

Abstract
DMOA-derived spiromeroterpenoids are a group of natural products with complex structures and varied biological activities. Recently, we reported the first enantioselective total synthesis of five spiromeroterpenoids based on a fragment coupling strategy. This full account describes details of a strategy evolution that culminated in successful syntheses of the targeted natural products. Although our alkylative dearomatization methodology was unable to deliver the desired spirocyclic products in our first-generation approach, our second-generation approach based on oxidative [3+2] cycloaddition produced the asnovolin H core along with several complex dimers. Challenges with the dearomatization approach finally led us to develop a third generation, non-dearomatization approach based on a fragment coupling strategy to construct the conserved, sterically hindered bis-neopentyl linkage of the spiromeroterpenoids through 1,2-addition. To enable scalable access of the natural products, a refined, multigram-scale synthesis of the coupling partners was developed. A series of stereoselective transformations were developed through judicious choice of reagents and conditions. Finally, modular spirocycle construction logic was demonstrated through the synthesis of a small library of spiromeroterpenoid analogues.
Graphical Abstract

INTRODUCTION
3,5-Dimethylorsellinic acid (DMOA)-derived meroterpenoids are a group of fascinating natural products with complex structures and diverse biological activities. As shown in Figure 1, asperterpene A (1) exhibits potent BACE1 inhibitory activity with an IC50 of 78 nM which is more active than LY2811376 (IC50 = 260 nM), a clinical BACE1 inhibitor produced by Eli Lilly. Compound 1 also displays a highly strained twist-boat, twist-boat, boat, half-chair conformation.1 Tropolactone A (2), exhibits in vitro cytotoxicity against human colon carcinoma (HCT-116) cells with an IC50 value of 13.2 μg/mL. Compound 2 also features a rare, highly-substituted tropone ring which is fused to the B ring boat through a tetrahydropyran moiety.2 The congener emervaridone A (3) was shown to inhibit ESBL-producing E. coli with a MIC value of 2 μg/mL which is comparable to that of the clinically used antibiotic amikacin. Compound 3 features a highly complex caged structure with two continuous all-carbon quaternary centers.3 Despite impressive biological activities and challenging structures, the chemical synthesis of DMOA-derived meroterpenoids only appeared recently; surprisingly, all completed syntheses have focused on berkeleyone A (4) and its congeners.4-7 Compared to other subfamilies, spiromeroterpenoids feature a conserved spiro-tetrahydrofuran linkage connecting the terpenoid and polyketide moieties. The congener asnovolin E (5)8 is one of a very few fibronectin-suppressing factors found to date. Some spiromeroterpenoids feature astonishingly complex skeletons with very high oxidation states such as the orthoester fumigatonin (6).9
Figure 1.

Representative DMOA-derived spiromeroterpenoids
The biosyntheses of DMOA-derived meroterpenoids have been extensively studied and have revealed that all members share a conserved biosynthetic origin. In the cyclization phase, enzymatic dearomatization of DMOA with farnesyl pyrophosphate (FPP) followed by selective terminal epoxidation generated the dearomatized polyene precursor 7 which bears the critical C1’ all-carbon quaternary center (Scheme 1). Cyclase NvfL catalyzed the cationic polyene cyclization and the intermediate 8 underwent an interesting cationic hydride shift/cyclization process to produce the precursor asnovolin H (9). 10 This cascade reaction rapidly forged the spirocyclic skeleton of the natural product and the axial C12-methyl stereochemistry. Subsequent redox modification of the A and D rings generated the gateway compound asnovolin A (13). In the oxidation phase, the endoperoxidase NvfI catalyzed C-H activation of the angular C13-methyl group leading to the incorporation of three oxygen atoms to generate the congener fumigatonoid A (14), which significantly increased the oxidation and complexity of the natural product family. Further enzymatic tailoring produced fumigatonoid B (15), fumigatonoid C (16) and novofumigatonin (17). The array of enzymes catalyzes unprecedented reactions; their detailed cata-lytic mechanisms remain to be elucidated and will rely on efficient access to synthetic natural product substrates.
Scheme 1.

Complete biosynthesis for novofumigatonin
The C1’ all-carbon quaternary center of spiromeroterpenoids such as 5 and 6 derived from dearomatization posed a significant synthetic challenge. Taking completed syntheses of berkelyone A (4) as examples, enolate chemistry has been used in the formation of the quaternary carbon centers (Figure 2). In the Maimone group’s pioneering synthesis of 4,4 the enolate generated from tricyclic ketone 18 reacted with diketene to form tetracycle 19. In the Newhouse group’s synthesis,5 enol allyl ether 20 was formed through O-allylation and isomerization. Claisen rearrangement forged the key quaternary carbon found in 21. In Li’s synthesis,6 alkylative dearomatization of acylphloroglucinol 23 with a primary alkyl triflate electrophile 22 established the quaternary carbon center. In 2011, a dissertation from Baran group described the first synthetic studies towards DMOA-derived meroterpenoids with a focus on the spiromeroterpenoid simplicissin (cf. 29, Figure 4).11 In 2019, our group reported the first biomimetic dearomatization of DMOA and prepared a number of non-natural meroterpenoid isomers.12 This breakthrough paved the way for a collaborative study with the Abe group to exploit the promiscuity of meroterpenoid cyclases where we obtained a number of unprecedented meroterpenoid analogues.13 However, we were not able to access any natural products through biomimetic chemistry. In 2022, we reported the first abiotic and enantioselective total syntheses of five DMOA-derived spiromeroterpenoids.14 Herein, we detail our efforts on the strategy evolution of the syntheses of DMOA-derived spiromeroterpenoids. The chemistry developed and lessons learned during our investigations may serve as inspiration for the synthesis of other targets in this family.15
Figure 2.

Key reactions to form the C1’ all-carbon quaternary carbon center of select meroterpenoids
Figure 4.

Second generation approach to asnovolin H
RESULTS AND DISCUSSION
First generation approach:
At the outset of our studies, asnovolin H (9) was chosen as our synthetic target with the plan that a scalable synthesis of this compound would allow us to prepare asnovolins D, J, and A sequentially according to the biosynthetic scheme. The spirocyle of asnovolin H may be formed through acid-mediated spirocyclization of substrate 25 which may be formed using dearomatization of DMOA with electrophile 26 (Figure 3). However, when this reaction was applied to model electrophiles 27, the desired dearomatized product 28 was never obtained. Careful monitoring of the reaction indicated possible product formation through TLC and UPLC-MS analysis, but the desired product could not be isolated after various workup procedures. The desired product 28 may be unstable due to facile rearomatization to form a stable allylic cation. Moreover, we later learned that the conformation of 25 may constrain the formation of undesired stereochemistry of the spirocycle stereocenter and equatorial C12-methyl group due to Furster-Plattner considerations.16 Accordingly, this approach was abandoned.
Figure 3.

First generation dearomatization approach to asnovolin H and subsequent model study
Second generation approach:
In our second-generation approach, we observed that the tetrahydrofuran is a conserved motif connecting the polyketide and terpene fragments which called our attention to use of direct oxidative, dearomative [3+2] cycloaddition to construct a spirocycle between olefin 30 and DMOA (Figure 4). Decalin 30 is also a known compound with a defined axial C12-methyl group which was reported by Corey and Shenvi.17 This type of oxidative [3+2] cycloaddition was also used in a concise synthesis of brevione A (33).18
Using DMOA-methyl ester 34 and methylene cyclohexane 35 as a model substrate, we explored oxidative [3+2] cycloaddition reactivity (Table 1). Use of Ag2O as oxidant resulted in complete decomposition. To our surprise, AgOAc produced the complex dimer 36 in 13% yield, the structure of which was confirmed by single X-ray crystal structure analysis (Figure 5). Ag2CO3/Celite® was found to be less effective and only a trace amount of dimeric product was observed. In 1968, Moussa and coworkers reported the dimerization of an acylphloroglucinol substrate using FeCl3 in refluxing ethanol.19 However, this condition was not successful on our substrate (entry 4). A combination of ceric ammonium nitrate (CAN) and Cu(OAc)2 resulted in full conversion and a 38% isolated yield of 36. Mn(OAc)3 and Cu(OAc)2 produced dimer 36 in 30% yield. Other single electron oxidants such as FeCl3/DDQ, CuBr2, K3Fe(CN)6, Frémy’s salt, and acridinium photocatalysts did not produce the desired spirocyclic product.
Table 1.
Optimization of the dimerization of DMOA methyl ester 34

| ||
|---|---|---|
| entry | conditions | results |
| 1 | Ag2O (2 equiv.), CH3CN, r.t. | Decomposition of 34 |
| 2 | AgOAc (2 equiv.), CH3CN, 84 °C | 13% |
| 3 | Ag2CO3 (2 equiv.), CH3CN, 60 °C | Trace 36 |
| 4 | FeCl3 (2.2 equiv), H2O/EtOH, 100 °C | Trace 36 |
| 5 | CAN (3 equiv.), Cu(OAc)2 (1.5 equiv.), AcOH, r.t. | 38% |
| 6 | Mn(OAc)3 (2 equiv.), Cu(OAc)2 (1.5 equiv.), AcOH, r.t. | 30% |
| 7 | K3Fe(CN)6 (2.2 equiv), CH3CN/water, r.t. | Decomposition of 34 recovery of 34 |
| 8 | Fremy’s salt (2.2 equiv), CH3CN,50 °C | no observation of 36 recovery of 34 |
| 9 | Acridinium photocataiyst, Cu(OTFA)2 | no observation of 36 |
Figure 5.
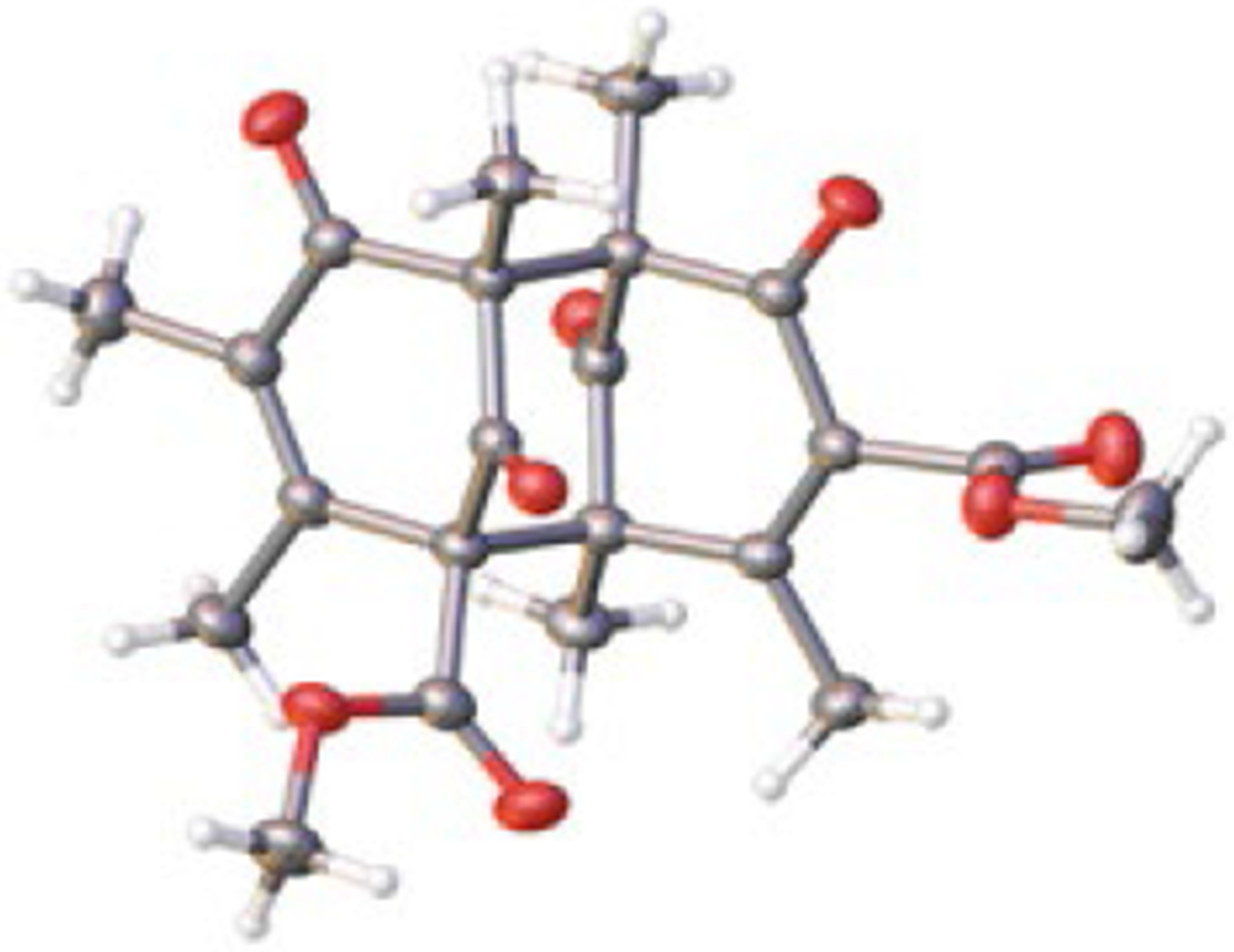
X-Ray crystal structure of dimer 36 with ellipsoids at the 50% probability level
The formation of dimer 36 indicated the generation of a phenolic radical intermediate which preferred dimerization rather than radical addition to the model exocyclic alkene 35. For other non-fully substituted phenols, it is well documented that oxidative [3+2] processes generally produce benzodihydrofuran products.20 In order to probe reactivity on a substrate similar to DMOA methyl ester, methyl atratate 37 was subjected to CAN oxidation (Scheme 2). The expected benzodihydrofuran was not observed but a new dimer 38 was isolated in 20% yield. It is interesting to note that the latter dimer contains the core structure of the natural product tridentorubin 39.21 Oxidative dimerization reactivity discovered for these highly substituted phenols provides an inspiration for the future synthesis of complex meroterpenoids such as 39.
Scheme 2.

Oxidative dimerization reactivity of methyl atratate
As single electron oxidants afforded dimerization of DMOA methyl ester, two electron oxidants were next evaluated. To our delight, using phenyliodine (III) diacetate (PIDA) as oxidant, the desired [3+2] cycloaddition product 40 was observed and isolated in 17% yield along with two [5+2] cycloaddition regioisomers 41 and 42 in 18% and 11% yields, respectively, from the phenoxonium cation intermediates I and II when 10 equiv. of alkene 35 was used (Scheme 3). During preparation of this manuscript, Zhang and coworkers reported related oxidative [3+2] cycloadditions with concomitant dearomatization towards their synthesis of simplicissin.22 The George23 and Trauner groups24 also reported biomimetic, oxidative intramolecular [3+2] and [5+2] cycloadditions in their syntheses of merochlorins A and B, respectively. To improve the yield and selectivity, other cation-stabilizing solvents including trifluoroethanol, nitromethane, chloroform, and dichloromethane in combination with HFIP were evaluated, but only extensive decomposition was observed. Lowering the amount of alkene to two equiv. only produced trace products along with extensive decomposition. Other I(III) and I(V) oxidants such as Dess-Martin Periodinane (DMP), 2-iodoxybenzoic acid (IBX), Koser’s reagent, and Pb(OAc)4 were found to be ineffective.
Scheme 3.

PIDA-mediated oxidative [3+2] cycloaddition.
It was noticed in the syntheses of merochlorins A and B22, 23 that different protecting groups for the phenols tuned the reactivity towards either oxidative [5+2] or [3+2] cycloaddition. Thus, we prepared several modified substrates and found that the para-OTBS protected substrate 43 produced the [5+2] product 41 in 22% yield as single isomer and that the ortho-Me-protected substrate 44 produced 40 as single product in 35% yield on 50 mg scale (Scheme 4). We then applied this optimized condition to the decalin system 45. However, unfortunately in this case the desired [3+2] cycloaddition product was not observed, and the only identifiable side product was the dearomatized, acetoxylation product 46, a compound which was also synthesized and confirmed through subjection of 44 to PIDA in acetic acid.
Scheme 4.

Divergent oxidative cyclization reactivity of differentially protected DMOA substrates
The mechanism of the PIDA-mediated oxidation of 44 appears to involve a phenoxonium cation intermediate which is trapped by excess olefin 35 to form the cycloaddition product or by acetate group to form 46. The presumed phenoxonium cation intermediate can also lead to decomposition through a quinone methide intermediate. To further increase the yield and obviate the formation of side product 46, we proposed an alternate phenoxonium cation precursor for [3+2] cycloaddition (Scheme 5). For example, cyclic carbonate 48 may generate a zwitterion 49 through CO2 extrusion. [3+2] Cycloaddition of oxyallyl cations have been documented.25 Compound 48 was prepared from PIDA oxidation of the para-Boc protected substrate 47.26 A series of photochemical conditions were tested, but in these experiments rearomatization occurred. Screening of various Lewis acids and thermal conditions led to the finding that microwave heating of 48 produced 2,4-dihydroxy-3,5-dimethylphthalide (50, DHDMP). Mechanistically, we propose that deprotonation of the C6-Me produced dienol intermediate 51 which may be followed by SN2’ attack of the allylic carbonate by the methyl ester followed by decarboxylation to produce 52. Final demethylation would afford 50.
Scheme 5.

Attempted [3+2] cycloaddition using a cyclic carbonate as a possible phenoxonium precursor.
The reaction producing 50 was only reproducible on a 20 mg scale using microwave heating. DHDMP is the biosynthetic precursor of meroterpenoids such as emervaridone A and a scalable synthesis of DHDMP will be helpful to explore future alkylative dearomatization reactivity.12 Thus, we developed an alternative scalable synthesis of DHDMP (Scheme 6). Deprotonation of the bis-MOM-protected DMOA methyl ester 53 with LDA produced intermediate 54 which was oxidized by the Davis oxaziridine reagent to produce phthalide 55. The reaction worked well on a small scale (75% yield, 200 mg scale), but on a gram scale the yield was lower, and the imine byproduct generated from Davis oxaziridine also reacted with the anion 54. Accordingly, we altered the protocol to trap anion 54 with B(OMe)3 followed by an oxidative quench with basic H2O2. The latter transformation was cleaner and more reproducible on a gram scale. After acid-mediated hydrolysis of the MOM group, DHDMP (50) was obtained on a gram scale.
Scheme 6.

Scalable synthesis of DHDMP
The difficulties encountered with PIDA oxidation prompted us to explore an alternative phenol umpolung method. In 2019, the Procter group reported a phenol/2-naphthol oxidative coupling using an interrupted Pummerer reaction.27 Application of this methodology to our system could mitigate nucleophilic addition of acetate which was observed in our PIDA oxidation attempts. When benzothiophene S-oxide 56 was activated by TFAA followed by addition of 44, a deep yellow color appeared which we initially assumed corresponded to the production of aryloxysulfonium salt 57 as a novel umpolung intermediate (Scheme 7). After addition of alkene 35, we isolated two products, 58 and 59, in 37 and 34% yields, respectively. It was clear from 1H-NMR spectral analysis that the double bond on the benzothiophene heterocycle had participated in [2,3]-rearrangement to produce dearomatized products 58 and 59. In our experiments, aliphatic sulfoxides such as 60, DMSO, and 61 did not show reactivity while aromatic sulfoxides 62 and 63 showed similar rearrangement chemistry.
Scheme 7.

Interrupted Pummerer reaction for phenol umpolung and application to a DMOA methyl ester substrate.
Despite the expectation that dearomatization approaches could build up the fully substituted dienone system directly from abundant DMOA as we demonstrated on the model system, our preliminary success could not be extended to a more elaborate system. Moreover, all efforts attempting to optimize and improve the oxidative cycloaddition approach met with failure. Lastly, the aforementioned approach did not address the stereochemistry control problem at the C1’-quaternary carbon center and C12-axial methyl. Accordingly, we postulated that a non-dearomatization, stepwise construction of the highly substituted D-ring of the spiromeroterpenoids could be a feasible and practical solution and next turned our attention to this approach.
Third generation approach:
For a non-dearomatization approach, we noticed that several spiromeroterpenoids such as asnovolins A and J have reduced D-ring moieties. Asnovolin A (19) is a gateway compound connecting both lower and higher oxidation state congeners and was thus selected as our target molecule. Both A and D ring simplification of 19 afforded intermediate 64 (Figure 6). We considered that the axial C12-methyl could be established through hydrogenation of the alkene, although the facial selectivity for this transformation at the outset was uncertain. The C-O bond of 64 could be formed through oxa-Michael addition. Precursor 65 revealed a hindered bis-neopentyl C-C bond which was disconnected to afford fragments 66 and 67. The syn-dimethyl relationship of 66 indicated an application of a Diels-Alder cycloaddition for its preparation.
Figure 6.

Third generation retrosynthesis of asnovolin A
Large scale synthesis of fragments:
Enone alcohol 71 is a known compound which was reported by Cole and coworkers in their synthesis of (−)-phomactin A.28 The asymmetric Diels-Alder reaction between tiglic aldehyde and Rawal’s diene established the crucial chiral quaternary carbon center (Scheme 8a). The derived cycloadduct 70 was converted to enone 72 in two steps. Luche reduction of 72 produced 66 as a single diastereomer on a gram scale. The decalin fragment 67 was prepared in five steps from methyl carvone according to Nagamitsu and Xie’s procedure.29, 30 Although we were able to secure decalin 67 on a gram scale, the route suffered from low atom and redox economy and several steps required refinement. For example, alkylation of 73 required 3.0 equiv. of 3-bromopropionitrile, a non-activated electrophile, to produce 40-60% yields of 74 along with 20-30% of unreacted starting material. Under strongly basic conditions, undesired double alkylation, polymerization of an in situ-generated acrylonitrile reagent were observed. The reported high literature yield for 74 (66%, 98% brsm) was not reproducible in our hands. We found that epoxidation of the propenyl moiety required purified m-CPBA for high yields (93% yield), otherwise only a 60 % yield was obtained in our hands using commercially available m-CPBA. However, purification of the reagent involves washing an ether solution of commercial m-CPBA with pH 7 buffer which is a potential hazard. We found that an environmentally friendly epoxidation of 74 using in situ generated dimethyl dioxirane (DMDO) obtained from Oxone® and acetone produced epoxide 75 in 77–85% yield on a multigram scale without need for column purification.31 Reductive cyclization of epoxy nitrile 75 (Cp2TiCl2, Zn) was found to work well in degassed THF. Final deoxygenation of the derived product 76 involved mesylation, iodination, and reduction which produced decalin 77 in a reproducible 75% yield. Conversion of 74 to 77 suffered from low redox and atom economy. Ideally, direct, reductive alkenyl nitrile cyclization may form 77 very efficiently from substrate 74. A thorough survey of the literature revealed only a few HAT and transition metal-mediated, reductive alkenyl nitrile cyclizations. In 2018, the Murphy group reported a Fe(acac)3-catalyzed HAT cyclization an amine-tethered alkenyl nitrile system.32 In 2011, the Sensuki group reported a mechanistic study using Ni(0)-PCy3 to effect oxidative cyclization of alkenyl-aryl nitriles to form a metallacycle with triflic acid as additive to activate the nitrile moiety.33 In two other examples, Fe(acac)3 and Mn(dmp)3-catalyzed alkenyl nitrile cyclizations involving five-membered ring formation34 or in a rigid framework.35 With these precedents in mind, we tested numerous HAT and transition metal-catalyzed conditions on substrate 74, but these attempts were not successful. In all cases, either no reaction or conjugate reduction of the enone was observed. We were curious about these results and conducted a preliminary conformational search which indicated that alkenyl nitrile 74 prefers a conformation with the isopropenyl and alkyl nitrile substituents both in the diaxial position which makes the direct radical cyclization difficult (cf. Scheme 8 (c)). Thus, we believe that in the reductive epoxy nitrile cyclization step, a doubly coordinated intermediate such as 78 is required for C-C bond formation. Otherwise, the produced iminyl radical will be less stable than the tertiary radical and the retro-process will be favored.36
Scheme 8.

Synthesis of fragments 66 and 67.
With the two chiral fragments in hand, we explored the crucial 1,2-addition process (Table 2). To our delight, sequential treatment of 66 with n-BuLi (1.0 equiv., 2 mins at −78 °C) and t-BuLi (2.2 equiv., 60 mins at −78 °C), followed by addition of decalin 67 afforded the desired product 79 in 5% yield with a significant amount of diol 80 as side product likely derived from oxidation of the anion by residue oxygen (entry 1). We found the Li-I exchange time and temperature affected the yield of adduct 79 significantly. Shortening the Li-I exchange time and increasing reaction temperature improved the yield dramatically (entries 2 and 3). The highest yield (83%) was achieved when the Li-I exchange was shortened to 10 min. and reaction temperature for 1,2-addition was increased to 25 °C (entry 4). The higher temperature required for this reaction indicated a relatively high energy barrier for this sterically hindered, neopentyl coupling. When the reaction was conducted on a 500 mg scale, the yield dropped significantly. For this case, it appeared that Li-I exchange was not complete and t-BuLi addition to 67 occurred. Moreover, adduct 79 was found to unstable; we also identified the SN2’ side product 81 after quenching the reaction with 1.0 equiv. of TMSC1. As a result, rapid column purification was performed, and the product was used immediately in the next step (Scheme 9). The expected MnO2 oxidation/oxa-Michael addition of 79 worked smoothly to afford spirocycle 64 in good yield (67–72%). Other variants of fragment 66 such as 83, the silyl enol ether of ketone 82, and the ring-opened form of decalin 74 were not successful reaction partners for 1,2-addition.
Table 2.
Optimization of the neopentyl 1,2-addition.

| |||
|---|---|---|---|
| entry | Li-I exchange | 1,2-addition | yield |
| 1 a | −78 °C, 60 min | −78 °C, 60 min | 5% 79, 30% 80 |
| 2 a | −78 °C, 10 min 0°C, 10 min | −78 °C to 0 °C , 60 min | 42% |
| 3 a | −78 °C, 10 min 0°C, 10 min | 0 °C, 60 min | 57% |
| 4 b | −78 °C, 10 min 25 °C, 15 min | 25 °C, 60 min | 83%, 200 mg scale |
| 5 b | −78 °C, 10 min 25 °C, 15 min | 25 °C, 60 min | 66%, 500 mg scale |
| 6 b | entry 4, quenched with 1 equiv. TMSCI | 25% 81 | |
| a: 1.0 equiv. 66 was used. b: 1.2 equiv. 66 was used. | |||

| |||
Scheme 9.
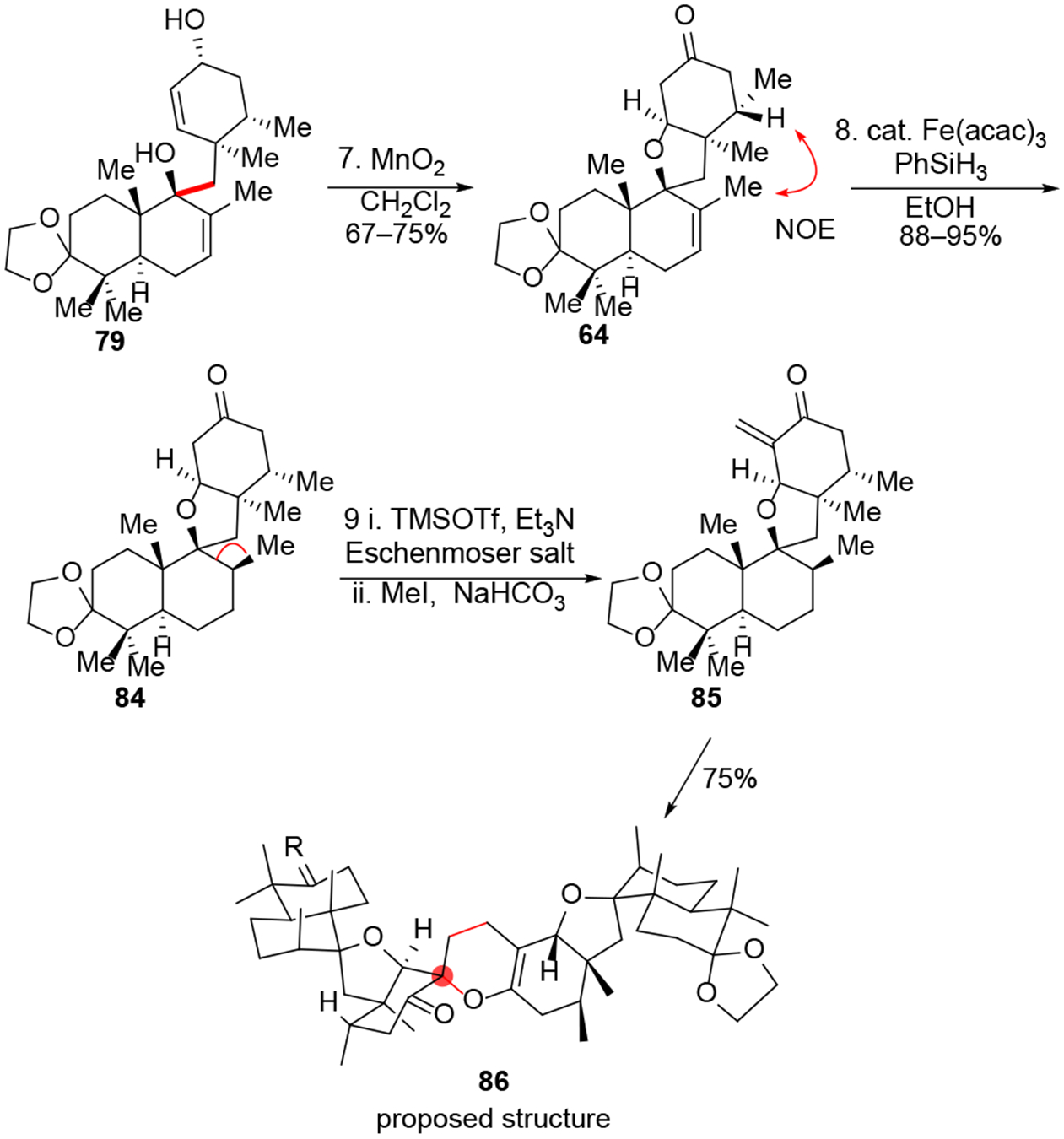
Forward synthesis of spirocycle 84 and attempted alkene isomerization.
The next key issue we encountered was to hydrogenate the double bond to establish the axial C12-methyl group. We noticed a strong NOE correlation for spirocycle 64 between the C12 vinyl methyl and C6’ methine (Scheme 9). We proposed that if the hydrogenation occurred from the top face, the resulting equatorial methyl group would be in strong interference with C6’, thus the bottom face approach may be favored to deliver the axial methyl group. Accordingly, we explored heterogeneous hydrogenation using catalytic Pd/C and Pd(OH)2/C and H2 in methanol, but in this case no reaction was observed presumably as both faces are hindered. We also attempted to hydrogenate enone 67 to preinstall the axial methyl group, but heterogeneous hydrogenation using Pd/C afforded an inseparable 1:1.5 mixture of diastereomers.
Recently, the Yang group reported an elegant synthesis of (+)-waihoensene wherein an axial methyl group was installed via MHAT hydrogenation of an exocyclic alkene.37 In their study, the initially formed tertiary radical abstracted a nearby ketone α-H to establish axial methyl stereochemistry. We proposed that in our system, the initially formed tertiary radical may be able to abstract the C6’-H, thus forging the axial C12-methyl stereochemistry. To our delight, iron (III) hydride (MHAT) reduction of substrate 64 worked well and produced the desired product 84 as a single diastereomer (Scheme 9), the mechanism of which was explored through deuterium-labeling experiments and computational studies as outlined in our previous work.14 In the current study, we further found that the conformational barrier from the chair radical conformer A to the boat conformer B was 7.76 kcal/mol (Figure 7).
Figure 7.

DFT calculations for the Curtin–Hammett-driven HAT reduction at M06-2X/6-31G(d,p)/PBF (EtOH).
Conversion of 84 to the vinylogous ester 87 proved to be a non-trivial transformation. We first designed an Eschenmoser methylenation/olefin isomerization sequence to fulfill this goal due to step economy (Scheme 9). Although the Eschenmoser reaction afforded exocyclic enone 85 efficiently and endocyclic olefin 87 is much more stable thermodynamically, we observed that the production of hetero-Diels-Alder dimer 86 was a facile process leading to the isolation of this compound in 75% yield after treatment of 85 in CDCE overnight.
The high yield of dimer 86 also indicated potential high efficiency to obtain 87 if alkene isomerization could be achieved which led us to evaluate several conditions (Table 3). Hydrogen activated Pd/C has been reported to isomerize an exocyclic enone to an endocyclic isomer,38 but our substrate was found to be inert to these conditions (cf. entry 1). Palladium (II) catalysts such as (PhCN)2PdCl2 are known to isomerize terminal to internal olefins;39 however, these conditions only promoted dimerization and acetal hydrolysis (entry 2). Catalytic (PPh3)3RhCl/Et3SiH has been reported to isomerize a series of 5-membered α-alkylidene carbonyls to cyclic α,β-unsaturated carbonyls,40 but the high temperature required in this case promoted dimer formation (entry 3). A trace amount of product was formed and a 1,4-hydrosilation product was also observed. Recently, radical-type alkene isomerization using metal hydride atom transfer chemistry has been applied to several total syntheses.41 These conditions required mild heating in benzene at 60°C. Use of a Co(salen)Cl catalyst system was not effective at r.t. and heat only promoted dimerization for our substrate (entry 4). Norton’s radical-type olefin isomerization chemistry using Co(dmgH)2 as catalyst42 was also evaluated, but dimer formation was still the major observed pathway (entry 5). Exciting success was found when catalytic RhCl3 in EtOH under reflux conditions was employed for isomerization of 85.43 However, the high temperature required resulted in competing dimerization, acetal hydrolysis, and Michael addition of EtOH (entry 6). Since the thermal stability of exocyclic olefin 85 was a significant problem, we considered whether its slow release may help reduce undesired dimerization and promote isomerization. Refluxing of RhCl3 in EtOH also produces HCl which can hydrolyze the ketal protecting group of substrate 85. Accordingly, we added K2CO3 or pH 7 buffer using the Mannich base precursor to 85 and indeed observed slightly improved yields of the desired product 87 (33%), but the basic conditions employed also accelerated Michael addition of EtOH to 85. Although we have also evaluated many other Ir-H and Ni-H conditions, no promising results were observed, and all reactions were plagued by dimerization. We also noticed that the yield of dimer 86 was variable in different batches and 86 exists as an inseparable mixture of isomers. As a model of 85 shows that the two faces of the exocyclic enone are very similar, Diels-Alder dimerization may occur from both diastereo faces. After numerous attempted purifications and structural characterizations, we propose the dimeric core structure shown 86 with the spirocyclic stereochemistry undefined.
Table 3.
Exploration of isomerization of 85

| ||
|---|---|---|
| conditions | results | |
| 1 | Pd/C, H2 (activation), EtOAc, r.t. | no reaction, 85 recovered, no dimer |
| 2 | (PhCN)2PdCl2, benezene, 60 °C, | dimer and trace 87, acetal deprotection |
| 3 | (Ph3P)3RhCl, Et3SiH, 100 °C | dimer, hydrosilation, trace 87 |
| 4 | Co(salen)Cl, PhSiH3, r.t. to 60 °C | dimer |
| 5 | Co(dmgH)2 •2 H2O, PhSiH3, 60 °C | dimer, reduction, and trace 87 |
| 6 | RhCl3 •xH2O (0.2 equiv.), EtOH, 100 °C | 20% yield of 87. dimer, acetal hydrolysis |
| 7 | using Mannich base as substrate K2CO3 (2 equiv.) | 33% yield of 87. EtOH addition no acetal hydrolysis |
Considering the insurmountable problem of exocyclic alkene isomerzation, we developed an alternative desaturation/cross -coupling sequence to prepare spirocycle 87 (Scheme 10). IBX was found to be an optimal desaturation reagent for substrate 84 to access enone 88. Ito-Saegusa oxidation of 84 was found to be sluggish and incomplete in which case we also observed desaturation of the C5’-C6’ site. Subsequent iodination and Suzuki coupling produced the divergent intermediate 87 in high yield. This three-step sequence was found to be more scalable than the elusive olefin isomerization and produced 350 mg of 87 in a single pass. The potassium enolate derived from 87 was acylated in high yield using the Mander reagent to produce ketoester 90. A single crystal X-ray structure of 83 served to confirm all stereochemistry established thus far in the sequence. After hydrolysis, we achieved the first synthesis of asnovolin J 12. The route was found to be scalable in producing 133 mg of asnovolin J in a single pass. For the synthesis of asnovolin A 19, addition of m-CPBA to 12 produced a complex mixture; we reasoned that the electron-rich C2’-C3’ bond and enolizable C5’ position could also undergo undesired oxidation. We considered that the electron density of the C2’-C3’ double bond could be reduced through addition of Lewis acids and found that Sc(OTf)3 in conjunction with m-CPBA could indeed promote the desired transformation to produce asnovolin A in 76% yield.44 We conducted a 1H NMR study to monitor the effect of Sc(OTf)3 on the electronics of substrate 12 (Figure 8) for Bayer-Villiger oxidation. Premixing 12 with 3 equiv. of Sc(OTf)3 produced a spectrum where the C11’-Me, C5’-H, and C8’-Me signals shifted downfield, whereas the chemical shift of C2-H did not change. The spectrum supported the idea that Sc(OTf)3 chelation to the bidentate β-keto ester moiety occurred which reduced the electron density of the D-ring, while Sc(OTf)3 activation of C3-ketone towards Baeyer-Villiger oxidation was not definitive likely as the fast association/dissociation of Sc(III) Lewis acid is shorter than the NMR timescale. We were ultimately able to produce 30 mg of asnovolin A 19 which will be used in future studies to provide material to access highly oxidized spiromeroterpenoids through chemoenzymatic synthesis. Acid-mediated methanolysis of asnovolin A 19 produced asnovolin E 5 in 41% yield. To our surprise, desaturation of the C5’-C6’ position of substrate 87 using IBX was no longer effective even under more harsh conditions. We had postulated that the vinylogous oxygen may stabilize a putative radical cation and thus no proton was eliminated. After considerable experiments, we found that benzeneseleninic acid anhydride45 enabled efficient desaturation of enone 87. The dienone chermesin B (91) was isolated in 86% yield after hydrolysis. A similar desaturation protocol also worked well on β-keto ester 90 to produce dienone 92 after hydrolysis. The final C3-ketone reduction of 92 to simplicissin was not “simple” at all. Canonical bulky reductants including L-Selectride and LiAlH(t-Bu)(i-Bu)2 primarily reduced the C5’-C6’ double bond and then reduced the C3-ketone to a mixture of axial and equatorial alcohols. Other strategies including enone protection via Diels–Alder cycloaddition with CpTMS46 or Ph3P/TMSOTf47 and stereoinversion of the C3- equatorial hydroxyl group via Mitsunobu reaction were not successful. Finally we found that Meerwein-Ponndorf-Verley (MPV) reduction48 of 92 using in situ-generated (i-PrO)2AlCl was a highly selective method to reduce the C3-ketone with useful levels of diastereoselectivity. Simplicissin (93) was purified and isolated in 59% yield as a single diastereomer. In total, we achieved the collective synthesis of five DMOA-derived spiromeroterpenoids in our successful third generation route.
Scheme 10.

Synthesis of key intermediate 80 and collective synthesis of five spiromeroterpenoids.
Figure 8.

1H-NMR study of the Sc(OTf)3 chelation effect on as-novolin J. top: asnovolin J + Sc(OTf)3 (3 equiv.), bottom: asnovolin J.
Synthesis of a Spirocycle Library:
Spirocycles are ubiquitous structural motif in drugs and natural products.49 Recently the theme of “escape from flatland”50 has spurred extensive research to make sp3-rich scaffolds for drug discovery. The spirocycle assembly logic in our spiromeroterpenoid synthesis provided an excellent platform to construct various sp3-rich spirocycles. There are many abundant, commercially available terpenes containing a ketone functionality which may be engineered to spirocycles through the 1,2-addition/oxidative oxa-Michael addition sequence without purification of the intermediate diol (Scheme 11). In general, we found that enolizable ketones work poorly and proto-deiodination of 66 occurred. For example, the terpenes (+)-nopinone, thujone, menthone, camphor, and fenchone did not produce the desired 1,2-addition to afford the corresponding diol product. When carvone was used, an separable diastereomeric mixture of 94-S and 94-R were isolated in reasonable yield after two steps. Nootkatone only produced spirocycle 95 in 5% yield. Cyclobutanone afforded the novel spirocycle 96 in 30% yield, presumably as enolization would introduce strain and was thus not favored. Several other ketones without enolizable protons were evaluated and gave good results. 3-Methylene 2-norbornanone produced an inseparable 1:1 diastereomer mixture 97 in 28% yield. In this case, no 1,4-addition was observed. The substrate 2-adamantanone produced spirocycle 98 in 40% yield as a crystalline solid. 9-Fluorenone generated spirocycle 99 in 48% yield. Thioxanthone was sparingly soluble in ether and in this case a dilute THF solution was used in which case spirocycle 100 was isolated in good yield. We also tested aldehyde-containing substrates including 1-methyl-1H-indole-2-carbaldehyde which produced a 1:1 diastereomeric mixture of 101-S and 101-R in 60% total yield. The unsaturated aldehyde reaction partner cyclocitral generated 102-S and 102-R in 67% combined yield.
Scheme 11.
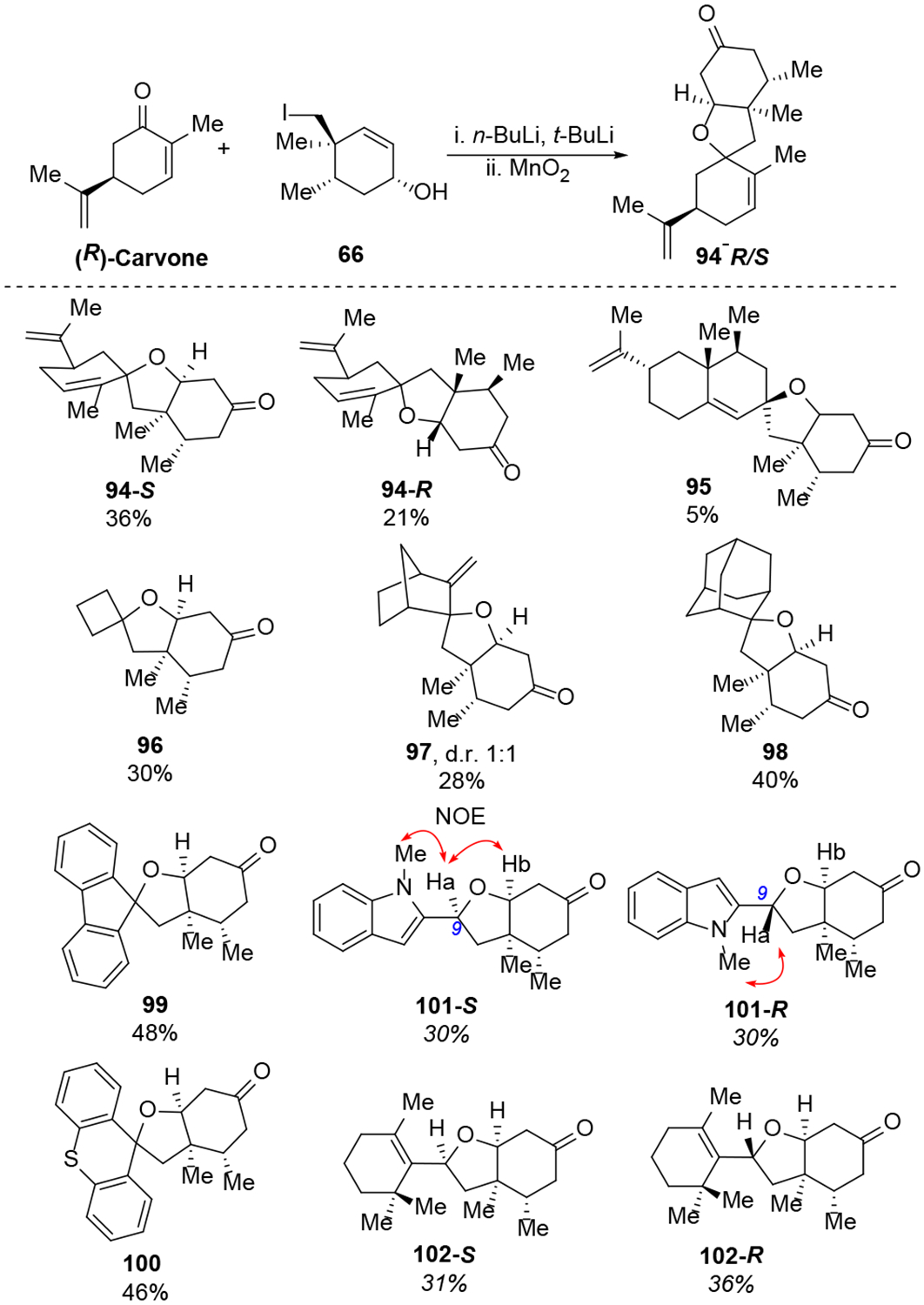
Synthesis of a small spirocycle library
CONCLUSIONS
In summary, we have detailed a full account of our strategy development leading to the first unified asymmetric total synthesis of DMOA-derived spiromeroterpenoids. Although the first- and second-generation routes based on dearomatization strategies were not successful, the lessons learned subsequently oriented our efforts to undertake stepwise construction of the highly substituted D-ring and a convergent fragment coupling strategy that proved to be advantageous in terms of stereochemical control. The oxidative dearomatization reactivity of highly substituted phenols discovered in our study can be leveraged in future studies. Several complex dimers were produced along with a scalable synthesis of DHDMP whose dearomatization reactivity will be explored in future work. An improved process synthesis of decalin 67 was revisited. Our third-generation route provided a robust and unified approach to DMOA-derived spiromeroterpenoids. Several stereoselective reactions were developed including MHAT chemistry to establish the axial C12-methyl group, Meerwein-Ponndorf-Verley (MPV) reduction to accomplish a challenging C3-ketone reduction to produce simplicissin, orthogonal desaturation of a vinylogous ester substrate, and a Lewis acid-assisted, regioselective Baeyer-Villiger oxidation to produce the congener asnovolin A. Finally, the 1,2-addition/oxidative cyclization sequence was extended to prepare a small spirocyclic library which in the future should provide novel leads for drug discovery.
EXPERIMENTAL SECTION
General Methods.
Unless stated otherwise, reactions were performed in flame-dried glassware, 10 mL test-tubes, or 4 mL capped reaction vials under an argon or nitrogen atmosphere using dry, deoxygenated solvents (distilled or passed over a column of activated alumina) and stirred with Teflon-coated magnetic stirring bars. HPLC grade methylene chloride, tetrahydrofuran, diethyl ether, toluene, benzene, and acetonitrile were purchased from Fisher Scientific and were purified and dried by passing through a PURE SOLV® solvent purification system (Innovative Technology, Inc.). All other reagents and catalysts were purchased and used as received from Strem, Sigma Aldrich, Oakwood Chemical, and Alfa Aesar. All NMR spectra were obtained in CDCl3, CD3OD, C6D6 (Cambridge Isotope Laboratories, Inc.) at ambient temperatures using a Varian 400 MHz, 500 MHz or 600 MHz spectrometer. Chemical shifts are reported in parts per million relative to the internal solvent peak (CDCl3: δ 7.26 for 1H; δ 77.16 for 13C. CD3OD: δ 3.31 for 1H; Δ 49.00 for 13C, C6D6: δ 7.16 for 1H; 128.06 for 13C). Data for 1H NMR are reported as follows: chemical shift, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constants, and integration. All 13C NMR spectra were recorded with complete proton decoupling. Structural assignments were made with additional information from gNOESY, gHSQC and gHMBC experiments. Crystal structures were determined using a Bruker D8 Venture diffractometer equipped with a Cu IμS DIAMOND X-ray source, HELIOS-Cu monochrometer and PHOTON-III M28 detector. Infrared spectra were recorded on a Bruker ALPHA P FT-IR spectrometer equipped with a diamond ATR module. High-resolution mass spectra were obtained at the Boston University Chemical Instrumentation Center using a Waters Q-TOF mass spectrometer. Melting points were recorded on a Mel-temp apparatus (Laboratory Devices) and are uncorrected. Analytical LC-MS was performed on a Waters Acquity UPC2 system (Ultra Performance Convergence Chromatography (Waters Mass-Lynx Version 4.2) with a Binary solvent manager (Supercritical CO2 and MeOH), a SQ mass spectrometer, a Waters 2996 PDA (PhotoDiode Array) detector, and an ELSD (Evaporative Light Scattering Detector). An Acquity UPC2-MS Torus 2-PIC 1.7 μm column was used for analytical UPC2-MS. Analytical thin layer chromatography (TLC) was performed using 0.25 mm silica gel 60-F plates (Merck KGaA). Flash chromatography was performed using ZEOprep 60 Eco 40-63 μm silica gel (Zeochem AG). Preparative TLC was conducted with glass-backed 250 μm silica gel 60-F plates (Merck KGaA). Yields refer to chromatographically and spectroscopically pure compounds, unless otherwise stated. All reactions were carried out in oven-dried glassware under an argon/nitrogen atmosphere unless otherwise noted. The Scilligence ELN Reaction Planner (Scilligence Corp.) was used for experimental procedure planning.
Experimental procedure and characterization data:
The detailed experimental and characterization data for new compounds are list below. For experimental details and characterization data for compounds 5, 12, 19, 64, 66, 72, 79, 84, 85, 86, 87, 88, 89, 90, 91, 92, and 93, please see reference 14.

Dimer 36:
To a 4 mL reaction vial was added DMOA methyl ester 34 (10 mg, 47 μmol, 1.0 equiv.) in AcOH (1 mL). The reaction was degassed with Ar for 30 mins. Then CAN (78 mg, 143 μmol, 3.0 equiv.) and Cu(OAc)2 (13 mg, 71 μmol, 1.5 equiv.) were added. The reaction was stirred at r.t. for 12 h. The reaction was finally quenched with sat. NaHCO3 and extracted with ethyl acetate (3 mL X 3). The organic layer was dried over Na2SO4, filtered, and concentrated. The crude material was purified by silica gel chromatography (hexane: ethyl acetate = 4:1) to afford the dimer 36 (3.8 mg, 38% yield). Physical state: white solid. Rf = 0.21(4:1 hexane : EtOAc). M.p.: 198–202 °C (CHCl3/isooctane). 1H{13C} NMR (500 MHz, CDCl3): δ 3.83 (s, 3H), 3.78 (s, 3H), 2.35 (s, 3H), 1.99 (s, 3H), 1.85 (s, 3H), 1.66 (s, 3H), 1.23 (s, 3H), 1.13 (s, 3H). 13C{1H} NMR(126 MHz, CDCl3): 200.1, 198.8, 191.0, 188.6, 167.4, 165.6, 159.2, 148.1, 136.4, 135.6, 69.1, 68.0, 60.5, 53.1, 52.9, 22.4, 20.1, 15.4, 12.8, 10.9, 10.6 IRυmax (Diamond ATR): 2952, 1736, 1674, 1320, 1241 cm−1. HRMS (ESI): [M + H]+ Calc.’d for C22H25O8: 417.1549, found: 417.1554

Dimer 38:
A 50 mL flask containing methyl atratate 37 (200 mg, 1.02 mol, 1.0 equiv.) in CH3CN (10 mL) was added CAN (448.9 mg, 2.04 mmol, 1.0 equiv.) at r.t. The mixture was stirred for 30 mins and was then quenched with sat. Na2S2O3. The mixture was extracted with ethyl acetate (10 mL X 3). The combined organic layer was washed with brine, dried with Na2SO4, filtered, and concentrated to afford a crude yellow solid. Purification by silica gel chromatography (hexane: ethyl acetate = 20:1 to 5:1) afforded the dimer 38 (29 mg, 15% yield.). Physical state: yellow solid. Rf = 0.33 (4:1 hexane : EtOAc). M.p.: 185–187 °C (hexanes/EtOAc). 1H{13C} NMR(500 MHz, CDCl3): δ 11.9 (s, 1H), 5.07 (s, 1H), 3.93 (s, 3H), 3.83 (s, 3H), 2.54 (s, 3H), 2.19 (s, 3H), 2.04 (s, 3H), 1.72 (s, 3H). 13C{1H} NMR(126 MHz, CDCl3): 191.4, 190.7, 172.7, 164.0, 164.0, 159.1, 145.3, 143.6, 137.8, 118.2, 108.2, 106.8, 103.1, 61.5, 53.0, 52.3, 17.7, 17.0, 14.3, 8.6. IRυmax (Diamond ATR): 2960, 1708, 1512, 1254 cm−1. HRMS (DART+): [M + H]+ Calc. ’d for C20H21O9: 405.1180, found: 405.1181

To solution of DMOA methyl ester 34 (10 mg, 47 μmol, 1.0 equiv.) in 0.3 mL of HFIP and methylene cyclohexane (45 mg, 57μL, 475 μmol, 10 equiv.) in a 4 mL vial at 0 °C was added a solution of PIDA (23 mg, 71 μmol, 1.5 equiv.) in HFIP (0.2 mL) dropwise. The colorless solution became yellow immediately. After 30 mins, the reaction was quenched with sat. Na2S2O3. The reaction mixture was extracted with ethyl acetate (3 mL x 3). The combined organic layer was washed with brine, dried with Na2SO4, filtered, and concentrated to afford a crude yellow oil. Purification by preparative thin layer chromatography (hexane: ethyl acetate = 4:1) afforded 40 (2.6 mg, 17% yield.) as the desired [3+2] cycloaddition product as slightly yellow wax and 4.3 mg of a yellow oil which was determined to be an inseparable mixture of the two [5+2] cycloadduct regioisomers. The same reaction was repeated 3 times in which case the yellow oil was combined and purified by preparative HPLC to afford [5+2] isomer 41 (7.9 mg, 18% yield) as a white wax and [5+2] isomer 42 (4.8 mg, 11% yield) as a white wax.
[3+2] cycloadduct 40:
Physical state: slight yellow solid. Rf = 0.185 (4:1 hexane : EtOAc). 1H{13C} NMR (500 MHz, CDCl3): δ 3.85 (s, 3H), 2.03 (d, J = 12.40 Hz, 1H), 2.01 (overlap, 1H), 1.78 (d, J = 13.03 Hz, 1H), 1.79–1.84 (overlap, 2H), 1.77 (s, 3H), 1.73 (overlap, 1H), 1.63 (m, 1H), 1.55 (s, 3H), 1.47–1.52 (overlap, 3H), 1.37–1.45 (overlap, 2H). 13C{1H} NMR(126 MHz, CDCl3): 184.4, 177.7, 168.1, 153.9, 131.8, 108.2, 90.7, 52.4, 48.2, 43.0, 40.7, 38.0, 34.4, 32.2, 25.0, 23.1, 21.8, 16.5, 7.6. IRυmax (Diamond ATR): 2932, 2858, 1736, 1676, 1615, 1240, 1203 cm−1. HRMS (ESI): [M + H]+ Calc.’d for C18H25O4: 305.1753, found: 305.1742.
[5+2] cycloadduct 41:
Physical state: white wax. Rf = 0.37 (4:1 hexane: EtOAc). 1H{13C } NMR 1500 MHz, CDCl3): δ 3.82 (s, 3H), 2.06 (s, 3H), 1.97 (d, J = 12.95 Hz, 1H), 1.78 (d, J = 12.95 Hz, 1H), 1.58–1.68 (overlap, 3H), 1.34 (s, 3H), 1.19–1.32 (overlap, 5H), 1.18 (s, 3H), 1.00–1.1 (overlap, 2H). 13C{1H} NMR 126 MHz, CDCl3): 207.0, 195.0, 166.6, 166.3, 133.1, 72.8, 52.4, 52.0, 45.4, 40.6, 34.3, 33.8, 24.9, 22.5, 22.0, 17.2, 16.0, 9.0. IRυmax (Diamond ATR): 2934, 1735, 1667, 1436, 1333, 1230 cm−1. HRMS (ESI): [M + H]+ Calc.’d for C18H25O4: 305.1753, found: 305.1750.
[5+2] cycloadduct 42:
Physical state: white wax. Rf = 0.41(4:1 hexane : EtOAc). 1H{13C} NMR (500 MHz, CDCl3): δ 3.84 (s, 3H), 2.02 (s, 3H),1.91 (d, J = 13.74 Hz, 1H), 1.80 (d, J = 14.41 Hz, 1H), 1.61–1.72 (overlap, 3H), 1.31–1.45 (overlap, 4H), 1.29 (s, 3H), 1.21 (s, 3H), 1.26 (m, 1H), 0.99–1.15 (overlap, 2H). 13C[1H] NMR (126 MHz, CDCl3): 207.0, 195.0, 166.7, 163.0, 133.3, 62.9, 62.0, 52.6, 44.3, 41.1, 33.1, 32.5, 25.4, 22.7, 21.9, 19.7, 13.5, 11.0. IRυmax (Diamond ATR): 2935, 1760, 1736, 1677, 1445, 1216 cm−1. HRMS (ESI): [M + H]+ Calc.’d for C18H25O4: 305.1753, found: 305.1745

43:
To a 100 mL flask was added THF (25 mL), DMOA methyl ester 34 (500 mg, 2.38 mmol, 1 equiv.), TBSC1 (468 mg, 3.11 mmol, 1.3 equiv.), and EtN(i-Pr)2 (614.7 mg, 828 μL, 4.76 mmol, 2.0 equiv.). After 6 h, the reaction was quenched with sat. NH4Cl and was extracted with EtOAc (10 mL x 3). The combined organic layers were then dried over Na2SO4 and concentrated under vacuum. The crude product was purified using silica gel chromatography (hexane: EtOAc = 20: 1) to afford 43 (655 mg, 84% yield). Physical state: white solid. Rf = 0.83 (4: 1 hexane: EtOAc). M.p.: 78–80 °C (hexanes/EtOAc). 1H{13C} NMR 1500 MHz, CDCl3): δ 11.24 (s, 1H), 3.93 (s, 3H), 2.40 (s, 3H), 2.09 (s, 3H), 1.09 (s, 9H), 0.16 (s, 6H). 13C{1H} NMR (126 MHz, CDCl3): 172.81, 159.81, 156.90, 137.14, 120.37, 114.21, 107.39, 52.01, 29.85, 26.15, 19.11, 18.81, 14.33, 10.78, −3.08. IRυmax (Diamond ATR): 2955, 2930, 1654, 1321, 1207, 827 cm−1. HRMS (ESI): [M + H]+ Calc.’d for C17H29O4Si: 325.1835, found: 325.1839
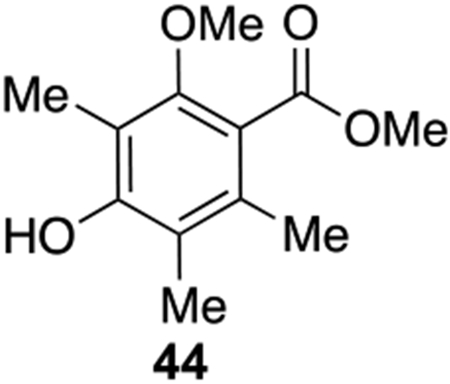
To a 500 ml flask was added DMOA methyl ester 34 (4.0 g, 19 mmol, 1.0 equiv.), benzyl bromide (3.58 g, 20.9 mmol, 1.1 equiv.), K2CO3 (3.94 g, 28 mmol, 1.5 equiv.), and acetone (150 mL). The mixture was heated at 60 °C for 3 h at which time TLC analysis showed that all starting material was consumed. Then MeI (5.4 g, 2.37 mL, 38 mmol, 2.0 equiv.) and K2CO3 (7.89 g, 57 mmol, 3.0 equiv.) were added and the reaction was refluxed for 18 h. The reaction mixture was diluted with water (100 mL) and acetone was removed under reduced pressure. Then the mixture was extracted with ethyl acetate (50 mL x 3) and combined organic layer was washed with brine, dried with Na2SO4, filtered, and concentrated. The crude product was purified by silica gel chromatography (hexane: EtOAc = 30: 1 to 20: 1) to afford the bis-ether intermediate (5.6 g, 93% yield). 5.6 g of the intermediate was dissolved in methanol (60 mL) and Pd/C (216 mg, 0.1 equiv.) and AcOH (0.5 mL) were added. The mixture was bubbled with a H2 balloon for 5 min and the reaction was stirred for 24 h. The mixture was filtered over Celite® and concentrated in vacuo. Purification by silica gel chromatography (hexane: EtOAc = 20: 1 to 15: 1) afforded the product 44 (2.77 g, 69% yield). Physical state: white solid. Rf = 0.38 (4:1 hexane : EtOAc). M.p.: 58–60 °C (hexane/EtOAc). 1H{13C} NMR (500 MHz, CDCl3): δ 4.80 (s, 1H), 3.91 (s, 3H), 3.73 (s, 3H), 2.17 (s, 3H), 2.16 (s, 3H), 2.13(s, 3H). 13C{1H} NMR (126 MHz, CDCl3): 169.9, 153.9, 153.8, 132.4, 121.6, 118.6, 114.4, 62.2, 52.3, 16.8, 11.8, 8.9. IRυmax (Diamond ATR): 3467, 2949, 1726, 1583, 1293, 1201cm−1. HRMS (ESI): [M + Na]+ Calc.’d for C12H16O4Na: 247.0946, found: 247.0937

To a stirred solution of 44 (85 mg, 0.379 mmol, 1.0 equiv.) in AcOH (2 mL) was added PIDA (159 mg, 0.492 mmol, 1.3 equiv.) and the reaction was stirred at r.t. and monitored until all starting material was consumed. The reaction was diluted with water and EtOAc, then quenched by careful addition of sat. NaHCO3. The reaction mixture was extracted with ethyl acetate (5 mL x 3) and combined organic layer was washed with brine, dried with Na2SO4, filtered, and concentrated. The crude product was purified by silica gel chromatography (hexane: EtOAc = 4:1) to afford 46 (107 mg, 93% yield). Physical state: yellow oil. Rf = 0.22 (4:1 hexane : EtOAc). 1H{13C} NMR(500 MHz, CDCl3): δ 3.85 (s, 3H), 3.84 (s, 3H), 2.12 (s, 3H), 1.93 (s, 3H), 1.82 (s, 3H), 1.42 (s, 3H). 13C{1H} NMR(126 MHz, CDCl3): 198.6, 169.4, 166.8, 163.5, 147.7, 126.6, 117.2, 80.5, 61.1, 52.4, 24.4, 20.4, 14.4, 9.06. IRυmax (Diamond ATR): 2952, 1735, 1665, 1236 cm−1. HRMS (ESI): [M + H]+ Calc.’d for C14H19O6: 283.1182, found: 283.1176

47:
To a 50 mL flask was added CH2Cl2 (9 mL), 34 (200 mg, 0.95 mmol, 1 equiv.), Boc2O (249 mg, 1.14 mmol, 1.2 equiv.), DMAP (2.32 mg, 19 μmol, 0.02 equiv.), and EtN(i-Pr)2 (9.4 mg, 10 μL, 57 μmol, 0.06 equiv.). The mixture was stirred for 4 h and was then concentrated. Direct purification by silica gel chromatography (hexane: EtOAc = 20:1) afforded compound 47 as a white solid (150 mg, 50% yield.). Physical state: white solid. Rf = 0.77 (4 : 1 hexane : EtOAc). M.p.: 53-55 °C. 1H{13C} NMR (500 MHz, CDCl3): δ 11.04 (s, 1H), 3.95 (s, 3H), 2.42 (s, 3H), 2.09 (s, 3H), 2.08 (s, 3H), 1.55 (s, 9H). 13C{1H} NMR(126 MHz, CDCl3): 172.3, 158.9, 152.2, 150.9, 137.4, 121.1, 116.6, 111.5, 83.6, 52.3, 27.7, 18.9, 12.7, 9.2. IRυmax (Diamond ATR): 2955, 2930, 1654, 1321, 1207, 827 cm−1. HRMS (ESI): [M + Na]+ Calc.’d for C16H22O6Na 333.1314, found: 333.2193

Cyclic carbonate 48:
To a 250 mL flask was added 47 (1.76 g, 5.67 mmol, 1.0 equiv.) and HFIP (56 mL) at r.t. and the mixture was stirred for 5 mins to ensure that all solids were dissolved. To another 50 mL flask was added PIDA (2.01 g, 6.24 mmol, 1.1 equiv.) in HFIP (30 mL). The solution of PIDA was added to the solution of 47 through a syringe pump over 60 mins and the reaction was stirred for another 15 mins. The reaction was concentrated directly and the crude product was purified by silica gel chromatography (hexane: EtOAc = 5: 1) to afford cyclic carbonate 48 (1.16 g, 81% yield). Physical state: white solid. Rf= 0.23 (4:1 hexane : EtOAc). M.p.: 78–80 °C (hexane/EtOAc): 1H{13C} NMR (500 MHz, CDCl3): δ 3.88 (s, 3H), 2.16 (s, 3H), 1.91 (s, 3H), 1.82 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3): 181.6, 165.2, 162.7, 150.0, 149.5, 132.3, 111.7, 82.3, 52.8, 28.8, 14.6, 8.3. IRυmax (Diamond ATR): 2954, 1842, 1727, 1658, 1105 cm−1. HRMS (ESI): [M + H]+ Calc.’d for C12H13O6: 253.0712 , found: 253.0719

Compound 48 (20 mg, 0.079 mmol, 1.0 equiv.) was dissolved in NMP (0.4 mL) in a 10 mL microwave tube. The tube was sealed and heated to 200 °C for 10 mins in the microwave reactor. The resulting dark solution was diluted with water and ethyl acetate. The mixture was extracted with ethyl acetate (3 x 4 ml) and the combined organic layer was washed with brine, dried with Na2SO4, filtered, and concentrated. The crude product was purified by silica gel chromatography (hexane: EtOAc = 4: 1) to afford 50 (7.2 mg, 46% yield). The data matched reported data by Abe and coworkers.51 1H{13C} NMR(500 MHz, CDCl3): δ 7.70 (s, 1H), 5.35 (s, 1H), 5.19 (s, 2H), 2.16 (s, 3H), 2.20 (s, 3H).

Di-MOM-DMOA methyl ester 53:
To a stirred solution of DMOA methyl ester 34 (4.0 g, 19 mmol) in THF (180 mL) at 0 °C was added NaH (60%, 1.9 g, 47 mmol, 2.5 equiv.). The resulting mixture was stirred at 0 °C for 20 min before MOM-Cl (3.83 g, 47 mmol, 2.5 equiv.) was slowly added. The resulting mixture was warmed to r.t. and was stirred for 3 h before being quenched with NH4Cl (200 mL, sat. aq.). The layers were separated, and the aqueous layer was extracted with EtOAc (3 x 50 mL). The combined organic layers were washed with NaHCO3 (50 mL, sat. aq.), dried, and concentrated in vacuo. Flash column chromatography (silica gel, hexane: EtOAc = 9:1) afforded the ester 53 (3.81 g, 67%) as a colorless oil. Physical state: colorless oil. Rf = 0.35 (4:1 hexane : EtOAc). 1H{13C} NMR(500 MHz, CDCl3): δ 4.95 (s, 2H), 4.91 (s, 2H), 3.90 (s, 3H), 3.60 (s, 3H), 3.54 (s, 3H), 2.22 (s, 3H), 2.17 (s, 3H), 2.16 (s, 3H). 13C{1H} NMR(126 MHz, CDCl3): 169.2, 156.2, 151.2, 132.7, 126.7, 126.0, 122.8, 100.3, 99.5, 57.7, 57.5, 52.2, 17.0, 13.1, 10.9. IRυmax (Diamond ATR): 2952, 1730, 1158, 1048, 957cm−1. HRMS (ESI): [M + H]+ Calc.’d for C15H23O5: 283.1545, found: 283.1559

Di-MOM DHDMP 55:
Procedure 1:
To a stirred solution of diisopropylamine (1.01 mL, 7.16 mmol, 1.2 equiv.) in dry THF (13 ml) at −78 °C under was added n-BuLi (2.86 mL, 2.5 M in hexane, 1.2 equiv.) dropwise. The mixture was stirred for 30 min at −78 °C for the generation of LDA. Then a solution of azeotropically dried 53 (1.78 g, 5.97 mmol, 1.0 equiv.) in THF (10 mL) was added to the solution of LDA at −78 °C dropwise. Subsequently, 3.3 mL of anhydrous THF was used to rinse the flask. The mixture was stirred for 30 mins. Then a solution of Davis oxaziridine (1.87 g, 7.2 mmol, 1.2 equiv.) in THF (18 mL) was added quickly. 4.4 mL of THF was used to rinse the flask containing the oxidant. The reaction mixture was stirred for 40 mins and was then quenched by NH4Cl. The reaction was warmed to r.t. and extracted with EtOAc (3 x 50 mL). The combined organic layer was washed with brine and dried over Na2SO4. The dried solution was filtered and concentrated. CH2Cl2 was then added to the crude solid in which case a white slurry formed which was filtered. The filtrate was concentrated and was purified by silica flash column chromatography (hexane: EtOAc = 10:1 to 4:1) to afford phthalide 55 (0.88 g, 52%). Physical state: white solid. Rf = 0.35 (2:1 hexane/EtOAc). M.p.: 72–75 °C (hexane/EtOAc). 1H{13C} NMR(500 MHz, CDCl3): δ 5.35 (s, 2H), 5.14 (s, 2H), 5.02 (s, 2H), 3.63 (s, 3H), 3.59 (s, 3H), 2.30 (s, 3H), 2.19 (s, 3H). 13C{1H} NMR(126 MHz, CDCl3): 169.35, 161.03, 154.15, 146.10, 126.16, 120.66, 112.48, 101.15, 99.66, 68.42, 57.87, 57.82, 12.23, 10.91. IRυmax (Diamond ATR): 2928, 1758, 1134, 949 cm−1 HRMS (DART+): [M + H]+ Calc.’d for C14H19O6: 283.11761, found: 283.11757
Procedure 2:
To a stirred solution of diisopropylamine (0.90 mL, 6.44 mmol, 1.2 equiv.) in dry THF (10 ml) at −78 °C under was added n-BuLi (2.56 mL, 2.5 M in hexane, 1.2 equiv.) dropwise. The reaction mixture was stirred for 30 min at −78 °C for the generation of LDA. Then a solution of azeotropically dried substrate 53 (1.60 g, 5.37 mmol, 1.0 equiv.) in THF (10 mL) was added to the solution of LDA at −78 °C dropwise and stirred for 30 mins. B(OMe)3 (0.9 mL, 8.05 mmol, 1.5 equiv.) was added dropwise and the reaction was stirred at −78 °C for 30 mins and 0 °C for an additional 30 mins. At 0 °C, H2O2 (3 equiv.) and NaOH (3 equiv.) was added and the reaction was stirred for 30 mins. The reaction was then quenched by NH4Cl and was extracted with EtOAc. The combined organic layer was washed with brine and dried over Na2SO4. The crude product was purified by silica flash column chromatography (hexane: EtOAc = 10:1 to 4:1) to afford 55 (1.52 g, 100%).

DHDMP 50:
To a stirred solution of 55 (1.6 g, 5.67 mmol, 1.0 equiv) in MeOH (27 mL) and THF (27 mL) was added conc. HCl (0.42 mL). The mixture was stirred at 50 °C for 4 h and was then quenched with sat. NaHCO3. The mixture was extracted with EtOAc (3 x 30 mL). The combined organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo to afford DHDMP 50 (1.05 g, 95%).
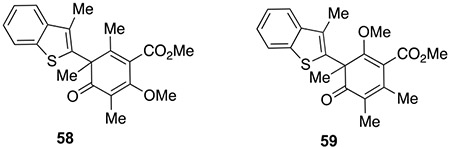
Sulfoxide 56 (8 mg, 49 μmol, 1.1 equiv.) was dissolved in CH2Cl2 (0.2 mL, 0.1 M) in an oven-dried tube flushed with Ar. TFAA (14 mg, 9.43 μL, 1.5 equiv.) was then added at −40 °C. After 5 min at the same temperature, DMOA methyl ester 34 (10 mg, 44 μmol, 1.0 equiv.) in CH2Cl2 (0.2 mL) was added dropwise. The colorless solution rapidly became deep orange. Alkene 35 (10.7 mg, 13 μL, 2.5 equiv.) was then added in one portion. The reaction was stirred at −40 °C for 30 mins; the cooling bath was removed, and the reaction was stirred for 30 mins. The solvents were removed, and the crude product was purified by preparative TLC (hexane: EtOAc = 5: 1) to afford 59 (5.7 mg, 34%) and 58 (6.3 mg, 37%).
C5-product: 58:
Physical state: slight yellow solid. M.p.: 85–90 °C (hexane/EtOAc). Rf = 0.34 (hexane : EtOAc = 4 : 1). 1H{13C} NMR(500 MHz, CDCl3): 67.79 (ddd, J = 7.69, 1.31, 1.06 Hz, 1H), 7.59 (ddd, J = 7.79, 1.89, 0.73 Hz, 1H), 7.34 (ddd, J = 7.06, 7.94, 1.35 Hz, 1H), 7.30 (ddd, J = 7.13, 7.17, 1.46 Hz, 1H),3.91 (s, 3H), 3.88 (s, 3H), 1.98 (d, J = 0.83 Hz, 3H), 1.96 (s, 3H), 1.77 (s, 3H), 1.74(d, J = 0.69 Hz, 3H).13C{1H} NMR(126 MHz, CDCl3): 201.96, 167.40, 163.97, 150.71, 141.12, 140.35, 137.94, 128.42, 126.68, 124.14, 124.12, 122.19, 121.71, 117.36, 61.34, 54.91, 52.53, 26.97, 16.96, 11.68, 9.08. IRυmax (Diamond ATR): 2964, 1735, 1654, 1587, 1434, 1303, 1252cm−1. HRMS (ESI): [M + H]+ Calc.’d for C21H23O4S: 371.1317, found: 371.1304
C3-product: 59-
Physical state: slight yellow solid. M.p.: 117– 120 °C (hexane/EtOAc). Rf =0.41(4:1 hexane : EtOAc).1H{13C} NMR(500 MHz, CDCl3): δ 7.78 (ddd, J = 8.08, 1.38, 0.57 Hz, 1H), 7.58 (ddd, J = 7.51, 1.61, 1.03 Hz, 1H), 7.32 (ddd, J = 7.07, 7.20, 1.24 Hz, 1H), 7.29 (ddd, J = 7.07, 7.69, 1.53 Hz, 1H), 3.87 (s, 3H), 3.56 (s, 3H),2.10(s, J = 0.98 Hz, 3H), 1.77 (s, 3H), 1.96 (s, J = 0.98 Hz, 3H), 1.85 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3): 198.25, 168.65, 165.16, 145.79, 140.90, 139.90, 138.12, 128.00, 125.34, 124.02, 123.97, 122.15, 121.72, 112.81, 60.27, 54.29, 52.49, 26.70, 18.36, 11.94, 11.48. IRυmax (Diamond ATR): 2948, 1728, 1654, 1303cm−1. HRMS(ESI): [M + H]+ Calc.’d for C21H23O4S: 371.1317, found: 371.1311

Diol 80:
Allylic alcohol 66 (60 mg, 0.225 mmol, 1.0 equiv.) in a flame-dried 10 mL test-tube with a stir bar was azeotropically dried with benzene (0.3 mL x 3) and further dried using high vacuum for 30 mins. Then the vial was vacuumed and refilled with Ar three times. Then distilled grade pentane (1.2 mL) and dry ether (0.96 mL) were added. In a separate 10 mL test-tube, decalin 67 (59 mg, 0.225 mmol, 1.0 equiv.) was weighed and azeotropically dried with benzene (0.5 mL x 3) and was further dried in vacuo for 30 mins. The flask containing allylic alcohol 66 was placed in a −78 °C dry ice/ acetone bath. n-BuLi (2.5 M in hexane, 90 μL, 1.0 equiv.) was added dropwise. 2 mins later after addition, t-BuLi (1.7 M in pentane, 0.292 mL, 2.2 equiv.) was added dropwise and a white slurry formed. The slurry was stirred at −78 °C for 60 mins. The dried decalin 67 was dissolved in 0.5 mL ether and the solution was added to the lithium dianion slurry dropwise. The test tube containing the decalin was rinsed twice with ether (0.25 mL each time). After 1.5 h, the reaction was quenched by slow addition of DI water (10 mL). The reaction was extracted with EtOAc (3 x 5 mL), and the combined organic layer was washed with brine and dried over Na2SO4, filtered and concentrated and was purified by silica gel column chromatography (10:1 to 2:1 hexane/EtOAc) to afford the diol product 79 as sticky, colorless oil which became a white foam under high vacuum (4.9 mg, 5.3%) and the diol side product 80 (10.8 mg, 30%). Physical state: colorless oil. Rf = 0.08 (2:1 hexane : EtOAc). Rotation: (c 0.1, CHCl3). 1H{13C} NMR (500 MHz, CDCl3): δ 5.78 (dt, J = 10.76, 1.75 Hz, 1H), 5.46 (dd, J = 10.16, 2.00 Hz, 1H), 4.27 (dddd, J = 9.69, 6.00, 1.85, 1.85 Hz, 1H), 3.45 (d, J = 10.04 Hz, 1H), 3.33 (d, J = 11.09 Hz, 1H), 1.95 (ddq, J = 12.54, 7.0, 2.44 Hz, 1H), 1.86 (dddd, J = 1.58, 2.45, 6.00, 12.54 Hz, 1H), 1.37 (td, J = 10.24, 12.94 Hz, 1H), 0.89 (d, J = 6.53 Hz, 3H), 0.79 (s, 3H). 13C{1H} NMR(126 MHz, CDCl3): 136.32, 133.21, 68.73, 68.07, 41.12, 37.44, 29.55, 17.58, 16.04. IRυmax (Diamond ATR): 3439, 2959, 1701, 1280, 1042, 755 cm−1. HRMS (ESI): [M + Hl+ Calc.’d for C9H17O2: 157.1229, found: 157.0665

The allylic alcohol 66 (56 mg, 0.210 mmol, 1.0 equiv.) in a flamedried 10 mL test-tube with a stir bar was azeotropically dried with benzene (3 x 0.3 mL) and further dried in high vacuum for 30 mins. Then the vial was vacuumed and refilled with Ar three times. Then distilled grade pentane (1.2 mL) and dry ether (0.8 mL) were added. In a separated 10 mL test-tube, the decalin 67 (44 mg, 0.225 mmol, 0.8 equiv.) was azeotropically dried with benzene (3 x 0.5 mL) and further dried in high vacuum for 30 mins. The flask containing allylic alcohol 66 was placed in a −78 °C dry ice/ acetone bath. Then n-BuLi (2.5 M in hexane, 84 μL, 1.0 equiv.) was added dropwise. 2 mins later, t-BuLi (1.7 M in pentane, 0.272 mL, 2.2 equiv.) was added dropwise and an orange slurry formed. The slurry was stirred at −78 °C for 10 mins, then r.t. for 15 mins. Then the dried decalin 67 was dissolved in 0.5 mL ether and the solution was added to the lithium dianion slurry dropwise. The test-tube containing decalin was rinse twice with ether (0.25 mL each time). After 1.5 h, the reaction was quenched by slow addition of TMSCl (23 mg, 0.210 mmol, 1.0 equiv.) and the mixture was stirred at r.t. for 30 mins. The reaction was quenched by water and extracted with EtOAc (3 x 3 mL). The combined organic layer was washed with brine and dried over Na2SO4, filtered and concentrated and repeated purification with silica gel column afforded the SN2’ product 81 (20 mg, 24% yield). Physical state: white foam. Rf = 0.25 (10:1 hexane: EtOAc). Rotation: (c 0.2, CHCl3). 1H{13C} NMR (500 MHz, CDCl3): δ 5.71 (dd, J = 10.30, 2.15 Hz, 1H), 5.62 (ddd, J = 10.30, 1.70, 1.70 Hz, 1H), 5.21 (m, 1H), 4.32 (dddd, J = 10.04, 5.75, 1.70, 1.70 Hz, 1H), 3.91-3.98 (overlap, 4H), 2.38 (m, 1H), 2.31(d, J = 15.34 Hz, 1H), 2.01(dd, J = 10.56, 7.07 Hz, 1H), 1.90 (d, J = 14.31, 4.56 Hz, 1H), 1.85-1.89 (overlap, 2H), 1.80 (td, J = 13.61, 3.78 Hz, 1H), 1.72 (d, J = 1.61Hz, 3H), 1.70 (m, 1H), 1.53 (dt, J = 13.05, 3.72 Hz, 1H), 1.38-1.44 (overlap, 2H), 1.34 (d, J = 15.46 Hz, 1H), 1.03 (s, 3H), 0.939 (d, J = 6.91 Hz, 1H), 0.90 (s, 3H), 0.87 (s, 3H), 0.82 (s, 3H). 13C{1H} NMR(126 MHz, CDCl3): 141.2, 139.5, 131.9, 121.0, 113.2, 80.5, 68.0, 64.92, 64.88, 45.8, 42.1, 41.98, 41.1, 39.2, 38.6, 31.2, 28.5, 26.7, 24.4, 24.1, 22.5, 19.56, 19.43, 16.7, 15.5. IRυmax (Diamond ATR): 2962, 2934, 1608, 1510, 1246, 1036 cm−1. HRMS (DART+): [M + Hl+ Calc.’d for C25H39O3: 387.2893, found: 387.2878

Triethyl orthoformate (0.53 mL, 0.89 mmol, 7.0 equiv.) was added to a solution of ethylene glycol (0.127 mL, 2.27 mmol, 5.0 equiv.), p-toluenesulfonic acid monohydrate (4.32 mg, 3.5 μmol, 5.0 mol %), and neopentyl iodide 72 (120 mg, 0.454 mmol, 1 equiv.) in ether (1.0 mL) at 23 °C. The reaction vessel was sealed, and the reaction vessel was placed in an oil bath that had been preheated to 50 °C. The reaction mixture was stirred for 29 h at 50 °C. The product mixture was cooled to 23 °C over 30 min. The cooled product mixture was diluted with ethyl acetate (25 mL), and transferred to a separatory funnel and washed sequentially with sat. NaHCO3 (15 mL) and brine (15 mL). The organic layer was dried over Na2SO4, filtered, concentrated, and purified by flash-column chromatography (eluting with 5% ether/hexane) to provide the ketal 83 as a colorless oil (117 mg, 83%). Physical state: colorless oil. Rf = 0.6 (10:1 hexane: EtOAc). 1H{13C} NMR(500 MHz, CDCl3): δ 5.60 (d, J = 10.22 Hz, 1H), 5.54 (dd, J =10.05, 1.24 Hz, 1H), 4.02–3.84 (overlap, 4H), 3.24 (d, J = 10.52 Hz, 1H), 3.18 (d, J = 9.43 Hz, 1H), 2.17 (m, 1H), 1.68–1.72 (overlap, 2H), 1.0 (s, 3H), 0.86 (d, J = 7.14 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ 140.64, 127.02, 105.77, 64.80, 64.60, 38.51, 33.39, 20.50, 19.18, 15.57. IRυmax (Diamond ATR): 2962, 2876, 1112, 1061 cm−1 HRMS (ESI): [M + H]+ Calc.’d for C11H18IO2: 309.0352, found: 309.0426
General procedure for spirocycle library synthesis:
The allylic alcohol 66 (50 mg, 0.188 mmol, 1.0 equiv.) in a flame-dried 10 mL test-tube with a stir bar was azeotropically dried with benzene (3 x 0.3 mL) and further dried in high vacuum for 30 mins. Then the vial was vacuumed and refilled with Ar three times. Then distilled grade pentane (1.0 mL) and dry ether (0.80 mL) were added. The test-tube was placed in a −78 °C dry ice/ acetone bath. Then n-BuLi (2.5 M in hexane, 75 μL, 1.0 equiv.) was added drop-wise. 2 mins later after addition, t-BuLi (1.7 M in pentane, 0.243 mL, 2.2 equiv.) was added dropwise and an orange-yellow slurry formed. The slurry was stirred at −78 °C for 10 mins. Then the reaction was moved to r.t. and stirred for 15 mins. Then a solution of the ketone or aldehyde (1.5 equiv.) in ether (1.0 mL) was added and the mixture was stirred for 60 mins. The reaction was quenched by slow addition of DI water (5 mL) and extracted with EtOAc (3 x 5 mL) and the combined organic layer was washed with brine and dried over Na2SO4. The dried solution was concentrated, and the residue was dissolved in CH2Cl2 (2.0 mL) and MnO2 (200 mg, 12 equiv.) was added. The reaction was stirred for 24 h and then the mixture was filtered through celite and concentrated. The mixture was purified using an ISCO Combiflash system (normal phase, 0% EtOAc to 10% EtOAc/hexane gradient elution over 10 mins, then 10% EtOAc/hexane elution for 5 mins) to afford the desired the spirocycle.

94-S: 20 mg, 36% yield. Physical state: colorless oil. Rf = 0.15 (10 :1 hexane : EtOAc). Rotation: (c 0.2, CHCl3). 1H{13C} NMR (500 MHz, CDCl3): δ 5.50 (m, 1H), 4.72 (s, 2H), 4.10 (t, J = 5.28 Hz, 1H), 2.66 (d, J = 1.80 Hz, 1H), 2.65 (d, J = 0.78 Hz, 1H), 2.23-2.30 (overlap, 2H), 2.19 (dddd, J = 13.13, 4.98, 2.53 Hz, 1H), 2.02-2.14 (overlap, 3H), 1.97 (d, J = 13.55 Hz, 1H), 1.92 (dddd, J = 16.6, 11.6, 2.5, 2.5 Hz, 1H), 1.72 (s, 3H), 1.68 (m, 3H), 1.69-1.72(overlap, 1H), 1.54 (t, J = 12.33 Hz, 1H), 1.08 (s, 3H), 0.96(d, J = 6.83 Hz, 3H). 13C{1H} NMR (150 MHz, CDCl3): δ 210.51, 149.13, 136.23, 136.23, 125.39, 109.23, 83.31, 82.96, 49.53, 44.79, 44.21, 43.10, 40.73, 39.68, 35.69, 31.06, 20.53, 19.02, 17.49, 15.92. IRυmax (Diamond ATR): 2962, 2878, 1719, 1278 cm−1. HR MS (ESI): [M + H]+ Calc.’d for C19H29O3: 289.2168, found: 289.2160.
94-R: 12 mg, 21% yield. Physical state: colorless oil. Rf = 0.09 (10:1 hexane : EtOAc). Rotation: (c 0.2, CHCl3). 1H{13C} NMR (500 MHz, CDCl3): δ 5.60 (d, J = 5.33 Hz, 1H), 4.71(t, J = 1.57 Hz, 1H), 4.67 (s, 1H), 4.23 (dd, J = 5.51, 2.54 Hz, 1H), 2.67 (d, J = 16.8 Hz, 1H), 2.50 (dd, J = 15.44, 5.48 Hz, 1H), 2.30-2.40 (overlap, 2H), 2.14-2.20 (overlap, 3H), 2.10 (m, 1H), 2.06 (td, J = 13.05, 1.98 Hz, 1H), 1.88 (d, J = 13.8 Hz, 1H), 1.81 (s, 3H), 1.71 (s, 3H), 1.61(t, J = 12.35Hz, 1H), 1.10 (s, 3H), 0.99 (d, J = 6.7 Hz, 3H). 13C{1H} NMR (150 MHz, CDCl3): δ 209.95, 149.5, 136.1, 127.1, 108.9, 87.2, 81.3, 47.0, 45.3, 44.7, 44.4, 37.95, 35.57, 31.33, 21.1, 20.5, 16.7, 16.4. IRυmax (Diamond ATR): 2961, 2923, 2877, 1719 cm−1.

4 mg, 5% yield. Physical state: colorless foam. Rf = 0.15 (10:1 hexane : EtOAc). Rotation: (c 0.2, CHCl3). 1H{13C} NMR (500 MHz, CDCl3): δ 5.22 (s, 1H), 4.68 (s, 1H), 4.66 (s, 1H), 4.04 (dd, J = 5.22, 2.88 Hz, 1H), 2.62 (dd, J = 16.24, 3.18 Hz, 1H), 2.53 (dd, J = 15.51, 4.77 Hz, 1H), 2.28 (dd, J = 12.87 Hz, 1H), 2.14-2.24 (overlap, 4H), 2.05 (d, J = 16.54 Hz, 1H), 2.02 (d, J = 13.58 Hz, 1H), 1.85 (d, J = 12.66 Hz, 1H), 1.79 (d, J = 10.62 Hz, 1H), 1.74 (d, J = 12.66 Hz, 1H), 1.70 (s, 3H), 1.66 (dd, J = 13.2, 5.88 Hz, 1H), 1.62 (d, J = 11.85 Hz, 1H), 1.45 (m, 1H), 1.17 (dd, J = 4.03, 11.16 Hz, 1H), 1.07 (s, 3H), 0.96 (d, J = 7.00 Hz, 3H), 0.95 (s, 3H), 0.88 (d, J = 7.22 Hz. 3H). 13C{1H} NMR (150 MHz, CDCl3): δ 209.89, 150.42, 144.84, 127.08, 108.59, 83.91, 80.39, 52.60, 44.79, 44.76, 42.13, 41.19, 38.60, 38.17, 35.82, 32.93, 32.28, 21.05, 17.60, 17.07, 16.3, 15.85. IRυmax (Diamond ATR): 2962, 2929, 2873, 1720, 1455, 997 cm−1. HRMS (DART+): [M + H]+ Calc.’d for C24H37O2: 357.2788, found: 357.2780

12 mg, 30% yield. Physical state: colorless oil. Rf = 0.44 (3:1 hexane : EtOAc). Rotation: (c 0.133, CHCl3). 1H{13C} NMR (500 MHz, CDCl3): 3.88 (dd, J = 3.38, 4.69 Hz, 1H), 2.58 (dd, J = 16.51, 3.56 Hz, 1H), 2.49 (dd, J = 15.43, 4.64 Hz, 1H), 2.22-2.28 (overlap, 3H), 2.13-2.16 (overlap, 2H), 2.00-2.10 (overlap, 3H), 1.83(d, J = 13.35 Hz, 1H), 1.66-1.73 (m, 1H), 1.52 (m, 1H), 1.03 (s, 3H), 0.95 (d, J = 6.51 Hz, 3H). 13C{1H} NMR (150 MHz, CDCl3): δ 210.08, 84.98, 81.72, 50.24, 44.54, 44.31, 42.34, 38.25, 37.52, 34.05, 16.34, 12.92. IRυmax (Diamond ATR): 2961, 2930, 2875, 1719, 1112 cm−1. HRMS(DART+): [M + H]+ Calc.’d for C13H21O2: 209.1536, found: 209.1530

14 mg, 28% yield. Physical state: colorless oil. Rf = 0.59 (3:1 hexane : EtOAc). 1H{13C} NMR (500 MHz, CDCl3): 4.93 (s, 1H), 4.89 (s, 1H), 4.85 (s, 2H), 4.23 (dd, J = 4.65, 2.60 Hz, 1H), 3.76 (dd, J = 5.62, 4.11 Hz, 1H), 2.67-2.72 (overlap, 3H), 2.58-2.65 (overlap, 2H), 2.48 (dd, J = 14.96, 4.43 Hz, 1H), 2.33 (bs, 1H), 2.22-2.31 (overlap, 3H), 2.15-2.19 (overlap, 4H), 1.94 (d, J = 13.23 Hz, 1H), 1.83 (d, J = 13.23 Hz, 1H), 1.79 (d, J = 12.63 Hz, 1H), 1.61-1.67 (overlap, 3H), 1.28-1.45 (overlap, 8H), 1.09 (s, 3H), 1.06 (s, 3H), 0.97 (d, J = 6.39 Hz, 3H), 0.95 (d, J = 6.76 Hz, 3H). 13C{1H} NMR (150 MHz, CDCl3): δ 210, 209.9, 165.1, 159.9, 104.4, 102.7, 86.69, 86.65, 83.7, 53.22, 51.4, 49.0, 45.7, 45.1, 44.7, 44.24, 44.20, 43.59, 42.58, 41.92, 36.58, 35.81, 35.72, 33.61, 30.25, 29.38, 20.97, 20.71, 18.05, 16.2, 16.06, 15.87. IRυmax (Diamond ATR): 2959, 2872, 1722, 1063 cm−1. HRMS(DART): [M + H]+ Calc.’d for C17H25O2: 261.1849, found: 261.1853
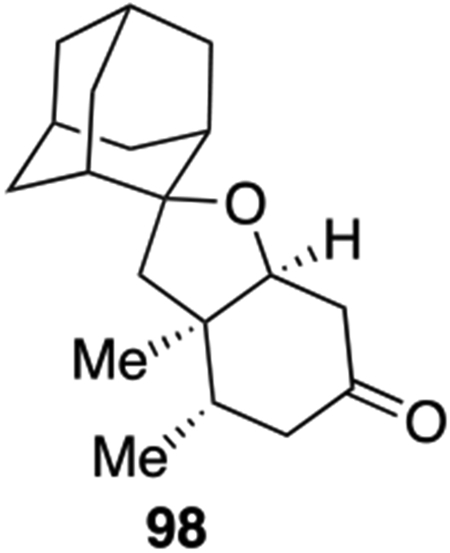
22 mg, 40% yield. Physical state: white solid. Rf = 0.5 (3:1 hexane : EtOAc). Rotation: (c 0.2, CHCl3). M.p: 90–92 °C (hexane/EtOAc). 1H{13C} NMR (600 MHz, CDCl3): δ 3.92 (dd, J = 4.97, 2.77 Hz, 1H), 2.61 (dt, J = 15.70, 1.91 Hz, 1H), 2.48 (dd, J = 15.70, 4.50 Hz, 1H), 2.12-2.18 (overlap, 5H), 2.07 (d, J = 11.05 Hz, 1H), 2.0 (d, J = 12.97 Hz, 1H), 1.76 (overlap, 1H), 1.72-1.75 (overlap, 3H), 1.68-1.70 (overlap, 2H), 1.54 (d, J = 13.09 Hz, 1H), 1.41-1.46 (overlap, 2H), 1.03 (s, 3H), 0.92 (d, J = 6.58 Hz, 3H). 13C{1H} NMR (150 MHz, CDCl3): δ 210.65, 84.02, 83.03, 48.28, 46.99, 44.90, 43.99, 41.85, 39.39, 39.27, 37.56, 36.57, 36.15, 35.46, 34.28, 33.63, 33.10, 27.09, 26.53, 16.63, 16.09. IRυmax (Diamond ATR): 2899, 2852, 1718, 1450, 1109, 928 cm−1. HRMS(DART+): [M + H]+ Calc.’d for C19H29O2: 289.2162, found: 289.2158
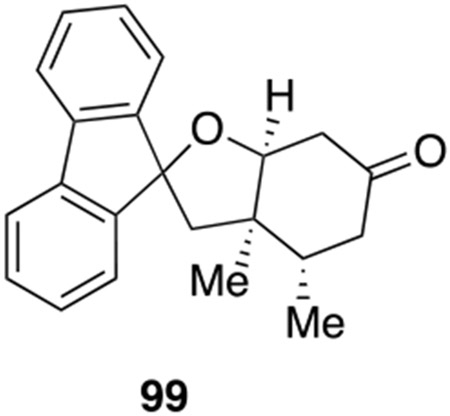
29 mg, 48% yield. Physical state: yellow solid. Rf = 0.33 (3:1 hexane : EtOAc). Rotation: (c 0.2, CHCl3). M.p: 205–210 °C (hexane/EtOAc). 1H{13C} NMR (600 MHz, CDCl3): δ 7.59 (t, J = 6.83 Hz, 2H), 7.55 (d, J = 7.41 Hz, 2H), 7.43 (d, J = 7.67 Hz, 2H), 7.34 (td, J = 7.67, 0.90 Hz, 2H), 7.29 (td, J = 7.09, 1.23 Hz, 2H), 4.67 (dd, J = 5.23, 2.78 Hz, 1H), 2.71-2.76 (overlap, 2H), 2.69 (dd, J = 15.4, 5.56 Hz, 1H), 2.67 (d, J = 14.4 Hz, 1H), 2.42 (d, J = 14.4 Hz, 1H), 2.40 (ddd, J = 15.08, 4.37, 1.44 Hz, 1H), 2.35 (dd, J = 15.4, 13.6 Hz, 1H), 1.33 (s, 3H), 1.125 (d, J = 7.00 Hz, 3H). 13C{1H} NMR (150 MHz, CDCl3): δ 209.25, 150.75, 147.30, 140.16, 139.65, 129.24, 128.97, 128.38, 128.17, 124.53, 123.79, 120.06, 119.91, 88.54, 86.03, 48.55, 44.82, 44.68, 42.01, 35.5, 16.93, 16.29. IRυmax (Diamond ATR): 2961, 1718, 1448, 760, 731 cm−1. HRMS(DART+): [M + H]+ Calc.’d for C22H23O2: 319.1692, found: 319.1698

Note: Thioxanthone dissolved in 3 mL of THF was added. 31 mg, 46 % yield. Physical state: slight yellow solid. Rf = 0.21 (3:1 hexane: EtOAc). Rotation: (c 0.073, CHCl3). M.p: 110–112 °C (hexane/EtOAc). 1H{13C} NMR (600 MHz, CDCl3): δ 7.69 (dd, J = 8.24, 1.20 Hz, 1H), 7.56 (dd, J = 7.81, 5.63 Hz, 1H), 7.49 (dd, J = 7.59, 0.79 Hz, 1H), 7.47 (dd, J = 7.70, 0.91 Hz, 1H), 4.56 (dd, J = 4.60, 2.43 Hz, 1H), 3.08 (dt, J = 15.77, 2.12 Hz, 1H), 2.74 (dd, J = 15.77, 4.47 Hz, 1H), 2.59 (d, J = 13.42 Hz, 1H), 2.19-2.25 (overlap, 3H), 1.94( d, J = 13.65 Hz, 1H), 1.01 (s, 3H), 0.73 (d, J = 6.13 Hz, 1H). 13C{1H} NMR (150 MHz, CDCl3): δ 209.7, 143.51, 143.02, 131.84, 131.76, 128.03, 127.71, 126.85, 126.76, 126.44, 126.39, 123.82, 122.13, 87.48, 82.89, 50.60, 44.94, 44.67, 41.79, 35.05, 16.42, 16.12. IRυmax (Diamond ATR): 2961, 2932, 2870, 1719, 1452, 1043, 758 cm−1. HRMS(DART+): [M + H]+ Calc.’d for C22H23O2S: 351.1413, found: 351.1411

101-R 17 mg, 30% yield. Physical state: yellow foam. Rf = 0.43 (3:1 hexane : EtOAc). Rotation: (c 0.2, CHCl3). 1H{13C} NMR (600 MHz, CDCl3): δ 7.58 (d, J = 7.22 Hz, 1H), 7.31 (d, J = 9.12 Hz, 1H), 7.22 (t, J = 7.22 Hz, 1H), 7.09 (d, J = 7.22 Hz, 1H), 6.46 (s, 1H), 5.28 (t, J = 7.55 Hz, 1H), 4.02 (dd, J = 2.55, 4.43 Hz, 1H), 3.76 (s, 3H), 2.62 (dd, J = 15.36, 2.35 Hz, 1H), 2.52 (dd, J = 13.87, 7.31 Hz, 1H), 2.50 (dd, J = 13.49, 4.34 Hz, 1H), 2.26 (overlap, 3H), 2.19 (dd, J = 13.18, 8.76 Hz, 1H), 1.20 (s, 3H), 1.08 (d, J = 7.13 Hz, 3H). 13C{1H} NMR (150 MHz, CDCl3): δ 209.87, 140.18, 138.43, 127.02, 121.93, 120.72, 119.58, 109.25, 99.22, 84.69, 70.12, 44.86, 44.12, 41.81, 41.43, 34.63, 30.36, 16.7, 15.48. IRυmax (Diamond ATR): 2961, 2922, 1718, 1468, 1310, 749cm−1. HRMS(DART+): [M + H]+ Calc.’d for C19H24NO2: 298.1801, found: 298.1797

101-S, 17 mg, 30% yield. Physical state: yellow solid. M.p: 198 °C (Hexane/EtOAc). Rf = 0.28 (3:1 hexane : EtOAc). Rotation: (c 0.2, CHCl3). 1H{13C} NMR (500 MHz, CDCl3): δ 7.58 (d, J = 7.08 Hz, 1H), 7.31 (d, J = 7.59 Hz, 1H), 7.21 (t, J = 6.98 Hz, 1H), 7.09 (d, J = 6.98 Hz, 1H), 6.42 (s, 1H), 5.15 (t, J = 7.26 Hz, 1H), 4.00 (t, J = 5.06 Hz, 1H), 3.76 (s, 3H), 2.72 (dd, J = 15.81, 4.39 Hz, 1H), 2.63 (dd, J = 15.95, 5.18 Hz, 1H), 2.38 (dd, J = 12.76, 7.04 Hz, 1H), 2.27 (m, 1H), 2.22 (d, J = 8.9 Hz, 1H), 2.20 (d, J = 5.71 Hz, 1H), 2.15 (d, J = 13.75 Hz, 1H), 1.17 (s, 3H), 1.01 (d, J = 6.72Hz, 3H). 13C{1H} NMR (150 MHz, CDCl3): δ 209.71, 139.90, 138.17, 127.17, 121.81, 120.79, 119.61, 109.16, 98.89, 86.06, 71.75, 44.24, 43.75, 43.41, 42.50, 35.37, 30.39, 17.14, 16.15. IRυmax (Diamond ATR): 2960, 2934, 1718, 1469, 1313, 749 cm−1.

102-S, 17.1 mg, 31% yield. Physical state: colorless oil. Rf = 0.59 (4:1 hexane : EtOAc). Rotation: (c 0.2, CHCl3). 1H{13C} NMR (600 MHz, CDCl3): δ 4.37 (dd, J = 10.67, 5.68 Hz, 1H), 3.62 (dd, J = 8.26, 6.54 Hz, 1H), 2.86 (dd, J = 14.24, 6.86 Hz, 1H), 2.74 (dd, J = 14.76, 8.68 Hz, 1H), 2.24-2.32 (overlap, 2H), 1.97-2.03 (m, 1H), 1.91-1.96 (overlap, 3H), 1.81 (s, 3H), 1.81 (dd, J = 12.42, 5.70 Hz, 1H), 1.52-1.60 (overlap, 2H), 1.46 (ddd, J = 13.1, 6.91, 3.10 Hz, 1H), 1.37 (ddd, J = 13.1, 10.66, 3.82 Hz, 1H), 1.10 (s, 3H), 0.99 (s, 3H), 0.98 (d, J = 6.86 Hz, 3H), 0.96 (s, 3H). 13C{1H} NMR (150 MHz, CDCl3): δ 211.27, 134.92, 132.12, 83.45, 77.03, 47.22, 44.93, 44.77, 43.25, 40.01, 36.23, 34.58, 34.42, 29.11, 27.82, 20.73, 19.73, 19.36, 15.83. IRυmax (Diamond ATR): 2958, 2927, 2904, 1719, 1074 cm−1. HRMS(DART+): [M + H]+ Calc.’d for C19H31O2: 291.2318, found: 291.2322
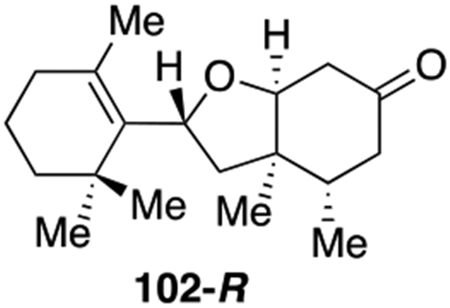
102-R, 19.8 mg, 36% yield. Physical state: colorless oil. Rf = 0.48 (3:1 hexane : EtOAc). Rotation: (c 0.2, CHCl3). 1H{13C} NMR (500 MHz, CDCl3): δ 4.68 (dd, J = 11.94, 6.45 Hz, 1H), 4.23 (t, J = 4.85 Hz, 1H), 2.68 (dd, J = 15.5, 5.22 Hz, 1H), 2.54 (dd, J = 15.5, 5.22 Hz, 1H), 2.27 (m, 1H), 2.24 (ddd, J = 15.6, 3.9, 0.81 Hz, 1H), 2.17 (dd, J = 15.6, 13.64 Hz, 1H), 2.08 (dd, J = 13.15, 6.17 Hz, 1H), 1.94-1.97 (overlap, 2H), 1.85 (dd, J = 13.2, 11.73 Hz, 1H), 1.73 (s, 3H), 1.51-1.59 (overlap, 2H), 1.43 (ddd, J = 12.98, 6.35, 3.20 Hz, 1H), 1.35 (ddd, J = 14.25, 10.83, 3.24 Hz, 1H), 1.12 (s, 3H), 1.07 (s, 3H), 1.018 (d, J = 6.76 Hz, 3H), 1.93 (s, 3H). 13C{1H} NMR (150 MHz, CDCl3): δ 210.22, 136.76, 131.19, 84.83, 75.93, 45.03, 44.93, 44.34, 42.98, 40.18, 34.61, 34.58, 33.86, 29.30, 27.88, 20.53, 19.37, 17.46, 16.87. IRυmax (Diamond ATR): 2960, 2927, 2865, 1721, 1458, 1072, 914 cm−1.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health (NIH) (R35 GM 118173) and Prelude Biosciences for financial support. We thank Dr. James McNeely (Boston University) for assistance with com-putations and Dr. Jeffrey Bacon (Boston University) for assistance with X-ray crystal structure analysis. We thank the National Sci-ence Foundation (NSF) for support of NMR (CHE-0619339) and MS (CHE-0443618) facilities at BU and the NIH (S10OD028585) for support of a single crystal XRD system.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Copies of 1H and 13C NMR spectra and X-ray crystal structure analyses (PDF) and computational data.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
REFERENCES
- (1).Qi C; Bao J; Wang J; Zhu H; Xue Y; Wang X; Li H; Sun W; Gao W; Lai Y; Chen JG; Zhang Y Asperterpenes A and B, Two Unprecedented Meroterpenoids from: Aspergillus Terreus with BACE1 Inhibitory Activities. Chem. Sci 2016, 7(10), 6563–6572. 10.1039/c6sc02464e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Cueto M; MacMillan JB; Jensen PR; Fenical W Tropolactones A-D, Four Meroterpenoids from a Marine-Derived Fungus of the Genus Aspergillus. Phytochemistry 2006, 67 (16), 1826–1831. 10.1016/j.phytochem.2006.01.008. [DOI] [PubMed] [Google Scholar]
- (3).He Y; Hu Z; Li Q; Huang J; Li XN; Zhu H; Liu J; Wang J; Xue Y; Zhang Y Bioassay-Guided Isolation of Antibacterial Metabolites from Emericella Sp. TJ29. J. Nat. Prod 2017, 80 (9), 2399–2405. 10.1021/acs.jnatprod.7b00077. [DOI] [PubMed] [Google Scholar]
- (4).Ting CP; Xu G; Zeng X; Maimone TJ Annulative Methods Enable a Total Synthesis of the Complex Meroterpene Berkeleyone A. J. Am. Chem. Soc 2016, 138, 55. 10.1021/jacs.6b10397. [DOI] [PubMed] [Google Scholar]
- (5).Elkin M; Szewczyk SM; Scruse AC; Newhouse TR Total Synthesis of (±)-Berkeleyone A. J. Am. Chem. Soc 2017, 139, 1790–1793. 10.1021/jacs.6b12914. [DOI] [PubMed] [Google Scholar]
- (6).Zhang Y; Ji Y; Franzoni I; Guo C; Jia H; Hong B; Li H Enantioselective Total Synthesis of Berkeleyone A and Preaustinoids. Angew. Chemie - Int. Ed 2021, 60 (27), 14869–14874. 10.1002/anie.202104014. [DOI] [PubMed] [Google Scholar]
- (7).Xu G; Elkin M; Tantillo DJ; Newhouse TR; Maimone TJ Traversing Biosynthetic Carbocation Landscapes in the Total Synthesis of Andrastin and Terretonin Meroterpenes. Angew. Chem. Int. Ed 2017, 56 (41), 12498–12502. 10.1002/anie.201705654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ishikawa K; Sato F; Itabashi T; Wachi H; Takeda H; Wakana D; Yaguchi T; Kawai KI; Hosoe T Asnovolins A-G, Spiromeroterpenoids Isolated from the Fungus Aspergillus Novofumigatus, and Suppression of Fibronectin Expression by Asnovolin E. J. Nat. Prod 2016, 79 (9), 2167–2174. 10.1021/acs.jnatprod.6b00013. [DOI] [PubMed] [Google Scholar]
- (9).Emi Okuyama MY Fumigatonin, a New Meroterpenoid from Aspergillus Fumigatus. Tetrahedron Lett. 1984, 25 (30), 3233–3234. [Google Scholar]
- (10).Matsuda Y; Bai T; Phippen CBW; Nødvig CS; Kjærbølling I; Vesth TC; Andersen MR; Mortensen UH; Gotfredsen CH; Abe I; Larsen TO Novofumigatonin Biosynthesis Involves a Non-Heme Iron-Dependent Endoperoxide Isomerase for Orthoester Formation. Nat. Commun 2018, 9 (1), 2587. 10.1038/s41467-018-04983-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lockner JW Studies in Meroterpenoid Synthesis. 2011, No. November. [Google Scholar]
- (12).Powers Z; Scharf A; Cheng A; Yang F; Himmelbauer M; Mitsuhashi T; Barra L; Taniguchi Y; Kikuchi T; Fujita M; Abe I; Porco JA Biomimetic Synthesis of Meroterpenoids by Dearomatization-Driven Polycyclization. Angew. Chem. Int. Ed 2019, 58, 16141–16146. 10.1002/anie.201910710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mitsuhashi T; Barra L; Powers Z; Kojasoy V; Cheng A; Yang F; Taniguchi Y; Kikuchi T; Fujita M; Tantillo DJ; Porco JA; Abe I Exploiting the Potential of Meroterpenoid Cyclases to Expand the Chemical Space of Fungal Meroterpenoids. Angew. Chemie 2020, 132 (52), 23980–23989. 10.1002/ange.202011171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yang F; Porco JA Unified, Asymmetric Total Synthesis of the Asnovolins and Related Spiromeroterpenoids: A Fragment Coupling Approach. J. Am. Chem. Soc 2022, 144 (28), 12970–12978. 10.1021/jacs.2c05366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).This manuscript has appeared as a preprint: Yang F; Oladokun O; Porco JA Jr. Evolution of a Strategy for the Unified, Asymmetric Total Syntheses of DMOA-Derived Spiromeroterpenoids. ChemRxiv. 2024; doi: 10.26434/chemrxiv-2024-0m8pp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Fürst A; Plattner PA Über Steroide Und Sexualhormone. 2A,3A- Und 2B,3B-Oxido-Chlolestane; Konfiguration Der 2-Oxy-Cholestane. Helv. Chim. Acta 1949, 32 (37), 275–283. [DOI] [PubMed] [Google Scholar]
- (17).Shenvi RA; Corey EJ Synthetic Access to Bent Polycycles by Cation-π Cyclization. Org. Lett 2010, 12, 3548–3551. 10.1021/ol101410g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Yokoe H; Mitsuhashi C; Matsuoka Y; Yoshimura T; Yoshida M; Shishido K Enantiocontrolled Total Syntheses of Breviones A, B, and C. J. Am. Chem. Soc 2011, 133 (23), 8854–8857. 10.1021/ja202874d. [DOI] [PubMed] [Google Scholar]
- (19).Moussa GE Phenol Dehydrogenations 12. Oxidative Coupling of 3,5-Dimethyl-2,4,6-Trihydroxybenzophenone. Acta Chem. Scand 1968, 10, 3329. [Google Scholar]
- (20).Chiba K; Fukuda M; Kim S; Kitano Y; Tada M Dihydrobenzofuran Synthesis by an Anodic [3 + 2] Cycloaddition of Phenols and Unactivated Alkenes. J. Org. Chem 1999, 64, 7654. 10.1021/jo9908243. [DOI] [Google Scholar]
- (21).Lang M; Mühlbauer A; Gräf C; Beyer J; Lang-Fugmann S; Polborn K; Steglich W Studies on the Structure and Biosynthesis of Tridentoquinone and Related Meroterpenoids from the Mushroom Suillus Tridentinus (Boletales). European J. Org. Chem 2008, No. 5, 816–825. 10.1002/ejoc.200700908. [DOI] [Google Scholar]
- (22).Zhang Z; Sun Z; Song J; Guo H; Wang Y; Hu X Construction of the A-B-C Ring of Simplicissin through an Oxidative Dearomatization/Iodination/[3+2] Annulation Cascade. Org. Lett 2023, 25 (48), 8645–8649. 10.1021/acs.orglett.3c03464. [DOI] [PubMed] [Google Scholar]
- (23).Pepper HP; George JH Biomimetic Total Synthesis of (±)-Merochlorin A. Angew. Chem. Int. Ed 2013, 52 (46), 12170–12173. 10.1002/anie.201307200. [DOI] [PubMed] [Google Scholar]
- (24).Meier R; Strych S; Trauner D Oxidative 3+2: Biomimetic Synthesis of (±)-Merochlorin B. Org. Lett 2014, 16, 2634–2637. 10.1021/ol500800z. [DOI] [PubMed] [Google Scholar]
- (25).Li H; Wu J (3+2)-Cycloaddition Reactions of Oxyallyl Cations. Synthesis (Germany). 2014, pp 22–33. 10.1055/s-0034-1378918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wenderski TA; Hoarau C; Mejorado L; Pettus TRR Dearomatization Applications of I(III) Reagents and Some Unusual Reactivity amongst Resorcinol Derived Cyclohexadienones. Tetrahedron 2010, 66 (31), 5873–5883. 10.1016/j.tet.2010.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).He Z; Pulis AP; Procter DJ The Interrupted Pummerer Reaction in a Sulfoxide-Catalyzed Oxidative Coupling of 2-Naphthols. Angew. Chemie - Int. Ed 2019, 58 (23), 7813–7817. 10.1002/anie.201903492. [DOI] [PubMed] [Google Scholar]
- (28).You LF; Hsung RP; Bedermann AA; Kurdyumov AV; Tang Y; Buchanan GS; Cole KP An Enantioselective Synthesis of the ABD Tricycle for (−)-Phoinactin A Featuring Rawal’s Asymmetric Diels-Alder Cycloaddition. Adv. Synth. Catal 2008, 350 (18), 2885–2891. 10.1002/adsc.200800552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Odani A; Ishihara K; Ohtawa M; Tomoda H; Omura S; Nagamitsu T Total Synthesis of Pyripyropene A. Tetrahedron 2011, 67 (42), 8195–8203. 10.1016/j.tet.2011.06.084. [DOI] [Google Scholar]
- (30).Zhong Z; Zhao G; Xu D; Dong B; Song D; Xie X; She X Bioinspired Total Syntheses of Isospongian-Type Diterpenoids (−)-Kravanhins A and C. Chem. - An Asian J 2016, 11 (10), 1542–1547. 10.1002/asia.201600398. [DOI] [PubMed] [Google Scholar]
- (31).Charbonneau L; Foster X; Kaliaguine S Ultrasonic and Catalyst-Free Epoxidation of Limonene and Other Terpenes Using Dimethyl Dioxirane in Semibatch Conditions. ACS Sustain. Chem. Eng 2018, 6 (9), 12224–12231. 10.1021/acssuschemeng.8b02578. [DOI] [Google Scholar]
- (32).Turner OJ; Murphy JA; Hirst DJ; Talbot EPA Hydrogen Atom Transfer-Mediated Cyclisations of Nitriles. Chem. - A Eur. J 2018, 24 (70), 18658–18662. 10.1002/chem.201805236. [DOI] [PubMed] [Google Scholar]
- (33).Ohashi M; Ikawa M; Ogoshi S Intramolecular Oxidative Cyclization of Alkenes and Nitriles with Nickel(0). Organometallics 2011, 30 (10), 2765–2774. 10.1021/om2001603. [DOI] [Google Scholar]
- (34).Xu B; Xun W; Su S; Zhai H Total Syntheses of (−)-Conidiogenone B, (−)-Conidiogenone, and (−)-Conidiogenol. Angew. Chemie Int. Ed 2020, 59 (38), 16475–16479. 10.1002/anie.202007247. [DOI] [PubMed] [Google Scholar]
- (35).Liu J; Ma D A Unified Approach for the Assembly of Atisine- and Hetidine-type Diterpenoid Alkaloids: Total Syntheses of Azitine and the Proposed Structure of Navirine C. Angew. Chem. Int. Ed 2018, 57 (22), 6676–6680. 10.1002/anie.201803018. [DOI] [PubMed] [Google Scholar]
- (36).Yamamoto Y; Matsumi D; Hattori R; Itoh K The Cp2TiPh-Mediated Reductive Radical Cyclization of Cyanoketones and Related Reactions. Efficient Trapping of Ketyl Radicals by Cp2TiPh- Coordinated Polar Multiple Bonds. J. Org. Chem 1999, 64 (9), 3224–3229. 10.1021/jo982492s. [DOI] [PubMed] [Google Scholar]
- (37).Qu Y; Wang Z; Zhang Z; Zhang W; Huang J; Yang Z Asymmetric Total Synthesis of (+)-Waihoensene. J. Am. Chem. Soc 2020, 142 (14), 6511–6515. 10.1021/jacs.0c02143. [DOI] [PubMed] [Google Scholar]
- (38).Okamura H; Shimizu H; Yamashita N; Iwagawa T; Nakatani M Total Synthesis of (+)-Epiepoformin and (−)-Phyllostine. Tetrahedron 2003, 59 (51), 10159–10164. 10.1016/j.tet.2003.10.065. [DOI] [Google Scholar]
- (39).Sen Ayusman; Lai T-W Mechanism of Palladium(II)-Catalyzed C═C Bond Isomerization in Olefins. J. Org. Chem, 1984, 23, 3257–3258. [Google Scholar]
- (40).Wakamatsu T; Tanaka M; Mitsuhashi H; Maruno M The Migration of Double Bond under the Neutral Conditions. The Transformation of Alpha-Alkylidene Cyclic Carbonyl Compounds to Alpha-Unsaturated Cyclic Carbonyl Compounds. Chem. Lett 1994, 1455–1458. [Google Scholar]
- (41).Crossley SWM; Barabé F; Shenvi RA Simple, Chemoselective, Catalytic Olefin Isomerization. J. Am. Chem. Soc 2014, 136 (48), 16788–16791. 10.1021/ja5105602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Delgado KR; Youmans DD; Diver ST Mild Isomerization of Conjugated Dienes Using Co-Mediated Hydrogen Atom Transfer. Org. Lett 2020, 22 (2), 750–754. 10.1021/acs.orglett.9b04594. [DOI] [PubMed] [Google Scholar]
- (43).Disanayaka Bmsara W; Weedon AC A Convenient Synthesis of 2-Methyl-2-Cyclopentene-1-One. Synthesis. 1983, 952. [Google Scholar]
- (44).Kotsuki H; Arimura K; Araki T; Shinohara T Sc (OTf) 3 - and TfOH-Catalyzed Baeyer-Villiger Oxidation of Carbonyl Compounds with m -Chloroperbenzoic Acid. Synlett 1999, 4, 462–464. [Google Scholar]
- (45).Barton DHR; Lester DJ; Ley SV Dehydrogenation of Steroidal Ketones Using Benzeneseleninic Anhydride. J. Chem. Soc. Chem. Commun 1978, 3, 130–131. 10.1039/C39780000130. [DOI] [Google Scholar]
- (46).Herzon SB; Calandra NA; King SM Efficient Entry to the Hasubanan Alkaloids: First Enantioselective Total Syntheses of (−)-Hasubanonine, (−)-Runanine, (−)-Delavayine, and (+)-Periglaucine B. Angew. Chemie - Int. Ed 2011, 50 (38), 8863–8866. 10.1002/anie.201102226. [DOI] [PubMed] [Google Scholar]
- (47).Yahata K; Minami M; Watanabe K; Fujioka H Selective Transformations of Carbonyl Functions in the Presence of α,β-Unsaturated Ketones: Concise Asymmetric Total Synthesis of Decytospolides A and B. Org. Lett 2014, 16 (14), 3680–3683. 10.1021/ol501463p. [DOI] [PubMed] [Google Scholar]
- (48).Campbell EJ; Zhou H; Nguyen SBT Catalytic Meerwein-Pondorf-Verley Reduction by Simple Aluminum Complexes. Org. Lett 2001, 3 (15), 2391–2393. 10.1021/ol0162116. [DOI] [PubMed] [Google Scholar]
- (49).Hiesinger K; Dar’In D; Proschak E; Krasavin M Spirocyclic Scaffolds in Medicinal Chemistry. J. Med. Chem 2021, 64 (1), 150–183. 10.1021/acs.jmedchem.0c01473. [DOI] [PubMed] [Google Scholar]
- (50).Lovering F; Bikker J; Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem 2009, 52 (21), 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- (51).Matsuda Y; Wakimoto T; Mori T; Awakawa T; Abe I Complete Biosynthetic Pathway of Anditomin: Nature’s Sophisticated Synthetic Route to a Complex Fungal Meroterpenoid. J. Am. Chem. Soc 2014, 136 (43), 15326–15336. 10.1021/ja508127q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.