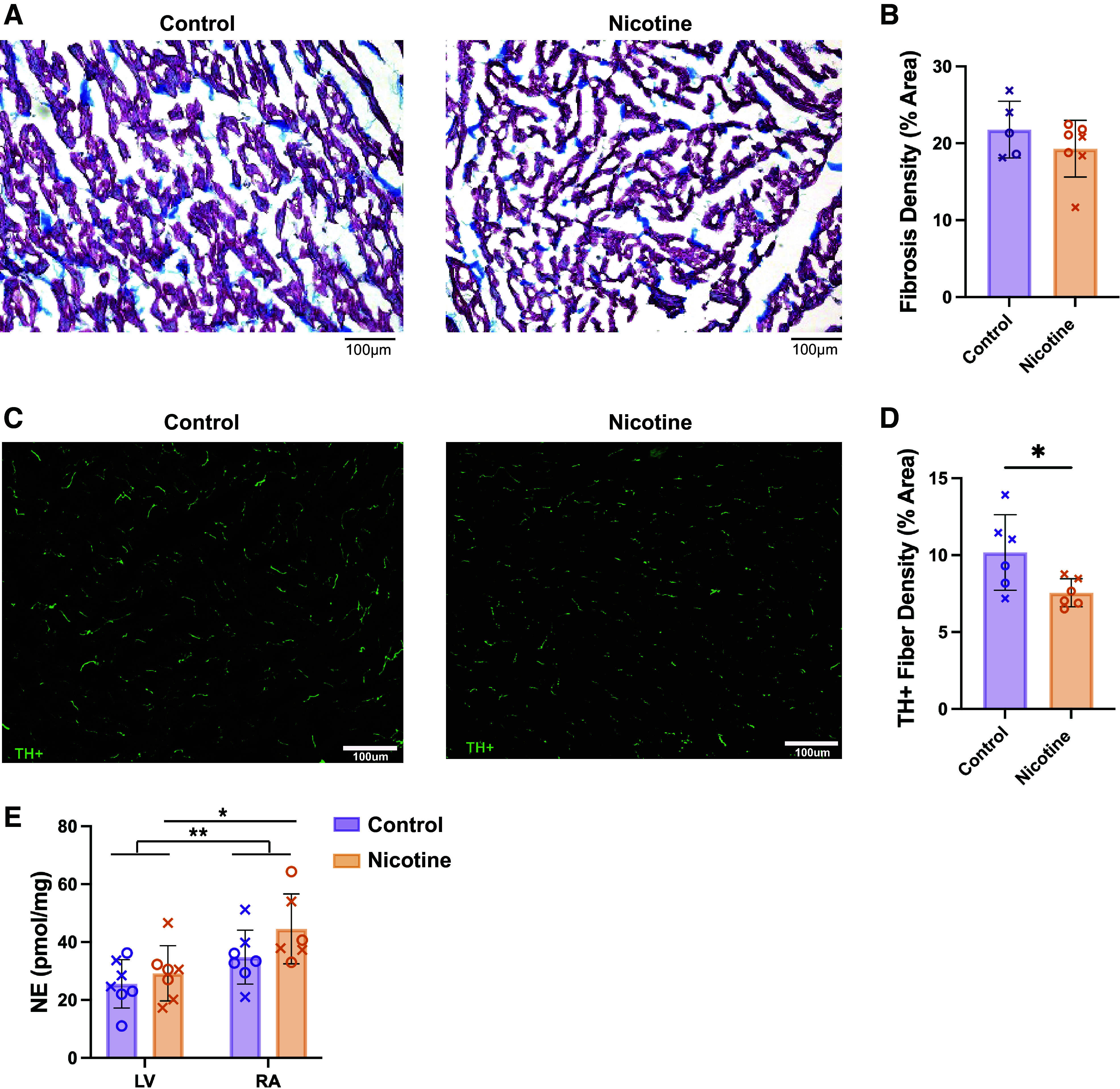
Fibrosis, sympathetic nerve density, and norepinephrine (NE) content in control and nicotine-exposed hearts. A: representative images of Masson’s trichrome staining; fibrosis indicated in blue and myocardial tissue in red. B: average fibrosis density in left ventricle. n = 5–7/group. C: representative immunofluorescence images of tyrosine hydroxylase (TH) labeling. D: sympathetic nerve fiber density in left ventricle. n = 6/group. E: NE content of left ventricle (LV) and right atria (RA). n = 7/group. Data are represented as means ± SD. *P < 0.05 and **P < 0.01 by Mann–Whitney test (B), two-tailed, unpaired t test (D), or two-way ANOVA with repeated measures (E). Scale bar = 100 µm. X, males; O, females.
DISCUSSION
The objective of this study was to investigate the effects of chronic nicotine exposure on cardiac sympathetic responses, electrophysiological and structural remodeling, and potential arrhythmogenic effects. To our knowledge, this study is the first to use the innervated rabbit heart model to evaluate sympathetic responses in hearts chronically exposed to nicotine. Here, we show that NIC hearts had 1) higher ex vivo baseline HRs, 2) elevated SNS thresholds and reduced LV sympathetic nerve density, 3) increased susceptibility to Ca2+ and APD alternans that does not improve with SNS, 4) maintained β-adrenergic responsiveness in the ventricle but reduced responsiveness of the SA node, and 5) no change in arrhythmia propensity. Taken together, these results suggest that chronic nicotine exposure leads to sympathetic remodeling and altered cardiac electrophysiological responses, but that these changes are not necessarily arrhythmogenic following 28 days of exposure.
Chronic Nicotine Exposure Increases Ex Vivo Baseline Heart Rate
Perfusion of a low dose of nicotine has previously been shown to result in an acute increase in HR in isolated rabbit hearts (54). In addition, it has been shown that chronic nicotine administration led to a reduction in HRV suggesting a shift to sympathetic dominance and decreased vagal tone (17, 55). Here, we show that the hearts of rabbits exposed to nicotine for 28 days had elevated baseline ex vivo HR in the innervated heart preparation (Fig. 2B). Since sympathetic innervation to the heart remains intact in this preparation, this result may suggest an elevation in tonic sympathetic nerve activity under resting conditions (without electrical stimulation) following nicotine exposure. Alternatively, the increase in baseline HR may also be due to a reduction in tonic parasympathetic activity (postganglionic parasympathetic neurons are present in the ex vivo heart) and/or due to functional or structural alterations within the SA node itself, which may represent important areas for future study.
Electrophysiological Responses to SNS after Chronic Nicotine Exposure
Although we found that NIC hearts had significantly increased electrical SNS thresholds (measured by the stimulation frequency required to produce a 15% increase in HR, Fig. 2C), similar changes in APD, CaTD, and HR were observed with SNS at suprathreshold frequency stimulation (Fig. 3). The elevated SNS thresholds could be due to many factors, including changes in sympathetic nerve density or function, altered β-adrenergic responsiveness, or structural remodeling of the myocardium. Chronic nicotine administration is associated with increased atrial fibrosis (24–26); however, we found no difference in ventricular fibrosis between groups in this study (Fig. 7A), indicating potential chamber differences in the fibrotic remodeling induced by nicotine. We did observe a reduction in sympathetic nerve density in NIC hearts (Fig. 7C) that could explain the blunted responsiveness to SNS. However, despite the decrease in nerve density, we observed no difference in NE content in the RA and LV of NIC hearts compared with CT (Fig. 7E). Typically, NE content is associated with local sympathetic nerve density whereby more nerves = more NE. However, sustained electrical activation of postganglionic sympathetic neurons has been shown to stimulate the activity of TH, increasing NE synthesis allowing the neuron to maintain elevated transmitter release during extended periods of activation (56, 57). Related data were obtained in human and isolated rabbit heart studies showing that acute nicotine administration leads to an increase in NE release (58–60). Finally, studies in rats with chronic administration of nicotine showed an increase in cardiac NE turnover, but no change in cardiac NE content (61). Thus, our data connecting decreased nerve density with normal NE content are consistent with studies showing that nicotine stimulates NE synthesis in sympathetic neurons independent of nerve density. It is possible that increased NE production may partially compensate for reduced sympathetic nerve density, but changes in the spatial localization and distribution of NE release, NE reuptake, or proportion of myocytes or receptors stimulated because of altered nerve density may significantly impact functional effects.
Chronic Nicotine Exposure Increases the Susceptibility for APD and CaT Alternans
We found that NIC hearts had APD and CaT alternans that emerged at longer PCLs (slower pacing frequencies) compared with CT (Fig. 4, A and B), indicating increased alternans susceptibility. Our result is corroborated by a study in post-MI dogs that showed an increase in depolarizing and repolarizing alternans after nicotine infusion (28) and our previous work showing an increase in the magnitude of APD and CaT alternans after SHS exposure (8). Although others have shown increased monophasic APD50 alternans with SNS (62), we expected that the PCL at which alternans emerges would be shorter with SNS given that β-adrenergic stimulation accelerates Ca2+ cycling (e.g., faster SERCA reuptake of Ca2+ and shorter period of RyR refractoriness) (42, 52). Indeed, we found that alternans emerged at shorter PCLs (higher pacing frequencies) with SNS in CT but not in NIC hearts (Fig. 4, A and B). This result could be explained by decreased responsiveness to acute SNS following NIC exposure (potentially because of diminished sympathetic innervation) that may be particularly evident at faster PCLs. Interestingly, we observed similar findings in aged mouse hearts, where chronically elevated sympathetic tone (that naturally occurs with age) was also associated with increased alternans susceptibility that was not improved with SNS (36). It is known that APD and Ca2+ alternans are strongly associated with increased risk for arrhythmias (42, 63–65); however, we did not observe an increase in arrhythmia susceptibility in NIC hearts in this study. Although the levels of cotinine recorded in the study are comparable with light-to-heavy smokers (51), it is possible that increased arrhythmogenic activity may be observed with longer duration nicotine exposure and later-stage remodeling (e.g., fibrosis) that may occur. Indeed, we did not observe a change in fibrosis in the present study, which may be an important additional contributor to arrhythmogenesis.
β-Adrenergic Responsiveness to NE after Chronic Nicotine Exposure
Because SNS may result in variable NE release throughout the heart (because of variations in nerve density and/or function), NE was directly added to the perfusate to assess β-adrenergic responsiveness of the myocardium. NIC hearts tended to have smaller and slower changes in HR compared with CT, however, comparable β-adrenergic responses were observed between groups in the ventricular myocardium (Fig. 5). Reduced HR responses to NE in NIC hearts may be a result of elevated HR at time 0 (therefore any HR increases may be more modest) but could also suggest a somewhat blunted β-adrenergic response of the SA node. This blunted β-adrenergic response could be due to loss of coupling between nerves and β-adrenergic receptors (66) or β-adrenergic receptor downregulation, as prior studies previously showed decreased β-adrenergic receptor density and reduced catecholamine response in lymphocytes from habitual smokers versus nonsmokers (67). On the other hand, because we saw no difference in NE-mediated APD and CaTD shortening between groups, our data suggest that ventricular β-adrenergic responsiveness may be preserved following 4 wk of nicotine treatment. As such, the loss of the protective effect of SNS in shortening the PCL at which alternans emerges in NIC hearts is likely due to differences in nerve density and potential altered localization of NE release, rather than impaired responses to NE. The differences in SA nodal versus ventricular responsiveness to NE may be indicative of regional divergence in nicotine-induced sympathetic remodeling, and future studies are required to further elucidate these findings.
Limitations
With 28 days of nicotine exposure, we saw a change in some, but not all, electrophysiological parameters. It is possible that with longer exposure duration, there may be more profound changes in electrophysiology, Ca2+ handling, and arrhythmia susceptibility. This could form part of a follow-up study that could consider whether nicotine-induced remodeling is reversible and may be important considering the use of nicotine patches as a smoking withdrawal strategy. We specifically focused on ventricular action potentials, Ca2+ transients, and nerve density in this study and therefore did not assess corresponding atrial parameters, which may be more severely impacted by nicotine exposure due to greater innervation of the atria. Although we included both sexes in our study, we were insufficiently powered to assess sex differences in the context of chronic nicotine exposure, and this remains an important area for future study as sex differences in repolarization following e-cigarette use have recently been reported (68).
Conclusions
Using the innervated rabbit heart model, we determined the effects of chronic nicotine exposure on cardiac electrophysiology, Ca2+ handling, and sympathetic remodeling and responses. We found that chronic nicotine exposure results in reduced response to SNS, as indicated by elevated electrical stimulation thresholds. Following nicotine exposure, SNS also failed to slow the emergence of cardiac alternans, which may be proarrhythmic. This is likely due to sympathetic hypoinnervation of the myocardium, given that ventricular β-adrenergic responses to NE were unaltered following nicotine exposure. Taken together, our data indicate that chronic nicotine exposure for just 28 days results in potentially detrimental sympathetic and electrophysiological remodeling that we suggest may be further exacerbated with continued longer-term usage.
DATA AVAILABILITY
Full data sets are available from the corresponding author upon reasonable request.
GRANTS
This study was funded by University of California Tobacco-Related Disease Research Program Grant T29IP0365C; National Institutes of Health Grants R01 HL111600, R01 HL093056, and T32 GM144303; and National Natural Science Foundation of China Grant NSFC, 82200346. G.A.N. has been supported by British Heart Foundation Programme Grant RG/17/3/32,774 and Medical Research Council Biomedical Catalyst Developmental Pathway Funding Scheme Grant MR/S037306/1.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
Crystal Ripplinger is an editor of American Journal of Physiology-Heart and Circulatory Physiology and was not involved and did not have access to information regarding the peer-review process or final disposition of this article. An alternate editor oversaw the peer-review and decision-making process for this article.
AUTHOR CONTRIBUTIONS
A.G., G.A.N., B.A.H., and C.M.R. conceived and designed research; A.G., C.E.R.S., J.L.C., L.N., L.R.M., I.-J.L., S.T., Z.W., L.W., and W.R.W. performed experiments; A.G., C.E.R.S., J.L.C., L.N., L.R.M., I.-J.L., S.T., Z.W., L.W., and W.R.W. analyzed data; A.G., C.E.R.S., J.L.C., L.N., Z.W. W.R.W., G.A.N., B.A.H., and C.M.R. interpreted results of experiments; A.G., C.E.R.S., and C.M.R. prepared figures; A.G., C.E.R.S., and C.M.R. drafted manuscript; A.G., C.E.R.S., J.L.C., B.A.H., and C.M.R. edited and revised manuscript; A.G., C.E.R.S., J.L.C., L.N., L.R.M., I.-J.L., S.T., Z.W., L.W., W.R.W., G.A.N., B.A.H., and C.M.R. approved final version of manuscript.
REFERENCES
- 1. He H, Pan Z, Wu J, Hu C, Bai L, Lyu J. Health effects of tobacco at the global, regional, and national levels: results from the 2019 Global Burden of Disease Study. Nicotine Tob Res 24: 864–870, 2022. doi: 10.1093/ntr/ntab265. [DOI] [PubMed] [Google Scholar]
- 2. Gallucci G, Tartarone A, Lerose R, Lalinga AV, Capobianco AM. Cardiovascular risk of smoking and benefits of smoking cessation. J Thorac Dis 12: 3866–3876, 2020. doi: 10.21037/jtd.2020.02.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mensah GA, Brown DW. An overview of cardiovascular disease burden in the United States. Health Aff (Millwood) 26: 38–48, 2007. doi: 10.1377/hlthaff.26.1.38. [DOI] [PubMed] [Google Scholar]
- 4. Aune D, Schlesinger S, Norat T, Riboli E. Tobacco smoking and the risk of sudden cardiac death: a systematic review and meta-analysis of prospective studies. Eur J Epidemiol 33: 509–521, 2018. doi: 10.1007/s10654-017-0351-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deo R, Norby FL, Katz R, Sotoodehnia N, Adabag S, DeFilippi CR, Kestenbaum B, Chen LY, Heckbert SR, Folsom AR, Kronmal RA, Konety S, Patton KK, Siscovick D, Shlipak MG, Alonso A. Development and validation of a sudden cardiac death prediction model for the general population. Circulation 134: 806–816, 2016. doi: 10.1161/CIRCULATIONAHA.116.023042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barua RS, Ambrose JA. Mechanisms of coronary thrombosis in cigarette smoke exposure. Arterioscler Thromb Vasc Biol 33: 1460–1467, 2013. doi: 10.1161/ATVBAHA.112.300154. [DOI] [PubMed] [Google Scholar]
- 7. Chen C-Y, Chow D, Chiamvimonvat N, Glatter KA, Li N, He Y, Pinkerton KE, Bonham AC. Short-term secondhand smoke exposure decreases heart rate variability and increases arrhythmia susceptibility in mice. Am J Physiol Heart Circ Physiol 295: H632–H639, 2008. doi: 10.1152/ajpheart.91535.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Z, Wang L, Tapa S, Pinkerton KE, Chen CY, Ripplinger CM. Exposure to secondhand smoke and arrhythmogenic cardiac alternans in a mouse model. Environ Health Perspect 126: 127001, 2018. doi: 10.1289/EHP3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ali FRM, Seidenberg AB, Crane E, Seaman E, Tynan MA, Marynak K. E-cigarette unit sales by product and flavor type, and top-selling brands, United States, 2020–2022. MMWR Morb Mortal Wkly Rep 72: 672–677, 2023. doi: 10.15585/mmwr.mm7225a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lisko JG, Tran H, Stanfill SB, Blount BC, Watson CH. Chemical composition and evaluation of nicotine, tobacco alkaloids, pH, and selected flavors in E-cigarette cartridges and refill solutions. Nicotine Tob Res 17: 1270–1278, 2015. doi: 10.1093/ntr/ntu279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim H-J, Shin H-S. Determination of tobacco-specific nitrosamines in replacement liquids of electronic cigarettes by liquid chromatography–tandem mass spectrometry. J Chromatogr A 1291: 48–55, 2013. doi: 10.1016/j.chroma.2013.03.035. [DOI] [PubMed] [Google Scholar]
- 12. Lim HH, Shin HS. Measurement of aldehydes in replacement liquids of electronic cigarettes by headspace gas chromatography-mass spectrometry. Bull Korean Chem Soc 34: 2691–2696, 2013. doi: 10.5012/bkcs.2013.34.9.2691. [DOI] [Google Scholar]
- 13. Pellegrino R, Tinghino B, Mangiaracina G, Marani A, Vitali M, Protano C, Osborn J, Cattaruzza M. Electronic cigarettes: an evaluation of exposure to chemicals and fine particulate matter (PM). Ann Ig 24: 279–288, 2011. [PubMed] [Google Scholar]
- 14. Goniewicz ML, Boykan R, Messina CR, Eliscu A, Tolentino J. High exposure to nicotine among adolescents who use Juul and other vape pod systems (‘pods’). Tob Control 28: 676–677, 2019. doi: 10.1136/tobaccocontrol-2018-054565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pankow JF, Kim K, McWhirter KJ, Luo W, Escobedo JO, Strongin RM, Duell AK, Peyton DH. Benzene formation in electronic cigarettes. PLoS One 12: e0173055, 2017. doi: 10.1371/journal.pone.0173055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grilli M, Parodi M, Raiteri M, Marchi M. Chronic nicotine differentially affects the function of nicotinic receptor subtypes regulating neurotransmitter release. J Neurochem 93: 1353–1360, 2005. doi: 10.1111/j.1471-4159.2005.03126.x. [DOI] [PubMed] [Google Scholar]
- 17. Moheimani RS, Bhetraratana M, Yin F, Peters KM, Gornbein J, Araujo JA, Middlekauff HR. Increased cardiac sympathetic activity and oxidative stress in habitual electronic cigarette users: implications for cardiovascular risk. JAMA Cardiol 2: 278–284, 2017. doi: 10.1001/jamacardio.2016.5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shekha K, Ghosh J, Thekkoott D, Greenberg Y. Risk stratification for sudden cardiac death in patients with non-ischemic dilated cardiomyopathy. Indian Pacing Electrophysiol J 5: 122–138, 2005. [PMC free article] [PubMed] [Google Scholar]
- 19. Malliani A, Lombardi F, Pagani M, Cerutti S. Power spectral analysis of cardiovascular variability in patients at risk for sudden cardiac death. J Cardiovasc Electrophysiol 5: 274–286, 1994. doi: 10.1111/j.1540-8167.1994.tb01164.x. [DOI] [PubMed] [Google Scholar]
- 20. Dvorakova M, Lips KS, Brüggmann D, Slavikova J, Kuncova J, Kummer W. Developmental changes in the expression of nicotinic acetylcholine receptor α-subunits in the rat heart. Cell Tissue Res 319: 201–209, 2005. doi: 10.1007/s00441-004-1008-1. [DOI] [PubMed] [Google Scholar]
- 21. Wang H, Yang B, Zhang L, Xu D, Wang Z. Direct block of inward rectifier potassium channels by nicotine. Toxicol Appl Pharmacol 164: 97–101, 2000. doi: 10.1006/taap.2000.8896. [DOI] [PubMed] [Google Scholar]
- 22. Wang H, Shi H, Zhang L, Pourrier M, Yang B, Nattel S, Wang Z. Nicotine is a potent blocker of the cardiac A-type K+ channels. Circulation 102: 1165–1171, 2000. doi: 10.1161/01.cir.102.10.1165. [DOI] [PubMed] [Google Scholar]
- 23. Myles RC, Wang L, Bers DM, Ripplinger CM. Decreased inward rectifying K+ current and increased ryanodine receptor sensitivity synergistically contribute to sustained focal arrhythmia in the intact rabbit heart. J Physiol 593: 1479–1493, 2015. doi: 10.1113/jphysiol.2014.279638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shan H, Zhang Y, Lu Y, Zhang Y, Pan Z, Cai B, Wang N, Li X, Feng T, Hong Y, Yang B. Downregulation of miR-133 and miR-590 contributes to nicotine-induced atrial remodelling in canines. Cardiovasc Res 83: 465–472, 2009. doi: 10.1093/cvr/cvp130. [DOI] [PubMed] [Google Scholar]
- 25. Goette A, Lendeckel U, Kuchenbecker A, Bukowska A, Peters B, Klein HU, Huth C, Röcken C. Cigarette smoking induces atrial fibrosis in humans via nicotine. Heart 93: 1056–1063, 2007. doi: 10.1136/hrt.2005.087171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miyauchi M, Qu Z, Miyauchi Y, Zhou S-M, Pak H, Mandel WJ, Fishbein MC, Chen P-S, Karagueuzian HS. Chronic nicotine in hearts with healed ventricular myocardial infarction promotes atrial flutter that resembles typical human atrial flutter. Am J Physiol Heart Circ Physiol 288: H2878–H2886, 2005. doi: 10.1152/ajpheart.01165.2004. [DOI] [PubMed] [Google Scholar]
- 27. Kléber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev 84: 431–488, 2004. doi: 10.1152/physrev.00025.2003. [DOI] [PubMed] [Google Scholar]
- 28. Yashima M, Ohara T, Cao J-M, Kim Y-H, Fishbein MC, Mandel WJ, Chen P-S, Karagueuzian HS. Nicotine increases ventricular vulnerability to fibrillation in hearts with healed myocardial infarction. Am J Physiol Heart Circ Physiol 278: H2124–H2133, 2000. doi: 10.1152/ajpheart.2000.278.6.H2124. [DOI] [PubMed] [Google Scholar]
- 29. Mehta MC, Jain AC, Mehta A, Billie M. Cardiac arrhythmias following intravenous nicotine: experimental study in dogs. J Cardiovasc Pharmacol Ther 2: 291–298, 1997. doi: 10.1177/107424849700200407. [DOI] [PubMed] [Google Scholar]
- 30. Mills EJ, Wu P, Lockhart I, Wilson K, Ebbert JO. Adverse events associated with nicotine replacement therapy (NRT) for smoking cessation. A systematic review and meta-analysis of one hundred and twenty studies involving 177,390 individuals. Tob Induc Dis 8: 8, 2010. doi: 10.1186/1617-9625-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mills EJ, Thorlund K, Eapen S, Wu P, Prochaska JJ. Cardiovascular events associated with smoking cessation pharmacotherapies. Circulation 129: 28–41, 2014. doi: 10.1161/CIRCULATIONAHA.113.003961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jacob P 3rd, Yu L, Duan M, Ramos L, Yturralde O, Benowitz NL. Determination of the nicotine metabolites cotinine and trans-3′-hydroxycotinine in biologic fluids of smokers and non-smokers using liquid chromatography-tandem mass spectrometry: Biomarkers for tobacco smoke exposure and for phenotyping cytochrome P450 2A6 activity. J Chromatogr B Analyt Technol Biomed Life Sci 879: 267–276, 2011. doi: 10.1016/j.jchromb.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang L, Morotti S, Tapa S, Francis Stuart SD, Jiang Y, Wang Z, Myles RC, Brack KE, Ng GA, Bers DM, Grandi E, Ripplinger CM. Different paths, same destination: divergent action potential responses produce conserved cardiac fight-or-flight response in mouse and rabbit hearts. J Physiol 597: 3867–3883, 2019. doi: 10.1113/JP278016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ng GA, Brack KE, Coote JH. Effects of direct sympathetic and vagus nerve stimulation on the physiology of the whole heart—a novel model of isolated Langendorff perfused rabbit heart with intact dual autonomic innervation. Exp Physiol 86: 319–329, 2001. doi: 10.1113/eph8602146. [DOI] [PubMed] [Google Scholar]
- 35. Swift LM, Kay MW, Ripplinger CM, Posnack NG. Stop the beat to see the rhythm: excitation-contraction uncoupling in cardiac research. Am J Physiol Heart Circ Physiol 321: H1005–H1013, 2021. doi: 10.1152/ajpheart.00477.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Francis Stuart SD, Wang L, Woodward WR, Ng GA, Habecker BA, Ripplinger CM. Age-related changes in cardiac electrophysiology and calcium handling in response to sympathetic nerve stimulation. J Physiol 596: 3977–3991, 2018. doi: 10.1113/JP276396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Myles RC, Wang L, Kang C, Bers DM, Ripplinger CM. Local β-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ Res 110: 1454–1464, 2012. doi: 10.1161/CIRCRESAHA.111.262345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ripplinger CM, Glukhov AV, Kay MW, Boukens BJ, Chiamvimonvat N, Delisle BP, Fabritz L, Hund TJ, Knollmann BC, Li N, Murray KT, Poelzing S, Quinn TA, Remme CA, Rentschler SL, Rose RA, Posnack NG. Guidelines for assessment of cardiac electrophysiology and arrhythmias in small animals. Am J Physiol Heart Circ Physiol 323: H1137–H1166, 2022. doi: 10.1152/ajpheart.00439.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. O'Shea C, Holmes AP, Yu TY, Winter J, Wells SP, Correia J, Boukens BJ, De Groot JR, Chu GS, Li X, Ng GA, Kirchhof P, Fabritz L, Rajpoot K, Pavlovic D. ElectroMap: high-throughput open-source software for analysis and mapping of cardiac electrophysiology. Sci Rep 9: 1389, 2019. doi: 10.1038/s41598-018-38263-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Caldwell JL, Lee IJ, Ngo L, Wang L, Bahriz S, Xu B, Bers DM, Navedo MF, Bossuyt J, Xiang YK, Ripplinger CM. Whole-heart multiparametric optical imaging reveals sex-dependent heterogeneity in cAMP signaling and repolarization kinetics. Sci Adv 9: eadd5799, 2023. doi: 10.1126/sciadv.add5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tapa S, Wang L, Francis Stuart SD, Wang Z, Jiang Y, Habecker BA, Ripplinger CM. Adrenergic supersensitivity and impaired neural control of cardiac electrophysiology following regional cardiac sympathetic nerve loss. Sci Rep 10: 18801, 2020. doi: 10.1038/s41598-020-75903-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang L, Myles RC, De Jesus NM, Ohlendorf AK, Bers DM, Ripplinger CM. Optical mapping of sarcoplasmic reticulum Ca2+ in the intact heart: ryanodine receptor refractoriness during alternans and fibrillation. Circ Res 114: 1410–1421, 2014. doi: 10.1161/CIRCRESAHA.114.302505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang L, Myles RC, Lee IJ, Bers DM, Ripplinger CM. Role of reduced sarco-endoplasmic reticulum Ca2+-ATPase function on sarcoplasmic reticulum Ca2+ alternans in the intact rabbit heart. Front Physiol 12: 656516, 2021. doi: 10.3389/fphys.2021.656516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. De Jesus NM, Wang L, Lai J, Rigor RR, Francis Stuart SD, Bers DM, Lindsey ML, Ripplinger CM. Antiarrhythmic effects of interleukin 1 inhibition after myocardial infarction. Heart Rhythm 14: 727–736, 2017. doi: 10.1016/j.hrthm.2017.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Curtis MJ, Walker MJ. Quantification of arrhythmias using scoring systems: an examination of seven scores in an in vivo model of regional myocardial ischaemia. Cardiovasc Res 22: 656–665, 1988. doi: 10.1093/cvr/22.9.656. [DOI] [PubMed] [Google Scholar]
- 46. Gardner RT, Habecker BA. Infarct-derived chondroitin sulfate proteoglycans prevent sympathetic reinnervation after cardiac ischemia-reperfusion injury. J Neurosci 33: 7175–7183, 2013. doi: 10.1523/JNEUROSCI.5866-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Parrish DC, Alston EN, Rohrer H, Nkadi P, Woodward WR, Schütz G, Habecker BA. Infarction-induced cytokines cause local depletion of tyrosine hydroxylase in cardiac sympathetic nerves. Exp Physiol 95: 304–314, 2010. doi: 10.1113/expphysiol.2009.049965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lindsey ML, LeBlanc AJ, Ripplinger CM, Carter JR, Kirk JA, Hansell Keehan K, Brunt KR, Kleinbongard P, Kassiri Z. Reinforcing rigor and reproducibility expectations for use of sex and gender in cardiovascular research. Am J Physiol Heart Circ Physiol 321: H819–H824, 2021. doi: 10.1152/ajpheart.00418.2021. [DOI] [PubMed] [Google Scholar]
- 49. Denfeld QE, Lee CS, Habecker BA. A primer on incorporating sex as a biological variable into the conduct and reporting of basic and clinical research studies. Am J Physiol Heart Circ Physiol 322: H350–H354, 2022. doi: 10.1152/ajpheart.00605.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Usselman CW, Lindsey ML, Robinson AT, Habecker BA, Taylor CE, Merryman WD, Kimmerly D, Bender JR, Regensteiner JG, Moreau KL, Pilote L, Wenner MM, O’Brien M, Yarovinsky TO, Stachenfeld NS, Charkoudian N, Denfeld QE, Moreira-Bouchard JD, Pyle WG, DeLeon-Pennell KY. Guidelines on the use of sex and gender in cardiovascular research. Am J Physiol Heart Circ Physiol 326: H238–H255, 2024. doi: 10.1152/ajpheart.00535.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kunutsor SK, Spee JM, Kieneker LM, Gansevoort RT, Dullaart RPF, Voerman AJ, Touw DJ, Bakker SJL. Self-reported smoking, urine cotinine, and risk of cardiovascular disease: findings from the PREVEND (Prevention of Renal and Vascular End-Stage Disease) prospective cohort study. J Am Heart Assoc 7: e008726, 2018. doi: 10.1161/JAHA.118.008726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tomek J, Rodriguez B, Bub G, Heijman J. β-Adrenergic receptor stimulation inhibits proarrhythmic alternans in postinfarction border zone cardiomyocytes: a computational analysis. Am J Physiol Heart Circ Physiol 313: H338–H353, 2017. doi: 10.1152/ajpheart.00094.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kawano H, Okada R, Yano K. Histological study on the distribution of autonomic nerves in the human heart. Heart Vessels 18: 32–39, 2003. doi: 10.1007/s003800300005. [DOI] [PubMed] [Google Scholar]
- 54. Wennmalm A. Nicotine stimulates prostaglandin formation in the rabbit heart. Br J Pharmacol 59: 95–100, 1977. doi: 10.1111/j.1476-5381.1977.tb06981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moheimani RS, Bhetraratana M, Peters KM, Yang BK, Yin F, Gornbein J, Araujo JA, Middlekauff HR. Sympathomimetic effects of acute e-cigarette use: role of nicotine and non-nicotine constituents. J Am Heart Assoc 6: e006579, 2017. doi: 10.1161/JAHA.117.006579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zigmond RE, Ben-Ari Y. Electrical stimulation of preganglionic nerve increases tyrosine hydroxylase activity in sympathetic ganglia. Proc Natl Acad Sci USA 74: 3078–3080, 1977. doi: 10.1073/pnas.74.7.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zigmond RE, Chalazonitis A. Long-term effects of preganglionic nerve stimulation on tyrosine hydroxylase activity in the rat superior cervical ganglion. Brain Res 164: 137–152, 1979. doi: 10.1016/0006-8993(79)90011-8. [DOI] [PubMed] [Google Scholar]
- 58. Gourlay SG, Benowitz NL. Arteriovenous differences in plasma concentration of nicotine and catecholamines and related cardiovascular effects after smoking, nicotine nasal spray, and intravenous nicotine. Clin Pharmacol Ther 62: 453–463, 1997. doi: 10.1016/S0009-9236(97)90124-7. [DOI] [PubMed] [Google Scholar]
- 59. Gillespie MN, Owasoyo JO, Kiritsy-Roy JA, O'Connor WN, Van Loon GR. Exaggerated nicotine-induced norepinephrine release from atherosclerotic rabbit hearts. Toxicology 37: 147–157, 1985. doi: 10.1016/0300-483x(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 60. Löffelholz K. Autoinhibition of nicotinic release of noradrenaline from postganglionic sympathetic nerves. Naunyn-Schmiedebergs Arch Pharmak 267: 49–63, 1970. doi: 10.1007/BF00997114. [DOI] [PubMed] [Google Scholar]
- 61. Westfall TC. Influence of chronic nicotine administration on blood pressure and heart norepinephrine turnover. Eur J Pharmacol 10: 19–24, 1970. doi: 10.1016/0014-2999(70)90152-4. [DOI] [PubMed] [Google Scholar]
- 62. Ng GA, Brack KE, Patel VH, Coote JH. Autonomic modulation of electrical restitution, alternans and ventricular fibrillation initiation in the isolated heart. Cardiovasc Res 73: 750–760, 2007. doi: 10.1016/j.cardiores.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 63. Laurita KR, Rosenbaum DS. Cellular mechanisms of arrhythmogenic cardiac alternans. Prog Biophys Mol Biol 97: 332–347, 2008. doi: 10.1016/j.pbiomolbio.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chudin E, Goldhaber J, Garfinkel A, Weiss J, Kogan B. Intracellular Ca(2+) dynamics and the stability of ventricular tachycardia. Biophys J 77: 2930–2941, 1999. doi: 10.1016/S0006-3495(99)77126-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pearman CM, Madders GWP, Radcliffe EJ, Kirkwood GJ, Lawless M, Watkins A, Smith CER, Trafford AW, Eisner DA, Dibb KM. Increased vulnerability to atrial fibrillation is associated with increased susceptibility to alternans in old sheep. J Am Heart Assoc 7: e009972, 2018. [Erratum in J Am Heart Assoc 8: e04679, 2019]. doi: 10.1161/JAHA.118.009972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Prando V, Da Broi F, Franzoso M, Plazzo AP, Pianca N, Francolini M, Basso C, Kay MW, Zaglia T, Mongillo M. Dynamics of neuroeffector coupling at cardiac sympathetic synapses. J Physiol 596: 2055–2075, 2018. doi: 10.1113/JP275693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Laustiola KE, Lassila R, Kaprio J, Koskenvuo M. Decreased beta-adrenergic receptor density and catecholamine response in male cigarette smokers. A study of monozygotic twin pairs discordant for smoking. Circulation 78: 1234–1240, 1988. doi: 10.1161/01.cir.78.5.1234. [DOI] [PubMed] [Google Scholar]
- 68. Ruedisueli I, Lakhani K, Nguyen R, Gornbein J, Middlekauff HR. Electronic cigarettes prolong ventricular repolarization in people who smoke tobacco cigarettes: implications for harm reduction. Am J Physiol Heart Circ Physiol 324: H821–H832, 2023. doi: 10.1152/ajpheart.00057.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Full data sets are available from the corresponding author upon reasonable request.